Back to Journals » The Application of Clinical Genetics » Volume 14
Autoimmunity in Wiskott–Aldrich Syndrome: Updated Perspectives
Authors Sudhakar M, Rikhi R, Loganathan SK, Suri D, Singh S ![]()
Received 22 May 2021
Accepted for publication 18 July 2021
Published 20 August 2021 Volume 2021:14 Pages 363—388
DOI https://doi.org/10.2147/TACG.S213920
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Murugan Sudhakar, Rashmi Rikhi, Sathish Kumar Loganathan, Deepti Suri, Surjit Singh
Department of Pediatrics, Advanced Pediatrics Center, Postgraduate Institute of Medical Education and Research, Chandigarh, India
Correspondence: Deepti Suri
Department of Pediatrics, Advanced Pediatrics Centre, Postgraduate Institute of Medical Education and Research, Chandigarh, 160012, India
Email [email protected]
Abstract: Wiskott–Aldrich syndrome (WAS) is an uncommon X-linked combined-immunodeficiency disorder characterized by a triad of thrombocytopenia, eczema, and immunodeficiency. Patients with WAS are also predisposed to autoimmunity and malignancy. Autoimmune manifestations have been reported in 26%– 72% of patients with WAS. Autoimmunity is an independent predictor of poor prognosis and predisposes to malignancy. Development of autoimmunity is also an early pointer of the need for hematopoietic stem–cell transplantation. In this manuscript, we have collated the published data and present a narrative review on autoimmune manifestations in WAS. A summary of currently proposed immunopathogenic mechanisms and genetic variants associated with development of autoimmunity in WAS is also included.
Keywords: thrombocytopenia, vasculitis, genetics, hematopoietic stem–cell transplant, bleeding, malignancy
Introduction
Wiskott–Aldrich syndrome (WAS) is an uncommon X-linked combined immunodeficiency disorder that has a heterogeneous clinical spectrum.1–3 Manifestations vary from a relatively milder form of the disease (intermittent X-linked thrombocytopenia [XLT]) characterized by thrombocytopenia with little or no immunodeficiency to severe WAS, with profound immunodeficiency, bleeding episodes, autoimmunity, and increased risk of malignancy.3–5 Many patients with WAS have intermediate grades of severity. It is this heterogeneity in the clinical spectrum that makes the initial diagnosis of WAS so challenging.
In a US study, WAS incidence was reported to be 3.3–5.2 per million live male births.6 National registry data on primary immunodeficiency diseases from Sweden and Switzerland estimated WAS incidence to be 3.7 and 4.1 per million live births, respectively.7,8
In 1994, almost six decades after the initial description of the condition, Derry et al identified the gene responsible for the defect:9 WAS, which comprises 12 exons and is located on the short arm of the X chromosome (Xp11.23).10 It encodes for WAS protein (WASp), a 502–amino acid cytosolic protein and a key molecule for actin-cytoskeleton polymerization.11–14 WASp is ubiquitously expressed in all nonerythroid hematopoietic cells.15 It consists of a pleckstrin homology (PH) domain and an enabledvasodilator-stimulated phosphoprotein homology (EVH1, also known as WH1) domain at the amino terminal, a short basic domain (B), a Cdc42- and Rac-interactive binding (CRIB) domain, a large proline-rich region (PRR), and a verprolin/central/acidic (VCA) domain at the carboxyl terminal. (Figure 1)
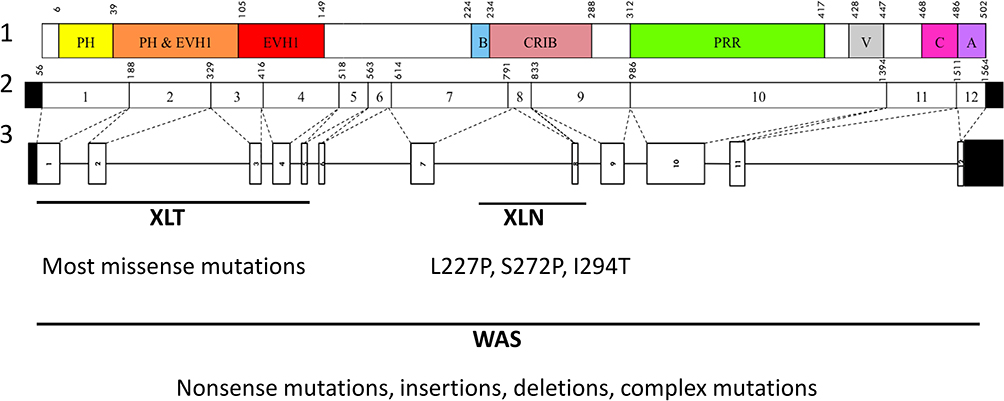 |
Figure 1 Structure of WAS gene, cDNA transcript, and Wiskott–Aldrich syndrome protein. Lane 1, Wiskott–Aldrich syndrome protein structure; lane 2, WAS cDNA structure and positions; lane 3, WAS gene structure. Variants associated with XLN: L227P, S272P, and I294T. Abbreviations: PH, pleckstrin homology; EVH1, vasodilator-stimulated phosphoprotein homology; B, basic; CRIB, Cdc42- and Rac-interactive binding; PRR, proline-rich region; V, verprolin; C, central; A, acidic; XLT, X-linked thrombocytopenia; XLN, X-linked neutropenia; WAS, Wiskott–Aldrich syndrome. |
WAS mutations affect actin cytoskeleton–dependent cellular processes, immunological synapse formation,16–20 cell migration, and signaling,21,22 and result in impaired functioning of WASp, causing XLT/WAS. Gain-of-function mutations WAS gene manifest as severe congenital X-linked neutropenia, which is characterized by recurrent bacterial infections, neutropenia, and monocytopenia without thrombocytopenia.23,24
The clinical course of WAS is complicated by frequent bleeding episodes, eczema, and recurrent infections. Patients with WAS are also predisposed to autoimmune manifestations and malignancies.3,4 Autoimmunity is an independent independent predictor of poor prognosis and predisposes to malignancy.25 As such, development of autoimmunity is an early indicator of the need for hematopoietic stem–cell transplantation (HSCT) in patients with XLT/WAS. Though HSCT is curative, it does not completely ameliorate the risk of later development of autoimmunity in WAS. Recent reports on the emergence of post-HSCT autoimmunity in these patients have rekindled interest in this field, and suggest the need for a better understanding of autoimmunity.26–30 Despite being one of the earliest immunodeficiency syndromes described in the literature, pathophysiological mechanisms of underlying autoimmunity in WAS are still unclear.31 This review focuses on putative pathogenetic mechanisms, genetic predisposition, and clinical manifestations of XLT/WAS with autoimmunity.
Methods
We carried out a literature search on PubMed, Scopus, Web of Knowledge, Google, and Google Scholar using the keywords “Wiskott Aldrich syndrome,” “autoimmunity,” “eczema,” “anemia,” “nephritis,” “arthritis,” “neutropenia,” “vasculitis,” “Wiskott Aldrich syndrome protein,” and “malignancy.” A supplementary manual search to identify additional primary studies was conducted and papers published up to December 2020 collated. Data on autoimmune manifestations were extracted from all single/multicenter cohorts and clinical case reports.
Demographic details, clinical and genetic profiles, WAS clinical scores, WASp expression, and outcomes were tabulated.
Incidence of Autoimmune Manifestations
One of the earliest reports of autoimmunity in WAS was published in 1976 by Gershwin et al, who evaluated patients with primary immunodeficiency diseases for the presence of autoantibodies.32 Three of eleven children with WAS had autoantibodies against nucleic acids. However, a US multicentre study of a cohort of patients with WAS made no mention of autoimmunity.6
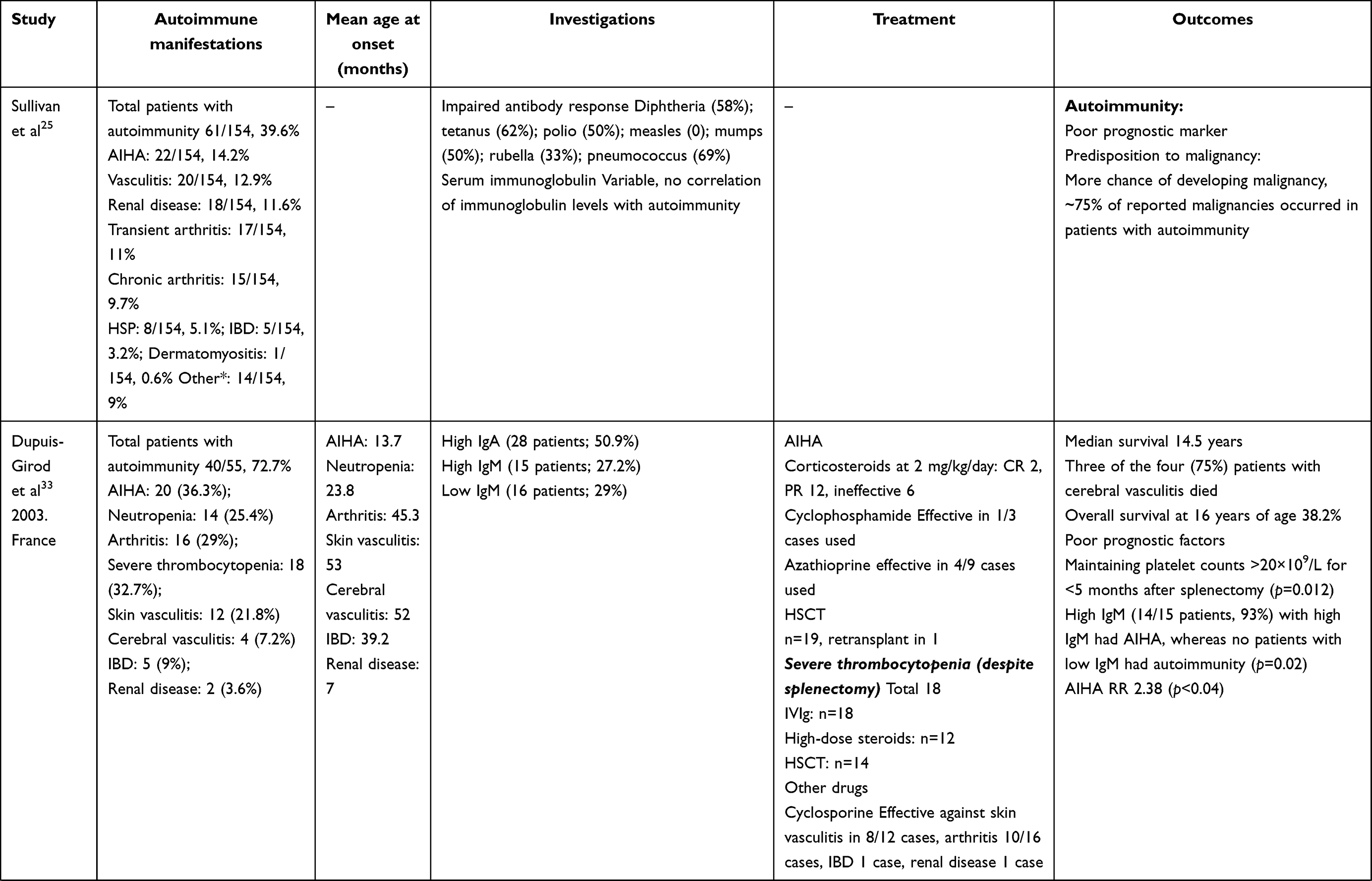
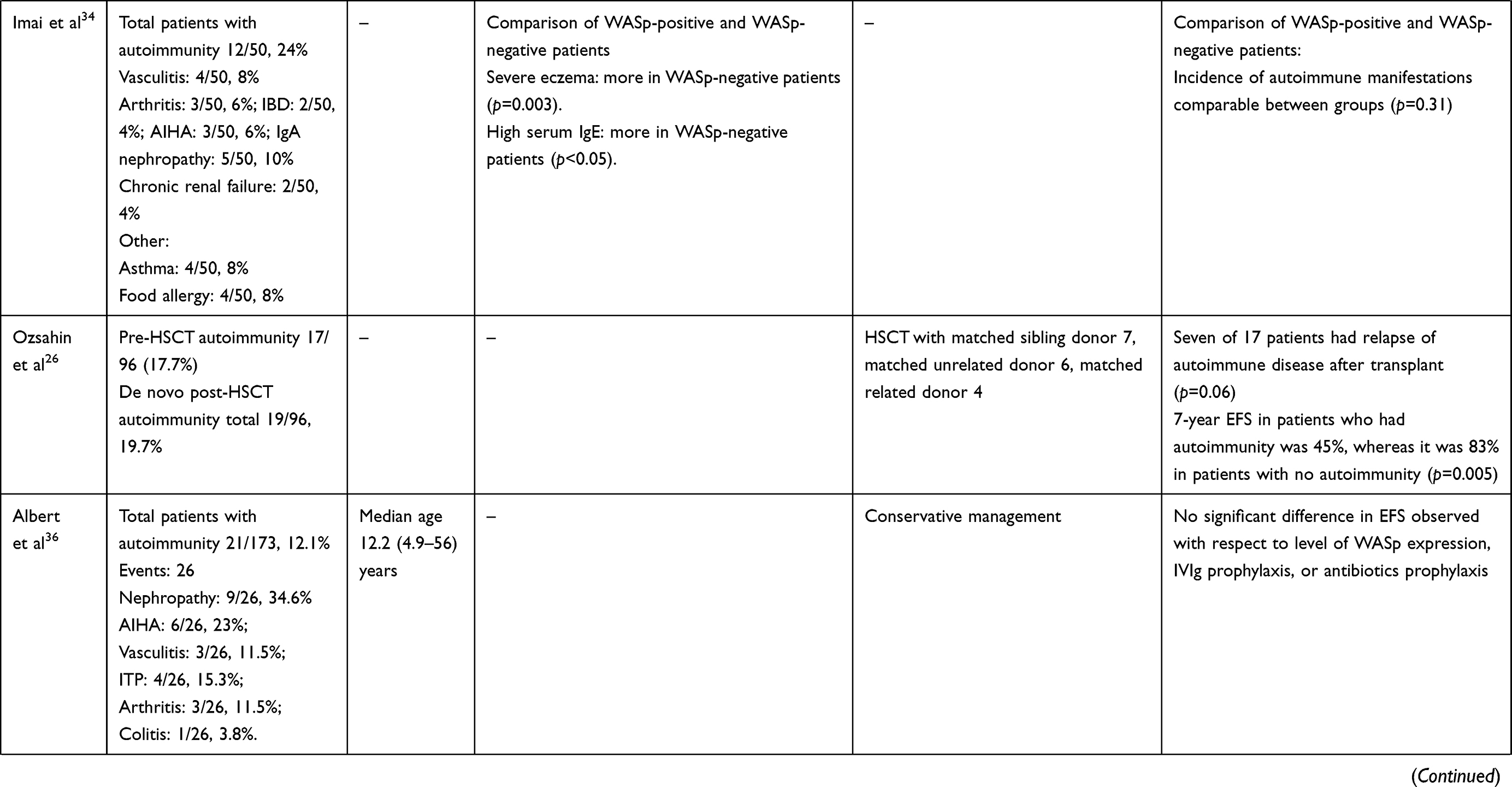
In 1994, Sullivan et al reported autoimmune manifestations in 40% of patients with WAS.25 The study highlighted autoimmunity to be a risk factor of the development of malignancy. Since then, autoimmunity has been a well-recognized entity in patients with WAS and an important indicator in predicting clinical outcomes and prognoses. Subsequently, several studies from different centers have reported variable figures (26%–72%) for the occurrence of autoimmune manifestations in patients with WAS (Table 1).25,28–30,33–40
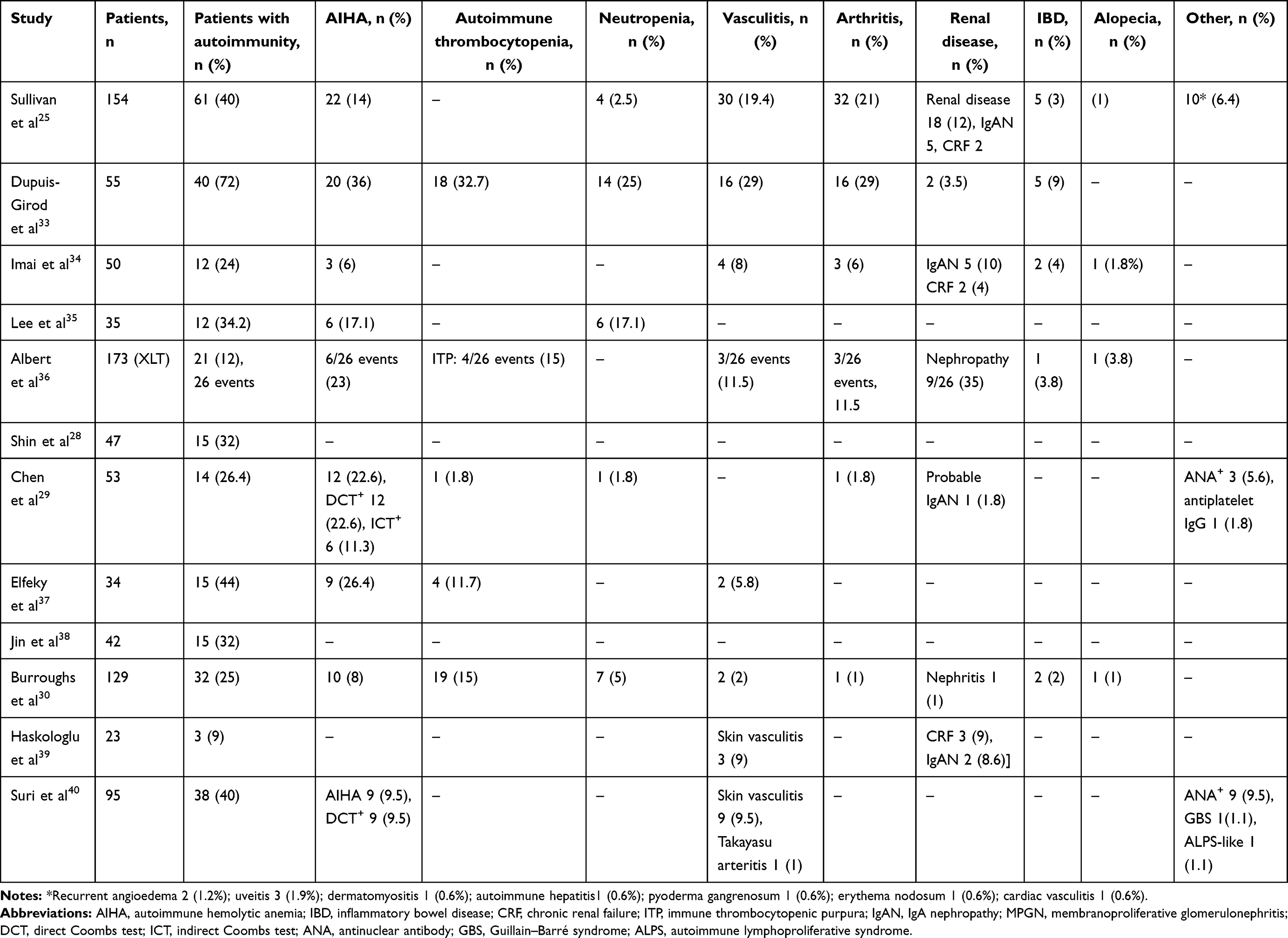 |
Table 1 Frequency of autoimmune manifestations reported in large cohorts of XLT/WAS |
Proposed Mechanisms of Autoimmunity
WASp is involved in cytoskeleton remodeling, and absence of or residual WASp-expression defects cause- functional defects in all immune-system cells. Formation of immunological synapses in T cells and T-cell receptor (TCR)-dependent activation is impaired in WAS.41–43 Reduced cytotoxic activity of T cells, natural killer cells, and naturally occurring regulatory T (nTreg) cells contributes to poor pathogen clearance.41–45 Motility, adhesion, and migration of B cells is also defective.45 However, underlying mechanisms for occurrence of autoimmune manifestations are still not completely understood. Some hypotheses currently proposed for development of autoimmunity in WAS are summarized in the following sections.
Role of T Cells in Autoimmunity in WAS
Autoimmunity is caused by failure in self-tolerance mechanisms. Treg cells play a pivotal role in immunotolerance and prevent autoimmunity. They prevent autoimmunity by maintaining tolerance to self-antigens and suppression of excessive immunoresponse. Development and function of Treg cells requires effective TCR signaling with involvement of the CD28 costimulator, expression of master regulator FOXP3, and growth maintenance by IL2.46 Although peripheral blood Treg-cell numbers have been found to be comparable in patients with WAS and healthy controls, WASp-deficient Treg cells demonstrated impaired ability to suppress proliferation of activated T-effector cells.47,48 Distribution and phenotype of nTreg cells have also been found to be normal in the thymi and spleens of WASp-deficient mice.49 nTreg cells, however, have been found to be reduced in inflamed peripheral tissue and lymph nodes. Reduction in nTreg cells correlates with lack of tissue-homing markers like integrin a4β7, chemokine receptor CCR4, and P- and E-selectin ligands.44
A mouse model of autoimmunity has failed to control aberrant T-cell activation by WASp-deficient nTreg cells.45 WASp-deficient nTreg cells fail to suppress B-cell activation and proliferation. Defective granzyme-mediated B-cell killing by nTreg cells has been demonstrated in some studies.45,47
Role of B Cells in Autoimmunity in WAS
The role of B cells and autoantibodies in the pathogenesis of autoimmune diseases like lupus is well recognized. B-cell dysfunction in patients with WAS is evident from the variable distribution of serum immunoglobulins and demonstration of autoantibodies. Classically, WAS is associated with low serum IgM, normal IgG, and elevated IgE and IgA levels. Patients have impaired response to polysaccharides and other T cell–independent antigens.3,50
Elevated IgM levels correlate with development of autoimmunity. Around 90% of patients with elevated IgM levels have been found to have developed AIHA in comparison to none with low IgM.33
WASp deficiency affects adhesion, motility, and homing of B cells.45,51 Defective B-cell function results in insufficient pathogen clearance, chronic immunoactivation, and failure of peripheral B-cell tolerance. In addition, reduced surface expression of complement receptors CD21 (CR2) and CD35 (CR1) results in impaired opsonization and negative selection of self-reactive B cells, thereby breaking peripheral tolerance and helping in autoantibody production.52
Murine WASp-deficient B cells demonstrate increased proliferation with autoantibody production and differentiation into plasmablasts.53 Enhanced proliferation of transitional B cells in response to stimulation by antigen or MYD88 has been seen in both humans and mice.54–56
Breg cells influence the balance and recruitment of Treg cells and TH17 cells during inflammation. Recent studies have suggested that WASp is required for normal Breg-cell numbers and functions.57 Murine studies have also revealed reduced levels of IL10 secreting Breg cells (B10) in patients with WAS.44
Bouma et al found reduced numbers of IL10-producing Breg cells, reduced Treg cells, and increased TH17 cells in arthritic WAS-knockout mice. Adoptive transfer of wild-type Breg cells ameliorated arthritis and restored the balance between Treg and TH17 cells.57
Role of Invariant NKT Cells in Autoimmunity in WAS
Invariant NKT cells possess properties of both T and NK cells. They prevent autoimmunity by limiting development of TH17 cells, anti-DNA antibody production, and autoreactive B-cell production.58,59 Reduced numbers, defective invariant NKT cells, and increased IFNγ production have been reported in WASp-deficient mice.60,61
Role of IFN1 in Autoimmunity in WAS
Plasmacytoid dendritic cells (pDCs) are specific subsets that produce IFN1 in response to foreign nucleic acids. Viral infections and certain self-nucleic acids serve as stimulants of TLR7 and TLR9 and keep them persistently activated. Susceptibility to viral infections, impaired clearance, and exposure of self-antigens following cell death activate the IFN1 pathway.62 This pathogenic mechanism is associated with several autoimmune diseases, such as lupus, Sjögren’s syndrome, and psoriasis.62
WASp deficiency leads to exaggerated activation of TLR9 by its ligand and subsequent increased IFN1 signature.63 WASp-deficient mice show persistent activation of pDCs and elevated IFN1 levels. Ablation of IFN1 in WASp-deficient mice results in blockade of chronic activation of pDCs. This is accompanied by remission of colitis and reduction in spleen size.63
Defects in Apoptosis Pathway and Development of Autoimmunity in WAS
Restimulation-induced cell death is a process that augments Fas-mediated apoptosis of activated T cells. It helps eliminate T cells that are activated against chronically expressed antigens like autoantigens. Nikolov et al demonstrated defective FasL in WASp-deficient mice leading to defective elimination of activated T cells and predisposition to autoimmunity.64 Defective Fas has also been hypothesized to be a pathophysiological mechanism for lymphoproliferative lupus–like syndrome in Fas/FasL-negative mice.65,66
Role of Inflammasomes in Autoimmunity in WAS
Excessive inflammasome activation has been shown in human monocytes and mouse DCs.67 Some autoimmune manifestations (especially rash and arthralgias) are related to excessive NLRP3-inflammasome activation.
Correlation of Genetic Variants with Autoimmunity
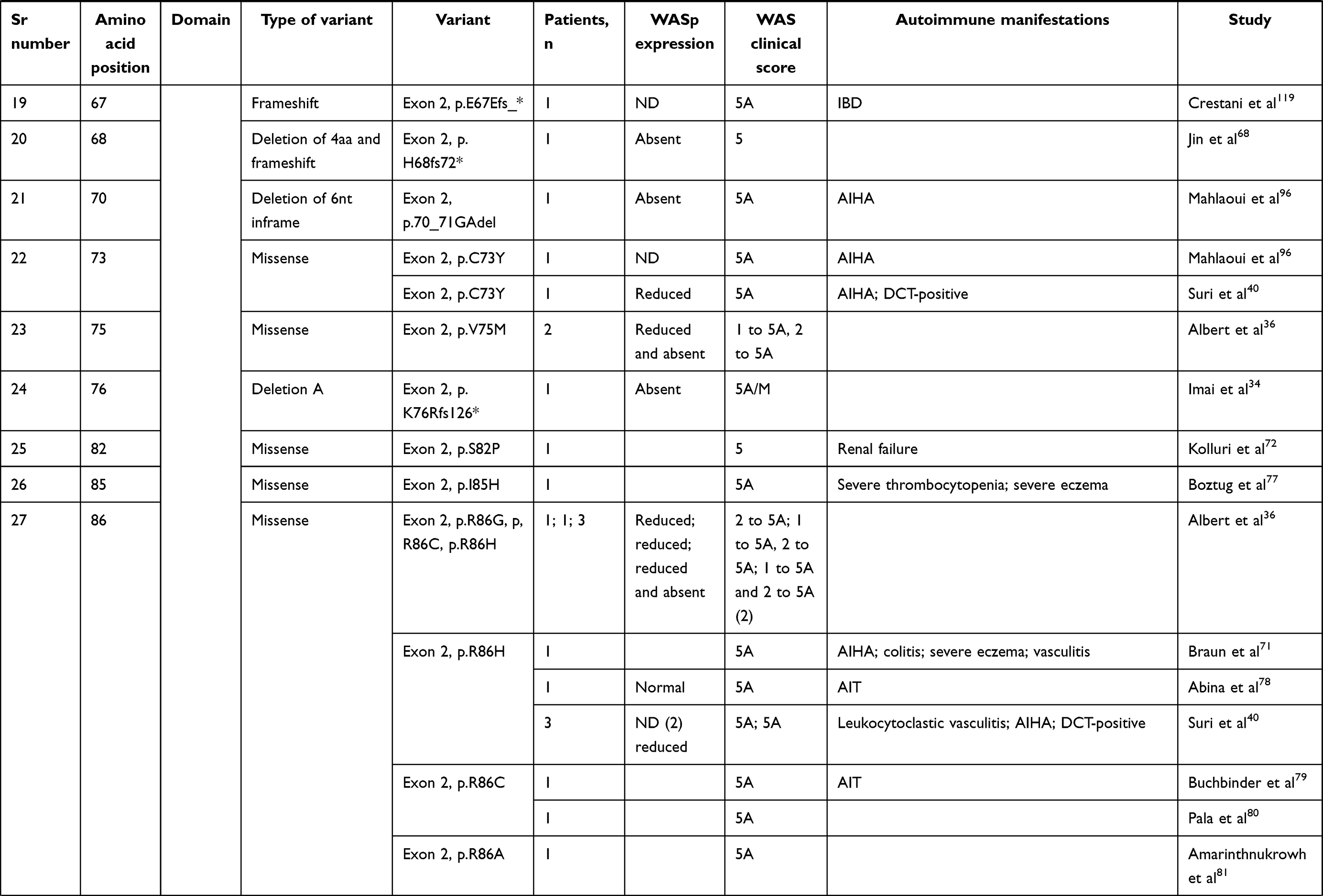
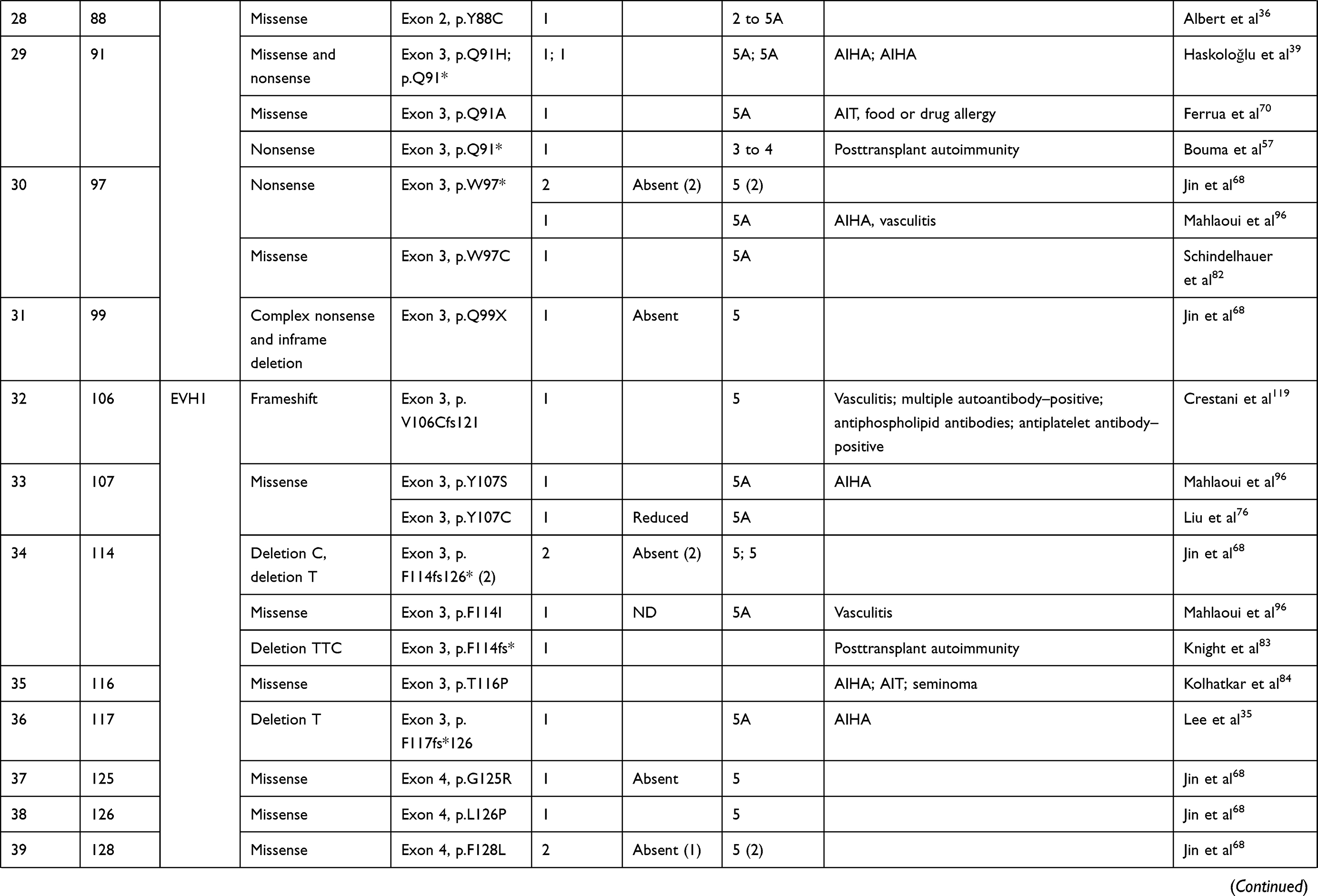
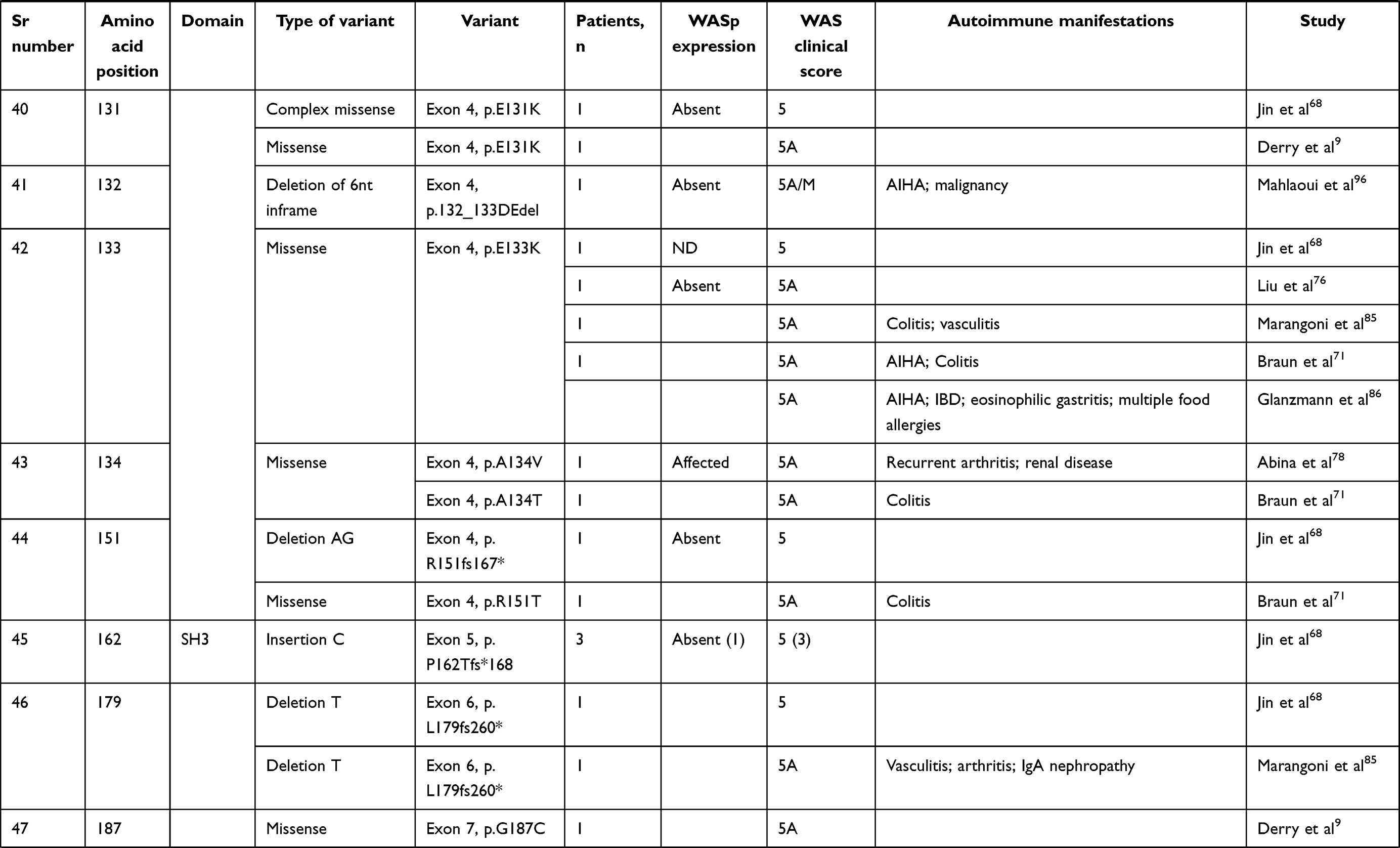
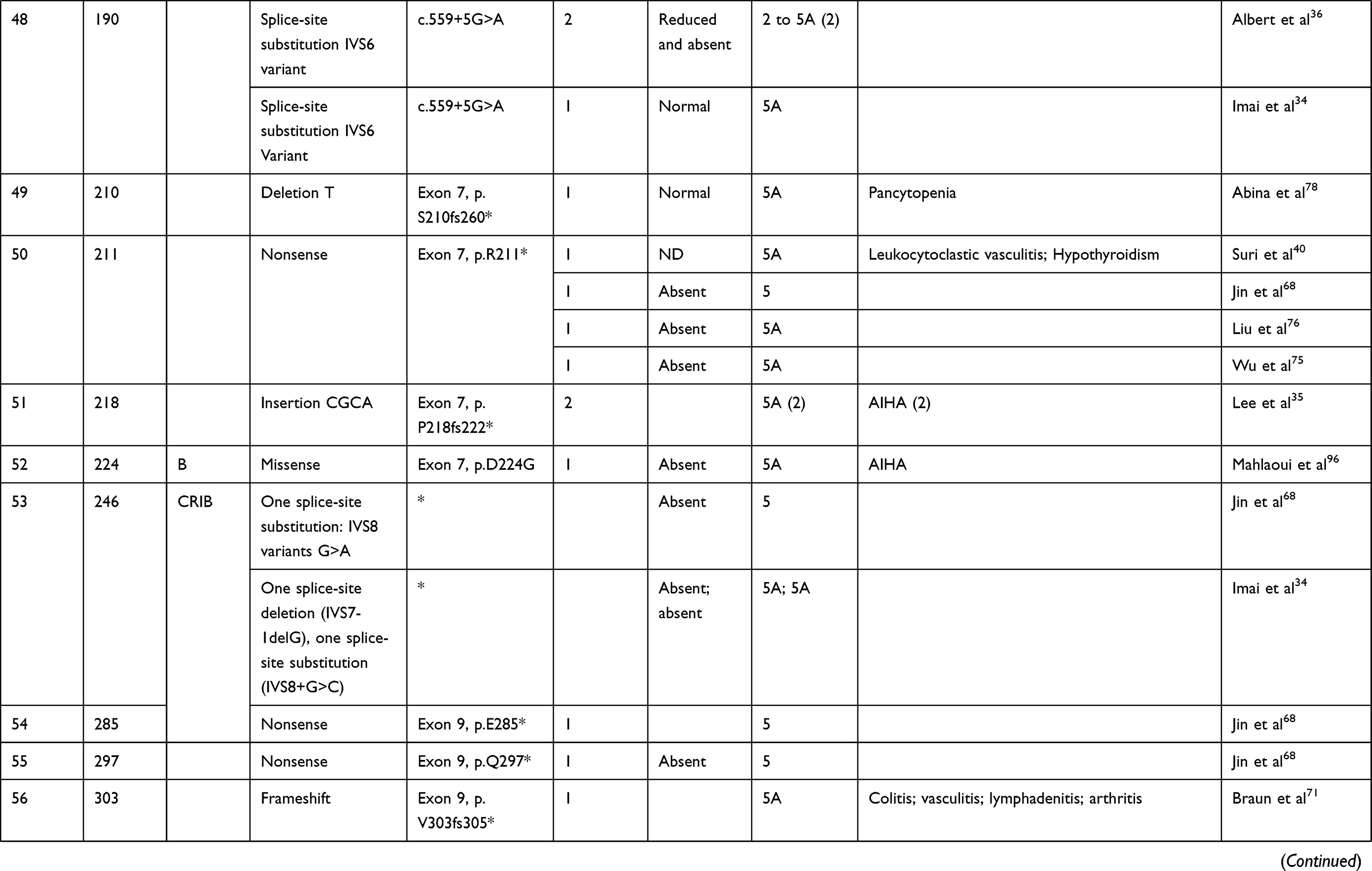
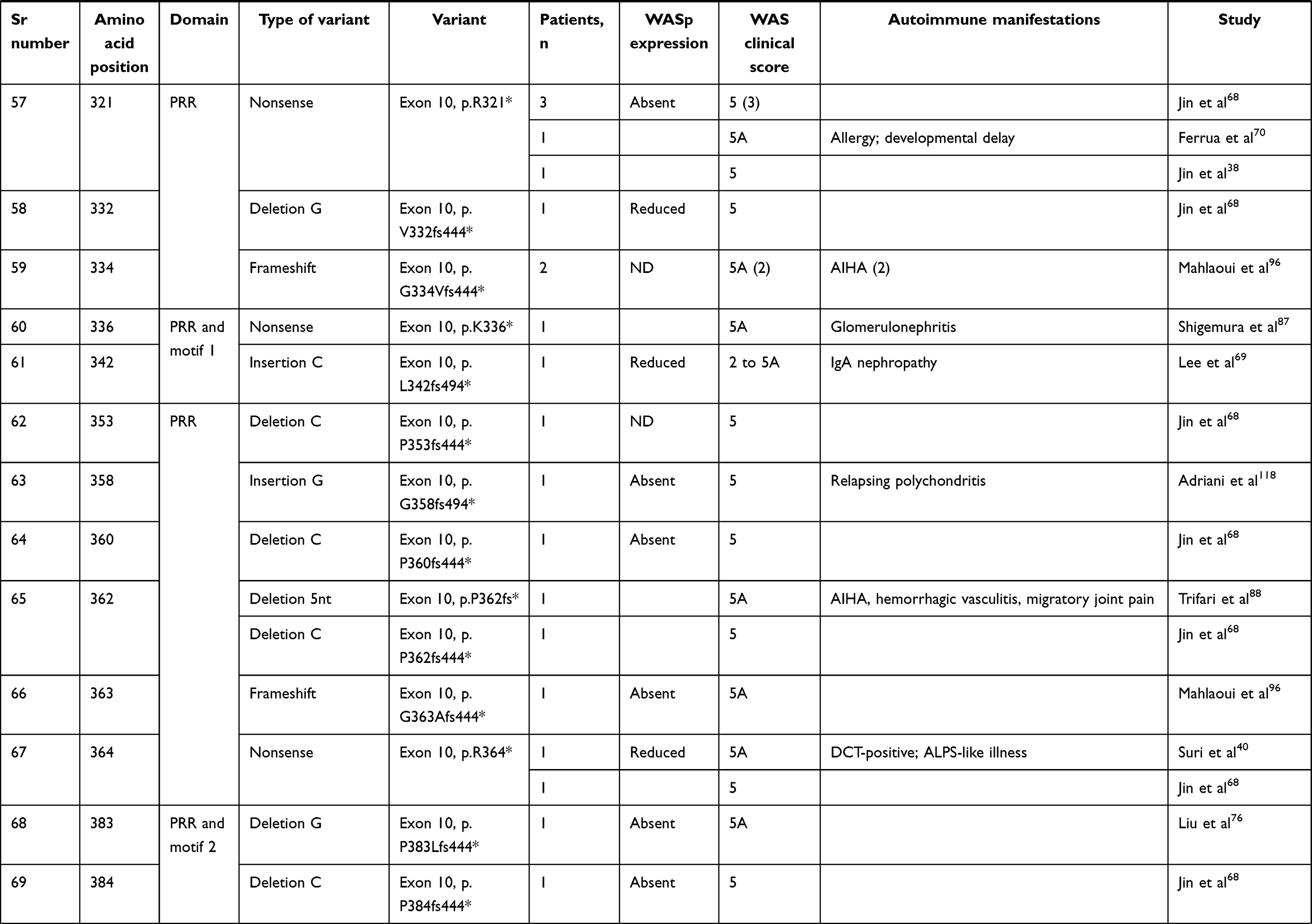
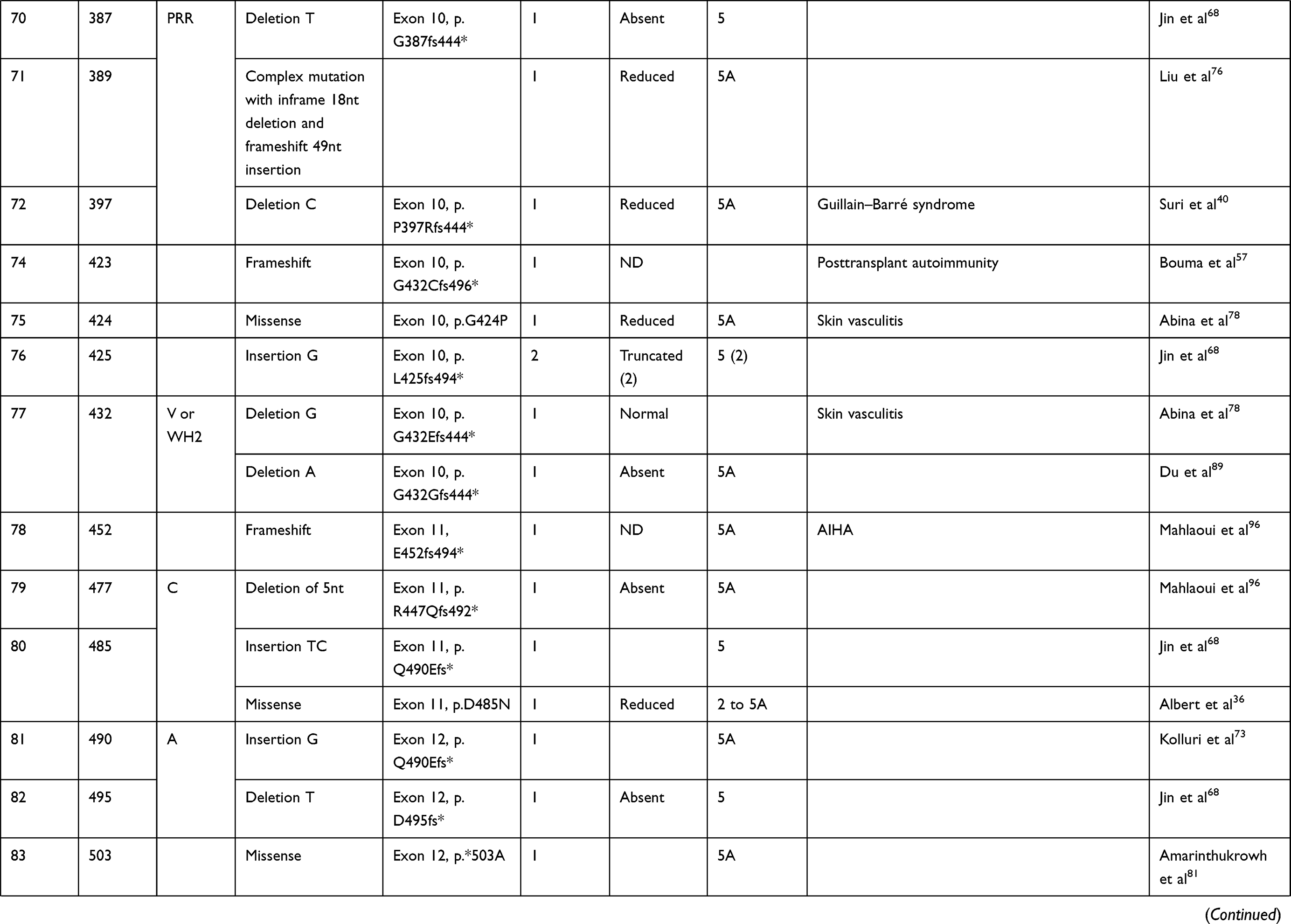
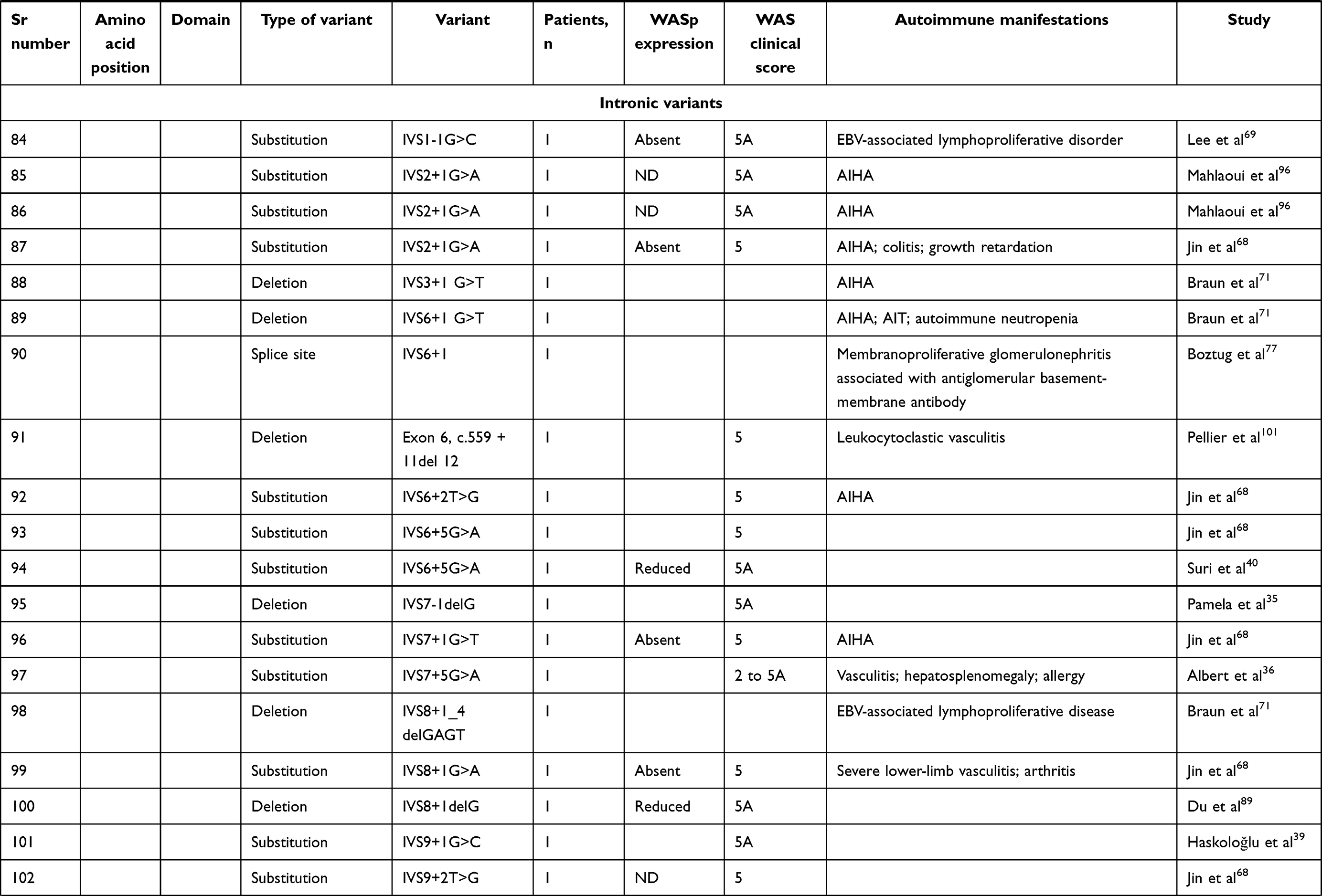
We compiled details of genetic variants reported in the literature in patients with WAS and autoimmune manifestations. In sum, 166 variants were associated with autoimmunity in 197 WAS patients (Table 2, Figure 2). These included 143 exonic (136 well-defined) and 23 intronic variants. Exonic variants were located at 83 amino acid positions. Most variants were found in the PH and EVH1 domains (n=84,50.6%) followed by the PRR domain (n=20, 12.2%), VCA domain (n=9, 5.5%), and B and CRIB domains (n=4, 2.4%). The variants described included missense (56), deletions (50, 41 exonic and nine intronic), nonsense (27), splicesite (25), insertions (7), unknown frameshift (five), and complex (four).
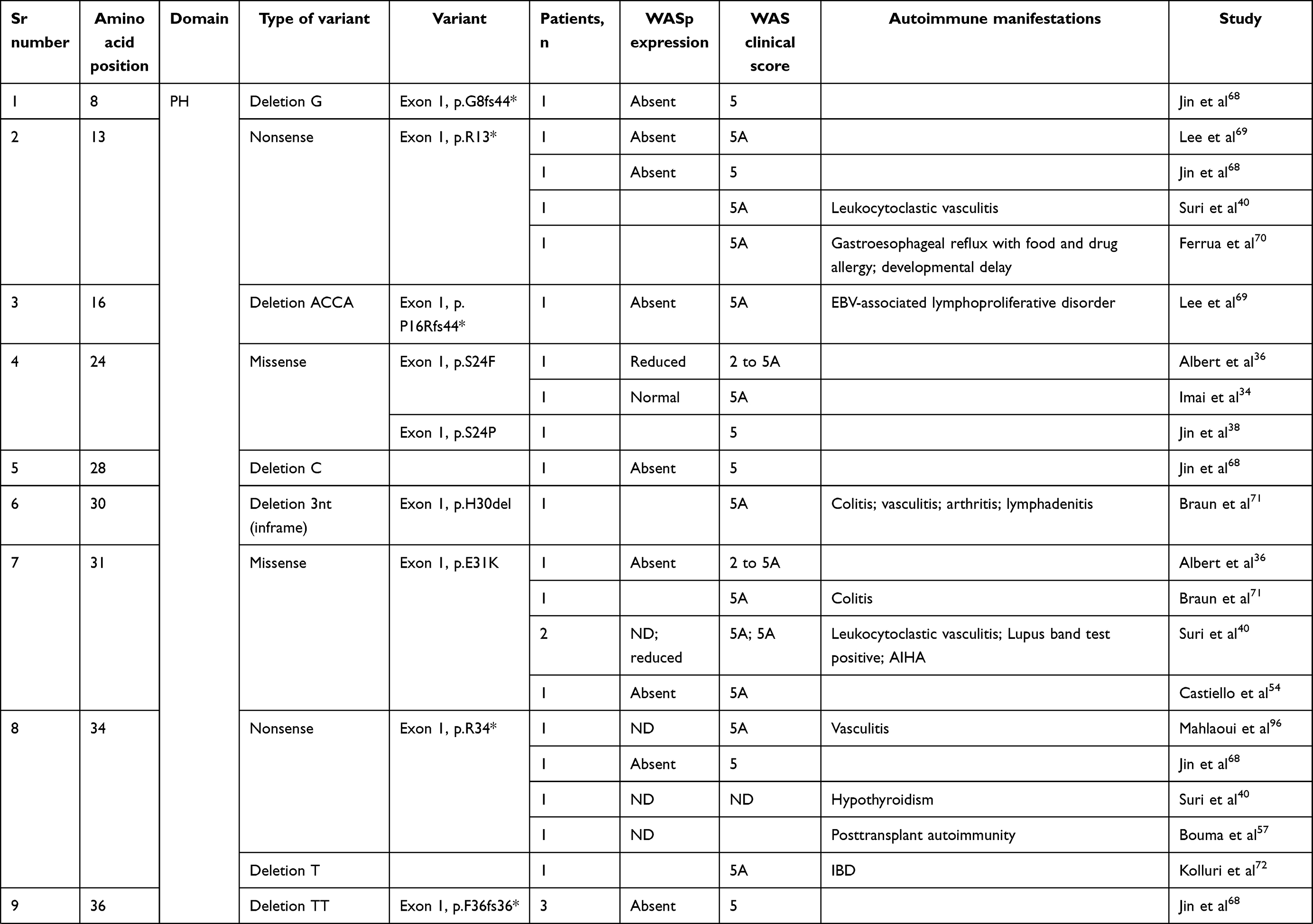 |
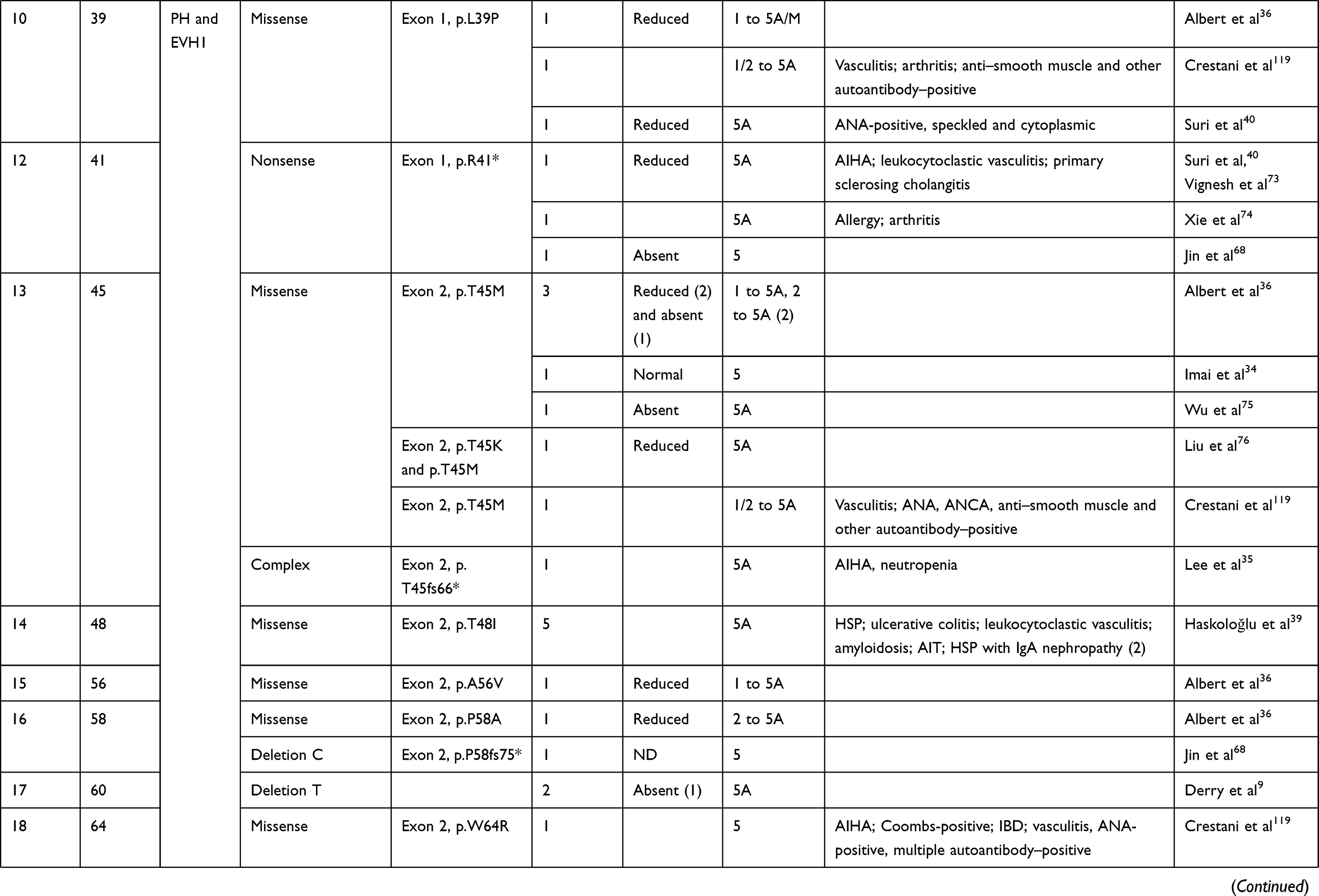 |
 |
 |
 |
 |
 |
 |
 |
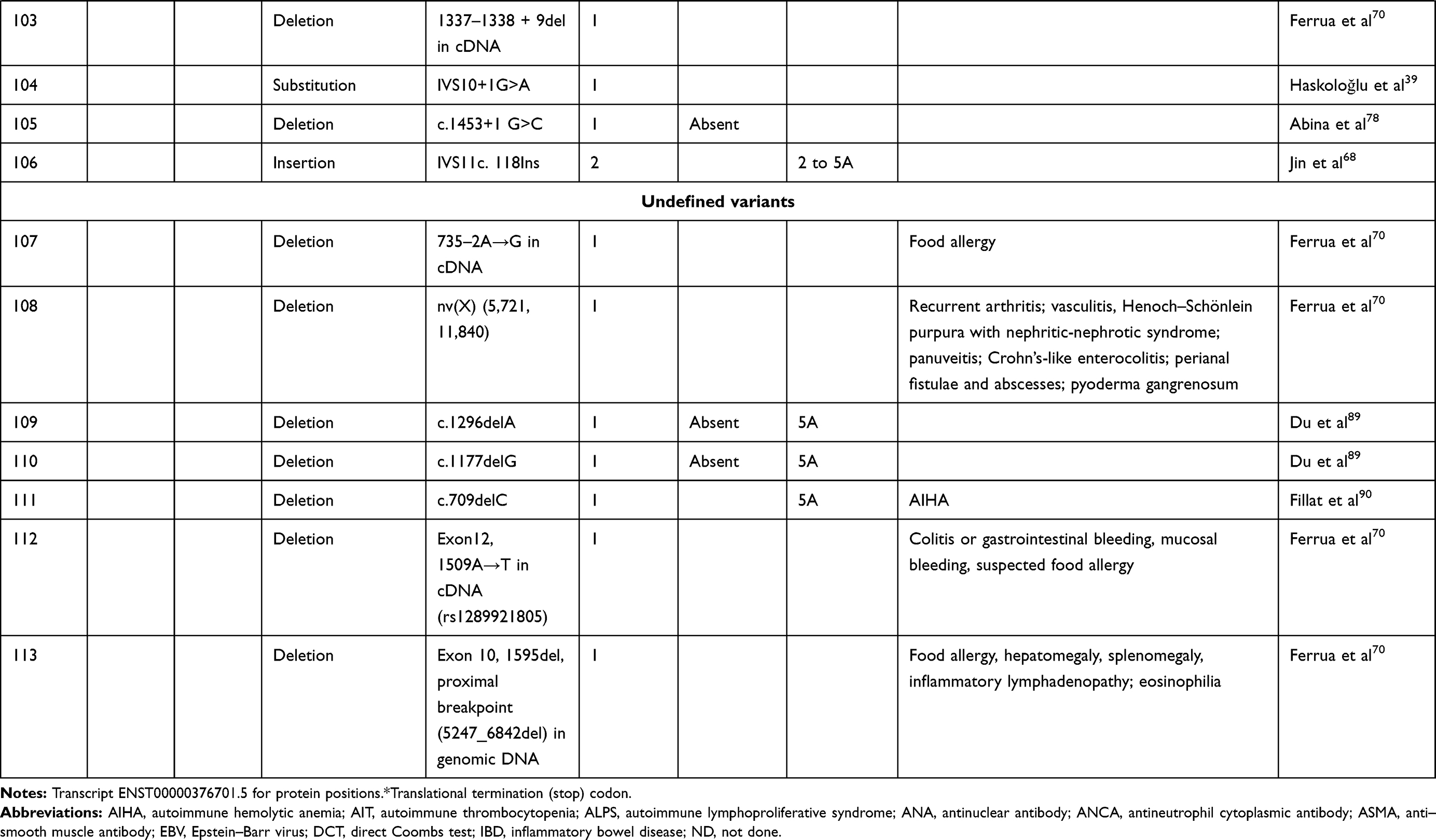 |
Table 2 Genetic variants associated with autoimmunity in patients of XLT/WAS |
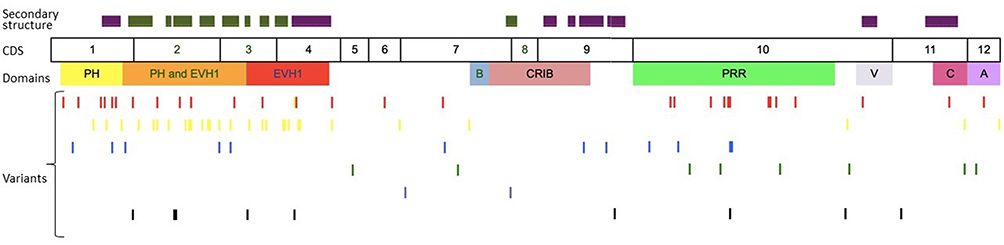 |
Figure 2 Variants reported in patients of XLT/WAS with autoimmunity. Secondary structure of WASp: α-helix (purple) and β-strands (green)., CDS, coding sequence; PH, pleckstrin homology; EVH1, vasodilator-stimulated phosphoprotein homology; B, basic; CRIB, Cdc42-and Rac-interactive binding; PRR, proline-rich region; V, verprolin; C, central; A, acidic; . Reported variants: deletions (red), missense (yellow), nonsense (blue), insertions (green), splice site (pink), complex and undefined frameshift variants (black). |
Deletions are spread across the gene, while most missense variants are seen in the PH and EVH1 domains. Missense variants (p.T45M and p.T45K) at position 45 in PH and EVH1 have been found to be associated with autoimmunity. Normal/absent WASp expression was found such patients.34,36,75,76 Twelve patients with missense variants at position 86 (R86G, R86C, R86H and R86A) developed autoimmunity. Both p.E31K and p.E133K were reported in five patients each that developed autoimmunity.
Patients with XLT can progress to autoimmune WAS. The XLT phenotype with missense variants in PH and EVH1 is most associated with development of autoimmune WAS. Albert et al identified 13 positions associated with XLT to autoimmune WAS progressions, 9 of which lie in PH and EVH1 domain.
Patients with XLT can also develop autoimmunity. Albert et al identified 13 amino acid positions that were prone to progress to autoimmune WAS.36
We identified 18 variants in 25 patients reported to demonstrate progression of XLT to autoimmune WAS. Of these, 13 (all missense) were located in PH and EVH1.
Clinical Manifestations
Autoimmune Hemolytic Anemia (AIHA)
AIHA is the commonest autoimmune manifestation seen in patients with WAS, accounting for 30%–85% of all autoimmune manifestations. The reported frequency of AIHA is 8%–36%.25,29,3033–36,40 The mean age of development of AIHA was 13.7–17.5 (1–58) months in two studies (Table 3).29,33 All patients had AIHA onset aged <5 years.33 The commonest clinical presentations of AIHA include anemia, jaundice, and hepatosplenomegaly.29,40 Positive antinuclear antibodies have been demonstrated in 5%–9% of patients with AIHA, while Coombs tests were positive in some patients.29,40
 |
 |
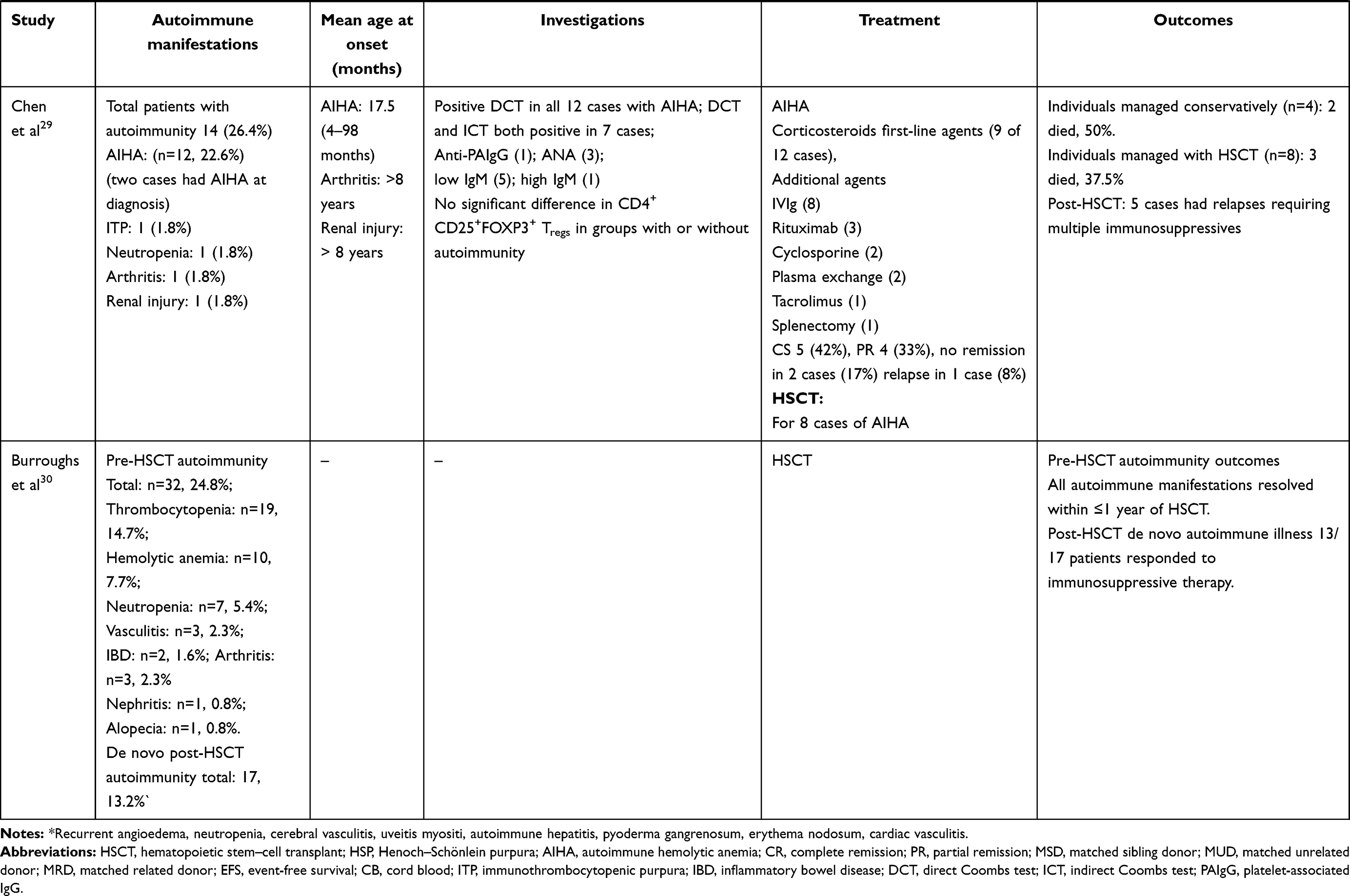 |
Table 3 Clinical profile, treatment, and outcomes of patients with autoimmunity in XLT/WAS |
Autoimmune Thrombocytopenia (AIT)
Microthrombocytopenia is a cardinal feature of WAS. Abnormality in WASp results in megakaryocyte dysfunction, leading to formation of small platelets. These abnormal microplatelets are recognized by self-antibodies and are prematurely cleared by the spleen.91–93 Immunomediated thrombocytopenia also plays a contributing role in 15%–32% of patients (Tables 1 and 2).29,30,33,36 Antiplatelet antibodies have been found in 40% of WASp-deficient mice.94 Assays for antiplatelet antibodies are difficult to standardize and may not be easily available, especially in developing countries. Moreover, these antibodies may have a poor correlation with AIT.95 AIT may evolve over and exacerbate the underlying thrombocytopenia that is characteristic of WAS. A sudden drop in baseline platelet counts (usually <10×109/L) with or without overt clinical bleeding is an important clinical clue to emergence of AIT. Failure to demonstrate a significant rise or fall in platelet count after platelet transfusion may herald the development of AIT.95 Most autoimmune manifestations evolve over time, but immunothrombocytopenia may have an early age of onset. In a cohort of patients with WAS aged <2 years with a clinical severity score of 5, ten of 26 (38.4%) had antiplatelet antibody–positive severe refractory thrombocytopenia.96 Intracranial hemorrhage is a major cause of mortality in patients with WAS.25
Differentiating AIT from baseline thrombocytopenia in WAS is crucial, as management strategies vary. Institution of appropriate immunosuppressive therapy is needed to maintain platelet counts.
Autoimmune Neutropenia
Autoimmune neutropenia is found in 2%–25% of patients with WAS.25,29,30,33,35
Vasculitides
Vasculitis is the second-commonest autoimmune manifestation, and has been found in 1.5%–29% of patients with WAS. It accounts for 6%–45% of all autoimmune manifestations.25,30,33,34,36,39,40 (Tables 1 and 2)
Two patterns of vasculitic abnormality have been reported in WAS: medium-sized and small-vessel vasculitis of skin, renal, coronary, cerebral, or hepatic arteries, and large-vessel vasculitis involving the aorta and its major branches.40,5097–101 Involvement of small vessels, especially those of the skin,25,33,39,102,103 is the commonest vasculitic abnormality (75%).33 IgA vasculitis (previously termed Henoch–Schönlein purpura) has been reported in 28.5% of patients with vasculitis.25 Kawakami et al described Kawasaki disease in a patient with WAS.104 Involvement of small- and medium-sized arteries of the gastrointestinal tract,97,105,106 heart,104,105 liver,98,104 gallbladder,105 kidneys,98,106 stomach,102 and cerebral blood vessels,98,107 has also been reported. In a single-center study of 55 patients of WAS, Dupuis-Girod et al noted cutaneous vasculitis at a mean age of 52.5 (11–184) months.33 Lao et al reported large-vessel vasculitis involving the aorta and renal artery in a 5-year-old boy with WAS.100 Pellier et al described five children with WAS who developed aortic aneurysms predominantly involving the thoracic and abdominal aortae.101 Four of these five patients with vasculitis were asymptomatic, and aneurysms were discovered only on screening.
Predisposition to developing vasculitis has been ascribed to immunodysregulation in WAS. Patients with WAS typically have depressed levels of IgM and elevated levels of IgA and IgE. It has been suggested that immunodeposition within the vessel wall can lead to necrotizing vasculitis.98 Alternatively, vasculitis could result from an infectious insult due to the underlying immunodeficiency. Pellier et al showed evidence of varicellazoster virus, Epstein–Barr virus, and human herpesvirus 6 in the aortic vessel wall on histopathology in a patient with WAS and aortic aneurysm.101
Arthritis
Arthritis has been reported in 1%–29% of patients with WAS, and accounts for 3%–52% of all autoimmune manifestations (Table 1).25,29,30,33,35,36 Sullivan et al observed arthritis in 32 of 152 patients (21%). Of these, 17 had transient arthritis and 15 persistent arthritis.25 Median age at presentation of arthritis was 45.3 (13–180) months.33
Renal Disease
Onset of renal disease in XLT/WAS occurs at a relatively later age: 7–20 years.36,37 Clinical manifestations include transient proteinuria,33 hematuria, azotemia, and nephritic–nephrotic syndrome.25,29,3335–37,100 Reported histopathological patterns include IgA nephropathy, membranoproliferative glomerulonephritis, mesangial proliferation, and interstitial nephritis.108–111 IgA nephropathy is the commonest renal disease, described in 27%–41% of patients in various long-term cohorts.25,35,36 Prevalence appears to be higher in patients with residual WASp expression.35,36 Screening for renal involvement has been recommended in all patients with XLT/WAS.
Aberrant glycosylation of IgA is attributed as a cause for renal disease associated with WAS. Elevated levels of β1,6-N-acetyl-glucosaminyl transferases have been documented in patients with WAS. This leads to aberrant O-glycosylation of sialophorin in patients with WAS.101,112
Inflammatory Bowel Disease (IBD)
Bleeding from the gastrointestinal tract is a common symptom in WAS and often secondary to thrombocytopenia. In our cohort, 49.4% of children presented with blood-stained stools.40 Autoimmune colitis has also been reported in 1%–9% of patients.25,29,30,35,36 Both ulcerative colitis and Crohn’s disease have been observed. WAS-associated colitis is challenging to treat and often refractory to immunosuppressants.113 In our experience, IBD can be a presenting feature of WAS. Ohya et al reported that 16.6% of patients with IBD had a WAS mutation.113 Similarly, Cannioto et al showed that 25% of patients aged <2 years with IBD were later shown to have WAS.114
WASp-deficient mice have been observed to have chronic colitis with mucosal thickening and lymphocytic and neutrophilic infiltrates in the lamina propria.115 Suggested pathogenic mechanisms for IBD in WAS include WASp deficiency-mediated dysfunction of Treg cells and anti-inflammatory macrophages, increased self-reactive B cells, and altered gut microbiota.113,116
Other Rheumatic Manifestations
Monteferrante et al described lupus nephritis in a patient with WAS.117 Similarly, other connective-tissue disorders like dermatomyositis,27 uveitis,27 autoimmune hepatitis,27 primary sclerosing cholangitis,40 amyloidosis,39 and relapsing polychondritis118 have been reported in the context of WAS.27 Crestani et al reported positive antineutrophil cytoplasmic antibodies, and positive antiphospholipid antibodies in patients with WAS.119
Autoimmune Skin Diseases
Eczematous dermatitis is a cardinal clinical manifestation of WAS. Eosinophilia and elevated levels of serum IgE are associated findings. DC dysfunction and skewed TH2 immunity may play a role in the development of eczema in this condition.120 Apart from atopy, other autoimmune skin manifestations have been reported in WAS. These include recurrent angioedema, pyoderma gangrenosum, and erythema nodosum.25 Alopecia has been reported in 1%–3% of patients with WAS.25,30,34,36
Posttransplant Autoimmune Manifestations
Development of autoimmunity is considered a predictor of severe disease, and often warrants the need for early HSCT in patients with XLT/WAS. HSCT is curative and is associated with 5-year survival of 91%.30 However, occurrence of autoimmunity in the posttransplant period has been observed in 13%–20% of patients (Table 4).26–30 Autoimmune cytopenia is the commonest autoimmune manifestation seen after HSCT.30 Burroughs et al reported that 75% of patients with autoimmune manifestations responded to immunosuppressive therapy and attained remission within a year of transplant.30 Mixed or split donor chimerism is an important predictor of development of post-HSCT autoimmunity.26,27,30 Autoimmunity is most frequently encountered in patients who undergo matched unrelated donor transplants. However, it is also seen in matched related-donor and matched sibling-donor transplants.26,30 Presence of pretransplant autoimmune manifestations does not increase the risk of development of post-HSCT autoimmunity.30
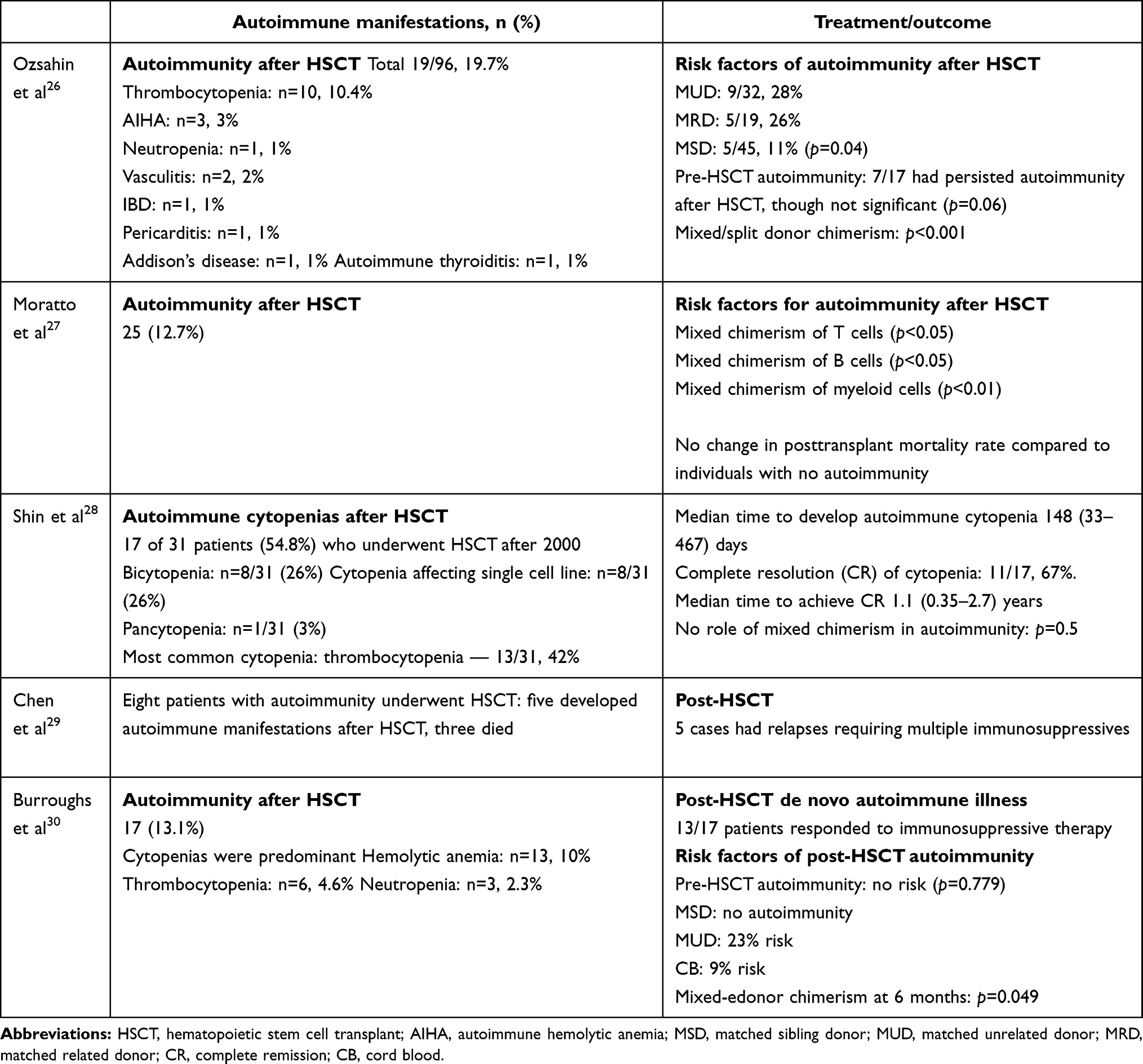 |
Table 4 Studies reporting posttransplant autoimmunity in XLT/WAS |
Is Autoimmunity in WAS a Poor Prognostic Marker?
Age at Onset of Autoimmunity
Mahalaoui et al evaluated a subgroup of 26 patients that had onset of autoimmunity before the age of 2 years in their cohort of 160 patients with WAS.96 The authors concluded that development of early autoimmunity predicted a severe refractory disease course and required early HSCT for survival.
Autoimmunity and Risk of Malignancy
Presence of autoimmunity also increases the risk for development of malignancy in patients with WAS. Sullivan et al observed that 25% of patients with a history of autoimmune disease developed malignancy compared to 5% of patients without autoimmunity.25 Sallah et al demonstrated that 18% of patients with AIHA developed lymphoreticular malignancy.121
Treatment of Autoimmunity
Currently, HSCT is the best curative therapy available for WAS. Early studies on HSCT in WAS reported effective reconstitution of lymphoid cells, but impaired platelet engraftment.122–124 Long-term overall survival in recipients of unrelated bone-marrow grafts is 70%–78%.26,125 However, recent studies have reported event-free survival with HLA-identical sibling bone-marrow grafts to be 88%, with overall survival of 90%–95%.27,30,126. Results of unrelated- and alternative-donor HSCT have also greatly improved to >90%.30,37 HSCT with TCRαβ/CD19 depletion or posttransplant cyclophosphamide therapy for haploidentical donors has shown promising results and new opportunities for successful curative therapy in WAS/XLT.127
Gene therapy is an evolving and promising alternative approach when a transplant is not feasible due to unavailability of matched donors. Gene therapy was attempted with a gibbon ape leukemia virus γ-retroviral vector in 2006.77,127 Though successful engraftment was reported, it was limited by development of leukemia due to insertional leukemogenesis. Subsequently, therapy with a lentiviral vector was attempted in 31 patients with WAS. This resulted in successful engraftment, discontinuation of intravenous immunoglobulin (IVIg), improvement in platelet counts, and reduction in infections. Resolution of autoimmune manifestations was also demonstrated with gene therapy; however, the rate of de novo autoimmunity posttransplant was comparable with HSCT.127
Chemoprophylaxis with antimicrobials and monthly VIg are often used for prevention of infections. Management is usually individualized based on disease severity. Treatment of autoimmune manifestations is challenging, and may require require immunosuppressive therapies. These agents can further increase the risk of infections. Corticosteroids remain the first-line therapy for all autoimmune manifestations. Corticosteroids can induce variable rates of remission in patients with AIHA (Table 2). Additional agents are needed in patients who do not respond or attain partial remission. Cyclophosphamide, azathioprine, IVIg, rituximab, cyclosporine, plasma exchange, and tacrolimus have been used with variable results.
High- dose IVIg and oral or parenteral corticosteroids are used as first-line agents in patients with AIT who have significant bleeds. Rituximab is used for refractory cases. Splenectomy significantly increases and often normalizes platelet counts in refractory thrombocytopenia.77,128,129 However, splenectomy is associated with increased risk of sepsis and necessitates lifelong antimicrobial prophylaxis.129 Moreover, thrombocytopenia may recur after splenectomy in patients with WAS.33 Splenectomy is reserved for very severe cases with no prospects from other curative interventions. Severe AIT after splenectomy is usually treated with IVIg, high-dose steroids, azathioprine, and cyclophosphamide. A majority of patients with skin vasculitis, arthritis, IBD, and renal disease associated with WAS respond to standard immunosuppressive regimens containing steroids and cyclosporine.33
Conclusion
Autoimmune manifestations are well-recognized complications in WAS. Varied clinical manifestations have been associated with the syndrome. Autoimmune cytopenia is the commonest. Development of autoimmunity is a poor prognostic marker and a predictor of development of malignancy. Pathogenic mechanisms for autoimmunity are not clearly defined. Corticosteroids with or without additional immunosuppressive agents are needed for treatment of autoimmune manifestations. HSCT is curative, but there is a risk of development of posttransplant autoimmunity.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wiskott A. Familiarer, angeborener morbus werlhofii? Monatsschr Kinderheilkd. 1937;68:212–216.
2. Aldrich RA, Steinberg AG, Campbell DC. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics. 1954;13:133–139.
3. Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117(4):725–738. doi:10.1016/j.jaci.2006.02.005
4. Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kapoor N. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. 2009;15(1):84–90. doi:10.1016/j.bbmt.2008.10.007
5. Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrich syndrome. Curr Opin Hematol. 2008;15(1):30–36. doi:10.1097/MOH.0b013e3282f30448
6. Perry GS
7. Fasth A. Primary immunodeficiency disorders in Sweden: cases among children, 1974–1979. J Clin Immunol. 1982;2(2):86–92. doi:10.1007/BF00916891
8. Ryser O, Morell A, Hitzig WH. Primary immunodeficiencies in Switzerland: first report of the national registry in adults and children. J Clin Immunol. 1988;8(6):479–485. doi:10.1007/BF00916954
9. Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78(4):635–644. doi:10.1016/0092-8674(94)90528-2
10. Kwan SP, Lehner T, Hagemann T, et al. Localization of the gene for the Wiskott-Aldrich syndrome between two flanking markers, TIMP and DXS255, on Xp11.22-Xp11.3. Genomics. 1991;10(1):29–33. doi:10.1016/0888-7543(91)90480-3
11. Rivero-Lezcano OM, Marcilla A, Sameshima JH, Robbins KC. Wiskott-Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol Cell Biol. 1995;15(10):5725–5731. doi:10.1128/mcb.15.10.5725
12. Symons M, Derry JM, Karlak B, et al. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. 1996;84(5):723–734. doi:10.1016/s0092-8674(00)81050-8
13. Miki H, Nonoyama S, Zhu Q, Aruffo A, Ochs HD, Takenawa T. Tyrosine kinase signaling regulates Wiskott-Aldrich syndrome protein function, which is essential for megakaryocyte differentiation. Cell Growth Differ. 1997;8:195–202.
14. Miki H, Takenawa T. Direct binding of the verprolin-homology domain in N-WASP to actin is essential for cytoskeletal reorganization. Biochem Biophys Res Commun. 1998;243(1):73–78. doi:10.1006/bbrc.1997.8064
15. Parolini O, Berardelli S, Riedl E, et al. Expression of Wiskott-Aldrich syndrome protein (WASP) gene during hematopoietic differentiation. Blood. 1997;1(90):70–75. doi:10.1182/blood.V90.1.70
16. Cory GO, MacCarthy-Morrogh L, Banin S, et al. Evidence that the Wiskott-Aldrich syndrome protein may be involved in lymphoid cell signaling pathways. J Immunol. 1996;157:3791–3795.
17. Gallego MD, Santamaría M, Peña J, Molina IJ. Defective actin reorganization and polymerization of Wiskott-Aldrich T cells in response to CD3-mediated stimulation. Blood. 1997;90(8):3089–3097. doi:10.1182/blood.V90.8.3089
18. Krawczyk C, Bachmaier K, Sasaki T, et al. Cbl-b is a negative regulator of receptor clustering and raft aggregation in T cells. Immunity. 2000;13(4):463–473. doi:10.1016/s1074-7613(00)00046-7
19. Sasahara Y, Rachid R, Byrne MJ, et al. Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation. Mol Cell. 2002;10(6):1269–1281. doi:10.1016/s1097-2765(02)00728-1
20. Dupré L, Aiuti A, Trifari S, et al. Wiskott-Aldrich syndrome protein regulates lipid raft dynamics during immunological synapse formation. Immunity. 2002;17(2):157–166. doi:10.1016/s1074-7613(02)00360-6
21. Linder S, Higgs H, Hüfner K, Schwarz K, Pannicke U, Aepfelbacher M. The polarization defect of Wiskott-Aldrich syndrome macrophages is linked to dislocalization of the Arp2/3 complex. J Immunol. 2000;165(1):221–225. doi:10.4049/jimmunol.165.1.221
22. Jones GE, Zicha D, Dunn GA, Blundell M, Thrasher A. Restoration of podosomes and chemotaxis in Wiskott-Aldrich syndrome macrophages following induced expression of WASp. Int J Biochem Cell Biol. 2002;34(7):806–815. doi:10.1016/s1357-2725(01)00162-5
23. Devriendt K, Kim AS, Mathijs G, et al. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27(3):313–317. doi:10.1038/85886
24. Ancliff PJ, Blundell MP, Cory GO, et al. Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood. 2006;108(7):2182–2189. doi:10.1182/blood-2006-01-010249
25. Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994;125(6):876–885. doi:10.1016/s0022-3476(05)82002-5
26. Ozsahin H, Le Deist F, Benkerrou M, et al. Bone marrow transplantation in 26 patients with Wiskott-Aldrich syndrome from a single center. J Pediatr. 1996;129(2):238–244. doi:10.1016/s0022-3476(96)70248-2
27. Moratto D, Giliani S, Bonfim C, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: an International Collaborative Study. Blood. 2011;118(6):1675–1684. doi:10.1182/blood-2010-11-319376
28. Shin CR, Kim MO, Li D, et al. Outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome. Bone Marrow Transplant. 2012;47(11):1428–1435. doi:10.1038/bmt.2012.31
29. Chen N, Zhang ZY, Liu DW, Liu W, Tang XM, Zhao XD. The clinical features of autoimmunity in 53 patients with Wiskott-Aldrich syndrome in China: a Single-Center Study. Eur J Pediatr. 2015;174(10):1311–1318. doi:10.1007/s00431-015-2527-3
30. Burroughs LM, Petrovic A, Brazauskas R, et al. Excellent outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome: a PIDTC report. Blood. 2020;135(23):2094–2105. doi:10.1182/blood.2019002939
31. Catucci M, Castiello MC, Pala F, Bosticardo M, Villa A. Autoimmunity in wiskott-Aldrich syndrome: an unsolved enigma. Front Immunol. 2012;3:209. doi:10.3389/fimmu.2012.00209
32. Gershwin ME, Blaese RM, Steinberg AD, Wistar R
33. Dupuis-Girod S, Medioni J, Haddad E, et al. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. 2003;111(5):e622–7. doi:10.1542/peds.111.5.e622
34. Imai K, Morio T, Zhu Y, et al. Clinical course of patients with WASP gene mutations. Blood. 2004;103(2):456–464. Epub 2003 Sep 11. PMID: 12969986. doi:10.1182/blood-2003-05-1480
35. Lee PP, Chen TX, Jiang L-P, et al. Clinical and molecular characteristics of 35 Chinese children with Wiskott-Aldrich syndrome. J Clin Immunol. 2009;29(4):490–500. doi:10.1007/s10875-009-9285-9
36. Albert MH, Bittner TC, Nonoyama S, et al. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood. 2010;115(16):3231–3238. doi:10.1182/blood-2009-09-239087
37. Elfeky RA, Furtado-Silva JM, Chiesa R, et al. One hundred percent survival after transplantation of 34 patients with Wiskott-Aldrich syndrome over 20 years. J Allergy Clin Immunol. 2018;142(5):1654–1656.e7. doi:10.1016/j.jaci.2018.06.042
38. Jin YY, Wu J, Chen TX, Chen J. When WAS gene diagnosis is needed: seeking clues through comparison between patients with wiskott-aldrich syndrome and idiopathic thrombocytopenic purpura? Front Immunol. 2019;10:1549. doi:10.3389/fimmu.2019.01549
39. Haskoloğlu Ş, Öztürk A, Öztürk G, et al. Clinical features and outcomes of 23 patients with Wiskott-Aldrich syndrome: a single-center experience. Turk J Haematol. 2020;37(4):271–281. doi:10.4274/tjh.galenos.2020.2020.0334
40. Suri D, Rikhi R, Jindal AK, et al. Wiskott Aldrich syndrome: a multi-institutional experience from India. Front Immunol. 2021;12:627651. doi:10.3389/fimmu.2021.627651
41. Malinova D, Fritzsche M, Nowosad CR, et al. WASp-dependent actin cytoskeleton stability at the dendritic cell immunological synapse is required for extensive, functional T cell contacts. J Leukoc Biol. 2016;99(5):699–710. doi:10.1189/jlb.2A0215-050RR
42. Bouma G, Mendoza-Naranjo A, Blundell MP, et al. Cytoskeletal remodeling mediated by WASp in dendritic cells is necessary for normal immune synapse formation and T-cell priming. Blood. 2011;118(9):2492–2501. doi:10.1182/blood-2011-03-340265
43. Stabile H, Carlino C, Mazza C, et al. Impaired NK-cell migration in WAS/XLT patients: role of Cdc42/WASp pathway in the control of chemokine-induced beta2 integrin high-affinity state. Blood. 2010;115(14):2818–2826. doi:10.1182/blood-2009-07-235804
44. Maillard MH, Cotta-de-almeida V, Takeshima F, et al. The Wiskott-Aldrich syndrome protein is required for the function of CD4+CD25+Foxp3+ regulatory T cells. J Exp Med. 2007;204(2):381–391. doi:10.1084/jem.20061338
45. Westerberg L, Larsson M, Hardy SJ, Fernández C, Thrasher AJ, Severinson E. Wiskott-Aldrich syndrome protein deficiency leads to reduced B-cell adhesion, migration, and homing, and a delayed humoral immune response. Blood. 2005;105(3):1144–1152. doi:10.1182/blood-2004-03-1003
46. Molina IJ, Kenney DM, Rosen FS, Remold-O’Donnell E. T cell lines characterize events in the pathogenesis of the Wiskott-Aldrich syndrome. J Exp Med. 1992;176(3):867–874. doi:10.1084/jem.176.3.867
47. Rivers E, Thrasher AJ. Wiskott-Aldrich syndrome protein: emerging mechanisms in immunity. Eur J Immunol. 2017;47(11):1857–1866. doi:10.1002/eji.201646715
48. Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci. 2013;1285(1):26–43. doi:10.1111/nyas.12049
49. Marangoni F, Bosticardo M, Charrier S, et al. Evidence for long-term efficacy and safety of gene therapy for Wiskott-Aldrich syndrome in preclinical models. Mol Ther. 2009;17(6):1073–1082. doi:10.1038/mt.2009.31
50. Golding B, Muchmore AV, Blaese RM. Newborn and Wiskott-Aldrich patient B cells can be activated by TNP-Brucella abortus: evidence that TNP-Brucella abortus behaves as a T-independent type 1 antigen in humans. J Immunol. 1984;133:2966–2971. PMID: 6436369.
51. Meyer-Bahlburg A, Becker-Herman S, Humblet-Baron S, et al. Wiskott-Aldrich syndrome protein deficiency in B cells results in impaired peripheral homeostasis. Blood. 2008;112(10):4158–4169. doi:10.1182/blood-2008-02-140814
52. Erdei A, Isaák A, Török K, et al. Expression and role of CR1 and CR2 on B and T lymphocytes under physiological and autoimmune conditions. Mol Immunol. 2009;46(14):2767–2773. doi:10.1016/j.molimm.2009.05.181
53. Recher M, Burns SO, de la Fuente MA, et al. B cell-intrinsic deficiency of the Wiskott-Aldrich syndrome protein (WASp) causes severe abnormalities of the peripheral B-cell compartment in mice. Blood. 2012;119(12):2819–2828. doi:10.1182/blood-2011-09-379412
54. Castiello MC, Bosticardo M, Pala F, et al. Wiskott-Aldrich Syndrome protein deficiency perturbs the homeostasis of B-cell compartment in humans. J Autoimmun. 2014;50:42–50. doi:10.1016/j.jaut.2013.10.006
55. Kolhatkar NS, Scharping NE, Sullivan JM, et al. B-cell intrinsic TLR7 signals promote depletion of the marginal zone in a murine model of Wiskott-Aldrich syndrome. Eur J Immunol. 2015;45(10):2773–2779. doi:10.1002/eji.201545644
56. Simon KL, Anderson SM, Garabedian EK, Moratto D, Sokolic RA, Candotti F. Molecular and phenotypic abnormalities of B lymphocytes in patients with Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2014;133(3):896–9.e4. doi:10.1016/j.jaci.2013.08.050
57. Bouma G, Carter NA, Recher M, et al. Exacerbated experimental arthritis in Wiskott-Aldrich syndrome protein deficiency: modulatory role of regulatory B cells. Eur J Immunol. 2014;44(9):2692–2702. doi:10.1002/eji.201344245
58. Mars LT, Araujo L, Kerschen P, et al. Invariant NKT cells inhibit development of the Th17 lineage. Proc Natl Acad Sci U S A. 2009;106(15):6238–6243. Epub 2009. doi:10.1073/pnas.0809317106
59. Yang JQ, Wen X, Kim PJ, Singh RR. Invariant NKT cells inhibit autoreactive B cells in a contact- and CD1d-dependent manner. J Immunol. 2011;186(3):1512–1520. doi:10.4049/jimmunol.1002373
60. Astrakhan A, Ochs HD, Rawlings DJ. Wiskott-Aldrich syndrome protein is required for homeostasis and function of invariant NKT cells. J Immunol. 2009;182(12):7370–7380. doi:10.4049/jimmunol.0804256
61. Locci M, Draghici E, Marangoni F, et al. The Wiskott-Aldrich syndrome protein is required for iNKT cell maturation and function. J Exp Med. 2009;206(4):735–742. Epub 2009 Mar 23. doi:10.1084/jem.20081773
62. Rönnblom L. The type I interferon system in the etiopathogenesis of autoimmune diseases. Ups J Med Sci. 2011;116(4):227–237. doi:10.3109/03009734.2011.624649
63. Prete F, Catucci M, Labrada M, et al. Wiskott-Aldrich syndrome protein-mediated actin dynamics control type-I interferon production in plasmacytoid dendritic cells. J Exp Med. 2013;210(2):355–374. doi:10.1084/jem.20120363
64. Nikolov NP, Shimizu M, Cleland S, et al. Systemic autoimmunity and defective Fas ligand secretion in the absence of the Wiskott-Aldrich syndrome protein. Blood. 2010;116(5):740–747. doi:10.1182/blood-2009-08-237560
65. Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356(6367):314–317. doi:10.1038/356314a0
66. Sobel ES, Kakkanaiah VN, Cohen PL, Eisenberg RA. Correction of gld autoimmunity by co-infusion of normal bone marrow suggests that gld is a mutation of the Fas ligand gene. Int Immunol. 1993;5(10):1275–1278. doi:10.1093/intimm/5.10.1275
67. Lee PP, Lobato-Márquez D, Pramanik N, et al. Wiskott-Aldrich syndrome protein regulates autophagy and inflammasome activity in innate immune cells. Nat Commun. 2017;8(1):1576. PMID: 29146903; PMCID: PMC5691069. doi:10.1038/s41467-017-01676-0
68. Jin Y, Mazza C, Christie JR, et al. Mutations of the Wiskott-Aldrich syndrome protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood. 2004;104(13):4010–4019. doi:10.1182/blood-2003-05-1592
69. Lee WI, Huang JL, Jaing TH, Wu KH, Chien YH, Chang KW. Clinical aspects and genetic analysis of taiwanese patients with wiskott-Aldrich syndrome protein mutation: the first identification of x-linked thrombocytopenia in the chinese with novel mutations. J Clin Immunol. 2010;30(4):593–601. doi:10.1007/s10875-010-9381-x
70. Ferrua F, Cicalese MP, Galimberti S, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, Phase 1/2 clinical study. Lancet Haematol. 2019;6(5):e239–e253. doi:10.1016/S2352-3026(19)30021-3
71. Braun CJ, Boztug K, Paruzynski A, et al. Gene therapy for Wiskott-Aldrich syndrome–long-term efficacy and genotoxicity. Sci Transl Med. 2014;6(227):227ra33. doi:10.1126/scitranslmed.3007280
72. Kolluri R, Shehabeldin A, Peacocke M, et al. Identification of WASP mutations in patients with Wiskott-Aldrich syndrome and isolated thrombocytopenia reveals allelic heterogeneity at the WAS locus. Hum Mol Genet. 1995;4(7):1119–1126. doi:10.1093/hmg/4.7.1119
73. Vignesh P, Suri D, Rawat A, et al. Sclerosing cholangitis and intracranial lymphoma in a child with classical Wiskott-Aldrich syndrome. Pediatr Blood Cancer. 2017;64(1):106–109. doi:10.1002/pbc.26196
74. Xie JW, Zhang ZY, Wu JF, et al. In vivo reversion of an inherited mutation in a Chinese patient with Wiskott-Aldrich syndrome. Hum Immunol. 2015;76(6):406–413. doi:10.1016/j.humimm.2015.04.001
75. Wu J, Liu D, Tu W, Song W, Zhao X. T-cell receptor diversity is selectively skewed in T-cell populations of patients with Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2015;135(1):209–216. doi:10.1016/j.jaci.2014.06.025
76. Liu DW, Zhang ZY, Zhao Q, et al. Wiskott-Aldrich syndrome/X-linked thrombocytopenia in China: clinical characteristic and genotype-phenotype correlation. Pediatr Blood Cancer. 2015;62(9):1601–1608. doi:10.1002/pbc.25559
77. Boztug K, Schmidt M, Schwarzer A, et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med. 2010;363(20):1918–1927. doi:10.1056/NEJMoa1003548
78. Hacein-Bey Abina S, Gaspar HB, Blondeau J, et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA. 2015;313(15):1550–1563. doi:10.1001/jama.2015.3253
79. Buchbinder D, Nugent DJ, Fillipovich AH. Wiskott-Aldrich syndrome: diagnosis, current management, and emerging treatments. Appl Clin Genet. 2014;7:55–66. doi:10.2147/TACG.S58444
80. Pala F, Morbach H, Castiello MC, et al. Lentiviral-mediated gene therapy restores B cell tolerance in Wiskott-Aldrich syndrome patients. J Clin Invest. 2015;125(10):3941–3951. doi:10.1172/JCI82249
81. Amarinthnukrowh P, Ittiporn S, Tongkobpetch S, et al. Clinical and molecular characterization of Thai patients with Wiskott-Aldrich syndrome. Scand J Immunol. 2013;77(1):69–74. doi:10.1111/sji.12004
82. Schindelhauer D, Weiss M, Hellebrand H, et al. Wiskott-Aldrich syndrome: no strict genotype-phenotype correlations but clustering of missense mutations in the amino-terminal part of the WASP gene product. Hum Genet. 1996;98(1):68–76. doi:10.1007/s004390050162
83. Knight T, Kotz K, Savaşan S. Autoimmune thyroiditis following HLA-matched sibling hematopoietic stem cell transplantation for Wiskott-Aldrich syndrome. Pediatr Transplant. 2018;22(5):e13222. doi:10.1111/petr.13222
84. Kolhatkar NS, Brahmandam A, Thouvenel CD, et al. Altered BCR and TLR signals promote enhanced positive selection of autoreactive transitional B cells in Wiskott-Aldrich syndrome. J Exp Med. 2015;212(10):1663–1677. doi:10.1084/jem.20150585
85. Marangoni F, Trifari S, Scaramuzza S, et al. WASP regulates suppressor activity of human and murine CD4(+) CD25(+) FOXP3(+) natural regulatory T cells. J Exp Med. 2007;204(2):369–380. doi:10.1084/jem.20061334
86. Glanzmann B, Möller M, Schoeman M, et al. Identification of a novel WAS mutation in a South African patient presenting with atypical Wiskott-Aldrich syndrome: a case report. BMC Med Genet. 2020;21(1):124. doi:10.1186/s12881-020-01054-6
87. Shigemura T, Nakazawa Y, Shimojo H, Kobayashi N, Agematsu K. Immune complex-mediated glomerulonephritis in a patient with Wiskott-Aldrich syndrome. J Clin Immunol. 2016;36(4):357–359. doi:10.1007/s10875-016-0258-5
88. Trifari S, Sitia G, Aiuti A, et al. Defective Th1 cytokine gene transcription in CD4+ and CD8+ T cells from wiskott-aldrich syndrome patients. J Immunol. 2006;177(10):7451–7461. doi:10.4049/jimmunol.177.10.7451
89. Du HQ, Zhang X, An YF, Ding Y, Zhao XD. Effects of Wiskott-Aldrich syndrome protein deficiency on IL-10-producing regulatory B cells in humans and mice. Scand J Immunol. 2015;81(6):483–493. doi:10.1111/sji.12282
90. Fillat C, Español T, Oset M, Ferrando M, Estivill X, Volpini V. Identification of WASP mutations in 14 Spanish families with Wiskott-Aldrich syndrome. Am J Med Genet. 2001;100(2):116–121. doi:10.1002/ajmg.1228
91. Ochs HD, Slichter SJ, Harker LA, Von Behrens WE, Clark RA, Wedgwood RJ. The Wiskott-Aldrich syndrome: studies of lymphocytes, granulocytes, and platelets. Blood. 1980;55(2):243–252. doi:10.1182/blood.V55.2.243.243
92. Gröttum KA, Hovig T, Holmsen H, Abrahamsen AF, Jeremic M, Seip M. Wiskott-Aldrich syndrome: qualitative platelet defects and short platelet survival. Br J Haematol. 1969;17(4):373–388. doi:10.1111/j.1365-2141.1969.tb01383.x
93. Baldini MG. Nature of the platelet defect in the Wiskott-Aldrich syndrome. Ann N Y Acad Sci. 1972;201(1):437–444. doi:10.1111/j.1749-6632.1972.tb16316.x
94. Marathe BM, Prislovsky A, Astrakhan A, Rawlings DJ, Wan JY, Strom TS. Antiplatelet antibodies in WASP (-) mice correlate with evidence of increased in vivo platelet consumption. Exp Hematol. 2009;37(11):1353–1363. doi:10.1016/j.exphem.2009.08.007
95. Rivers E, Worth A, Thrasher AJ, Burns SO. How I manage patients with Wiskott Aldrich syndrome. Br J Haematol. 2019;185(4):647–655. doi:10.1111/bjh.15831
96. Mahlaoui N, Pellier I, Mignot C, et al. Characteristics and outcome of early-onset, severe forms of Wiskott-Aldrich syndrome. Blood. 2013;121(9):1510–1516. Epub 2012 Dec 20. doi:10.1182/blood-2012-08-448118
97. Johnston SL, Unsworth DJ, Dwight JF, Kennedy CT. Wiskott-Aldrich syndrome, vasculitis and critical aortic dilatation. Acta Paediatr. 2001;90(11):1346–1348. doi:10.1080/080352501317130452
98. McCluggage WG, Armstrong DJ, Maxwell RJ, Ellis PK, McCluskey DR. Systemic vasculitis and aneurysm formation in the Wiskott-Aldrich syndrome. J Clin Pathol. 1999;52(5):390–392. doi:10.1136/jcp.52.5.390
99. Nozicka Z, Parízková E, Bednárová J, Lichý J. Izolovaná arteritida bazálních mozkových tepen u dítĕte s primární imunodeficiencí podmínĕnou Wiskottovým-Aldrichovým syndromem [Isolated arteritis in the basal cerebral arteries in a child with primary immunodeficiency due to the Wiskott-Aldrich syndrome]. Cesk Patol. 1987;23:215–221. Czech. PMID: 3442841.
100. Lao YL, Wong SN, Lawton WM. Takayasu’s arteritis associated with Wiskott-Aldrich syndrome. J Paediatr Child Health. 1992;28(5):407–409. doi:10.1111/j.1440-1754.1992.tb02703.x
101. Pellier I, Dupuis Girod S, Loisel D, et al. Occurrence of aortic aneurysms in 5 cases of Wiskott-Aldrich syndrome. Pediatrics. 2011;127(2):e498–504. doi:10.1542/peds.2009-2987
102. Watson RD, Gershwin ME, Smithwick E, Castles JJ, Ruebner B. Cutaneous T cell lymphoma and leukocytoclastic vasculitis in a long-term survivor of Wiskott-Aldrich syndrome. Ann Allergy. 1985;55:
103. Duzova A, Topaloglu R, Sanal O, et al. Henoch-Schönlein purpura in Wiskott-Aldrich syndrome. Pediatr Nephrol. 2001;16(6):500–502. doi:10.1007/s004670100583
104. Kawakami C, Miyake M, Tamai H. Kawasaki disease in a patient with Wiskott-Aldrich syndrome: an increase in the platelet count. Int J Hematol. 2003;77(2):199–200. doi:10.1007/BF02983223
105. Filipovich AH, Krivit W, Kersey JH, Burke BA. Fatal arteritis as a complication of Wiskott-Aldrich syndrome. J Pediatr. 1979;95(5 Pt 1):742–744. PMID: 490243. doi:10.1016/s0022-3476(79)80726-x
106. Longhurst HJ, Taussig D, Haque T, et al. Non-myeloablative bone marrow transplantation in an adult with Wiskott-Aldrich syndrome. Br J Haematol. 2002;116(2):497–499. doi:10.1046/j.1365-2141.2002.03269.x
107. Cooper MD, Chae HP, Lowman JT, Krivit W, Good RA. Wiskott-Aldrich syndrome. An immunologic deficiency disease involving the afferent limb of immunity. Am J Med. 1968;44(4):499–513. doi:10.1016/0002-9343(68)90051-x
108. Spitler LE, Wray BB, Mogerman S, Miller JJ
109. Gutenberger J, Trygstad CW, Stiehm ER, Opitz JM, Thatcher LG, Bloodworth JM
110. Webb MC, Andrews PA, Koffman CG, Cameron JS. Renal transplantation in Wiskott-Aldrich syndrome. Transplantation. 1993;56:1585. PMID: 8279047.
111. DeSanto NG, Sessa A, Capodicasa G, et al. IgA glomerulonephritis in Wiskott-Aldrich syndrome. Child Nephrol Urol. 1988–1989;9(1–2):118–120. PMID: 3251617.
112. Schurman SH, Candotti F. Autoimmunity in Wiskott-Aldrich syndrome. Curr Opin Rheumatol. 2003;15(4):446–453. doi:10.1097/00002281-200307000-00012
113. Ohya T, Yanagimachi M, Iwasawa K, et al. Childhood-onset inflammatory bowel diseases associated with mutation of Wiskott-Aldrich syndrome protein gene. World J Gastroenterol. 2017;23(48):8544–8552. doi:10.3748/wjg.v23.i48.8544
114. Cannioto Z, Berti I, Martelossi S, et al. IBD and IBD mimicking enterocolitis in children younger than 2 years of age. Eur J Pediatr. 2009;168(2):149–155. doi:10.1007/s00431-008-0721-2
115. Snapper SB, Rosen FS, Mizoguchi E, et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 1998;9(1):81–91. doi:10.1016/s1074-7613(00)80590-7
116. Biswas A, Shouval DS, Griffith A, et al. WASP-mediated regulation of anti-inflammatory macrophages is IL-10 dependent and is critical for intestinal homeostasis. Nat Commun. 2018;9(1):1779. doi:10.1038/s41467-018-03670-6
117. Monteferrante G, Giani M, van den Heuvel M. Systemic lupus erythematosus and Wiskott-Aldrich syndrome in an Italian patient. Lupus. 2009;18(3):273–277. doi:10.1177/0961203308095000
118. Adriani M, Aoki J, Horai R, et al. Impaired in vitro regulatory T cell function associated with Wiskott-Aldrich syndrome. Clin Immunol. 2007;124(1):41–48. doi:10.1016/j.clim.2007.02.001
119. Crestani E, Volpi S, Candotti F, et al. Broad spectrum of autoantibodies in patients with Wiskott-Aldrich syndrome and X-linked thrombocytopenia. J Allergy Clin Immunol. 2015;136(5):
120. Taylor MD, Sadhukhan S, Kottangada P, et al. Nuclear role of WASp in the pathogenesis of dysregulated TH1 immunity in human Wiskott-Aldrich syndrome. Sci Transl Med. 2010;2(37):37ra44. doi:10.1126/scitranslmed.3000813
121. Sallah S, Wan JY, Hanrahan LR. Future development of lymphoproliferative disorders in patients with autoimmune hemolytic anemia. Clin Cancer Res. 2001;7:791–794. PMID: 11309323.
122. Bach FH, Albertini RJ, Joo P, Anderson JL, Bortin MM. Bone-marrow transplantation in a patient with the Wiskott-Aldrich syndrome. Lancet. 1968;292(7583):1364–1366. doi:10.1016/s0140-6736(68)92672-x
123. Kapoor N, Kirkpatrick D, Blaese RM, et al. Reconstitution of normal megakaryocytopoiesis and immunologic functions in Wiskott-Aldrich syndrome by marrow transplantation following myeloablation and immunosuppression with busulfan and cyclophosphamide. Blood. 1981;57(4):692–696. PMID: 7008865. doi:10.1182/blood.V57.4.692.692
124. Ochs HD, Lum LG, Johnson FL, Schiffman G, Wedgwood RJ, Storb R. Bone marrow transplantation in the Wiskott-Aldrich syndrome. Complete hematological and immunological reconstitution. Transplantation. 1982;34(5):284–288. doi:10.1097/00007890-198211000-00009
125. Filipovich AH, Stone JV, Tomany SC, et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: Collaborative Study of the international bone marrow transplant registry and the national marrow donor program. Blood. 2001;97(6):1598–1603. doi:10.1182/blood.v97.6.1598
126. Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, et al. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European society for immunodeficiencies and European group for blood and marrow transplantation. Blood. 2008;111(1):439–445. doi:10.1182/blood-2007-03-076679
127. Mallhi KK, Petrovic A, Ochs HD. Hematopoietic stem cell therapy for Wiskott-Aldrich syndrome: improved outcome and quality of life. J Blood Med. 2021;12:435–447. doi:10.2147/JBM.S232650
128. Corash L, Shafer B, Blaese RM. Platelet-associated immunoglobulin, platelet size, and the effect of splenectomy in the Wiskott-Aldrich syndrome. Blood. 1985;65(6):1439–1443. PMID: 3995178. doi:10.1182/blood.V65.6.1439.bloodjournal6561439
129. Mullen CA, Anderson KD, Blaese RM. Splenectomy and/or bone marrow transplantation in the management of the Wiskott-Aldrich syndrome: long-term follow-up of 62 cases. Blood. 1993;82(10):2961–2966. PMID: 8219187. doi:10.1182/blood.V82.10.2961.2961
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.