Back to Journals » Infection and Drug Resistance » Volume 12
Bacteria Exploit Autophagy For Their Own Benefit
Authors Xiong Q, Yang M, Li P, Wu C
Received 21 June 2019
Accepted for publication 25 September 2019
Published 11 October 2019 Volume 2019:12 Pages 3205—3215
DOI https://doi.org/10.2147/IDR.S220376
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sahil Khanna
Qiuhong Xiong, Min Yang, Ping Li, Changxin Wu
Institutes of Biomedical Sciences, Shanxi University, Taiyuan 030006, People’s Republic of China
Correspondence: Qiuhong Xiong; Changxin Wu
Institutes of Biomedical Sciences, Shanxi University, No.92 Wucheng Road, Taiyuan 030006, People’s Republic of China
Tel +86 351 701 8958
Email [email protected]; [email protected]
Abstract: Autophagy is a lysosomal degradation pathway to clear long-lived proteins, protein aggregates, and damaged organelles. Certain microorganisms can be eliminated by an autophagic degradation process termed xenophagy. However, many pathogens deploy highly evolved mechanisms to evade autophagic degradation. What is more, series of pathogens have developed different strategies to exploit autophagy to ensure their survival. These bacteria could induce autophagy and/or prevent autophagosomes fusion with lysosomes through secreted effector proteins or utilizing host components, thereby maintaining the localization of the bacteria within the autophagosomes where they replicate. Here, we review the current knowledge of the mechanisms developed by the bacteria to benefit from autophagy for their survival.
Keywords: bacteria, autophagy, exploit, benefit
Introduction
Autophagy was first described as a response to starvation by the liver, with the term “autophagy” derived from the Greek words for “self” and “eating”. It involves the sequestration and transport of complete regions of the cytoplasm, including both soluble proteins and entire organelles within double-membrane vacuoles called autophagosomes, to the lysosomal system for degradation and recycling by lysosomal hydrolases.1 Autophagy is the lysosomal degradation process which regulates levels of long-lived proteins and organelles.1 The general autophagy is bulk autophagy which appears to randomly sequester cytosolic content, while selective autophagy requires cargo adaptors specifically enrich forming autophagosomes for certain cargos. So far, selective autophagy has given rise to terms such as mitophagy, ribophagy, aggrephagy, lipophagy, endoplasmic reticulum (ER)-phagy, and pexophagy according to the engulfed material.2
Autophagy pathways can be broken down into five basic phases: initiation, elongation, closure, maturation, and degradation.3 Autophagy is regulated at the molecular level by a family of dedicated genes called autophagy-related (ATG) genes.4 The first ATG gene was identified in yeast by Ohsumi in 1993.5 To date, at least 37 ATGs were identified.6 Among them, one subset has been referred to as the “core” autophagic machinery as they are required for autophagosome formation in all autophagy subtypes.7 These core ATGs can be subdivided into four subgroups: 1) the ATG1/ULK1 complex, composed of ATG1/ULK1, ATG13, ATG101 and FIP200; 2) the class III PI3K complex, composed of Vps34, Vps15, ATG6/Beclin1 and ATG14; 3) two ubiquitin-like protein conjugation systems which consist of ATG12, ATG5, ATG16L1, ATG8/LC3, ATG7, ATG10, ATG3 and ATG4; and 4) two transmembrane proteins, ATG9 (and associated proteins ATG2 and ATG18/WIPI2) and VMP1.7,8 The first and second subgroups of core ATGs regulate the initiation phase of autophagy, the third subgroup involves in autophagosome formation and membrane elongation, and the fourth subgroup is important for autophagosome formation and maturation.2,8
In 2005, Levine defined xenophagy as a process that host cells direct the cell’s digestive machinery to the breakdown of invading microorganisms to address the risk of pathogen invasion.9 Autophagy is the first line of host innate immune system to eliminate invasive pathogens, however, pathogens have evolved countermeasures to either evade or reconfigure the autophagy pathway for their own survival.10 Furthermore, autophagy or autophagy-related proteins also could be exploited by the pathogens. Studies have demonstrated that some viruses, such as poliovirus (PV), hepatitis C virus (HCV), dengue virus (DENV), human immunodeficiency virus (HIV), hepatitis B virus (HBV) and so on, could use autophagy for their replication, assembly and release.11–13 In 2011, Ogawa et al reviewed the strategies of some bacteria to manipulate autophagy for their own benefit, however, many bacterial effectors and host factors which were involved in the utilization of host autophagy by pathogens were identified during these years.10 Understanding how bacterial pathogens achieve this at the molecular level will provide new potential targets for therapeutic intervention. In this review, we will focus on the bacterial pathogens and summarize the current knowledge of the mechanisms developed by the bacteria to hijack autophagy for their own benefit.
Anaplasma phagocytophilum
A. phagocytophilum is a Gram-negative obligate intracellular bacterium that causes human granulocytic anaplasmosis. After invasion of the host cells, A. phagocytophilum replicates in a membrane-bound compartment contains autophagy-related proteins LC3 and Beclin1 but is endosomal or lysosomal markers are absent. Induction of autophagy facilitates Anaplasma infection while inhibition of autophagy arrests its growth.14 The secreted effector Anaplasma translocated substrate 1 (Ats-1) stimulates autophagy nucleation by interacting with Beclin1 therefore facilitates autophagosome formation and subsequently promotes its own growth by using the nutrients contained in the autophagosomes (Table 1, Figure 1).15,16
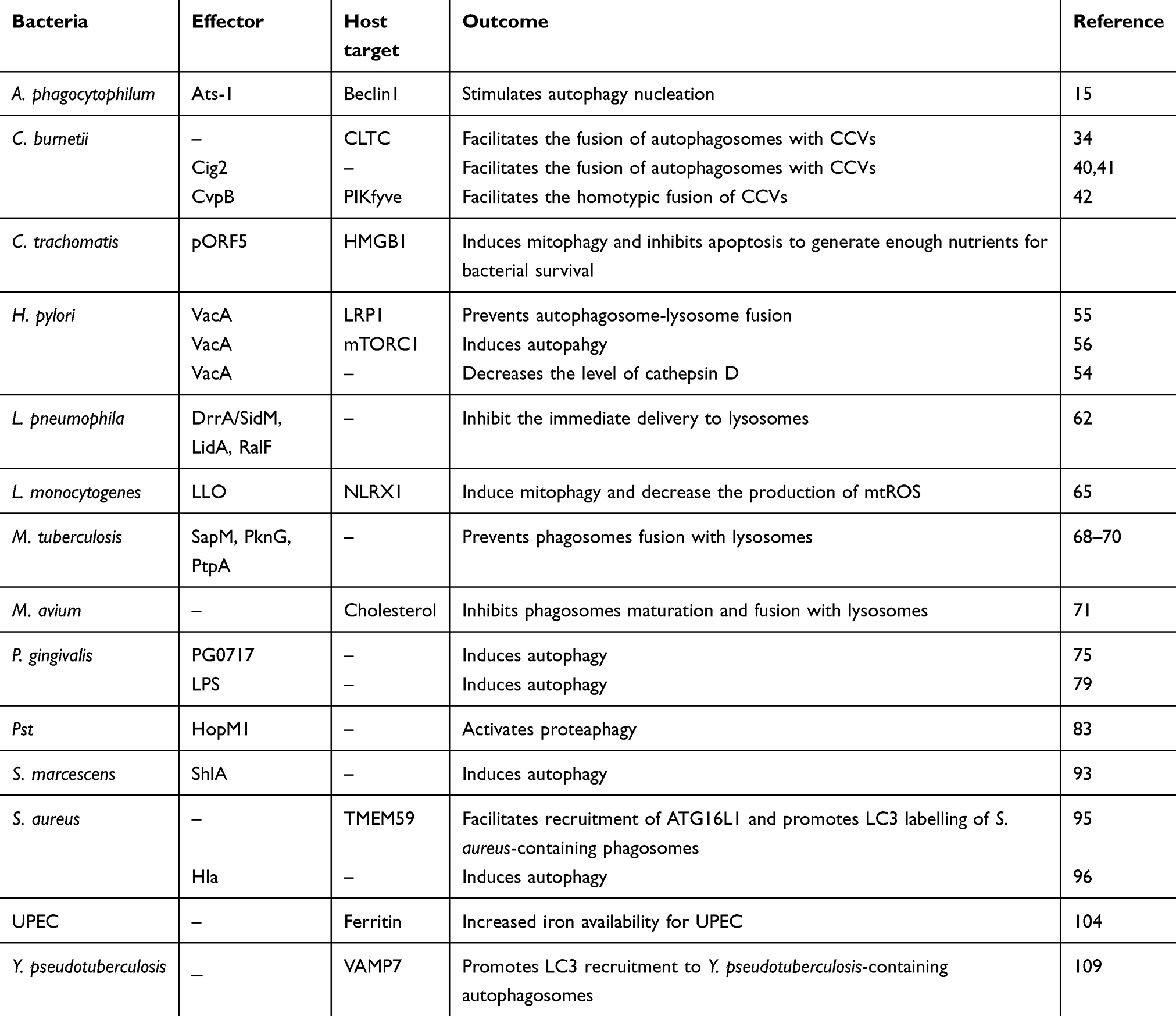 |
Table 1 Effectors And Host Targets Involve In Bacterial Exploitation Of Autophagy |
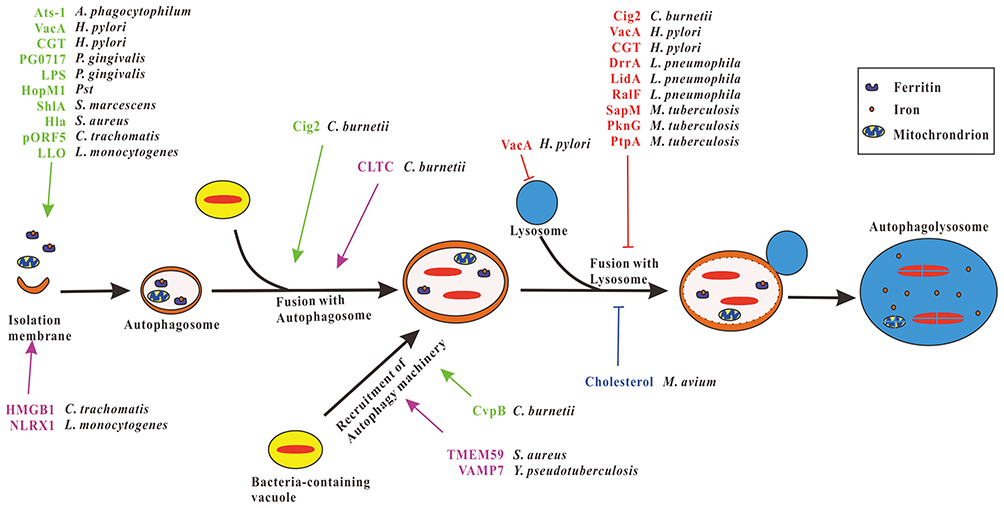 |
Figure 1 Exploitation of autophagy pathway by bacterial pathogens. After the invasion of the host cell, vacuoles containing intracellular bacteria fuse with autophagosomes or recruit autophagy machinery to form autophagic vacuoles favor the bacteria replication. Several bacteria have evolved different effector proteins that induce autophagy to form autophagosomes or promote bacteria-containing vacuoles fuse with autophagosomes or facilitate the recruitment of autophagy machinery to bacteria-containing vacuoles (green), thereby promoting the replication of bacteria. Some bacteria secrete effector proteins that impair the functions of lysosomes or inhibit bacteria-containing autophagosomes fuse with lysosomes (red) to block the lysosomal degradation of the bacteria. Furthermore, cholesterol of host cells prevents M. avium-containing autophagosomes fusion with lysosomes (blue), CLTC promotes C. burnetii-containing vacuoles fusion with autophagosomes, HMGB1 and NLRX1 induce mitophagy thus promote survival of C. trachomatis and L. monocytogenes respectively, TMEM59 and VAMP7 facilitate the recruitment of autophagy proteins to the S. aureus and Y. pseudotuberculosis-containing vacuoles, respectively (violet), and UPEC benefits from ferritinophagy. |
Brucella
Bacteria of the genus Brucella, contains six classic species, are the causative agent of brucellosis, a worldwide zoonosis with significant health and economic consequences.17 The internalized bacterium Brucella abortus traffics from the endocytic compartment to the ER to form Brucella-containing vacuole (rBCV), where the bacterium proliferates.18,19 Taguchi et al showed that the formation of rBCV required the autophagy protein ATG9, WIPI1 and rBCV conversion into a compartment with autophagic features (aBCV) accompany Brucella replication.20,21 The formation of aBCV required the autophagy-initiation proteins ULK1, Beclin1, ATG14L and PI3K but independent of the proteins involve in autophagosome membrane elongation such as ATG5, ATG16L1, ATG7, ATG4B, and LC3B. aBCV formation completes the Brucella intracellular cycle and promotes subsequent cell-to-cell spreading.21,22 Further, B. abortus induces autophagy and prevents Brucella-containing phagosomes fusion with lysosomes.22–24 Similar to B. abortus, B. melitensis infection triggers autophagosome formation, and autophagy favors B. melitensis survival.25,26 Deletion of host autophagy system ULK1, ATG9 and Beclin1 resulted in striking disruption of B. melitensis intracellular trafficking and replication.27 Thus, Brucella selectively co-opts autophagy-initiation complexes to subvert host clearance and facilitate the bacterium persist and replicate within aBCV, eventually promote infection.
Coxiella burnetii
C. burnetii, the causative agent of human Q fever, is a highly infectious Gram-negative bacterium. Coxiella hijacks the autophagosomes and redirect the nutrient by-products of the autophagolysosomes toward microbial replication rather than for the use by the host cell.28–30 After infection, C. burnetii establishes large acidic vacuoles containing multiple replicating bacteria that were labeled with autophagy protein LC3.31 Induction of autophagy by starvation or overexpression of LC3, Rab24 or Beclin1 promotes the formation of Coxiella-replicative vacuoles.32,33 The clathrin heavy chain (CLTC), a scaffolding protein of clathrin-coated vesicles, facilitating the fusion of autophagosomes with the Coxiella-containing vacuoles (CCVs) (Table 1, Figure 1).34 Intracellular Coxiella has two morphologically, compositionally and functionally distinct forms: the metabolically dormant, less replicating and environmentally stable small cell variants (SCVs) and the metabolically active, replicating and more fragile large cell variants (LCVs).35 It is believed that the differentiation from SCV to LCV is triggered by a nutrient-rich environment as induction of autophagy increases the number of LCVs in HeLa cells.36 Moreover, the CCVs acquire certain lysosomal characteristics, the low pH conditions favorable to Coxiella replication, while increasing the phagolysosomal pH inhibited the multiplication of the bacteria.37–39 The Coxiella effector proteins Cig2 and CvpB facilitate CCVs fusion with autophagosomes. Cig2 promotes fusion of the CCVs with autophagosomes by continuously maintaining LC3 on the CCVs membranes which delays autophagosome maturation and promotes constitutive fusion between autophagosome and CCVs (Table 1, Figure 1).40,41 CvpB can bind phosphatidylinositol 3-phosphate (PI3P) and perturb the activity of the phosphatidylinositol 5-kinase PIKfyve, thereby enriching PI3P on CCVs membranes which promote the recruitment of autophagosomal machinery to mediate homotypic fusion of CCVs (Table 1, Figure 1).42 Furthermore, autophagy can repair damaged membranes of CCVs to maintain the membrane integrity therefore promote bacteria replication.43
Chlamydia trachomatis
C. trachnomatis is an obligate intracellular bacterial pathogen which is associated with several human diseases, such as trachoma, pneumonia, and atherosclerosis.44 C. trachnomatis replicates within a membrane-bound compartment (the inclusion) which is not associated with autophagosomes. However, after inhibition of autophagy, the chlamydial inclusion size and progeny infectivity were decreased, morphology of chlamydial forms was aberrant suggest a potential supportive role of host autophagy in the pathogenesis of Chlamydia.44 In autophagy-deficient ATG5(-/-) fibroblasts, the growth of C. trachnomatis was increased, but in the presence of Bafilomycin A1, an inhibitor of vacuolar ATPase (vATPase), the growth was inhibited indicates that there should be at least two types of vATPase-bearing organelles that one defends against chlamydiae, while the other supports chlamydial growth.45 It was also showed that lysosomal degradation products were transferred to chlamydiae suggest that products generated within lysosomes contribute to the intracellular survival of C. trachnomatis.46 Moreover, Lei et al demonstrated that C. trachnomatis plasmid-encoded protein pORF5 up-regulated the expression of high mobility group box 1 (HMGB1) which induces mitophagy and inhibits apoptosis of host cells (Table 1, Figure 1).47 These findings suggest that C. trachnomatis could manipulate host cell death to usurp enough nutrients generated via autophagy for their survival and replication.
Francisella tularensis
F. tularensis is a Gram-negative, highly infectious, facultative intracellular pathogen that causes tularemia.48 After phagocytosed by host cells, Francisella escaped from the phagosome and underwent replication in Francisella-containing vacuoles (FCVs). FCV is a large, juxtanuclear, LC3- and LAMP1-positive vacuole whose formation is dependent on autophagy.49 Optimal intracellular bacterial growth requires autophagy that induces autophagic degradation of cellular proteins, thereby generating a surplus of amino acids to support intracellular growth of F. tularensis in macrophages as well as mouse embryo fibroblast (MEFs), but this process is independent of ATG5.50
Helicobacter pylori
H. pylori infection is associated with the development of chronic gastritis, peptic ulcer and gastric cancer. After internalization of human macrophage and gastric epithelial cells, autophagy was induced and H. pylori replicates in a double-layer vesicle which is characteristic of autophagosome.51,52 The secreted effector protein, vacuolating cytotoxin (VacA) can induce autophagy through binding to the low-density lipoprotein receptor-related protein-1 (LRP1) or through inhibition of mTORC1, but prevent autophagosome-lysosome fusion and decrease the level of cathepsin D thus impair the catalytic activity of lysosome (Table 1, Figure 1).53–56 The cholesterol-α-glucosyltransferase (CGT) of H. pylori also could trigger autophagy, and restrain autophagosome fusion with lysosomes to impair macrophage clearance of the bacteria (Table 1, Figure 1).57 Zhang et al demonstrated that gastric epithelial cells infected with H. pylori resulted in impaired lysosomal acidification and retrograde trafficking of mannose-6-phosphate receptors (MPRs). Inhibition of autophagosome formation and lysosomal functions promote intracellular survival of H. pylori.58 However, the induction of autophagy in turn limits the multiplication of H. pylori thereby conferring protection to host cells against H. pylori infection.51 In the early stage of infection of murine bone marrow derived-dendritic cells (BMDCs), H. pylori transiently replicates in autophagosomes, but in the late stage, H. pylori was degraded by autophagolysosomes.59
Legionella pneumophila
L. pneumophila, a Gram-negative bacterium, is an intracellular bacterial pathogen responsible for an acute form of pneumonia called Legionnaire’s disease. L. pneumophila could activate the autophagy pathway by a mechanism that does not require phagocytosis of the bacteria.60 After invasion, L. pneumophila resides within vacuoles whose biogenesis resembles autophagy, escapes the toxic phagosome-lysosome pathway,61 and perturb and delay the maturation of autophagosomes into autophagolysosomes.60,62 It has been demonstrated that the effector proteins such as DrrA/SidM, LidA and RalF prolong the association time of the Legionella-containing vacuoles with the ER and inhibit the immediate delivery to lysosomes (Table 1, Figure 1).62 Remarkably, Legionella continues to replicate within acidic lysosomal vacuoles.63 The replication of Legionella was inhibited when autophagosome formation is impaired, or vacuoles acidification and fusion with lysosomes is blocked.63,64 Thus, it seems that L. pneumophila persist in immature autophagosomal vacuoles for a period that is suitable for them to differentiate into an acid-resistant, replicative form. Subsequently, the adapted progeny continues to replicate within autophagolysosomes.62
Listeria monocytogenes
L. monocytogenes is Gram-positive bacterium causes enteritis, occasionally causes listeriosis. After infection of host cells, L. monocytogenes could secrete several effector proteins such as internalins, listeriolysin O (LLO), ActA and so on to evade killing by autophagy.10 Recently, Zhang and colleagues found that the bacterial effector LLO and host factor nucleotide-binding leucine-rich repeat-containing family member X1 (NLRX1) could induce mitophagy. Increased mitophagy decreased the production of mitochondrial reactive oxygen species (ROS) which controls L. monocytogenes infection, thereby facilitating its survival (Table 1, Figure 1).65
Mycobacterium
Mycobacterium tuberculosis causes tuberculosis which is one of the major causes of death from an infectious disease worldwide. In human lymphatic endothelial cells (LECs), M. tuberculosis was observed within autophagosomes, and autophagy promotes the growth of the bacterium in resting LECs.66 In human alveolar epithelial cells, M. tuberculosis-containing compartments surrounded by double membranes and labelled with autophagy marker LC3, inhibition of the autophagy impaired intracellular bacteria replication and improved host cell viability, and the bacteria-containing compartment fusion with lysosomes appears to be inhibited, suggesting that autophagy is involved in trafficking of M. tuberculosis bacilli and is required for its survival.67 It has already demonstrated that the secreted effector proteins, acid phosphatase M (SapM), the M. tuberculosis eukaryotic-like serine/threonine-protein kinase G (PknG) and the protein tyrosine phosphatase (PtpA) could prevent mycobacteria-containing phagosomes fusion with lysosomes (Table 1, Figure 1).68–70 The cholesterol, a component of host cell plasma membrane, could prevent M. avium-containing phagosomes maturation and fusion with lysosomes in mouse bone marrow-derived macrophages (BMDMs) (Table 1, Figure 1).71 Similar phenotypes were observed in mouse macrophages cell line Raw 264.7 infected with closely relative to M. tuberculosis, M. marinum.72 Furthermore, in the model organism Dictyostelium discoideum, autophagy is required for nonlytic ejection of M. marinum. Autophagic machinery was recruited at the distal pole of ejecting bacteria, disruption of autophagy causes the host cells to become leaky and die during ejection suggest that autophagy maintains the integrity of plasma membrane and promotes cell-to-cell transmission of M. marinum.73
Porphyromonas gingivalis
P. gingivalis is a Gram-negative bacterium causes periodontitis. It is also linked to several systemic chronic diseases such as rheumatoid arthritis, diabetes, and cancer.74 After internalization by host cells, the lipoprotein PG0717 of P. gingivalis activates autophagy (Table 1, Figure 1), then the bacterium evades the endocytic pathway to lysosomes and instead traffics to autophagosome, thereby establishing a replicative niche in an ATG7-dependent manner.75–77 When autophagy is inhibited in HCAEC and human gingival epithelial cells (GECs), the bacterium enter the endocytic pathway to lysosomes thus the viability of intracellular P. gingivalis is reduced.77,78 Recently, studies showed that lipopolysaccharide (LPS) from P. gingivalis can induce autophagy by suppressing PI3K/Akt/mTOR signaling pathway in human gingival fibroblasts (HGFs) (Table 1, Figure 1) and enhance the co-localization of bacterium with autophagosomes in human-cultured keratinocyte cells (HaCaT).79,80 Remarkably, P. gingivalis is an asaccharolytic pathogen, thus the bacterium traffic through the autophagic pathway can acquire essential nutrients from the autophagosomes, and also evade cell defense.60
Pseudomonas syringae pv. tomato DC3000
Autophagy plays an important role in maintaining a functional immune system in plants.81,82 Recently, Hofius and co-workers demonstrated that autophagy has both pro- and antibacterial functions upon infection of Arabidopsis thaliana with Pseudomonas syringae pv. tomato DC3000 (Pst).83 Autophagy was activated after infection with Pst and NEIGHBOR of BRCA1 (NBR1), a cargo receptor-mediated autophagic degradation limited the growth of Pst.84 In turn, as proteasome acts as a hub for plant immunity,85 Pst employs the effector protein Hrp outer protein M1 (HopM1) to suppress proteasomal activity and activate autophagic degradation of proteasomes (proteaphagy), thereby enhancing its pathogenicity (Table 1, Figure 1).83,85
Salmonellaenterica Serovar Typhimurium
It has been demonstrated that the ATG16L1 knock-out or T300A variant confers protection from cellular invasion by S. typhimuriunm in HCT116 cells, re-expression of wild-type ATG16L1 not T300A variant in ATG16L1 knock-out cells facilitates Salmonella invasion into the cells.86 Since ATG16L1 was recruited to the entry sites of Shigella flexneri, the ATG16L1 T300A variant may be less efficiently recruited to the plasma membrane, therefore reduce Salmonella invasion.87 Interestingly, Yu and colleagues found that depletion of autophagy components such as ATG5, LC3 and/or p62 inhibits the replication of cytosolic Salmonella but not affect the Salmonella invasion ability in HeLa cells suggest that autophagy facilitates Salmonella replication.88 Later, Kreibich et al demonstrated that, similar to Coxiella, autophagy proteins promote repair of Salmonella-containing vacuole (SCV) membrane damaged by the Salmonella type three secretion system 1 (T3SS-1) in an unknown mechanism, thereby allowing compartment maturation and subsequent expression of type three secretion system 2 (T3SS-2), which together promote intracellular survival.89–91 These findings suggest that Salmonella uses the autophagic process to its advantage and survives in cells.
Serratia marcescens
S. marcescens is a Gram-negative bacterium which is commonly involved in hospital-acquired infections (HAIs). After internalized by epithelial cells, Serratia replicates inside a large membrane-bound compartment which displays autophagic-like characteristics.92 However, the autophagic-like vacuoles are non-acidic and have no degradative properties suggest that Serratia utilizes autophagosomes for survival and proliferation by preventing the vacuoles fusion with lysosomes.92 Interestingly, the pore-forming toxin ShlA secreted by Serratia can induce autophagy prior to bacterial internalization which may pave the way for subsequent proliferation inside the autophagosomes (Table 1, Figure 1).93
Staphylococcus aureus
S. aureus can invade epithelial cells and then transit to an autophagosome-like vacuole which is characterized by double membrane and colocalization with LC3.94 In this process, human transmembrane protein TMEM59 which mainly localized in late endosomes/lysosomes may perform an in situ autophagic function that facilitates the recruitment of ATG16L1 and then promotes LC3 labelling of S. aureus-containing phagosomes.95 After the invasion of epithelial cells, S. aureus inhibits the bacteria-containing autophagosome maturation and fusion with lysosomes dependent on accessory gene regulator (Agr) system. Activation of autophagy by the inducer rapamycin significantly increases the intracellular load of S. aureus. In contrast, the growth of intracellular S. aureus was drastically impaired upon treatment with the autophagy inhibitor wortmannin. Similar results were obtained using atg5-deficient MEFs suggest that autophagy is indispensable for S. aureus replication.94 The virulence factor pore-forming toxin α-hemolysin (Hla) can trigger autophagy hence promote bacterial replication (Table 1, Figure 1).96 After replication, S. aureus eventually escape from autophagosomes into the cytoplasm and induce apoptosis-like cell death.94 Therefore, the autophagy machinery is essential for S. aureus replication and host cell killing.
Uropathogenic Escherichia coli (UPEC)
UPEC causes a frequent and important disease in humans, urinary tract infection (UTI). UPEC colonize the bladder and persist within the bladder epithelium as membrane-enclosed quiescent intracellular reservoirs (QIRs) that can seed recurrent UTI.97 The autophagy gene ATG16L1 plays an important role in inflammatory disease and intestinal cell abnormalities, however, studies showed that ATG16L1 deficiency confers protection in vivo to the host against both acute and latent uropathogenic E. coli (UPEC) infection,97–99 suggesting that UPEC can use autophagy to provide potential nutrient sources or a protected environment for their survival.100 ATG16L1 deficient mice cleared UPEC more rapidly and thoroughly which is associated with increased recruitment of innate immune cells to the infected bladders and a robust proinflammatory response,99 and this process is independent of the pathogen sensor nucleotide-binding oligomerization domain containing 2 (NOD2) but is dependent of IL-1β.101,102 Recent studies have demonstrated that T300A variant in ATG16L1 increases the expression level of small secretory RAB GTPases, such as RAB11A, RAB27B and RAB33B that are important for UPEC expulsion, thereby limiting the UPEC persistence in urothelium.103 Mechanistic studies revealed that UPEC shuttles with ferritin-bound iron into the autophagosomal and lysosomal compartments within the urothelium, autophagic degradation of iron-bound ferritin (ferritinophagy) led to increased iron availability for UPEC (Table 1, Figure 1), then triggered bacterial overproliferation and host cell death. Inhibition of autophagy or inhibition of iron-regulatory proteins, or chelation of iron reversed the bacterial overgrowth and promoted host cell survival suggests that UPEC exploit ferritinophagy for their own survival.104
Yersinia pestis
Y. pestis is a Gram-negative bacterium and causes plague.105 Y. pestis can survive and replicate in phagosomes of murine macrophages.106 It has been demonstrated that Y. pestis-containing vacuoles colocalized with autophagy protein LC3, further, the Y. pestis-containing vacuoles failed to acidity below pH 7 in mouse BMDMs. These findings suggest that Y. pestis could avoid xenophagy by preventing vacuole maturation to the autolysosome, thus promotes its survival in autophagosomes.84 However, the replication of bacterium was not decreased in ATG5-deficient BMDMs suggest that autophagy is not required for Y. pestis survival in macrophages.84 It is possible that the bacteria only recruits the membrane to enlarge the Y. pestis-containing vacuoles into a spacious compartment or interferes normal process of autophagy to promote cell death thus escape from the macrophage.107 The connection between autophagy and Y. pestis still need to be addressed in future work.
Yersinia pseudotuberculosis
Y. pseudotuberculosis is a Gram-negative enteropathogenic bacterium that causes mesenteric lymphadenitis. After ingestion, the bacterium activates autophagy and replicates within autophagosomes in mouse BMDMs and HeLa cells.108,109 The vesicle-associated membrane proteins (VAMPs) play pivotal roles in the membrane traffic during the internalization of Y. pseudotuberculosis. VAMP7 promotes LC3 recruitment to Y. pseudotuberculosis-containing autophagosomes (YCVs) (Table 1, Figure 1).109 Like Y. pestis, Y. pseudotuberculosis also prevents YCVs mature to autophagolysosomes. However, different to Y. pestis, autophagy is required for Y. pseudotuberculosis survive, Y. pseudotuberculosis traffics to lysosomes for degradation upon autophagy inhibition.108
Conclusions
The role of autophagy in host defense against bacteria has investigated in depth. Generally, autophagy is antipathogenic, however, several bacterial pathogens have evolved countermeasures to hijack the autophagic pathway for their own profit. In most cases, these bacteria actively induce autophagy and/or block autophagosome fusion with the lysosome through secreted effector proteins, then use the autophagosome as a replicative niche for their growth (Figure 1). Interestingly, several bacteria could utilize the host components such as cholesterol, TMEM59 and VAMP7, thereby favoring the survival of some bacteria. The role of autophagy in enhancing replication of these pathogens is unknown, but nutrient acquisition and escape from cell defense are two likely explanations for these phenotypes. Remarkably, the role of autophagy in microbial infection may depend on the type of invading microbe and the cell type. In an era of increasing antibiotic resistance, understanding how pathogens interact with and manipulate the host autophagy pathway to achieve this will hopefully provide a basis for combating infection and increase our understanding of the role and regulation of autophagy. Considering the variety of mechanisms that developed by different pathogens, we need to correctly use autophagy modulators in eliminating bacterial pathogens and it is necessary to determine an effective strategy in the clinical treatment.
Acknowledgments
This work was supported by grants from the National Key R&D Program of China (2017YFD0500300) to CW, the National Natural Science Foundation of China (31801972) and the Natural Science Foundation of Shanxi Province, China (201801D221248) to QX.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Martinet W, Agostinis P, Vanhoecke B, Dewaele M, De Meyer GR. Autophagy in disease: a double-edged sword with therapeutic potential. Clin Sci (Lond). 2009;116(9):697–712. doi:10.1042/CS20080508
2. Mancias JD, Kimmelman AC. Mechanisms of selective autophagy in normal physiology and cancer. J Mol Biol. 2016;428(9Pt A):1659–1680. doi:10.1016/j.jmb.2016.02.027
3. Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25(19):1999–2010. doi:10.1101/gad.17558811
4. Klionsky DJ, Cregg JM, Dunn WA
5. Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333(1–2):169–174. doi:10.1016/0014-5793(93)80398-e
6. Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24(1):9–23. doi:10.1038/cr.2013.169
7. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131. doi:10.1016/j.ceb.2009.11.014
8. Xiong Q, Li W, Li P, Yang M, Wu C, Eichinger L. The role of ATG16 in autophagy and the ubiquitin proteasome system. Cells. 2018;8:1. doi:10.3390/cells8010002
9. Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120(2):159–162. doi:10.1016/j.cell.2005.01.005
10. Ogawa M, Mimuro H, Yoshikawa Y, Ashida H, Sasakawa C. Manipulation of autophagy by bacteria for their own benefit. Microbiol Immunol. 2011;55(7):459–471. doi:10.1111/j.1348-0421.2011.00343.x
11. Robinson M, Schor S, Barouch-Bentov R, Einav S. Viral journeys on the intracellular highways. Cell Mol Life Sci. 2018;75(20):3693–3714. doi:10.1007/s00018-018-2882-0
12. Echavarria-Consuegra L, Smit JM, Reggiori F. Role of autophagy during the replication and pathogenesis of common mosquito-borne flavi- and alphaviruses. Open Biol. 2019;9(3):190009. doi:10.1098/rsob.190054
13. Choi Y, Bowman JW, Jung JU. Autophagy during viral infection – a double-edged sword. Nat Rev Microbiol. 2018;16(6):341–354. doi:10.1038/s41579-018-0003-6
14. Niu H, Yamaguchi M, Rikihisa Y. Subversion of cellular autophagy by Anaplasma phagocytophilum. Cell Microbiol. 2008;10(3):593–605. doi:10.1111/j.1462-5822.2007.01068.x
15. Niu H, Xiong Q, Yamamoto A, Hayashi-Nishino M, Rikihisa Y. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proc Natl Acad Sci U S A. 2012;109(51):20800–20807. doi:10.1073/pnas.1218674109
16. Niu H, Rikihisa Y. Ats-1: a novel bacterial molecule that links autophagy to bacterial nutrition. Autophagy. 2013;9(5):787–788. doi:10.4161/auto.23693
17. Pappas G, Akritidis N, Bosilkovski M, Tsianos E. Brucellosis. N Engl J Med. 2005;352(22):2325–2336. doi:10.1056/NEJMra050570
18. Celli J. Surviving inside a macrophage: the many ways of Brucella. Res Microbiol. 2006;157(2):93–98. doi:10.1016/j.resmic.2005.10.002
19. Pizarro-Cerda J, Meresse S, Parton RG, et al. Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infect Immun. 1998;66(12):5711–5724.
20. Taguchi Y, Imaoka K, Kataoka M, et al. Yip1A, a novel host factor for the activation of the IRE1 pathway of the unfolded protein response during Brucella infection. PLoS Pathog. 2015;11(3):e1004747. doi:10.1371/journal.ppat.1004747
21. Starr T, Child R, Wehrly TD, et al. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe. 2012;11(1):33–45. doi:10.1016/j.chom.2011.12.002
22. Pizarro-Cerda J, Moreno E, Sanguedolce V, Mege JL, Gorvel JP. Virulent Brucella abortus prevents lysosome fusion and is distributed within autophagosome-like compartments. Infect Immun. 1998;66(5):2387–2392.
23. Arriola Benitez PC, Pesce Viglietti AI, Herrmann CK, et al. Brucella abortus promotes a fibrotic phenotype in hepatic stellate cells, with concomitant activation of the autophagy pathway. Infect Immun. 2018;86:1.
24. Pesce Viglietti AI, Gentilini MV, Arriola Benitez PC, Giambartolomei GH, Delpino MV. B. abortus modulates osteoblast function through the induction of autophagy. Front Cell Infect Microbiol. 2018;8:425. doi:10.3389/fcimb.2018.00026
25. Guo F, Zhang H, Chen C, et al. Autophagy favors Brucella melitensis survival in infected macrophages. Cell Mol Biol Lett. 2012;17(2):249–257. doi:10.2478/s11658-012-0009-4
26. Li T, Xu Y, Liu L, et al. Brucella melitensis 16M regulates the effect of AIR domain on inflammatory factors, autophagy, and apoptosis in mouse macrophage through the ROS signaling pathway. PLoS One. 2016;11(12):e0167486. doi:10.1371/journal.pone.0167486
27. Pandey A, Lin F, Cabello AL, et al. Activation of host IRE1alpha-dependent signaling axis contributes the intracellular parasitism of Brucella melitensis. Front Cell Infect Microbiol. 2018;8:103. doi:10.3389/fcimb.2018.00026
28. Colombo MI, Gutierrez MG, Romano PS. The two faces of autophagy: Coxiella and Mycobacterium. Autophagy. 2006;2(3):162–164. doi:10.4161/auto.2827
29. Escoll P, Rolando M, Buchrieser C. Modulation of host autophagy during bacterial infection: sabotaging host munitions for pathogen nutrition. Front Immunol. 2016;7:81. doi:10.3389/fimmu.2016.00081
30. Steele S, Brunton J, Kawula T. The role of autophagy in intracellular pathogen nutrient acquisition. Front Cell Infect Microbiol. 2015;5:51. doi:10.3389/fcimb.2015.00051
31. Beron W, Gutierrez MG, Rabinovitch M, Colombo MI. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun. 2002;70(10):5816–5821. doi:10.1128/iai.70.10.5816-5821.2002
32. Gutierrez MG, Vazquez CL, Munafo DB, et al. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol. 2005;7(7):981–993. doi:10.1111/j.1462-5822.2005.00527.x
33. Vazquez CL, Colombo MI. Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ. 2010;17(3):421–438. doi:10.1038/cdd.2009.129
34. Latomanski EA, Newton HJ. Interaction between autophagic vesicles and the Coxiella-containing vacuole requires CLTC (clathrin heavy chain). Autophagy. 2018;14(10):1710–1725. doi:10.1080/15548627.2018.1483806
35. McCaul TF, Williams JC. Developmental cycle of Coxiella burnetii: structure and morphogenesis of vegetative and sporogenic differentiations. J Bacteriol. 1981;147(3):1063–1076.
36. Gutierrez MG, Colombo MI. Autophagosomes: a fast-food joint for unexpected guests. Autophagy. 2005;1(3):179–181. doi:10.4161/auto.1.3.2063
37. Baca OG, Li YP, Kumar H. Survival of the Q fever agent Coxiella burnetii in the phagolysosome. Trends Microbiol. 1994;2(12):476–480. doi:10.1016/0966-842x(94)90651-3
38. Hackstadt T, Williams JC. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc Natl Acad Sci U S A. 1981;78(5):3240–3244. doi:10.1073/pnas.78.5.3240
39. Heinzen RA, Hackstadt T, Samuel JE. Developmental biology of Coxiella burnettii. Trends Microbiol. 1999;7(4):149–154. doi:10.1016/s0966-842x(99)01475-4
40. Newton HJ, Kohler LJ, McDonough JA, et al. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 2014;10(7):e1004286. doi:10.1371/journal.ppat.1004286
41. Kohler LJ, Reed SC, Sarraf SA, Arteaga DD, Newton HJ, Roy CR. Effector protein Cig2 decreases host tolerance of infection by directing constitutive fusion of autophagosomes with the coxiella-containing vacuole. MBio. 2016;7:4.
42. Martinez E, Allombert J, Cantet F, et al. Coxiella burnetii effector CvpB modulates phosphoinositide metabolism for optimal vacuole development. Proc Natl Acad Sci U S A. 2016;113(23):E3260–E3269. doi:10.1073/pnas.1522811113
43. Mansilla Pareja ME, Bongiovanni A, Lafont F, Colombo MI. Alterations of the Coxiella burnetii replicative vacuole membrane integrity and interplay with the autophagy pathway. Front Cell Infect Microbiol. 2017;7:112. doi:10.3389/fcimb.2017.00517
44. Al-Younes HM, Brinkmann V, Meyer TF. Interaction of Chlamydia trachomatis serovar L2 with the host autophagic pathway. Infect Immun. 2004;72(8):4751–4762. doi:10.1128/IAI.72.8.4751-4762.2004
45. Yasir M, Pachikara ND, Bao X, Pan Z, Fan H. Regulation of chlamydial infection by host autophagy and vacuolar ATPase-bearing organelles. Infect Immun. 2011;79(10):4019–4028. doi:10.1128/IAI.05308-11
46. Ouellette SP, Dorsey FC, Moshiach S, Cleveland JL, Carabeo RA. Chlamydia species-dependent differences in the growth requirement for lysosomes. PLoS One. 2011;6(3):e16783. doi:10.1371/journal.pone.0016783
47. Lei W, Li Q, Su S, Bu J, Huang Q, Li Z. Chlamydia trachomatis plasmid-encoded protein pORF5 protects mitochondrial function by inducing mitophagy and increasing HMGB1 expression. Pathog Dis. 2017;75:9. doi:10.1093/femspd/ftx111
48. Oyston PC, Sjostedt A, Titball RW. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;2(12):967–978. doi:10.1038/nrmicro1045
49. Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A. 2006;103(39):14578–14583. doi:10.1073/pnas.0601838103
50. Steele S, Brunton J, Ziehr B, Taft-Benz S, Moorman N, Kawula T. Francisella tularensis harvests nutrients derived via ATG5-independent autophagy to support intracellular growth. PLoS Pathog. 2013;9(8):e1003562. doi:10.1371/journal.ppat.1003562
51. Wang YH, Wu JJ, Lei HY. The autophagic induction in Helicobacter pylori-infected macrophage. Exp Biol Med (Maywood). 2009;234(2):171–180. doi:10.3181/0808-RM-252
52. Hu W, Zhang L, Li MX, et al. Vitamin D3 activates the autolysosomal degradation function against Helicobacter pylori through the PDIA3 receptor in gastric epithelial cells. Autophagy. 2019;15(4):707–725. doi:10.1080/15548627.2018.1557835
53. Terebiznik MR, Raju D, Vazquez CL, et al. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy. 2009;5(3):370–379. doi:10.4161/auto.5.3.7663
54. Raju D, Hussey S, Ang M, et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology. 2012;142(5):1160–1171. doi:10.1053/j.gastro.2012.01.043
55. Yahiro K, Satoh M, Nakano M, et al. Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J Biol Chem. 2012;287(37):31104–31115. doi:10.1074/jbc.M112.387498
56. Kim IJ, Lee J, Oh SJ, et al. Helicobacter pylori infection modulates host cell metabolism through VacA-dependent inhibition of mTORC1. Cell Host Microbe. 2018;23(5):583–593 e588. doi:10.1016/j.chom.2018.04.006
57. Lai CH, Huang JC, Cheng HH, et al. Helicobacter pylori cholesterol glucosylation modulates autophagy for increasing intracellular survival in macrophages. Cell Microbiol. 2018;20(12):e12947. doi:10.1111/cmi.12947
58. Zhang L, Hu W, Cho CH, et al. Reduced lysosomal clearance of autophagosomes promotes survival and colonization of Helicobacter pylori. J Pathol. 2018;244(4):432–444. doi:10.1002/path.5033
59. Wang YH, Gorvel JP, Chu YT, Wu JJ, Lei HY. Helicobacter pylori impairs murine dendritic cell responses to infection. PLoS One. 2010;5(5):e10844. doi:10.1371/journal.pone.0010844
60. Campoy E, Colombo MI. Autophagy in intracellular bacterial infection. Biochim Biophys Acta. 2009;1793(9):1465–1477. doi:10.1016/j.bbamcr.2009.03.003
61. Dubuisson JF, Swanson MS. Mouse infection by Legionella, a model to analyze autophagy. Autophagy. 2006;2(3):179–182. doi:10.4161/auto.2831
62. Joshi AD, Swanson MS. Secrets of a successful pathogen: legionella resistance to progression along the autophagic pathway. Front Microbiol. 2011;2:138. doi:10.3389/fmicb.2011.00215
63. Sturgill-Koszycki S, Swanson MS. Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J Exp Med. 2000;192(9):1261–1272. doi:10.1084/jem.192.9.1261
64. Amer AO, Swanson MS. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol. 2005;7(6):765–778. doi:10.1111/j.1462-5822.2005.00509.x
65. Zhang Y, Yao Y, Qiu X, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol. 2019;20(4):433–446. doi:10.1038/s41590-019-0324-2
66. Lerner TR, de Souza Carvalho-Wodarz C, Repnik U, et al. Lymphatic endothelial cells are a replicative niche for Mycobacterium tuberculosis. J Clin Invest. 2016;126(3):1093–1108. doi:10.1172/JCI83379
67. Fine KL, Metcalfe MG, White E, Virji M, Karls RK, Quinn FD. Involvement of the autophagy pathway in trafficking of Mycobacterium tuberculosis bacilli through cultured human type II epithelial cells. Cell Microbiol. 2012;14(9):1402–1414. doi:10.1111/j.1462-5822.2012.01804.x
68. Scherr N, Honnappa S, Kunz G, et al. Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2007;104(29):12151–12156. doi:10.1073/pnas.0702842104
69. Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A. 2011;108(48):19371–19376. doi:10.1073/pnas.1109201108
70. Zulauf KE, Sullivan JT, Braunstein M. The SecA2 pathway of Mycobacterium tuberculosis exports effectors that work in concert to arrest phagosome and autophagosome maturation. PLoS Pathog. 2018;14(4):e1007011. doi:10.1371/journal.ppat.1007011
71. de Chastellier C, Thilo L. Cholesterol depletion in Mycobacterium avium-infected macrophages overcomes the block in phagosome maturation and leads to the reversible sequestration of viable mycobacteria in phagolysosome-derived autophagic vacuoles. Cell Microbiol. 2006;8(2):242–256. doi:10.1111/j.1462-5822.2005.00617.x
72. Lerena MC, Colombo MI. Mycobacterium marinum induces a marked LC3 recruitment to its containing phagosome that depends on a functional ESX-1 secretion system. Cell Microbiol. 2011;13(6):814–835. doi:10.1111/j.1462-5822.2011.01581.x
73. Gerstenmaier L, Pilla R, Herrmann L, et al. The autophagic machinery ensures nonlytic transmission of mycobacteria. Proc Natl Acad Sci U S A. 2015;112(7):E687–692. doi:10.1073/pnas.1423318112
74. Atanasova KR, Yilmaz O. Prelude to oral microbes and chronic diseases: past, present and future. Microbes Infect. 2015;17(7):473–483. doi:10.1016/j.micinf.2015.03.007
75. Reyes L, Eiler-Mcmanis E, Rodrigues PH, et al. Deletion of lipoprotein PG0717 in Porphyromonas gingivalis W83 reduces gingipain activity and alters trafficking in and response by host cells. PLoS One. 2013;8(9):e74230. doi:10.1371/journal.pone.0074230
76. Dorn BR, Dunn WA
77. Dorn BR, Dunn WA
78. Lee K, Roberts JS, Choi CH, Atanasova KR, Yilmaz O. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence. 2018;9(1):845–859. doi:10.1080/21505594.2018.1454171
79. Liu J, Wang X, Zheng M, Luan Q. Lipopolysaccharide from Porphyromonas gingivalis promotes autophagy of human gingival fibroblasts through the PI3K/Akt/mTOR signaling pathway. Life Sci. 2018;211:133–139. doi:10.1016/j.lfs.2018.09.023
80. Hagio-Izaki K, Yasunaga M, Yamaguchi M, et al. Lipopolysaccharide induces bacterial autophagy in epithelial keratinocytes of the gingival sulcus. BMC Cell Biol. 2018;19(1):18. doi:10.1186/s12860-018-0168-x
81. Yang F, Kimberlin AN, Elowsky CG, et al. A plant immune receptor degraded by selective autophagy. Mol Plant. 2019;12(1):113–123. doi:10.1016/j.molp.2018.11.011
82. Dong J, Chen W. The role of autophagy in chloroplast degradation and chlorophagy in immune defenses during Pst DC3000 (AvrRps4) infection. PLoS One. 2013;8(8):e73091. doi:10.1371/journal.pone.0073091
83. Ustun S, Hafren A, Liu Q, et al. Bacteria exploit autophagy for proteasome degradation and enhanced virulence in plants. Plant Cell. 2018;30(3):668–685. doi:10.1105/tpc.17.00815
84. Pujol C, Klein KA, Romanov GA, et al. Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect Immun. 2009;77(6):2251–2261. doi:10.1128/IAI.00068-09
85. Ustun S, Sheikh A, Gimenez-Ibanez S, Jones A, Ntoukakis V, Bornke F. The proteasome acts as a hub for plant immunity and is targeted by Pseudomonas type III effectors. Plant Physiol. 2016;172(3):1941–1958. doi:10.1104/pp.16.00808
86. Messer JS, Murphy SF, Logsdon MF, et al. The Crohn’s disease: associated ATG16L1 variant and Salmonella invasion. BMJ Open. 2013;3:6. doi:10.1136/bmjopen-2013-002790
87. Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11(1):55–62. doi:10.1038/ni.1823
88. Yu HB, Croxen MA, Marchiando AM, et al. Autophagy facilitates Salmonella replication in HeLa cells. MBio. 2014;5(2):e00865–00814. doi:10.1128/mBio.00865-14
89. Kreibich S, Emmenlauer M, Fredlund J, et al. Autophagy proteins promote repair of endosomal membranes damaged by the Salmonella type three secretion system 1. Cell Host Microbe. 2015;18(5):527–537. doi:10.1016/j.chom.2015.10.015
90. Owen KA, Casanova JE. Salmonella manipulates autophagy to “serve and protect”. Cell Host Microbe. 2015;18(5):517–519. doi:10.1016/j.chom.2015.10.020
91. Casanova JE. Bacterial autophagy: offense and defense at the host-pathogen interface. Cell Mol Gastroenterol Hepatol. 2017;4(2):237–243.
92. Fedrigo GV, Campoy EM, Di Venanzio G, Colombo MI, Garcia Vescovi E. Serratia marcescens is able to survive and proliferate in autophagic-like vacuoles inside non-phagocytic cells. PLoS One. 2011;6(8):e24054.
93. Di Venanzio G, Stepanenko TM, Garcia Vescovi E. Serratia marcescens ShlA pore-forming toxin is responsible for early induction of autophagy in host cells and is transcriptionally regulated by RcsB. Infect Immun. 2014;82(9):3542–3554.
94. Schnaith A, Kashkar H, Leggio SA, Addicks K, Kronke M, Krut O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem. 2007;282(4):2695–2706.
95. Boada-Romero E, Letek M, Fleischer A, Pallauf K, Ramon-Barros C, Pimentel-Muinos FX. TMEM59 defines a novel ATG16L1-binding motif that promotes local activation of LC3. Embo J. 2013;32(4):566–582.
96. Mestre MB, Colombo MI. cAMP and EPAC are key players in the regulation of the signal transduction pathway involved in the alpha-hemolysin autophagic response. PLoS Pathog. 2012;8(5):e1002664.
97. Wang C, Symington JW, Mysorekar IU. ATG16L1 and pathogenesis of urinary tract infections. Autophagy. 2012;8(11):1693–1694.
98. Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456(7219):259–263.
99. Wang C, Mendonsa GR, Symington JW, et al. Atg16L1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proc Natl Acad Sci U S A. 2012;109(27):11008–11013.
100. Clyne M. Urinary tract infections: autophagy gene mutation confers protection against uropathogenic E. coli infection. Nat Rev Urol. 2012;9(8):410. doi:10.1038/nrurol.2012.140
101. Wang C, Yuan X, Ma E, et al. NOD2 is dispensable for ATG16L1 deficiency-mediated resistance to urinary tract infection. Autophagy. 2014;10(2):331–338. doi:10.4161/auto.27196
102. Symington JW, Wang C, Twentyman J, et al. ATG16L1 deficiency in macrophages drives clearance of uropathogenic E. coli in an IL-1beta-dependent manner. Mucosal Immunol. 2015;8(6):1388–1399. doi:10.1038/mi.2015.7
103. Wang C, Bauckman KA, Ross ASB, et al. A non-canonical autophagy-dependent role of the ATG16L1(T300A) variant in urothelial vesicular trafficking and uropathogenic Escherichia coli persistence. Autophagy. 2019;15(3):527–542. doi:10.1080/15548627.2018.1535290
104. Bauckman KA, Mysorekar IU. Ferritinophagy drives uropathogenic Escherichia coli persistence in bladder epithelial cells. Autophagy. 2016;12(5):850–863. doi:10.1080/15548627.2016.1160176
105. Perry RD, Fetherston JD. Yersinia pestis – etiologic agent of plague. Clin Microbiol Rev. 1997;10(1):35–66.
106. Lukaszewski RA, Kenny DJ, Taylor R, Rees DG, Hartley MG, Oyston PC. Pathogenesis of Yersinia pestis infection in BALB/c mice: effects on host macrophages and neutrophils. Infect Immun. 2005;73(11):7142–7150. doi:10.1128/IAI.73.11.7142-7150.2005
107. Klein KA, Bliska JB. How Yersinia pestis becomes a foreign obstruction in the digestive system of the macrophage. Autophagy. 2009;5(6):882–883. doi:10.4161/auto.9095
108. Moreau K, Lacas-Gervais S, Fujita N, et al. Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell Microbiol. 2010;12(8):1108–1123. doi:10.1111/j.1462-5822.2010.01456.x
109. Ligeon LA, Moreau K, Barois N, et al. Role of VAMP3 and VAMP7 in the commitment of Yersinia pseudotuberculosis to LC3-associated pathways involving single- or double-membrane vacuoles. Autophagy. 2014;10(9):1588–1602. doi:10.4161/auto.29411
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.