Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Bacterial load and inflammatory response in sputum of alpha-1 antitrypsin deficiency patients with COPD
Authors Balbi B, Sangiorgi C, Gnemmi I ![]() , Ferrarotti I
, Ferrarotti I ![]() , Vallese D, Paracchini E, Delle Donne L, Corda L, Baderna P, Corsico A
, Vallese D, Paracchini E, Delle Donne L, Corda L, Baderna P, Corsico A ![]() , Carone M, Brun P
, Carone M, Brun P ![]() , Cappello F
, Cappello F ![]() , Ricciardolo FLM
, Ricciardolo FLM ![]() , Ruggeri P
, Ruggeri P ![]() , Mumby S, Adcock IM
, Mumby S, Adcock IM ![]() , Caramori G
, Caramori G ![]() , Di Stefano A
, Di Stefano A
Received 28 February 2019
Accepted for publication 10 July 2019
Published 21 August 2019 Volume 2019:14 Pages 1879—1893
DOI https://doi.org/10.2147/COPD.S207203
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Bruno Balbi,1 Claudia Sangiorgi,1 Isabella Gnemmi,1 Ilaria Ferrarotti,2 Davide Vallese,1 Elena Paracchini,1 Lorena Delle Donne,1 Luciano Corda,3 Paolo Baderna,4 Angelo Corsico,2 Mauro Carone,1 Paola Brun,5 Francesco Cappello,6,7 Fabio LM Ricciardolo,8 Paolo Ruggeri,9 Sharon Mumby,10 Ian M Adcock,10 Gaetano Caramori,9 Antonino Di Stefano1
1Istituti Clinici Scientifici Maugeri, IRCCS, Division of Pneumology and Laboratory of Cytoimmunopathology of the Heart and Lung, Veruno, Italy; 2Department of Internal Medicine and Medical Therapy, University of Pavia, Pavia, Italy; 3Medicina Respiratoria, Seconda Medicina Interna, Spedali Civili, Brescia, Italy; 4Division of Pneumology, Aosta Hospital, Aosta, Italy; 5Department of Molecular Medicine, University of Padova, Padova, Italy; 6Dipartimento di Biomedicina Sperimentale e Neuroscienze Cliniche, Sezione di Anatomia Umana, Università di Palermo, Palermo, Italy; 7Euro-mediterranean Institute of Science and Technology (IEMEST), Palermo, Italy; 8Department of Clinical and Biological Sciences, A.O.U., San Luigi Gonzaga, Orbassano, University of Turin, Turin, Italy; 9UOC Di Pneumologia, Dipartimento di Scienze Biomediche, Odontoiatriche e Delle Immagini Morfologiche e Funzionali (BIOMORF), Università di Messina, Messina, Italy; 10Airways Disease Section, National Heart and Lung Institute, Imperial College London, UK
Correspondence: Antonino Di Stefano
Istituti Clinici Scientifici Maugeri Irccs, Via Maugeri, 4, Pavia 27100, Italy
Tel +39 032 288 4711
Fax +39 032 288 4776
Email [email protected]
Background: Airway inflammation may drive the progression of chronic obstructive pulmonary disease (COPD) associated with alpha-1 antitrypsin deficiency (AATD), but the relationship between airway microbiota and inflammation has not been investigated.
Methods: We studied 21 non-treated AATD (AATD-noT) patients, 20 AATD-COPD patients under augmentation therapy (AATD-AT), 20 cigarette smoke-associated COPD patients, 20 control healthy smokers (CS) and 21 non-smokers (CON) with normal lung function. We quantified sputum inflammatory cells and inflammatory markers (IL-27, CCL3, CCL5, CXCL8, LTB4, MPO) by ELISA, total bacterial load (16S) and pathogenic bacteria by qRT-PCR.
Results: AATD-AT patients were younger but had similar spirometric and DLCO values compared to cigarette smoke-associated COPD, despite a lower burden of smoking history. Compared to cigarette smoke-associated COPD, AATD-noT and AATD-AT patients had lower sputum neutrophil levels (p=0.0446, p=0.0135), total bacterial load (16S) (p=0.0081, p=0.0223), M. catarrhalis (p=0.0115, p=0.0127) and S. pneumoniae (p=0.0013, p=0.0001). Sputum IL-27 was significantly elevated in CS and cigarette smoke-associated COPD. AATD-AT, but not AATD-noT patients, had IL-27 sputum levels (pg/ml) significantly lower than COPD (p=0.0297) and these positively correlated with FEV1% predicted values (r=0.578, p=0.0307).
Conclusions: Compared to cigarette smoke-associated COPD, AATD-AT (COPD) patients have a distinct airway inflammatory and microbiological profile. The decreased sputum bacterial load and IL-27 levels in AATD-AT patients suggests that augmentation therapy play a role in these changes.
Keywords: alpha-1 antitrypsin deficiency, COPD, chronic airway inflammation, respiratory disability, sputum
Introduction
Alpha-1 antitrypsin deficiency (AATD) is a genetic condition considered a risk factor for developing chronic obstructive pulmonary disease (COPD).1 An increased burden of neutrophils is characteristic of AATD lungs.2 AAT, which is coded by the SERPINA1 gene (or Protease inhibitor-PI) is a serine protease inhibitor mainly secreted by the hepatocytes whose major target is elastases but can also inhibit the function of thrombin, trypsin, chymotrypsin, plasmin and plasminogen activator.2
In patients without AATD, neutrophilic pulmonary inflammation is considered central to the development and progression of cigarette smoke-associated COPD.3–5 Neutrophilic inflammation correlates with stable COPD severity6,7 and it further increases during COPD exacerbations.8,9 Neutrophil-related inflammatory mediators such as leukotriene (LT)B4,10 chemokine (C-X-C motif) ligand 8 (CXCL-8, previously named interleukin-8), chemokine ligand 3 [CCL3 also known as macrophage inflammatory protein-1α (MIP-1α)],6 and chemokine ligand 5 [CCL5 also known as RANTES (regulated on activation, normal T cell expressed and secreted)]11 are increased in the lower airways of cigarette smoke-associated COPD patients particularly in subjects with stable severe disease.4 Increased neutrophilia is also associated to increased levels of IL-27 protein in the lung of COPD patients.12 In patients with COPD associated with AATD, the general functional and inflammatory picture is worse when compared to cigarette smoke-associated COPD of similar severity.2,10,13,14
A pathogenic role of bacterial load leading to bronchial inflammation is implicated in cigarette smoke-associated COPD.3,15–17 Augmentation therapy, ie weekly infusion of exogenous AAT, in patients with COPD associated with severe AATD restores AAT levels to normal in blood, sputum and bronchoalveolar lavage (BAL)18–20 slowing the progression of the disease, 19–21 decreasing the number of exacerbations,22 and sputum neutrophil numbers, LTB4 levels and MPO expression.14,23 The relationship between bacterial load and sputum inflammation in patients with AATD, whether under augmentation therapy or untreated, has not been studied.
The aim of the present study was to investigate the total and specific bacterial load in sputum of patients with COPD associated to AATD compared to patients with cigarette smoke-associated COPD and control healthy smokers and non-smokers with normal lung function, and to correlate these data with inflammatory cells, cytokines and physiological parameters.
Methods
Subjects
Twenty cigarette smoke-associated COPD patients, 20 COPD patients with alpha-1 antitrypsin deficiency (AATD) in augmentation therapy (AATD-AT), 21 AATD patients not in augmentation therapy (AATD-noT), 20 control smokers with normal lung function (CS) and 21 control non-smokers with normal lung function (CON) who underwent sputum collection were recruited from the Respiratory Medicine Unit of the Istituti Clinici Scientifici Maugeri’s Institute of Veruno, Institute of Cassano delle Murge, IRCCS Policlinico S. Matteo, Pavia (Italy), and from the Spedali Civili, Brescia (Italy), and Pneumology Unit of Aosta (Italy). COPD was defined, according to international guidelines.1,13,24 Diagnostic criteria for AATD were as from the current ATS/ERS statement.1,13,24 Indications for AT are for patients with severe AATD and compound heterozygotes or homozygotes of deficient/Null variants of SERPINA1 gene (not only PI*ZZ but also PI*SZ, PI*Null/Null etc.) and with a significant airflow obstruction and in the context of general disability and health impairment. In COPD patients and in patients with AATD the severity of the airflow obstruction was graded using current GOLD criteria.1,13,24 COPD and AATD-AT patients had a FEV1/FVC ratio lower than 70% of predicted values at post-bronchodilator spirometry and absence of history and symptoms of asthma. In all patients, the severity of emphysema was graded based on high-resolution computed tomography (HRCT) scan radiologic reports as follows: 0=absence of emphysema, 1=1–25% of lung with emphysema, 2=26–50% of lung with emphysema, 3=51–75% of lung with emphysema, 4=76–100% of lung with emphysema. COPD patients and patients with AATD with evidence of bronchiectasis13 were excluded. All former smokers had stopped smoking from at least one year before inclusion in the study. All COPD patients and AATD patients were stable with no previous exacerbation in the three months before collection of the sputum. None of the subjects was treated with theophylline, antibiotics, antioxidants, mucolytics, and/or glucocorticoids in the three months prior sputum collection. Cigarette smoke-associated COPD patients and patients with COPD and AATD were using short-acting inhaled β2-agonists (SABA) and/or regular long-acting inhaled β2- agonists (LABA) and/or regular inhaled long-acting inhaled anti-muscarinics (LAMA) at the time of their recruitment. AATD-AT patients had been under augmentation therapy for at least three months. Subjects were tested for peripheral blood levels of AAT and for genetic analysis of SERPINA1 gene. Testing was performed on dried blood spot samples at the National Reference Center for AATD at the University of Pavia. The study conformed to the Declaration of Helsinki and was approved by the ethics committee of the Istituti Clinici Scientifici Maugeri SpA, Società Benefit (n. 989 CE). (Protocol Registration: Clinical Trials.gov ID: NCT02547532). Written informed consent was obtained from each subject and sputum collection were performed according to the local ethical committee guidelines.
Sputum samples collection and processing
Spontaneous or induced sputum was obtained from all study subjects. Induced sputum was obtained after inhalation of hypertonic saline (4.5%) following international guidelines.17,24 Sputum obtained from each subject was processed by the use of dithiothreitol (1:1) to obtain a liquid sputum specimen. This liquid sputum was divided into 3 aliquots: 1) one aliquot was used for Colony Forming Units (CFU) quantitation; 2) a second aliquot was processed for cytospin and surnatant preparations. May-Grunwald-Giemsa stained cytospins were used for cell count and cellular type differentiation. In the cytospin preparations a minimum number of 200 cells (range: 200–300 cells for all cases) was counted for each subject. In all cases squamous epithelial cells number was <80% of total cells counted (squamous epithelial cells percentage for all patient groups, mean±SD: 41.23±11.9). Supernatants were used for ELISA analysis of secreted cytokines related to innate immunity response and to neutrophilic activation (CCL3, CCL5, CXCL8, IL-27, LTB4, MPO, Table S1); 3) a third aliquot was used for bacterial RNA/DNA extraction (Qiagen, Pathogen Mini Kit, Cat. N. 54104) and qRT-PCR analysis. Bacterial analysis was performed using RT-PCR and Syber Green technology.6,7,11,12 Bacterial analysis included: total bacterial load (16S), Moraxella catarrhalis, Haemophilus influenzae, Streptococcus pneumoniae, and Staphylococcus aureus.7 Total bacterial load (16S) was quantified after sputum RNA/DNA extraction. DNA standard for qRT-PCR was prepared from pure DNA cultures of Escherichia coli to generate a standard curve. The standard curve was performed in triplicate with regression coefficients close to 1 (range of R2 values: 0.984—0.994 for E. coli) and showed a linear increase within the range of DNA concentration utilized, as previously reported.15 Genesig standard kits (Genesig Standard kit handbook HB10.04.09, published date 13/11/2017), including positive and negative controls for each of the bacteria studied, were used for qRT-PCR analysis of P. aeruginosa (RegA), H. influenzae (OMP P6), M. catarrhalis (copB), S. pneumoniae (alpha-fucosidase gene), S. aureus (FEMB gene). Genesig’s Manufacturers’ instructions were carefully followed for bacterial RT-PCR quantification. RT-PCR cycling conditions were: 95 °C for 5 min (PCR initial activation step); 40 amplification cycles of 95 °C for 5 s (denaturation) and 60 °C for 10 s (combined annealing/extension), followed by melting curve analysis to ensure the specificity of the PCR amplification. For each reaction, negative controls were run in triplicate, consisting of primers, PCR Mastermix and sterile water instead of DNA template. Amplification, data acquisition, and cycle threshold (CT) values analysis was performed using the Rotor Gene Q software (Rotor-Gene Q Series Software 2.0.2). Data were expressed as copies/ml of collected sputum from each patient studied.
Routine bacteriological methods for identification of bacteria were used.25 The detection limit for sputum cultures was 103 colony forming units (CFU)xml−1 of each sampled sputum. A comparative analysis between the traditional methodology (CFU) and the RT-PCR bacterial quantitation was also performed.
Quantitation of inflammatory cells in sputum samples
The total number of inflammatory and buccal epithelial cells was quantified in sputum samples (cells/ml) and cellular types were counted in each cytospin preparation. The percentage of neutrophils, eosinophils, macrophages, lymphocytes and squamous epithelial cells was calculated for each subject studied.
ELISA tests for measurement of mediators
Sputum supernatants were aliquoted and stored at −80 °C until their use for the ELISA assays that have been summarized in Table S1. ELISA assays were performed in sixteen patients per group (n=80) consecutively enrolled in this study. These assays were performed according to the manufacturers’ instructions.
Statistical analysis
Group data were expressed as mean ± standard deviation (SD) for functional data and median (range) or interquartile range (IQR) for morphologic data. Differences between groups were analyzed using analysis of variance (ANOVA) for functional data. The ANOVA test was followed by the unpaired t-test for comparison between groups. The Kruskal-Wallis test applied for morphologic data, when significant, was followed by the Mann-Whitney U test for comparison between groups. Correlation coefficients were calculated using the Spearman rank method. Probability values of p<0.05 were considered significant. Data analysis was performed using the Stat View SE Graphics program (Abacus Concepts Inc., Berkeley, CA, USA).
Results
Clinical characteristics of subjects
We obtained and studied sputum samples from 102 subjects: 20 with cigarette smoke-associated stable COPD, 21 AATD-noT, 20 AATD-AT, 20 CS and 21 CON (Table 1). Cigarette smoke-associated COPD patients were older than control subjects (both CS and CON), had a similar history of smoking to CS, and their spirometry values showed mild to severe bronchial obstruction. Compared to cigarette smoke-associated COPD patients, AATD-AT patients were younger, had a reduced history of smoking, similar spirometry and DLCO values as well as emphysema grading on high resolution computed tomography (HRCT) but increased total lung capacity (TLC) values. AATD-noT patients had intermediate characteristics between cigarette smoke-associated COPD and AATD-AT patients. They were of similar age to AATD-AT patients and thus younger than cigarette smoke-associated COPD. Their smoking history was similar to AATD-AT patients, but lower than cigarette smoke-associated COPD. They did not show marked airflow obstruction at spirometry and relatively low emphysema grading on HRCT of the chest, thus differing from both cigarette smoke-associated COPD and AATD-AT patients, but they had increased TLC values similarly to AATD-AT patients. Finally, they had reduced DLCO values, but still higher than those of cigarette smoke-associated COPD and AATD-AT patients (Table 1).
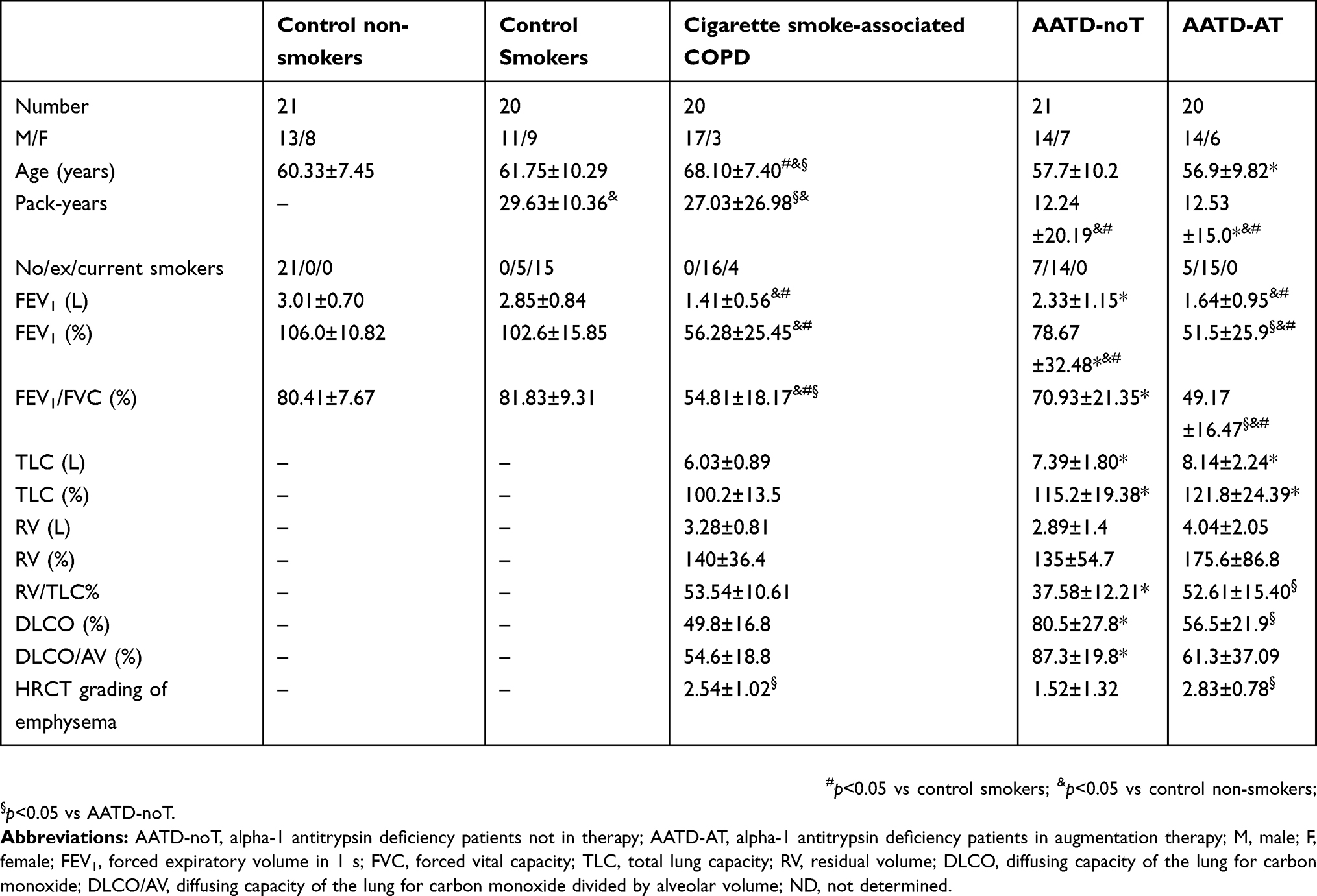 |
Table 1 Clinical characteristics of cigarette smoke-associated chronic obstructive pulmonary disease (COPD), alpha-1 antitrypsin deficiency patients (AATD) and control subjects who provided sputum samples |
All AATD patients had AAT plasma levels below the normal range. In AATD-AT, circulating AAT levels, measured just before augmentation therapy treatment, were lower compared to AATD-noT patients (28.03±18 vs 42.18±25 mg/dl; p=0.0545).
Genotype of AATD patients
The genotypes of AATD-noT patients were: PI*ZZ 8 cases, PI*SZ 6, PI*ZMwurzburg 1, PI*ZMvarallo 1, PI*ZQbrescia 1, PI*ZQparma 1, PI*SS 1, PI*Z new variant 1, PI*S new variant 1; the AAT genotypes of AATD-AT were: PI*ZZ 13, PI*SZ 2, PI*ZYorzinuovi 1, PI*SPlowel 1, PI*Q0 granitefallsQ0granitefalls 1, PI*Q0bresciaQ0brescia 1, PI*ZQ0parma 1.
Inflammatory cells in the sputum
The results of the histochemical analysis are summarized in Table 2 and Figure 1A–E. Total sputum inflammatory cells did not differ significantly across the five groups (Kruskal-Wallis test, p=0.6777). Differential cell counts reported as percentage, however, were different between the study groups. Neutrophils percentage (Kruskal-Wallis: 0.0137) was increased in cigarette smoke-associated COPD patients compared to CON and AATD-AT groups (Mann-Whitney: p=0.0015 and p=0.0135, respectively), and was also different from the AATD-noT group (p=0.0446). The percentage of eosinophils (Kruskal-Wallis: p=0.0508) tended to be higher in cigarette smoke-associated COPD, AATD-noT and AATD-AT compared to CON (Mann-Whitney: p=0.0229, p=0.0345 and p=0.0098, respectively). The percentage of lymphocytes (Kruskal-Wallis: p=0.0955) showed a higher trend in CS, cigarette smoke-associated COPD, and AATD-noT compared to CON (Mann-Whitney: p=0.0366, p=0.0141 and p=0.0439, respectively).
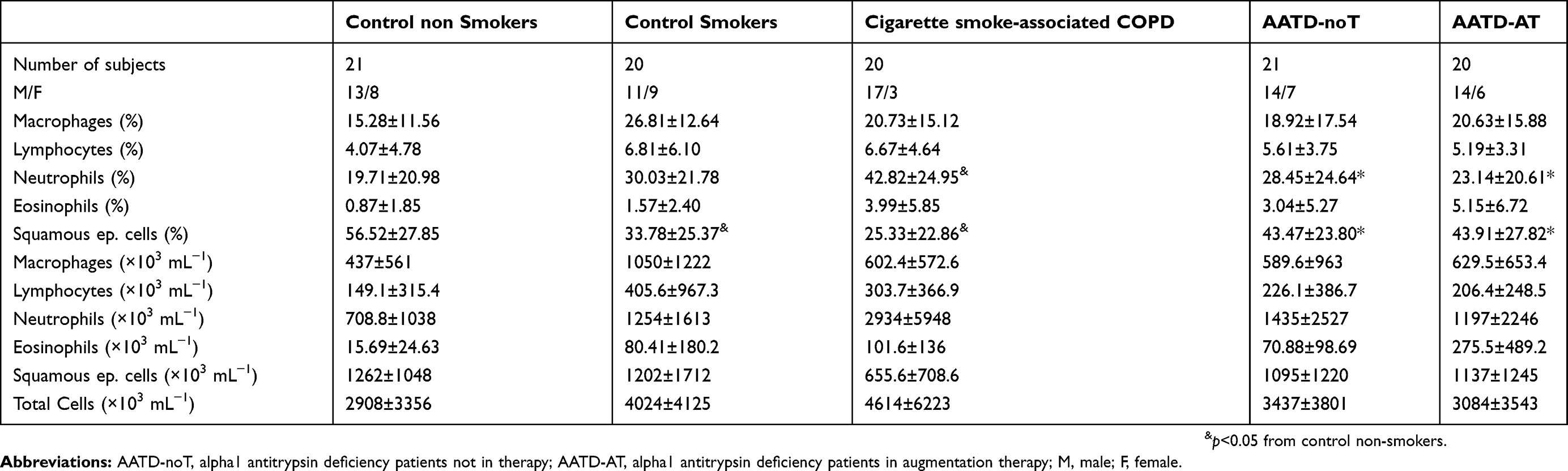 |
Table 2 Inflammatory cells in sputum samples of cigarette smoke-associated chronic obstructive pulmonary disease (COPD), alpha-1 anti-trypsin deficiency patients and control subjects |
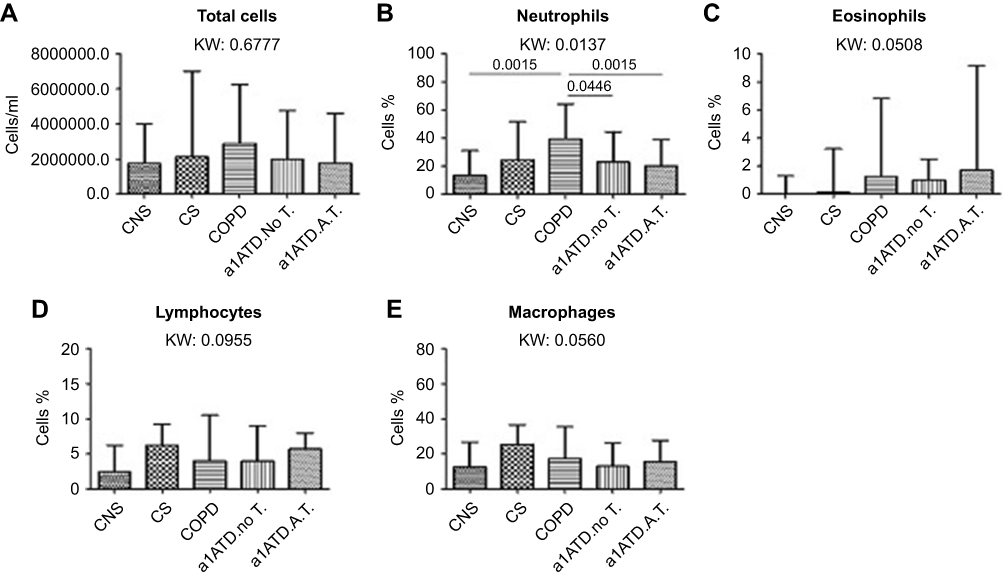 |
Figure 1 Total inflammatory cells (cells/ml) (A), and percentages of neutrophils (B), eosinophils (C), lymphocytes (D) and macrophages (E) in cytospin preparations from control non-smokers (CON, n=21), control smokers (CS, n=20), cigarette smoke-associated COPD (n=20), α1-anti trypsin deficiency not in therapy (AATD-noT, n=21) and AATD patients on augmentation therapy (AATD-AT, n=20). The total number of inflammatory cells, and the percentage of eosinophils, macrophages and lymphocytes did not differ (K. Wallis: p=0.6777, p=0.0508 and p=0.0560, p=0.0955, respectively) in the five groups studied. The percentage of neutrophils (K. Wallis: p=0.0137) was significantly higher in cigarette smoke-associated COPD compared to CNS (p=0.0015) and AATD-AT (p=0.0135) (B). Cigarette smoke-associated COPD also differed from AATD-NoT (p=0.0446). Data are presented as median and interquartile range. Statistical analysis between groups: Mann-Whitney U test, reported only when the Kruskal-Wallis (KW) test resulted significant. Abbreviations: CNS, control non-smokers; CS, control smokers; COPD, chronic obstructive pulmonary disease; α1ATDnoT, α1-anti trypsin deficiency patients not in therapy; α1ATDA.T., α1-anti trypsin deficiency patients on augmentation therapy. |
The percentage of macrophages was not statistically different across the five groups at the Kruskal-Wallis test (p=0.0560), even though a higher trend was observed in CS and AATD-noT compared to CON (Mann-Whitney: p=0.0050 and p=0.0203, respectively) (Table 2).
Inflammatory mediators measured by ELISA in the sputum supernatants
ELISA data are reported in Table 3 and Figure 2. Table S1 shows the ELISA tests used. CCL3 (MIP1α), CCL5 (RANTES), LTB4 and MPO levels did not differ across the five patient groups studied (Figure 2A–D). No significant differences were found in CXCL8 level between the five groups (Kruskal-Wallis: (p=0.0881) studied (Figure 2E). Sputum IL-27 levels in cigarette smoke-associated COPD were similar to that in AAT-noT, but significantly increased compared to AATD-AT and CON (p=0.0297 and p=0.0136, respectively).
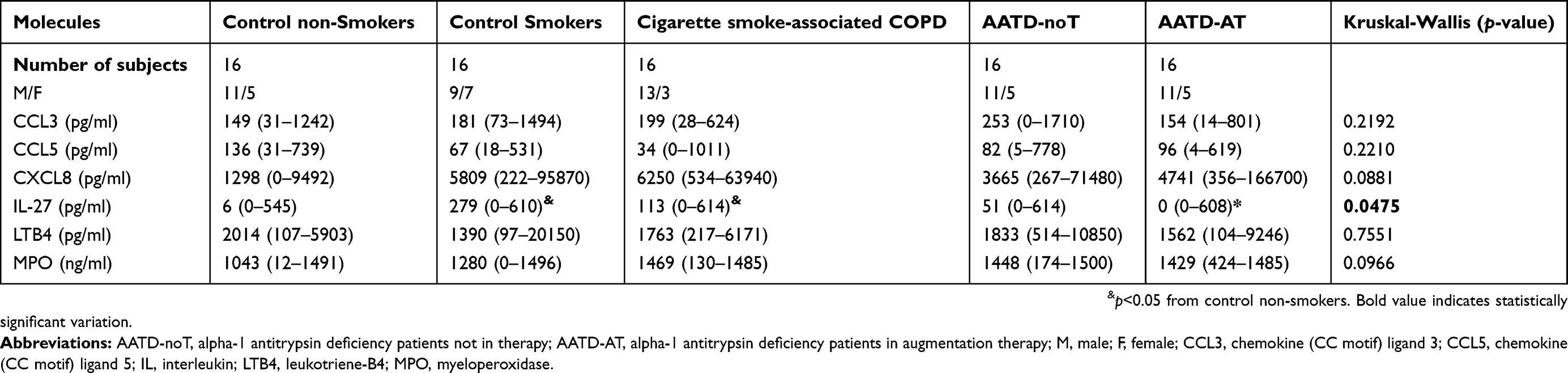 |
Table 3 Inflammatory mediators and cytokines in sputum supernatants samples of cigarette smoke-associate chronic obstructive pulmonary disease (COPD), alpha-1 anti-trypsin deficiency patients and control subjects |
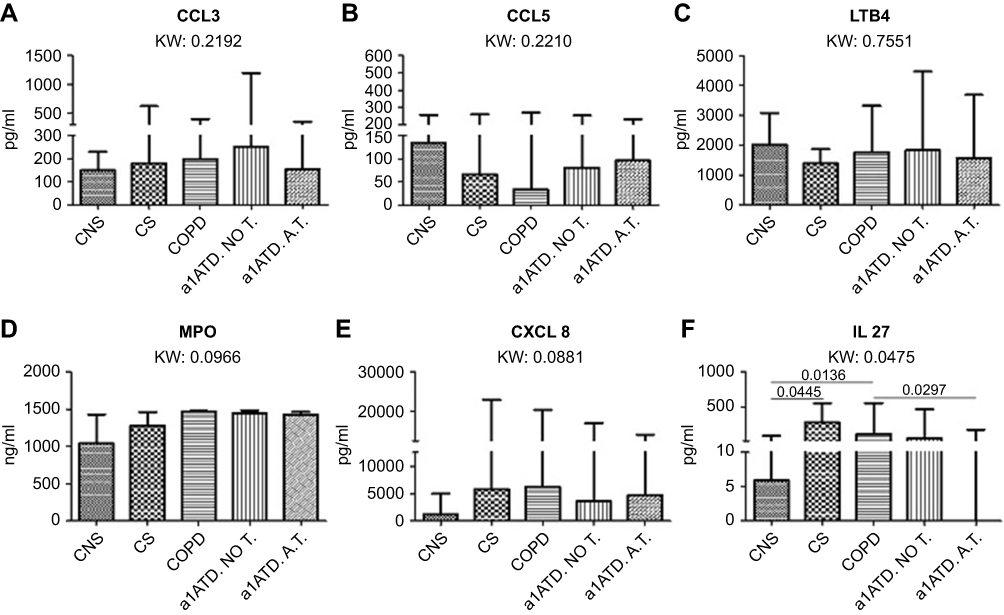 |
Figure 2 Chemokines and cytokines quantified (pg/ml) in the sputum supernatants of control non-smokers (CON, n=16), control smokers (CS, n=16), cigarette smoke-associated chronic obstructive pulmonary disease (COPD, n=16), α1-anti trypsin deficiency not in therapy (AATD-noT, n=16) and AATD in augmentation therapy (AATD-AT, n=16). Data are expressed as median and interquartile range. The Kruskal-Wallis (KW) test was used for multiple comparisons followed by the Mann-Whitney U-test for comparison between groups. CCL3, CCL5, LTB4 and MPO levels were similar in all five patient groups (A-D). CXCL8 (IL-8) was increased in CS, cigarette smoke-associated COPD and AATD-AT in comparison to CON, even though the difference did not reach statistical significance at the Kruskal Wallis test (E). IL-27 was significantly higher in cigarette smoke-associated COPD and in CS compared to CON. Cigarette smoke-associated COPD also showed higher values compared to AATD-AT (F). Abbreviations: CCL3, chemokine (CC motif) ligand 3; CCL5, chemokine (CC motif) ligand 5; IL, interleukin; LTB4, leukotriene-B4; MPO, myeloperoxidase. |
ELISA tests considerations
Considering patients and control subjects studied by ELISA tests (n=80, 16 cases for each group), all the significant changes reported when all cases have been studied for quantitation (n=102, 20–21 cases for each group) of functional data (Table 1), cell count (Table 2) and bacterial load (Figures 3 and 4) have been maintained. The percentage of neutrophils was as well significantly reduced in AATD-AT (n=16) patients compared to cigarette smoke-associated COPD (n=16); M. catarrhalis (copies/ml) and S. pneumoniae (copies/ml) were as well reduced in AATD-AT (n=16) patients compared to cigarette smoke-associated COPD (n=16) (p=0.050 and p=0.002, respectively).
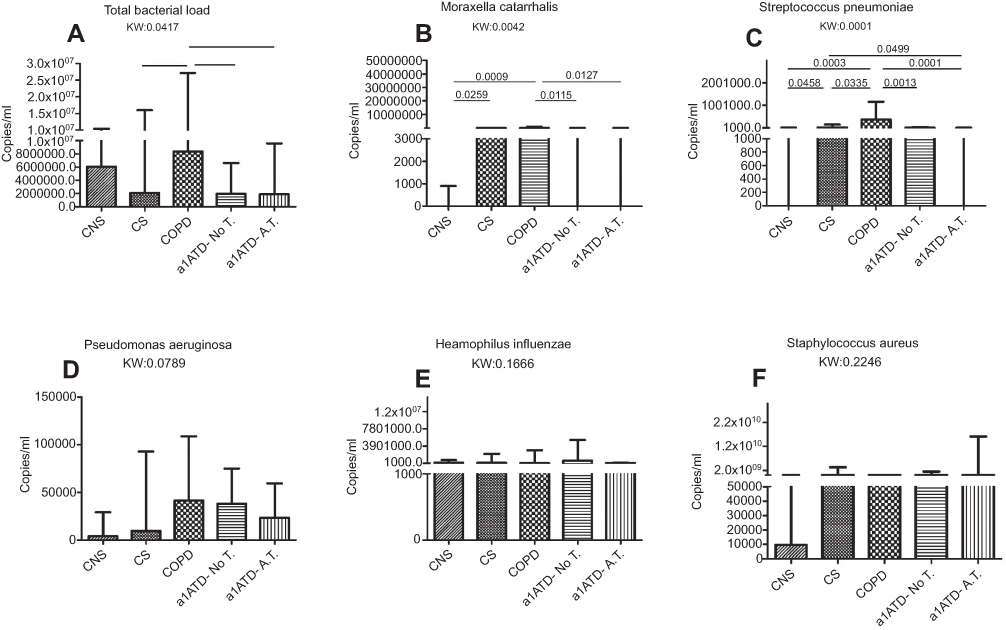 |
Figure 3 Total and specific bacterial load (copies/ml) quantified in the sputum of control non-smokers (CON, n=21), control smokers (CS, n=20), cigarette smoke-associated chronic obstructive pulmonary disease (COPD, n=20), α1-anti trypsin deficiency not in therapy (AATD-noT, n=21) and AATD in augmentation therapy (AATD-AT, n=20). Data are expressed as median and interquartile range. The Kruskal-Wallis (KW) test was used for multiple comparisons followed by the Mann-Whitney U-test for comparison between groups. The total bacterial load was significantly increased in cigarette smoke-associated COPD compared to CS, AATD-noT and AATD-AT. (A) M. catarrhalis was increased in cigarette smoke-associated COPD compared to CON, AATD-noT and AATD-AT. CS also had higher values compared to CON. (B) S. pneumoniae was significantly increased in cigarette smoke-associated COPD compared to CON, CS, AATD-noT and AATD-AT; CS also had higher values compared to CON and AATD-AT (C); P. aeruginosa (D); H. influenzae (E) and S. aureus (F) did not differ significantly at the Kruskal-Wallis test. Abbreviations: CNS, control non-smokers; CS, control smokers; COPD, chronic obstructive pulmonary disease; α1ATDnoT, α1-anti trypsin deficiency patients not in therapy; α1ATDA.T., α1-anti trypsin deficiency patients on augmentation therapy. |
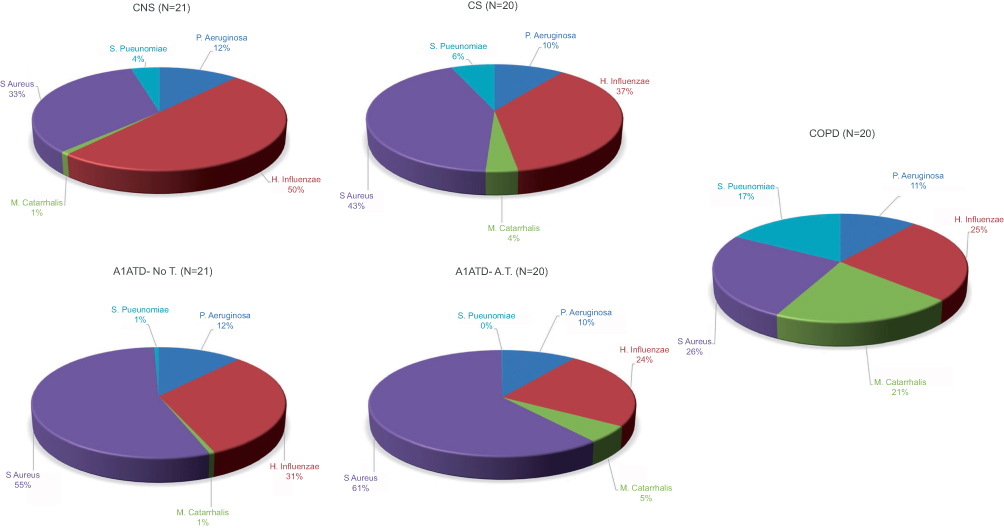 |
Figure 4 Relative proportions of P. aeruginosa, H. influenzae, M. catarrhalis, S. aureus and S. pneumoniae in sputum of control non-smokers (CON), control smokers (CS), cigarette smoke-associated chronic obstructive pulmonary diseased (COPD) patients, α1-anti trypsin deficiency not in therapy (AATD-noT) and AATD in augmentation therapy (AATD-AT) patients. The percentage of M. catarrhalis and S. pneumoniae was higher in cigarette smoke-associated COPD compared to CON. Interestingly, augmentation therapy in COPD patients with AATD significantly reduced the percentage of M. catarrhalis and S. pneumoniae to values close to those observed in CON subjects. Abbreviations: CNS, control non-smokers; CS, control smokers; COPD, chronic obstructive pulmonary disease; α1ATDnoT, α1-anti trypsin deficiency patients not in therapy; α1ATDA.T., α1-anti trypsin deficiency patients on augmentation therapy. |
Comparison of relevant inflammatory cells and biomarkers in patients with AATD-noT with bronchial obstruction and cigarette smoke-associated COPD
Since 42% of AATD patients not in the augmentation therapy (9 out of 21 AATD-noT) had a similar bronchial obstruction when compared to cigarette smoke-associated COPD patients, we compared them for the relevant inflammatory markers studied. The number of neutrophils (cells/ml and percentage), IL-27 and CCL5 proteins were not significantly different in the two groups studied (Mann-Whitney: p=0.186, p=0.194, p=0.053 and p=0.192, respectively). A slight but significant increase of M. catarrhalis (copies/ml) (p=0.016) and of S. pneumoniae (copies/ml) (p=0.0125) was observed in cigarette smoke-associated COPD compared to AATD-noT patients with bronchial obstruction (n=9). The reasons for this difference need a more extended analysis involving a higher number of AATD-noT patients.
Colony-forming units (CFU) in the sputum
In the group of CON, 20 subjects showed the presence of normal bacterial flora, and one showed growth of S. aureus. In the group of CS, 16 subjects showed normal bacterial flora, 2 showed H. parainfluenzae, and 2 showed H. influenzae. In cigarette smoke-associated COPD, 11 patients showed normal bacterial flora, 1 patient H. influenzae, 2 patients S. pneumoniae, 3 patients S. aureus, 1 patient S. aureus plus P. aeruginosa, 1 patient S. aureus plus H. influenzae, and 1 patient S. pneumoniae plus H. influenzae. In AATD-noT, 16 patients had normal bacterial flora, 2 patients P. aeruginosa, 1 patient S. pneumoniae, 1 patient S. aureus, and 1 patient S. aureus plus H. influenzae. In AATD-AT, 15 patients had normal bacterial flora, 1 patient P. aeruginosa, 1 patient H. influenzae, 1 patient S. aureus, 1 patient S. aureus plus H. influenzae, and 1 patient H. influenzae plus M. catarrhalis.
Quantification of bacterial load in the sputum
When data were expressed as absolute numbers (Figure 3) (copies/ml), AATD-NoT and AATD-AT patients had a significantly lower total bacterial load (16S) than cigarette smoke-associated COPD (p=0.0081 and p=0.0223) (Figure 3A). AATD-noT and AATD-AT had a lower number of copies/ml of Moraxella catarrhalis than cigarette smoke-associated COPD (p=0.0115 and p=0.0127) (Figure 3B). Cigarette smoke-associated COPD patients and CS had a higher number of copies/ml compared to CON (p=0.0009 and 0.0259). AATD-noT and AATD-AT also had significantly fewer copies/ml of Streptococcus pneumoniae compared to cigarette smoke-associated COPD (p=0.0013 and p=0.0001). CS also differed from AATD-AT (p=0.0499) (Figure 3C). No significant differences were observed for Pseudomonas aeruginosa (Kruskal-Wallis: p=0.0789), Haemophilus influenzae (Kruskal-Wallis: p=0.1666) and Staphylococcus aureus (Kruskal-Wallis: p=0.2246) across the five groups studied (Figure 3D–F).
To evaluate changes in the relative proportions of each bacterium studied, we expressed these data as a percentage of the total load value constituted by the sum of the five bacteria studied (Figure 4). In cigarette smoke-associated COPD, the percentage of M. catarrhalis was higher compared to CON, AATD-noT and AATD-AT (p=0.0015, p=0.0063 and p=0.0055, respectively). Again, in cigarette smoke-associated COPD, the percentage of S. pneumoniae was higher compared to CON, CS, AATD-noT and AATD-AT (p=0.0006, p=0.0094, p=0.0008 and p<0.0001, respectively). (Figure 4). The percentages of P. aeruginosa, H. influenzae and S. aureus did not significantly differ across the five groups (Kruskal-Wallis: (p=0.2720, p=0.1598 and p=0.1358, respectively) (Figure 4).
Correlations between clinical parameters, number of inflammatory cells, signaling mediators expression and bacterial load in the sputum
When we grouped patients with cigarette smoke-associated COPD (n=20), CS (n=20) and CON (n=21), the percentage of neutrophils in the sputum was inversely correlated with FEV1% predicted values (R=−0.301, p=0.0218) and positively correlated with M. catarrhalis (copies/ml) (R=0.27, p=0.0344), S. pneumoniae (copies/ml) (R=0.367, p=0.0045) and P. aeruginosa (copies/ml) (R=0.319, p=0.0133). The percentage of neutrophils (n=48) also correlated with CXCL8 (pg/ml) in the supernatants (R=0.395, p=0.0068) and MPO (ng/ml) (R=0.436, p=0.0047). However, multiple regression analysis considering neutrophils (%) as dependent variable and the six quantified biomarkers (CCL3, CCL5, CXCL8, IL-27, LTB4, MPO) as independent variables showed a significant positive association with MPO (ng/ml) (R=0.587, p=0.0007). When we grouped patients with cigarette smoke-associated COPD (n=20 or 16 for ELISA tests) and AATD-AT (n=20 or 16 for ELISA tests), M. catarrhalis (copies/ml) and S. pneumoniae (copies/ml) significantly and positively correlated with the percentage of neutrophils (R=0.489, p=0.0050 and R=0.570, p=0.0011, respectively) and IL-27 protein (pg/ml) (R=0.377, p=0.0423 and R=0.432, p=0.0199, respectively). Scored emphysema in all patients with AATD (n=41) correlated significantly with FEV1% (R=−0.68, p<0.0001), FEV1/FVC% (R=−0.58, p=0.0019), lung diffusion capacity of carbon monoxide (DLCO)% (R=−0.74, p=0.0006) and with residual volume (RV)% (R=047, p=0.022).
Spontaneous versus induced sputum analysis and day-to-day variability
In all patients and control subjects studied (n=102) spontaneous sputum was obtained from sixty patients and 42 patients underwent to the sputum induction procedure. We did not find significant differences between spontaneous and induced sputum samples for neutrophil % (Mann-Whitney: p=0.391), IL-27 protein (p=0.777), CCL5 protein (p=0.569), S. pneumoniae (copies/ml and percentage) (p=0.645 and p=0.485 respectively) and M. catarrhalis (copies/ml and percentage) (p=0.650 and 0.424 respectively). The absolute number of neutrophils (cells/ml) was slightly increased in spontaneous sputum compared to the induced one (Mann-Whitney: p=0.034). Spontaneous sputum was obtained from 15 cigarette smoke-associated COPD, 15 AATD-AT, 13 AATD-noT, 9 control smokers and 8 control non-smokers. These comparisons made in each individual group were all not significantly different.
Day-to-day variability has been reported for neutrophil and some cytokine quantitations in sputum of COPD and AATD patients.14 The study by Stone’s and coauthors suggest to optimize the analysis using 2–3 consecutive sputum samples collected in different days. Stone’s data were obtained in 12 COPD and 12 AATD patients. We performed our sputum analysis using a single sample from each studied patient or control subject using 20–21 patients for each group. Furthermore, data obtained were considered significant only when multiple and paired statistical tests were significant. We believe that the higher number of patients used in each group (20–21), the statistical analysis adopted and the similar technical approach (one collection for all cases studied, patients and control subjects) used for all the five groups studied may give reliable results.
Discussion
The main finding of our study is that the inflammatory and microbiological profiles in the lower airways of patients with AATD differ from those observed in cigarette smoke-associated COPD. Compared with cigarette smoke-associated COPD patients, patients with AATD, whether on AT or not, had lower amount of neutrophils in sputum and total load of bacteria (M. catarrhalis and S. pneumonia, in particular). Cigarette smoking per se affected the microbiota composition. Moreover, AATD-AT patients, but not AAT-noT patients, had decreased IL-27 levels in sputum compared with cigarette smoke-associated COPD. The observed differences in inflammatory profile among cigarette smoke-associated COPD and AATD-AT patients are noteworthy as the two groups have a similar clinical and functional profile.
Patients with AATD-AT were younger than the cigarette smoke-associated COPD group. This is not surprising considering that airflow obstruction and/or pulmonary emphysema usually appears at a younger age in these subjects.20 The inclusion criteria for AATD patients were not restricted to lung function impairment. Thus, in the AATD-noT group, 66% (13/21) of these patients had FEV1/FVC% values >70% of predicted, at variance with lung function data from AATD-AT patients. This and other characteristics of the two AATD groups indicate that they correspond to different time-points in the natural development of AATD-related lung disease, passing from the less severe airway and parenchymal inflammatory involvement in AAT-noT patients to severe COPD in AATD-AT patients.
Our data confirm an increased sputum neutrophilia in cigarette smoke-associated COPD in agreement with many other published reports3–11 whilst the decreased sputum neutrophil percentage and a trend to decline in absolute numbers in AATD-AT patients compared to cigarette smoke-associated COPD also confirm previous observations.14 However, the mechanisms causing a reduction in neutrophil accumulation in the lower airways and their decreased activation after augmentation therapy are controversial. Neutrophil elastase stimulates epithelial cells of the lower airways to release CXCL816,26–31,38 and increased BAL levels of CXCL8 were reported in AATD patients compared to cigarette smoke-associated COPD14 ; moreover, colonized AATD or colonized cigarette smoke-associated COPD patients have increased sputum CXCL8 concentrations compared to the same non-colonized group of patients.10 However, we did not find markedly significant differences in sputum CXCL8 levels between AATD-AT patients and cigarette smoke-associated COPD patients with a similar degree of airflow obstruction, suggesting that augmentation therapy reduces sputum neutrophilia in these patients, thus acting with other molecular mechanisms.
In the present paper, as an original contribution, AATD-AT patients compared with cigarette smoke-associated COPD have a significant reduction in sputum IL-27 protein levels, suggesting a role for this cytokine in reducing sputum neutrophilia in these patients. Interestingly, the sputum levels of IL-27 protein were also positively correlated with the specific bacterial load of M. catarrhalis and S. pneumoniae (copies/ml) in AATD-AT patients. In addition, when all AATD patients were grouped together, sputum IL-27 levels correlated positively with FEV1% predicted values, suggesting a role for the augmentation therapy in reducing sputum IL-27.
Monocytes/macrophages and neutrophils are the major source of IL-27 production during sepsis and blocking of IL-27 production in neutrophils in vitro reduces bacterial survival.32 Modulation of neutrophil function by IL-27 is also reported in intracerebral hemorrhage.33 Interestingly, IL-27 pre-treatment of neutrophils and monocytes in vitro decreases CXCL8 production34 and in a model of zymosan-induced peritonitis, the administration of IL-27 reduces neutrophil recruitment to the peritoneal cavity.35
We also observed a concomitant reduction of sputum neutrophil numbers and IL-27 protein levels in AATD-AT patients compared to cigarette smoke-associated COPD, with both study groups having a similar degree of airflow obstruction. For the above reasons we speculate that the reduction in sputum neutrophil numbers may be influenced, at least in part, by IL-27 activity. However, we cannot exclude that the concomitant decline in both sputum neutrophil and IL-27 levels could also be an effect of the neutrophil decrement per se since neutrophils synthesize IL-27 protein.32
CC-chemokines, in particular CCL3 and CCL5, have been reported to be involved in the pathogenesis of cigarette smoke-associated COPD and in the induction of neutrophil migration and activation.11,36 In the current study, at variance with our previous study performed on bronchial biopsies,11 we did not observed any significant change in sputum CCL3 and CCL5 levels among the five groups of subjects studied. Differences in the release of inflammatory mediators in different compartments of the lower airways have already been observed in many different chronic inflammatory diseases. Furthermore, we observed no significant difference in sputum MPO and LTB4 levels between AATD-AT patients (treated for at least 3 months), in comparison with cigarette smoke-associated COPD with a similar degree of obstruction (Figure 2C and D).
When comparing sputum MPO in cigarette smoke-associated COPD and AATD-noT patients with a similar degree of airflow obstruction there are conflicting results in the literature (Table 4). Whereas most of the previous studies in the same group of patients have shown a significant increase of sputum leukotriene B4 (Table 4). These differences may be explained by the lack of untreated AATD patients with severe/very severe COPD in our study, the differences in the ELISA assays used for measuring these mediators and the length of augmentation therapy treatment in the different studies (Table 4).
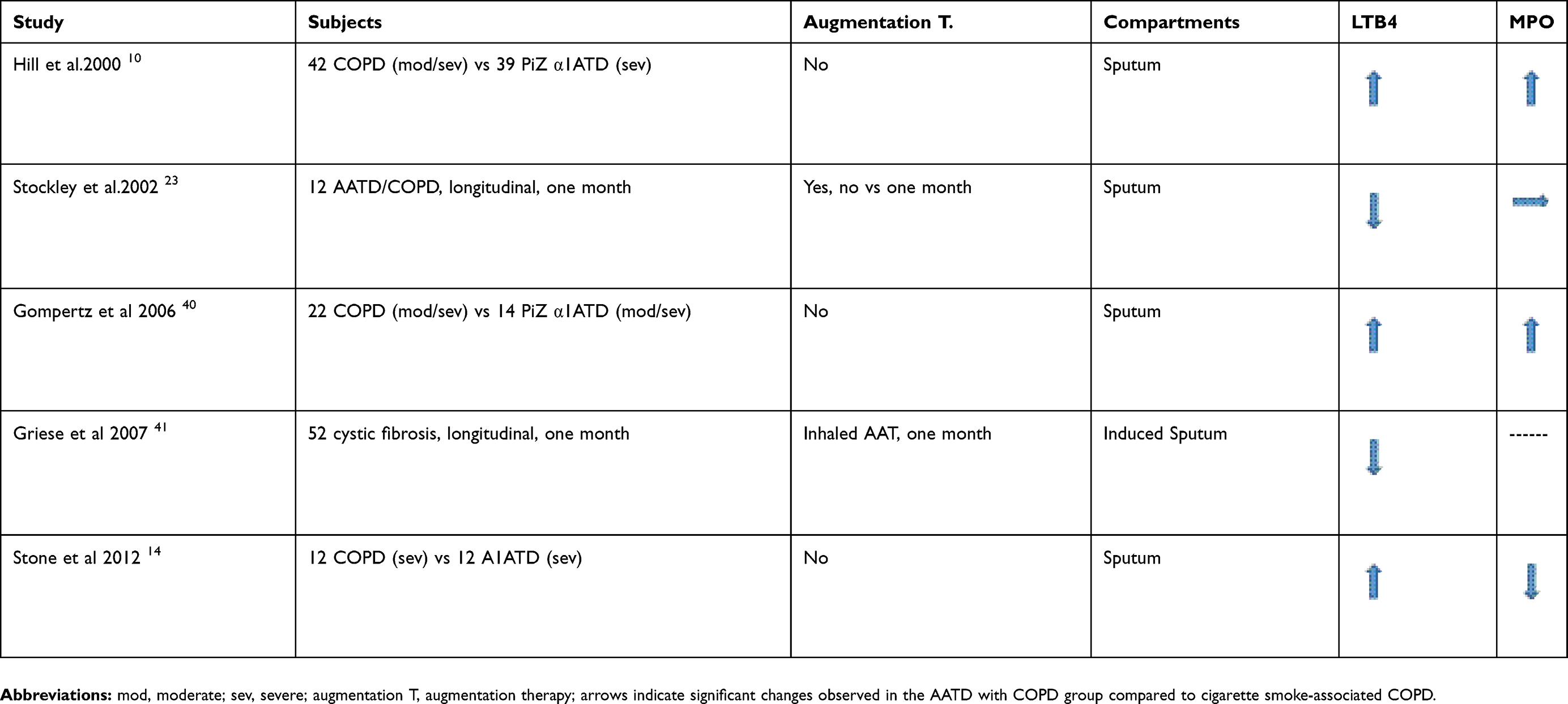 |
Table 4 Previous studies on the quantification of LTB4 and MPO in sputum samples of patients with AATD/COPD compared to cigarette smoke-associated COPD |
There are few published studies that have investigated the total and specific bacterial load and their correlations with sputum inflammation in patients with AATD-AT and AATD-noT. Both cigarette smoke-associated COPD and AATD patients in stable phase have increased sputum CXCL8 and LTB4 levels when colonized compared to non-colonized patients,10 suggesting a role for bacteria in increasing neutrophil-related inflammation.10 In our study, only a minority of patients showed sputum CFU for different bacterial species making impossible to directly compare inflammatory cells and mediators between colonized and non-colonized patients. Interestingly, we report here for the first time the absolute (expressed as copies/ml) and percentage values of the total and specific bacterial load in the five groups of subjects investigated.
We show here that cigarette smoking per se had a clear effect on microbiota composition, since CS were similar to CON for total bacterial load (16S) but showed significantly increased M catarrhalis and S. pneumoniae when compared to CON which is in keeping with previous studies.37 The highest values of total bacterial load were observed in cigarette smoke-associated COPD. P. aeruginosa showed a trend to be increased in cigarette smoke-associated COPD, AATD-noT and AATD-AT with respect to CON confirming earlier studies using different methods.16,26,38 M. catarrhalis and S. pneumoniae copies/ml were increased in cigarette smoke-associated COPD compared to CON and significantly decreased in AATD-AT, having a similar degree of airflow obstruction. This decrement in absolute numbers in AATD-AT was also associated to a decreased relative proportion of M. catarrhalis and S. pneumoniae when compared to cigarette smoke-associated COPD. Our observation of a positive correlation of M. catarrhalis and S. pneumoniae bacterial load with neutrophil percentage and IL-27 protein in the sputum of cigarette smoke-associated COPD and AATD-AT patients suggests a possible inter-relationship between specific bacterial load, IL-27 secretion and neutrophil recruitment. This is in keeping with recent observations that IL-27 impairs host innate immunity against pneumococcal infection.39 The mechanistic inter-relationship between neutrophil recruitment, IL-27 production and bacterial load, may be worth to be investigated in large controlled clinical trials of augmentation therapy in AATD.
In conclusion, compared to cigarette smoke-associated COPD, AATD-AT (COPD) patients have a distinct airway inflammatory and microbiological profile. The decreased sputum bacterial load and IL-27 levels in AATD-AT patients suggests that augmentation therapy play a role in these changes. The mechanistic inter-relationship between neutrophil recruitment, IL-27 production and bacterial load, may be worth to be investigated in large controlled clinical trials of augmentation therapy in AATD.
Data sharing statement
The authors intend to share individual deidentified participant data, including demographics, detailed clinical information and cell and supernatant measurements in sputum. The data will be accessible by emailing the authors after the manuscript is published. Subject’s identity information will not be available for privacy reasons.
Acknowledgments
This work was supported by grants from Grifols Inc. and from the Istituti Clinici Scientifici Maugeri, SpA Società Benefit, Ricerca Corrente. We are indebted to the Associazione Nazionale Alfa-1 AT Onlus (Italian AATD Patients’ Association) and Dr. Sara Bendinelli (former employee of Grifols Italia) for their support in this research.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
This study was funded by Istituti Clinici Scientifici Maugeri, IRCCS and by Grifols Inc. Grifols Inc. with a two-year grant. Grifols funding included laboratory supplies, travel expenses for collecting samples around the Country and a fellowship for a Ph.D. The authors report no other conflicts of interest in this work.
References
1. Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α1-antitrypsin deficiency. Eur Respir J. 2017;50. 1700610. doi: 10.1183/13993003.00610-2017.
2. Hubbard RC, Fells G, Gadek J, et al. Neutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophages. J Clin Invest. 1991;88:891–897.
3. Barnes PJ Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35:71–86. doi:10.1016/j.ccm.2013.10.004
4. Caramori G, Di Stefano A, Casolari P, et al. Chemokines and chemokine receptors blockers as new drugs for the treatment of chronic obstructive pulmonary disease. Curr Med Chem. 2013;20:4317–4349.
5. Caramori G, Kirkham P, Barczyk A, et al. Molecular pathogenesis of cigarette smoking-induced stable COPD. Ann N Y Acad Sci. 2015;1340:55–64. doi: 10.1111/nyas.12619.
6. Di Stefano A, Capelli A, Lusuardi M, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med. 1998;158:1277–1285. doi:10.1164/ajrccm.158.4.9802078
7. Parr DG, White AJ, Bayley DL, et al. Inflammation in sputum relates to progression of disease in subjects with COPD: a prospective descriptive study. Respir Res. 2006;7:136. doi:10.1186/1465-9921-7-136
8. Gompertz S, Bayley DL, Hill SL, et al. Relationship between airway inflammation and the frequency of exacerbations in patients with smoking related COPD. Thorax. 2001;56:36–41. doi:10.1136/thorax.56.1.36
9. Saetta M, Di Stefano A, Maestrelli P, et al. Airway eosinophilia in chronic bronchitis during exacerbations. Am J Respir Crit Care Med. 1994;150:1646–1652. doi:10.1164/ajrccm.150.6.7952628
10. Hill AT, Bayley DL, Campbell EJ, et al. Airways inflammation in chronic bronchitis: the effects of smoking and alpha1-antitrypsin deficiency. Eur Respir J. 2000;15:886–890.
11. Di Stefano A, Caramori G, Gnemmi I, et al. Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax. 2009;64:968–975. doi:10.1136/thx.2009.113647
12. Di Stefano A, Caramori G, Barczyk A, et al. Innate immunity but not NLRP3 inflammasome activation correlates with severity of stable COPD. Thorax. 2014;69(6):516–524. doi: 10.1136/thoraxjnl-2012-203062.
13. Parr DG, Guest PG, Reynolds JH, et al. Prevalence and impact of bronchiectasis in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2007;176:1215–1221. doi:10.1164/rccm.200703-489OC
14. Stone H, McNab G, Wood AM, et al. Variability of sputum inflammatory mediators in COPD and α1-antitrypsin deficiency. Eur Respir J. 2012;40:561–569. doi:10.1183/09031936.00162811
15. Di Stefano A, Ricciardolo FLM, Caramori G, et al. Bronchial inflammation and bacterial load in stable COPD is associated with TLR4 overexpression. Eur Respir J. 2017;49.1602006. doi: 10.1183/13993003.02006-2016.
16. Belkaid Y, Hand TW Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011
17. D’Anna SE, Balbi B, Cappello F, et al. Bacterial-viral load and the immune response in stable and exacerbated COPD: significance and therapeutic prospects. Int J Chron Obstruct Pulmon Dis. 2016;11:445–453. doi:10.2147/COPD.S93398
18. Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med. 1987;316:1055–1062. doi:10.1056/NEJM198704233161704
19. Balbi B, Ferrarotti I, Miravitlles M Efficacy of augmentation therapy for emphysema associated with α1-antitrypsin deficiency: enough is enough. Eur Respir J. 2016;47:35–38. doi: 10.1183/13993003.01145-2015
20. Luisetti M, Ferrarotti I, Corda L, et al. Italian registry of patients with alpha-1 antitrypsin deficiency: general data and quality of life evaluation. COPD. 2015;12 Suppl 1:52–57. doi: 10.3109/15412555.2015.1023393
21. Chapman KR, Burdon JG, Piitulainen E, et al. RAPID Trial Study Group. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991):360–368. doi: 10.1016/S0140-6736(15)60860-1.
22. Lieberman J Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest. 2000;118:1480–1485. doi:10.1378/chest.118.5.1480
23. Stockley RA, Bayley DL, Unsal I, et al. The effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2002;165:1494–1498. doi:10.1164/rccm.2109013
24. Erdman DD, Weinberg GA, Edwards KM, et al. GeneScan reverse transcription-PCR assay for detection of six common respiratory viruses in young children hospitalized with acute respiratory illness. J Clin Microbiol. 2003;41:4298–4303. doi:10.1128/jcm.41.9.4298-4303.2003
25. Ballows A, Hausler WJ, Herrmann KL, et al. Manual of Clinical Bacteriology.
26. Global Initiative for Chronic Obstructive Lung Disease (GOLD) [homepage on the Internet]. Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease; 2017. Available from: www.goldcopd.org.
27. Stockley RA, Turner AM α-1-antitrypsin deficiency: clinical variability, assessment, and treatment. Trends Mol Med. 2014;20:105–115. doi:10.1016/j.molmed.2013.11.006
28. Gottlieb DJ, Luisetti M, Stone PJ, et al. Short-term supplementation therapy does not affect elastin degradation in severe alpha(1)-antitrypsin deficiency. The American-Italian AATD Study Group. Am J Respir Crit Care Med 2000;162:2069–2072. doi:10.1164/ajrccm.162.6.2002032
29. Nakamura H, Yoshimura K, McElvaney NG, et al. Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. J Clin Invest. 1992;89:1478–1484. doi:10.1172/JCI115738
30. Sapey E, Stockley JA, Greenwood H, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183:1176–1186. doi:10.1164/rccm.201008-1285OC
31. Woolhouse IS, Bayley DL, Lalor P, et al. Endothelial interactions of neutrophils under flow in chronic obstructive pulmonary disease. Eur Respir J. 2005;25:612–617. doi:10.1183/09031936.05.00086304
32. Rinchai D, Khaenam P, Kewcharoenwong C, et al. Production of interleukin-27 by human neutrophils regulates their function during bacterial infection. Eur J Immunol. 2012;42:3280–3290. doi: 10.1002/eji.201242526.
33. Zhao X, SM T, CH L, et al. Neutrophil polarization by IL-27 as a therapeutic target for intracerebral hemorrhage. Nat Commun. 2017;8:602. doi: 10.1038/s41467-017-00770-7
34. Alagbe AE, Justo Junior AS, Ruas LP, et al. Interleukin-27 and interleukin-37 are elevated in sickle cell anemia patients and inhibit in vitro secretion of interleukin-8 in neutrophils and monocytes. Cytokine. 2018;107:85–92. doi:10.1016/j.cyto.2017.12.001
35. Watzlawick R, Kenngott EE, Liu FD, et al. Anti-inflammatory effects of IL-27 in zymosan-induced peritonitis: inhibition of neutrophil recruitment partially explained by impaired mobilization from bone marrow and reduced chemokine levels. PLoS One. 2015;10(9):e0137651. doi: 10.1371/journal.pone.0137651.
36. Caramori G, Di Stefano A, Casolari P, et al. Chemokines and chemokine receptors blockers as new drugs for the treatment of chronic obstructive pulmonary disease. Curr Med Chem. 2013;20:4317–4349.
37. Lim MY, Yoon HS, Rho M, et al. Analysis of the association between host genetics, smoking, and sputum microbiota in healthy humans. Sci Rep. 2016;6:23745. doi: 10.1038/srep23745.
38. Einarsson GG, Comer DM, McIlreavey L, et al. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax. 2016;71:795–803. doi:10.1136/thoraxjnl-2015-207235
39. Cao J, Wang D, Xu F, et al. Activation of IL-27 signalling promotes development of postinfluenza pneumococcal pneumonia. EMBO Mol Med. 2014;6:120–140. doi:10.1002/emmm.201302890
40. Gompertz S, Hill AT, Bayley DL, et al. Effect of expectoration on inflammation in induced sputum in alpha-1-antitrypsin deficiency.Respir Med. 2006;100:1094–1099. doi:10.1016/j.rmed.2005.09.024
41. Griese M, Latzin P, Kappler M, et al. alpha1-Antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients.Eur Respir J. 2007;29:240–250. doi:10.1183/09031936.00047306
Supplementary material
Methods
Lung function tests and volumes
Pulmonary function tests were performed as previously described1 according to guideline recommendations. Pulmonary function tests included measurements of FEV1 and FEV1/FVC under baseline conditions in all subjects (6200 Autobox Pulmonary Function Laboratory; Sensormedics Corp., Yorba Linda, CA, USA). To assess the reversibility of airflow obstruction and post bronchodilator functional values, the FEV1 and FEV1/FVC% measurements in the groups of subjects with FEV1/FVC% ≤70% pre-bronchodilator was repeated 20 min after the inhalation of 0.4 mg of salbutamol.
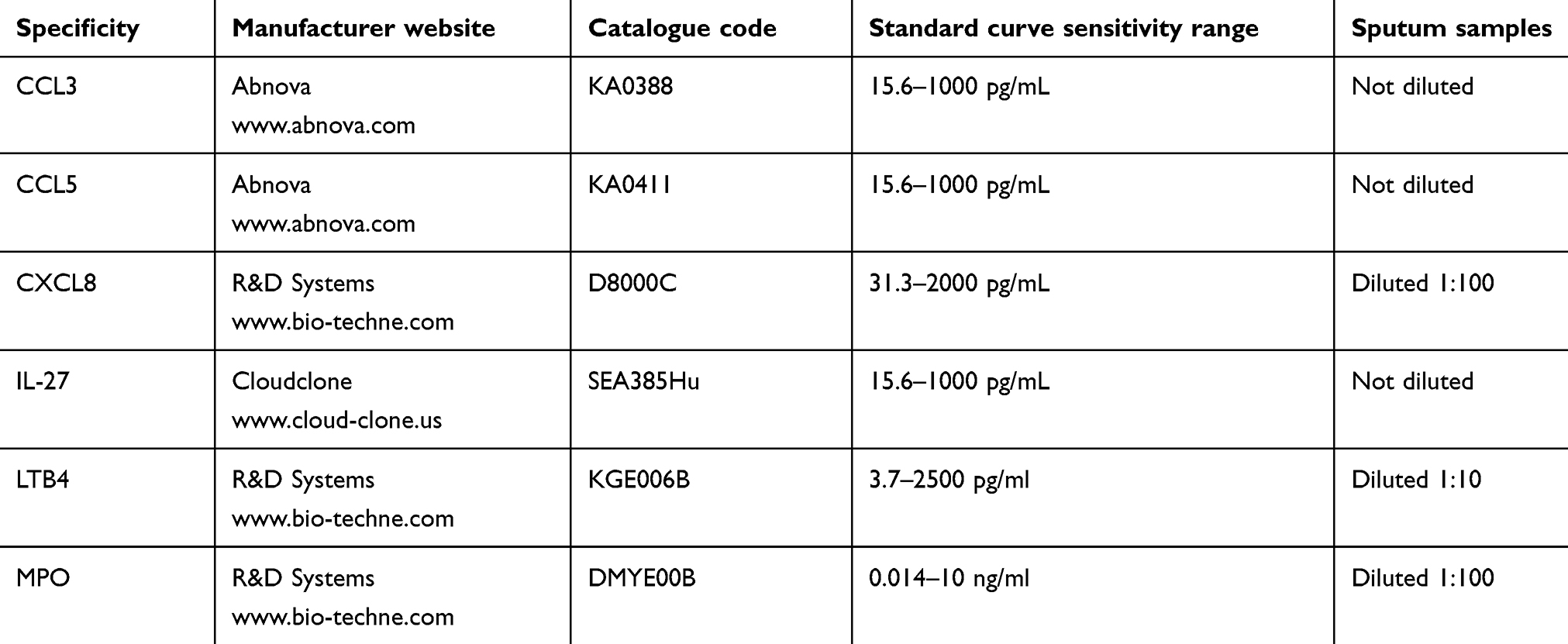 |
Table S1 Summary of the ELISA assays performed on sputum supernatants |
Reference
1. Di Stefano A, Caramori G, Gnemmi I, et al. Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax. 2009;64:968–975. doi:10.1136/thx.2009.113647
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.