Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Autoimmune encephalitis in psychiatric institutions: current perspectives
Authors Bost C, Pascual O, Honnorat J
Received 29 July 2016
Accepted for publication 22 September 2016
Published 27 October 2016 Volume 2016:12 Pages 2775—2787
DOI https://doi.org/10.2147/NDT.S82380
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Chloe Bost,1–3 Olivier Pascual,2,3 Jérôme Honnorat1–3
1French Reference Center of Paraneoplastic Neurological Syndrome, Hospices Civils de Lyon, Hôpital Neurologique, Bron, France; 2Synatac Team, NeuroMyoGene Institut, INSERM U1217/CNRS UMR5310, Lyon, France; 3University Claude Bernard Lyon 1, Lyon, France
Abstract: Autoimmune encephalitis is a rare and newly described group of diseases involving autoantibodies directed against synaptic and neuronal cell surface antigens. It comprises a wide range of neuropsychiatric symptoms. Sensitive and specific diagnostic tests such as cell-based assay are primordial for the detection of neuronal cell surface antibodies in patients’ cerebrospinal fluid or serum and determine the treatment and follow-up of the patients. As neurological symptoms are fairly well described in the literature, this review focuses on the nature of psychiatric symptoms occurring at the onset or during the course of the diseases. In order to help the diagnosis, the main neurological symptoms of the most representative synaptic and neuronal cell surface autoantibodies were detailed. Finally, the exploration of these autoantibodies for almost a decade allowed us to present an overview of autoimmune encephalitis incidence in psychiatric disease and the general guidelines for the management of psychiatric manifestations. For the majority of autoimmune encephalitis, the prognosis depends on the rapidity of the detection, identification, and the management of the disease. Because the presence of pronounced psychiatric symptoms drives patients to psychiatric institutions and can hinder the diagnosis, the aim of this work is to provide clues to help earlier detection by physicians and thus provide better medical care to patients.
Keywords: neuroimmunology, autoantibodies, organic psychosis, dementia, schizophrenia
Introduction
Autoimmune encephalitis is a new and rare disease, characterized by brain inflammation and circulating autoantibodies. Various autoimmune encephalitis have been described, and each of them linked to the presence of specific autoantibodies directed against synaptic and neuronal cell surface antigens. The main targets appear to be N-methyl-D-aspartate receptor (NMDAR), α-amino-3-hydroxy-5-methyl-4-isoxazolepropion acid receptor (AMPAR), leucine-rich glioma inactivated 1 (Lgi1), contactin-associated protein-like 2 (Caspr2), glutamate decarboxylase (GAD) or gamma-aminobutyric acid type B receptor (GABABR),1,2 but a significant number of autoimmune encephalitis are due to rarer or unidentified targets. Clinical symptoms usually correlate with the associated antibody subtype. Removal of these antibodies by plasma exchanges or immunotherapy generally induces clinical improvement.3,4 Neurological symptoms drastically vary according to epitope targeted by the autoantibody produced by the patients (Table 1). It is thus very important to know clinical symptoms and to recognize them in order to properly diagnose the patients and to give them adapted treatments.
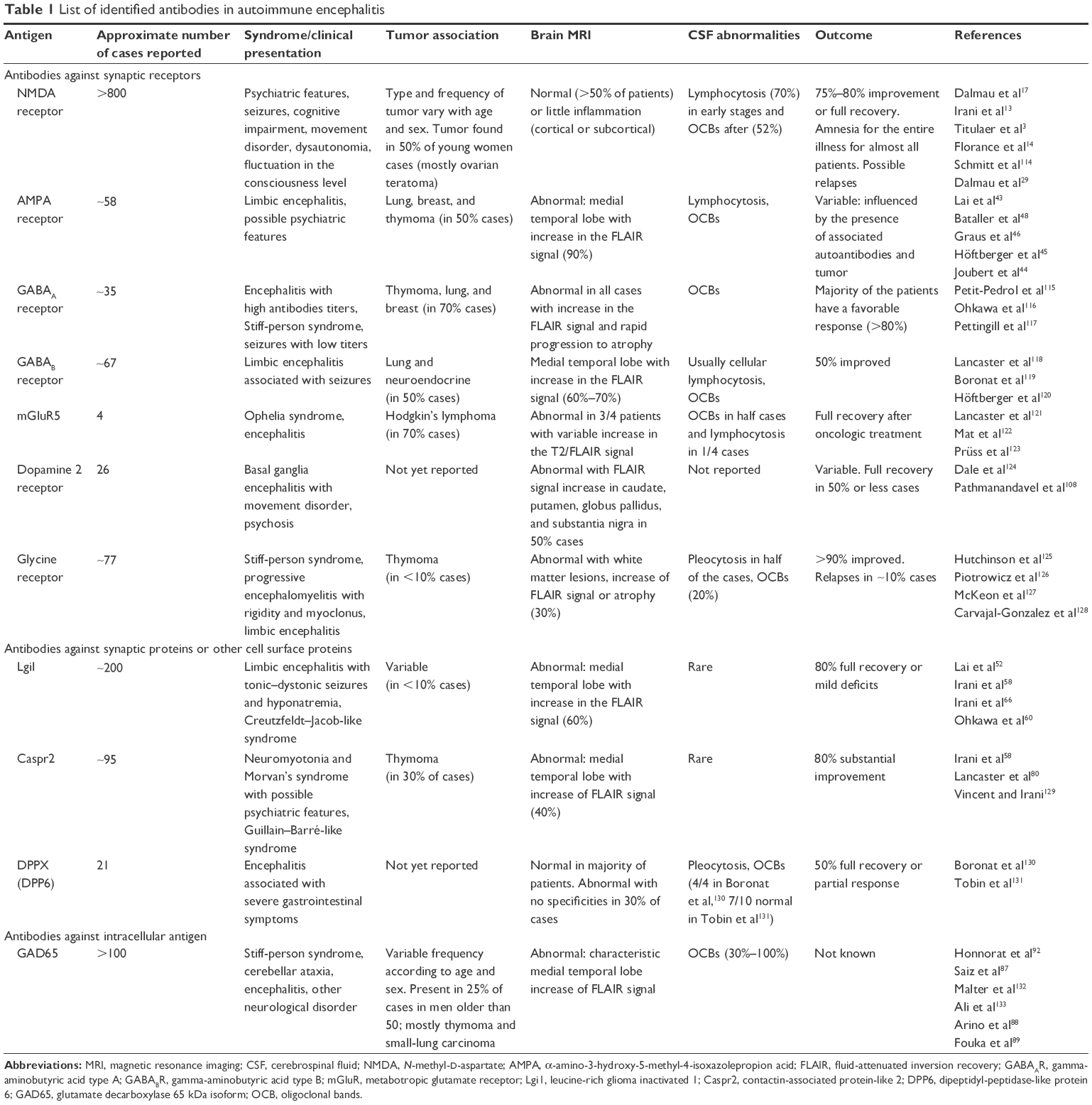 | Table 1 List of identified antibodies in autoimmune encephalitis |
Owing to the variety of antigens targeted by autoantibodies, autoimmune encephalitis is clinically heterogeneous, affecting both men and women, ranging from those with early age to those with older than 80 years. The common symptoms include a wide range of psychiatric and neurological symptoms.5,6 While most of the literature focuses on the neurological manifestations of these disorders, the initial presentation is often psychiatric.7 Psychiatric symptoms occur generally early in the progress of the disease but may also appear during the course of the disease.3,8 These psychiatric symptoms often slow down the diagnosis of the disease and alter the handling of the patient. This is a critical aspect as it is now clear that a rapid diagnosis is both necessary and limiting for a good outcome of the patients. In this regard, psychiatrists have a key role in the diagnosis process and orientation of the patients since they encounter many of them in their daily practice and often establish the first clinical diagnosis. This task is difficult as studies giving the specific symptomatology that would allow psychiatrists to establish their diagnosis and appropriate care are lacking.
Data are substantial for anti-NMDAR, anti-AMPAR, and anti-Lgi1 encephalitis but sparse for other cell surface antibody encephalitis such as anti-Caspr2 and anti-GAD encephalitis. This article reviews the psychiatric and behavioral manifestations of these various subtypes of autoimmune encephalitis.
Search strategy
Literature for this review was obtained by performing PubMed searches for each specific published neuronal surface antigen in the central nervous system (NMDA receptor, AMPA receptor, glycine receptor (GlyR), metabotropic glutamate receptors 1 and 5, gamma-aminobutyric acid type A receptor (GABAAR) and GABABR, dopamine receptor, Lgi1, Caspr2, dipeptidyl-peptidase-like protein 6 (DPP6; also named DPPX), voltage-gated calcium channels and Tr/Delta/Notch-like epidermal growth factor-related receptor (Tr/DNER). These terms were combined with the terms of “antibodies”, “autoimmune”, “autoimmunity”, or “encephalitis”, and/or “psychiatric”, “psychiatry”, “psychosis”, “schizophrenia”, and “dementia”. Non-English publications were excluded. Bibliographies of included studies were also hand searched. The search strategy included articles starting from the date of the first publication on antibodies to each specific antigen till June 30, 2016.
Anti-NMDAR encephalitis
Anti-NMDAR encephalitis is the most common autoimmune encephalitis described so far,9 with >900 cases identified worldwide since its first description in 2007.10,11 Even if it is still considered as a rare disease, the relatively high occurrence for this subtype of autoimmune encephalitis explains the focus of the literature on these antibodies in epidemiologic studies. Anti-NMDAR encephalitis represents 20% of immune-mediated encephalitis.12 It predominantly affects young women (60%), children (35%), and more rarely men and elderly patients.3,13–16
Psychiatric presentation
A Dutch retrospective study reported that 80% of patients diagnosed with anti-NMDAR encephalitis had initial psychiatric presentation8 and >60% were first admitted in a psychiatric unit. Other retrospective studies found similar results: psychiatric symptoms at the first presentation were reported for 80%–100% patients and the patients initially seen by psychiatrists represented 70%–80% of the cases.13,17 Most patients described did not have any psychiatric history;18 therefore, a first psychiatric episode should be considered as an argument to test the presence of anti-NMDAR antibodies in patient’s cerebrospinal fluid (CSF).
Psychiatric presentation is heterogeneous with grandiose and paranoid delusions, hallucinations (visual and auditory), bizarre behavior, agitation, fear, insomnia, confusion, and short-term memory loss.19 These manifestations are generally considered as acute psychosis, mania (with psychotic features), or onset of schizophrenia (Table 2).20 This period of the disease can be associated or not with major or discreet neurological signs, leading to an initial consultation in psychiatric institutions.18,21 If neurological signs, such as dystonia, oro-lingual-facial dyskinesias,17 or seizures are present, they should lead to a search for autoantibodies.21 Lejuste et al22 found that half of the patients with psychiatric presentation were patients with prior discrete neurologic symptoms that did not lead to further investigations (magnetic resonance imaging [MRI], CSF analysis) and were thus misdiagnosed. Autonomic manifestations such as hyperthermia and/or tachycardia are also frequent. Even if it is rare, some patients will not present any neurological symptoms during the disease (first episode and possible relapses).22,23 These patients present no particularities (fulfilled criteria for schizophrenia according to Diagnostic and Statistical Manual of Mental Disorders, fourth edition) and respond to classical immunomodulatory treatment but are difficult to diagnose.24,25
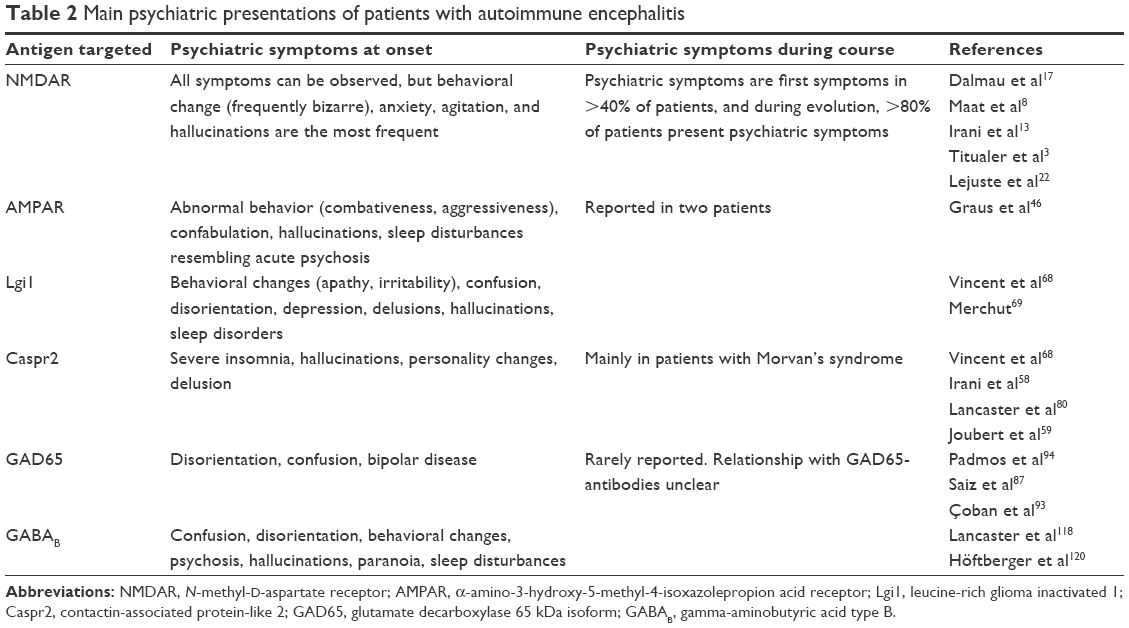 | Table 2 Main psychiatric presentations of patients with autoimmune encephalitis |
A study focusing on the presentation in pediatric population described more manic than psychotic symptoms in this population, including temper tantrums, behavioral change, agitation, aggression, and progressive speech deterioration, as well as hyperactivity and hypersexuality.14 Differential diagnosis generally arises between early onset schizophrenia, late onset autism, and childhood disintegrative disorder.26–28
Usual psychiatric drugs, including neuroleptics, benzodiazepines, and valproic acid, could occasionally help, but their effect is incomplete and transitory. Neuroleptics must be cautiously used because ~50% of patients with anti-NMDAR encephalitis treated by neuroleptics may develop intolerance characterized by high temperature, muscle rigidity, mutism or coma, and rhabdomyolisis biomarkers suggesting neuroleptic malignant syndrome.22
Neurological signs and symptoms
In anti-NMDAR encephalitis, a set of nonspecific symptoms comprises a characteristic syndrome.7,21,29 Presentation is variable depending on sex and age.29 However, in 70% of patients,18 clinical course begins with viral-like prodromes (fever, nausea, diarrhea) occurring ~1 or 2 weeks before psychiatric and/or neurologic symptoms leading to hospitalization. Acute psychiatric symptoms and cognitive impairments progress rapidly to severe neurological features (seizures, dyskinesias, dysautonomic symptoms) until a comatose phase.3,29 Fatal outcomes due to respiratory complications were frequent in the past. Autoimmune encephalitis is now well characterized with an easier clinical and biological diagnosis. The latter is based on the detection of polyclonal immunoglobulin G (IgG) directed against GluN1 subunit of the NMDAR in CSF. Although the presence of IgG is found in most cases, other immunoglobulin subtypes can also be found.30 The distinction between IgG, IgA and IgM immunoglobulins subtypes is essential as the prevalence,24,31 the physiopathology,32 and the clinical presentation33 are different. Anti-NMDAR encephalitis is a primary antibody-mediated disease, and the treatment is based on immunotherapy and tumor removal (if present). The frequency of an underlying teratoma, which is in 94% of cases of an ovarian teratoma,3 is dependent on age, sex, and ethnicity3,14,16,29 and occurs more frequently in young adult women (~50% in this subgroup).
Treatment and outcome
Guidelines have been published for the treatment of autoimmune encephalitis after antibody detection.29,34,35
Initially categorized in paraneoplastic disease, autoimmune encephalitis is also found in patients without tumors. Treatment is first based on tumor resection, when present, and first-line immunotherapy: corticosteroids associated with intravenous immunoglobulins (IVIg). Plasma exchanges are possible and showed efficacies but are more difficult to carry out in the context of autonomic instability or in poorly cooperative patients.29 If a tumor is not detected, tumor screening should be initiated, taking into consideration the frequency of underlying tumor with this antibody and patient’s age and sex.
Most patients respond within weeks to first-line treatments, but anti-NMDAR encephalitis patients are the slowest among autoimmune encephalitis. Early treatment allows good outcome in 80% of patients,16 but recovery is slow, >2 years.3
For the 47% of patients who do not respond to first-line treatments,3 a second-line immunotherapy was started with rituximab or cyclophosphamide or both. The outcome of the second-line immunotherapy in patients is improved in 65% of cases.3,29 Generally, the frequency of improvement is better for patients with tumor (80%) when compared to patients without tumor (48%). Patients without tumors consequently require more often a second-line immunotherapy.29
Despite this second-line treatment, relapse can occur in 20%–25% of the case.13,14,36 To prevent relapses, immunosuppressive treatment can be continued with mycophenolate mofetil or azathioprine during 1 year.3
Mechanisms
Antibodies found in patients are immunoglobulins G, classes IgG1 and IgG3.37 They target an ionotropic glutamate receptor, the NMDAR and more precisely GluN1, the obligatory subunit of the receptor.17,38 Syndromes observed in autoimmune encephalitis generally resemble those described in pharmacologic or genetic models of antigens’ disruption (eg, with ketamine39 and memory impairment and depressive-like behavior in mice models40). Detection of anti-NMDAR antibodies by immunohistochemistry on rodent brain slices indicates a high hippocampal staining, a moderate cortical staining and a limited cerebellar staining,10 correlating well with the symptomatology. Various studies performed using animal models and in vitro suggest that the antibody decreases the surface expression and total density17,41 of NMDARs, leading to an alteration in synaptic plasticity and synaptic transmission.41,42 These data point toward a direct pathogenic role of antibodies on the NMDAR itself.
Anti-AMPAR encephalitis
Anti-AMPAR encephalitis also belongs to autoimmune encephalitis with antibodies targeting ionotropic glutamate receptor. Initially described in a series of 10 patients 6 years ago,43 its frequency is lower than anti-NMDAR encephalitis but patients who have already been described bring relevant information. New cohorts were recently published, giving further details on the first description that has been made.44,45
Psychiatric presentation
Even if AMPAR and NMDAR are both ionotropic glutamate receptors and thereby are functionally related, clinical phenotypes are different. Patients who were described mostly presented limbic dysfunction and prominent psychiatric symptoms such as confusion, disorientation, confabulation, agitation, combativeness, and perseveration. In 2010, Graus et al46 found in an antibody screening study four patients (n=30 patients tested in total, including 17 with limbic encephalitis) with anti-AMPAR antibodies, including two with acute psychosis. These two women presented confusion and aggressive behavior. Confusion was in 70% of the cases, the first sign reported at the onset of the disease;44,45,47 this initial confusion can be associated with limbic encephalitis symptoms and seizures.
Neurological signs and symptoms
Anti-AMPAR encephalitis patients in addition to psychiatric symptoms frequently present classical limbic encephalitis features such as acute amnesia, confusion, and abnormal behavior. The variable presence of tumor44,45 did not seem to cause any differences in the clinical presentation, but had implications for treatment. Seizures are frequently present and may guide to set the diagnosis. Insomnia, lethargy, and decreased level of consciousness have also been described.45,46 Fulminant forms were also described4,43,44 with fever, coma, and hypertonia. Brain MRI is often abnormal. Severity of lesions, when they are present, appears to correlate with clinical outcome.9,43 Anti-AMPAR encephalitis should be considered in elderly patients, mainly women, with a median age of ~60 years (range 23–81 years).43,45
Treatment and evolution
Treatment consists of aggressive immunotherapy associated with tumor removal when present. IVIg treatment is generally followed by chemotherapy with cyclophosphamide or doxorubicin. Anti-AMPAR encephalitis is a treatable disorder but with poorer recovery rate when compared to Lgi1 or NMDAR autoimmune encephalitis. It is important to note that among patients who responded to the treatment, 48% remain with residual effects.45 Follow-up is crucial due to a propensity for relapse in these patients.43,46–48 Long-term outcomes hang on the appropriate management of relapses.43 Thus, chronic immunomodulatory treatment should be considered.
Special attention should be paid to possible associated autoantibodies (GAD, Sox1, amphiphysin) that correlate with a poorer prognosis.45 In these cases, symptoms and tumor can be more characteristic of the additional immune response. Detection of anti-AMPAR antibodies in patient’s CSF must lead to the administration of an aggressive and rapid therapy and to the search for a possible associated tumor or classical paraneoplastic antibodies.
Anti-AMPAR encephalitis diagnosis is based on the detection of polyclonal IgG against GluA1/2 subunits of AMPA receptor (formerly known as GluR1/R2) in patient’s CSF. Titer is generally correlated with clinical evolution.
Mechanisms
In AMPAR encephalitis, symptoms such as memory deficits, movement disorders, and cerebellar signs suggest the dysfunction of structures such as the hippocampus, the cerebellum, and the basal ganglia. The binding of patient’s anti-AMPAR autoantibodies with a high affinity to these structures in brain rat sections is another argument in agreement with the alteration in the limbic system and the cerebellum.43 Intrathecal synthesis of antibodies is strongly probable due to higher antibody titers in CSF than in serum43 and the finding of CSF positive but serum negative in some patients.45 Peng et al recently highlighted a rapid internalization of AMPA receptors in vitro, on cells treated with patient’s antibodies. While this mechanism is not clearly identified yet, internalization seems to be followed by degradation of the receptor.49
In vitro, cultured neurons recorded using whole-cell patch clamp and treated with patient’s CSF had reduced amplitude and frequency of miniature excitatory postsynaptic currents, demonstrating an impairment of AMPAR-dependant synaptic transmission in the presence of patient’s antibodies.49,50 There are few in vitro data, and most of them need to be reproduced in order to get enough credit. There is also a lack of in vivo studies allowing a better understanding of the mechanisms involved in the disease. Furthermore, differences in clinical presentation, from pure memory disturbances to fulminant encephalitis, suggest different mechanisms according to the patients.44
Anti-Lgi1 encephalitis
Lgi1 is a protein secreted by hippocampal neurons largely associated with epilepsy.51 In some cases of encephalitis, patients produce autoantibodies directed against Lgi1. There is still not a lot of data concerning Lgi1 autoantibodies as it was first assimilated to antibodies raised against the voltage-gated potassium channel.52 Indeed antibodies against voltage-gated potassium channels were first described in neuromyotonia,53 then in Morvan’s syndrome54,55 and finally in limbic encephalitis.56,57 More recently, studies demonstrated that this entity gathered various encephalitis due to distinct antibodies targeting Lgi1, Caspr2, and contactin 2.52,58 Identification of these antigens helped to clarify the apparent diversity of symptoms attributed to voltage-gated potassium channels antibodies. While anti-Lgi1 antibodies are preferentially associated with classical limbic encephalitis, anti-Capsr2 antibodies are associated with Morvan’s syndrome, neuromyotonia, and sometimes with neuropathies or limbic encephalitis.52,58,59
Psychiatric presentation
Patients with anti-Lgi1 encephalitis are predominantly men (sex ratio 4:3)60,61 with a median age of 54 years (range 32–67 years) at the onset.62 In recent years, case reports described heterogeneous psychiatric signs at the onset of the disease such as confusion, depression, paranoia, behavior disturbances, visual hallucinations, and dementia.63–65
Early onset of the disease is most often characterized by confusion and dementia without family history. These psychiatric signs are generally associated with neurological symptoms from the onset of the disease.
Neurological signs and symptoms
Patients with anti-Lgi1 antibodies mostly exhibit seizures and limbic encephalitis. Insomnia and paradoxical sleep disorders are typical features of the disease. Faciobrachial dystonic seizures (FDBS) are characteristics of anti-Lgi1 encephalitis and generally precede the limbic encephalitis.61,66 The term tonic–dystonic seizures is now preferred because the dystonic seizures can be located throughout the body.61,67 A recent study explored the anatomical origin of these tonic–dystonic seizures to help the diagnosis and found the motor cortex and the hippocampus as starting points.61 Complementary signs can help to comfort the diagnosis: hyponatremia is reported in 60%–80% cases58,68,69 and MRI is abnormal in 70%–80% of the cases with abnormalities in the temporal lobes.68–70 The frequency of associated cancers is low, <20%.9 As McQuillan and Bargman71 summed up, it should always be kept in mind that for a patient presenting confusion and hyponatremia, the confusion may not be secondary to hyponatremia and that an anti-Lgi1 autoantibody may be present.71
Treatment and evolution
In the literature, anti-Lgi1 encephalitis is described as responsive to immunotherapy.66,68,72–75 Guidelines suggest using high doses of corticosteroid, IVIg and plasmapheresis as the first-line therapy. In refractory cases, rituximab and cyclophosphamide should be used as second-line therapies.76 Anti-Lgi1 antibody encephalitis seems to be associated with poor cognitive outcome and evolves frequently with hippocampal atrophy. Relapses are reported in 10%–20% patients.57,76 Introduction of immunotherapy allows resolution of FDBS. The quickness to start immunotherapy is significantly correlated with the time to recover basal functions.77 Management of antibody titers seems to be the easiest way to manage clinical improvement.70 Residual symptoms after treatment consist of verbal memory impairment, and antibody titer at presentation seems to predict verbal memory index after treatment.
Mechanisms
The precise function of Lgi1 is still a matter of debate. Lgi1 is a neuronal secreted protein.78 Some reports indicate that Lgi1 prevents the inactivation of the Kv1 voltage-gated potassium channels through the cytoplasmic regulatory protein Kv.7,79,80 It has also been shown that by interacting with pre- and postsynaptic proteins, Lgi1 may also have a role in the regulation of neurotransmitter release81,82 and could control the function of AMPA receptors likely through its interaction with ADAM22.82 Anti-Lgi1 antibodies are mainly IgG4 subtype and IgG1 in a lesser extent.83
The pathogenic role of anti-Lgi1 antibodies was rapidly suggested by in vitro and in vivo evidences. First, antibody titers seem to be well correlated with clinical presentation as immunotherapy induces prompt improvement in patients.62 In the same order of idea, antibody titers seem to be linked with hippocampal atrophy. Indeed, in a prospective study, the two patients who developed hippocampal atrophy had the highest titers of antibodies. Anti-Lgi1 antibodies binded the N-terminal domain and the distal epitempin domain. This binding was found to disrupt the Lgi1–ADM22 protein interaction in an in vitro model.60 In mice models, symptoms induced by patient’s IgG infusion were similar to those obtained after potassium channel blockers’ administration (increase nerve excitability)84 but, to date, there is no other in vivo studies. Finally, anti-Lgi1 antibodies seem to be pathogenic, but pathophysiological mechanisms remain to be determined.
Others
Data concerning other antibodies involved in limbic encephalitis are partial and incomplete due to the low occurrence of these specific limbic encephalitis and the small amount of case reports. Here are the various psychiatric presentations concerning some known antibodies found in patients presenting limbic encephalitis.
Anti-Capsr2 encephalitis
Caspr2 is a protein of the neurexin family that is found in the brain and peripheral nerves. Its distribution is wide, but it seems to be concentrated at the juxtaparanodal region of myelinated axons.85 Anti-Caspr2 antibodies are mostly found in neuromyotonia, Morvan’s syndrome and limbic encephalitis. Anti-Caspr2 antibodies have been found in patients presenting psychiatric manifestations such as confusion and personality change with frontal lobe dysfunction. These psychiatric manifestations are generally associated with neurologic symptoms. FDBS, characteristic of Lgi1, is not found in patients presenting anti-Caspr2 antibodies, and confusion and cognitive impairment seem to be less frequent with anti-Caspr2 than with anti-Lgi1 antibodies.86 Detection of Capsr2 antibodies in patients with neuromyotonia is often correlated with an underlying tumor (frequently a thymoma),66 and patients having a tumor associated with anti-Caspr2 have a poorer prognosis. A recent work of Joubert et al59 reveals that the apparent clinical diversity of anti-Caspr2 encephalitis may be linked to the site of antibody synthesis. They compared patients with anti-Caspr2 antibodies in the CSF to patients presenting neuromyotonia or Morvan’s syndrome with anti-Caspr2 antibodies in the serum only. In this work, they showed that the presence of anti-Capsr2 antibodies in the CSF is associated with a much more homogeneous clinical pattern of autoimmune encephalitis, characterized by a prevalent limbic involvement and seizure occurrence.59 These results emphasize the lack of functional studies focusing on antibody specificities and the importance of testing both serum and CSF samples.
Encephalitis with anti-GAD antibodies
In more than 100 patients87–89 worldwide were found antibodies directed against GAD, a rate-limiting enzyme in the synthesis of gamma-aminobutyric acid, an inhibitory transmitter.90 Even if GAD is an intracellular protein, it can be exposed to antibodies during vesicular release.91 High titers of GAD65 antibodies are associated with different neurological disorders, including cerebellar ataxia,92 limbic encephalitis, and Stiff-person syndrome.91 A few psychiatric presentations are described such as disorientation and confusion but psychiatric troubles are less frequent than for other antibodies.87 Low level of GAD65 antibodies was also found to correlate with bipolar disorder presentations,93,94 but the causal link was not proven since a low level of GAD antibodies was described in other neurological pathologies, such as in epilepsy,95 myelopathy96 and myasthenia gravis.97 As a consequence, anti-GAD65 antibodies are rather considered as markers of autoimmune diseases than as pathogenic antibodies.87 Explanation could be found in a disease-specific epitope hypothesis as pointed out by a study revealing distinct epitopes between patients with Stiff-person syndrome and patients with limbic encephalitis.98
Many other antibodies directed against cell surface antigens such as GlyR, GABAAR and GABABR, metabotropic glutamate receptor 5, dopamine receptor D2, and DPP6 exist, but isolated psychiatric phenotypes are rarely described. If psychiatric symptoms can be present, they are generally associated with important neurological deficits or alterations that lead patients rapidly to neurological investigations rather than psychiatric ones.
How to diagnose autoimmune encephalitis in psychiatric departments?
As reported in the previous sections, limbic encephalitis patients often present initial psychiatric disorders that can lead them to psychiatric hospitalization (Table 2). Despite the fact that these diseases are rare and that studies on the frequency of anti-NMDAR antibodies failed to show particular subgroups in psychiatric illnesses such as schizophrenia, psychiatrists should pay close attention as they should statistically meet autoimmune encephalitis over the course of their career. Neurological investigations with neurological examination, lumbar puncture, and electroencephalogram (EEG) should be considered, especially when the patient has no history of psychiatric manifestations.
- Neurological examination: Particular attention should be paid on neurological symptoms that can be sometimes very discreet. They must be taken into account for the diagnosis. Memory deficits, seizures, dyskinesias and movement disorders, and headaches should be particularly questioned.99
- EEG: In a context of brain MRI and scan being normal, EEG is really useful and should be added to other investigations. Indeed EEG could present some abnormalities, even if a normal EEG does not exclude diagnosis of autoimmune encephalitis. In anti-NMDAR encephalitis, EEG is abnormal in >90% of cases, with rare but specific diffuse slow waves called extreme delta brush.17 Anti-Lgi1 encephalitis patients also present ictal EEG changes underlying severe movement’s abnormalities.66,100 Data are lacking for other type of encephalitis. Thus, for a first psychotic or schizophrenic episode, particularly in young patients, an EEG should be systematically realized.
- Autoantibodies characterization: The diagnosis of autoimmune encephalitis is based on the detection of specific IgG autoantibodies in the patients’ serum or CSF. Samples for diagnosis have to be obtained before immunotherapy. In anti-NMDAR encephalitis, CSF samples allow a better sensitivity (100% of patients are positive in CSF, against only 85% in serum).29 Detection of autoantibodies directed against neuronal cell surface antigens should be performed at least on CSF. In most cases, CSF samples will allow a better sensibility and specificity.101 Detection of Caspr2, Lgi1, or GlyR antibodies in serum samples only have rarely been reported, without proof of clinical relevance.
Global evaluation of the presence of antibodies against neuronal cell surface antigens can be initially evaluated on paraformaldehyde-fixed rodent brain sections. This technique suggests a shared epitope between human beings and rodents and trained and qualified staff. To confirm the target epitope, further investigations are needed, the gold standard consisting of cell-based assay (CBA). Contrary to Western blot or enzyme-linked immunosorbent assay (ELISA) techniques, CBA allows detection of conformational epitopes. After cell transfection and treatment by autoantibodies, immunofluorescence can be detected by flow cytometry or microscopy.5,102,103
Incidence in psychiatric diseases
The possible contribution of autoantibodies against neuronal cell surface antigens to psychiatric disease has drawn lots of interest over the past few years. Anti-NMDAR antibodies are of particular interest in these studies for many reasons. Among autoimmune encephalitis, anti-NMDAR encephalitis is the most frequent case reported so far, and patients often exhibit psychiatric manifestations. In addition, the central role of NMDAR hypofunction in psychotic symptoms’ origin particularly interested psychiatrists.
Following case reports of anti-NMDAR encephalitis mimicking psychosis (such as Lebon et al28) and following the hypothesis of an autoimmune basis to some idiopathic psychoses, studies started to evaluate the incidence of anti-NMDAR encephalitis in psychosis.104 The existence of anti-NMDAR antibodies in particular subgroups in psychiatric illnesses, such as schizophrenia, is still a matter of debate. Indeed, depending on patient’s selection and on detection methods used, the studies found between 0% and 10% of NMDAR antibody-positive patients.25,32,104–108
In conclusion, specific immunoglobulin G anti-GluN1109 is rarely found in psychiatric disease. If they have to be found, they will be mainly in schizophrenia.
Even if a minority of psychiatric patients presents NMDAR antibodies, psychiatrists have to be aware of this pathology because of the importance of a rapid diagnosis and because they are very often in the first-line place in the treatment process of these patients. In a fairly recent study, authors are now recommending the screening of NMDAR antibodies in all patients with first episode of schizophrenia.110
Studies including other neuronal autoantibodies detection are sparse. A Turkish study, which screened anti-NMDAR, anti-Caspr2, anti-Lgi1, and anti-GAD presence in primary dementia patients, chronic psychiatric patients and healthy controls, found only one patient with anti-NMDAR antibodies and one with anti-GAD antibodies.93 In the only study,32 to our knowledge, that tested the presence of AMPAR antibodies in psychiatric presentations (schizophrenia, borderline personality disorder, major depression and healthy controls), they did not find AMPAR antibodies in healthy controls or in patients (n=230 in each group). To date, there is no study in isolated psychosis testing anti-GlyR, GABAB, DPPX, and dopamine 2 receptors’ antibodies positivity.
Management of psychiatric manifestations
There are no published clinical trials on the best therapy for patients with autoimmune encephalitis. For all antibodies, patients received corticoid, plasmapheresis, and immunotherapy, based on anti-NMDAR encephalitis knowledge and some data of case reports. Management of psychiatric manifestations is clinically important and can impact the patient’s capacity to receive immunotherapy111 but lack practical feedback. Mainly, information comes from NMDAR encephalitis patients’ experiences. In 2014, Kuppuswamy et al112 reviewed management of psychotic symptoms, mood symptoms, and catatonic symptoms in NMDAR encephalitis. Treatments should not hide disease evolution neither worsen symptoms.22 They advised to choose atypical and more sedative antipsychotics rather than typical antipsychotics as dopamine antagonists that aggravate agitation, in order to treat psychotic symptoms. To treat mood symptoms, valproic acid was advised for sedation, sleep, and seizure benefits and thanks to the availability of an intravenous form. Uses of lithium and benzodiazepines are also reported in the literature but do not cause significant changes.112,113 Catatonic symptoms have to be treated in first line by benzodiazepines, and use of electroconvulsivotherapy is controversial. Neuroleptics can exacerbate neuropsychiatric symptoms and abnormal movements.14,22,113
Relapse possibilities combined with long-term persistence of behavioral and cognitive deficits highlight the importance of a medical follow-up, including psychiatric monitoring.
Conclusion
Autoimmune encephalitis is a rare and heterogeneous disease. Various psychiatric presentation can occur, associated or not with neurological symptoms. Most of the time, psychiatric symptoms appear subacutely, in patients without any psychosis history. The frequency and severity of these psychiatric features depend on antibodies. Even if case reports highlighted psychiatric features, in studies conducted in psychiatric patients, the level of autoimmune encephalitis was low, with variable results according to the studies. This apparent discrepancy is multifactorial and likely due to assays being used in the different studies, younger psychosis patients than autoimmune encephalitis patients and nonstandardized clinical definitions of psychosis and schizophrenia. Anyway, autoimmune encephalitis diagnosis is difficult and incidence in psychiatric disease is probably low. However, autoimmune encephalitis has to be in the differential diagnosis process of psychiatrists because they often have primary psychiatric presentations and a rapid treatment is essential.
For all patients, particular attention has to be paid on neurological signs, such as autonomic disability, disorientation, movement disorders, seizures, or hyponatremia. Neurological examination and early biological testing should be realized:
- for patients presenting neurological symptoms, even soft one;
- for young women with first psychiatric manifestations; and
- for all patients with atypical psychiatric presentation, evolution or treatment response.
In these specific cases, EEG and MRI can be performed but are generally non-indicative of the disease because they are not always abnormal and very rarely pathognomonic. The biological sample, CSF preferentially otherwise sera, should be sent to reference center and tested according to the gold standard guidelines. If patients tested are positive for autoimmune encephalitis antibodies, they should be referred to neurological centers. In any case, such biological samples associated with psychiatric history of the patients are interesting elements helping for prospective studies.
Disclosure
The authors report no conflicts of interest in this work.
References
van Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PAE. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev. 2014;13(3):299–312. | ||
Chefdeville A, Honnorat J, Hampe CS, Desestret V. Neuronal CNS syndromes likely mediated by autoantibodies. Eur J Neurosci. 2016;43:1532–1552. | ||
Titulaer MJ, Mccracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12(2):157–165. | ||
Wei YC, Liu CH, Lin JJ, et al. Rapid progression and brain atrophy in anti-AMPA receptor encephalitis. J Neuroimmunol. 2013;261(1–2):129–133. | ||
Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol. 2014;76(2):168–184. | ||
Leypoldt F, Armangue T, Dalmau J. Autoimmune encephalopathies. Ann N Y Acad Sci. 2014;1338:94–114. | ||
Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. 2011;10(8):759–772. | ||
Maat P, de Graaff E, van Beveren NM, et al. Psychiatric phenomena as initial manifestation of encephalitis by anti-NMDAR antibodies. Acta Neuropsychiatr. 2013;25(3):128–136. | ||
Joubert B, Honnorat J. Autoimmune channelopathies in paraneoplastic neurological syndromes. Biochim Biophys Acta. 2015;1848(10 Pt B):2665–2676. | ||
Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25–36. | ||
Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005;58(4):594–604. | ||
Granerod J, Ambrose HE, Davies NWS, et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis. 2010;10(12):835–844. | ||
Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic. Brain. 2010;133(Pt 6):1655–1667. | ||
Florance NR, Davis RL, Lam C, et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol. 2009;66(1):11–18. | ||
Armangue T, Leypoldt F, Dalmau J. Auto-immune encephalitis as differential diagnosis of infectious encephalitis. Curr Opin Neurol. 2014;27(3):361–368. | ||
Viaccoz A, Desestret V, Ducray F, et al. Clinical specificities of adult male patients with NMDA receptor antibodies encephalitis. Neurology. 2014;82(7):556–563. | ||
Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091–1098. | ||
Peery HE, Day GS, Dunn S, et al. Anti-NMDA receptor encephalitis. The disorder, the diagnosis and the immunobiology. Autoimmun Rev. 2012;11(12):863–872. | ||
Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr Res. 2016;176(1):36–40. | ||
Kayser MS, Dalmau J. The emerging link between autoimmune disorders and neuropsychiatric disease. J Neuropsychiatry Clin Neurosci. 2011;23(1):90–97. | ||
Rosenthal-Simons A, Durrant AR, Heresco-Levy U. Autoimmune-induced glutamatergic receptor dysfunctions: conceptual and psychiatric practice implications. Eur Neuropsychopharmacol. 2013;23(12):1659–1671. | ||
Lejuste F, Thomas L, Picard G, et al. Neuroleptic intolerance characterizes patients with anti-N-methyl-D-aspartate (NMDA) receptor encephalitis hospitalized in psychiatric institutions. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e280. | ||
Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti–N-methyl-d-aspartate receptor encephalitis. JAMA Neurol. 2013;70(9):1133–1139. | ||
Hammer C, Stepniak B, Schneider A, et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol Psychiatry. 2014;19:1–7. | ||
Zandi MS, Irani SR, Lang B, et al. Disease-relevant autoantibodies in first episode schizophrenia. J Neurol. 2011;258(4):686–688. | ||
Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis in psychiatry. Curr Psychiatry Rev. 2011;7(3):189–193. | ||
Creten C, Van Der Zwaan S, Blankespoor RJ, et al. Late onset autism and anti-NMDA-receptor encephalitis. Lancet. 2011;378:98. | ||
Lebon S, Mayor-Dubois C, Popea I, et al. Anti-N-methyl-D-aspartate (NMDA) receptor encephalitis mimicking a primary psychiatric disorder in an adolescent. J Child Neurol. 2012;27(12):1607–1610. | ||
Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63–74. | ||
Desestret V, Chefdeville A, Viaccoz A, et al. CSF IgA NMDAR antibodies are potential biomarkers for teratomas in anti-NMDAR encephalitis. Neurol Neuroimmunol Neuroinflamm. 2015;2(6):1–8. | ||
Busse S, Busse M, Brix B, et al. Seroprevalence of n-methyl-d-aspartate glutamate receptor (NMDA-R) autoantibodies in aging subjects without neuropsychiatric disorders and in dementia patients. Eur Arch Psychiatry Clin Neurosci. 2014;264(6):545–550. | ||
Steiner J, Walter M, Glanz W, et al. Increased prevalence of diverse N-methyl-D-aspartate glutamate receptor antibodies in patients with an initial diagnosis of schizophrenia: specific relevance of IgG NR1a antibodies for distinction from N-methyl-D-aspartate glutamate receptor encephalitis. JAMA Psychiatry. 2013;70(3):271–278. | ||
Prüss H, Höltje M, Maier N, et al. IgA NMDA receptor antibodies are markers of synaptic immunity in slow cognitive impairment. Neurology. 2012;78(22):1743–1753. | ||
Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry. 2012;83(6):638–645. | ||
Lancaster E, Dalmau J. Neuronal autoantigens – pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380–390. | ||
Gabilondo I, Saiz A, Galán L, et al. Analysis of relapses in anti-NMDAR encephalitis. Neurology. 2011;77(10):996–999. | ||
Tüzün E, Zhou L, Baehring JM, Bannykh S, Rosenfeld MR, Dalmau J. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol. 2009;118(6):737–743. | ||
Gleichman AJ, Spruce LA, Dalmau J, Seeholzer SH, Lynch DR. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci. 2012;32(32):11082–11094. | ||
Adell A, Jiménez-Sánchez L, López-Gil X, Romón T. Is the acute NMDA receptor hypofunction a valid model of schizophrenia? Schizophr Bull. 2012;38(1):9–14. | ||
Planaguma J, Leypoldt F, Mannara F, et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain. 2014;138(1):94–109. | ||
Mikasova L, De Rossi P, Bouchet D, et al. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain. 2012;135(pt 5):1606–1621. | ||
Jantzen SU, Ferrea S, Wach C, et al. In vitro neuronal network activity in NMDA receptor encephalitis. BMC Neurosci. 2013;14:17. | ||
Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65(4):424–434. | ||
Joubert B, Kerschen P, Zekeridou A, et al. Clinical spectrum of encephalitis associated with antibodies against the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor: case series and review of the literature. JAMA Neurol. 2015;72(10):1163–1169. | ||
Höftberger R, van Sonderen A, Leypoldt F, et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology. 2015;84(24):2403–2412. | ||
Graus F, Boronat A, Xifró X, et al. The expanding clinical profile of anti-AMPA receptor encephalitis. Neurology. 2010;74(10):857–859. | ||
Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. J Neurol. 2008;7(4):327–340. | ||
Bataller L, Galiano R, García-Escrig M, et al. Reversible paraneoplastic limbic encephalitis associated with antibodies to the AMPA receptor. Neurology. 2010;17(8):254–256. | ||
Peng X, Hughes EG, Moscato EH, Parsons TD, Dalmau J, Balice-Gordon RJ. Cellular plasticity induced by anti-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann Neurol. 2015;77(3):381–398. | ||
Gleichman AJ, Panzer JA, Baumann BH, Dalmau J, Lynch DR. Antigenic and mechanistic characterization of anti-AMPA receptor encephalitis. Ann Clin Transl Neurol. 2014;1(3):180–189. | ||
Gu W, Brodtkorb E, Steinlein OK. LGI1 is mutated in familial temporal lobe epilepsy characterized by aphasic seizures. Ann Neurol. 2002;52(3):364–367. | ||
Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9(8):776–785. | ||
Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol. 1997;41(2):238–246. | ||
Lee EK, Maselli RA, Ellis WG, Agius MA. Morvan’s fibrillary chorea: a paraneoplastic manifestation of thymoma. J Neurol Neurosurg Psychiatry. 1998;65(6):857–862. | ||
Barber PA, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology. 2000;54(3):771–772. | ||
Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol. 2001;50(1):73–78. | ||
Willis MD, Jones L, Vincent A, Wheeler R, O’Callaghan P, Hamandi K. VGKC-complex antibody encephalitis. QJM. 2014;107(8):657–659. | ||
Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain. 2010;133(9):2734–2748. | ||
Joubert B, Saint-Martin M, Noraz N, et al. Characterization of a subtype of autoimmune encephalitis with anti–contactin-associated protein-like 2 antibodies in the cerebrospinal fluid, prominent limbic symptoms, and seizures. JAMA Neurol. 2016;73(9):1115–1124. | ||
Ohkawa T, Fukata Y, Yamasaki M, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci. 2013;33(46):18161–18174. | ||
Navarro V, Kas A, Apartis E, et al. Motor cortex and hippocampus are the two main cortical targets in LGI1-antibody encephalitis. Brain. 2016;139(4):1079–1093. | ||
Malter MP, Frisch C, Schoene-Bake JC, et al. Outcome of limbic encephalitis with VGKC-complex antibodies: relation to antigenic specificity. J Neurol. 2014;261(9):1695–1705. | ||
Mayasi Y, Takhtani D, Garg N. Leucine-rich glioma-inactivated protein 1 antibody encephalitis: a case report. Neurol Neuroimmunol Neuroinflamm. 2014;1(4):e51. | ||
Casault C, Alikhani K, Pillay N, Koch M. Jerking & confused: leucine-rich glioma inactivated 1 receptor encephalitis. J Neuroimmunol. 2015;289:84–86. | ||
Messelmani M, Fekih-Mrissa N, Zaouali J, Mrissa R. Limbic encephalitis associated with leucine-rich glioma-inactivated 1 antibodies. Ann Saudi Med. 2015;35(1):76–79. | ||
Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69(5):892–900. | ||
Montojo MT, Petit-Pedrol M, Graus F, Dalmau J. Clinical spectrum and diagnostic value of antibodies against the potassium channel-related protein complex. Neurologia. 2015;6(2):356–372. | ||
Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127(3):701–712. | ||
Merchut M. Management of voltage-gated potassium channel antibody disorders. Neurol Clin. 2010;28(4):941–959. | ||
Radja GK, Cavanna AE. Treatment of VGKC complex antibody-associated limbic encephalitis: a systematic review. J Neuropsychiatry Clin Neurosci. 2013;25(4):264–271. | ||
McQuillan RF, Bargman JM. Hyponatraemia caused by LGI1-associated limbic encephalitis. NDT Plus. 2011;4(6):424–426. | ||
Thieben MJ, Lennon VA, Boeve BF, Aksamit AJ, Keegan M, Vernino S. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology. 2004;62(7):1177–1182. | ||
Irani SR, Buckley C, Vincent A, et al. Immunotherapy-responsive seizure-like episodes with potassium channel antibodies. Neurology. 2008;71(20):1647–1648. | ||
Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology. 2008;70(20):1883–1890. | ||
Quek AML, Britton JW, McKeon A, et al. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol. 2012;69(5):582–593. | ||
Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology. 2011;77(2):179–189. | ||
Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain. 2013;136(pt 10):3151–3162. | ||
Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet. 2005;14(12):1613–1620. | ||
Schulte U, Thumfart J, Klöcker N, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49(5):697–706. | ||
Lancaster E, Huijbers MGM, Bar V, et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol. 2011;69(2):303–311. | ||
Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313(5794):1792–1795. | ||
Fukata Y, Lovero KL, Iwanaga T, et al. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci U S A. 2010;107(8):3799–3804. | ||
Irani SR, Pettingill P, Kleopa KA, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol. 2012;72(2):241–255. | ||
Lalic T, Pettingill P, Vincent A, Capogna M. Human limbic encephalitis serum enhances hippocampal mossy fiber-CA3 pyramidal cell synaptic transmission. Epilepsia. 2011;52(1):121–131. | ||
Poliak S, Gollan L, Martinez R, et al. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 1999;24(4):1037–1047. | ||
Sunwoo J-S, Lee S-T, Byun J-I, et al. Clinical manifestations of patients with CASPR2 antibodies. J Neuroimmunol. 2015;281:17–22. | ||
Saiz A, Blanco Y, Sabater L, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain. 2008;131(pt 10):2553–2563. | ||
Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874–881. | ||
Fouka P, Alexopoulos H, Akrivou S, Trohatou O, Politis PK, Dalakas MC. GAD65 epitope mapping and search for novel autoantibodies in GAD-associated neurological disorders. J Neuroimmunol. 2015;281:73–77. | ||
Buddhala C, Hsu CC, Wu JY. A novel mechanism for GABA synthesis and packaging into synaptic vesicles. Neurochem Int. 2009;55(1–3):9–12. | ||
Gresa-Arribas N, Ariño H, Martínez-Hernández E, et al. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS One. 2015;10(3):e0121364. | ||
Honnorat J, Saiz A, Giometto B, et al. Cerebellar ataxia with anti-glutamic acid decarboxylase antibodies: study of 14 patients. Arch Neurol. 2001;58(2):225–230. | ||
Çoban A, Ismail Küçükali C, Bilgiç B, et al. Evaluation of incidence and clinical features of antibody-associated autoimmune encephalitis mimicking dementia. Behav Neurol. 2014;2014:935379. | ||
Padmos RC, Bekris L, Knijff EM, et al. A high prevalence of organ-specific autoimmunity in patients with bipolar disorder. Biol Psychiatry. 2004;56(7):476–482. | ||
Meinck HM, Faber L, Morgenthaler N, et al. Antibodies against glutamic acid decarboxylase: prevalence in neurological diseases. J Neurol Neurosurg Psychiatry. 2001;71(1):100–103. | ||
Pittock SJ, Lucchinetti CF, Parisi JE, et al. Amphiphysin autoimmunity: paraneoplastic accompaniments. Ann Neurol. 2005;58(1):96–107. | ||
Vernino S, Lennon VA. Autoantibody profiles and neurological correlations of thymoma. Clin Cancer Res. 2004;10(21):7270–7275. | ||
Manto M, Honnorat J, Hampe CS, et al. Disease-specific monoclonal antibodies targeting glutamate decarboxylase impair GABAergic neurotransmission and affect motor learning and behavioral functions. Front Behav Neurosci. 2015;9:78. | ||
Carvalho F, Massano J, Coelho R. Neuropsychiatric symptoms in autoimmune encephalopathies: a clinician’s guide. Int J Clin Neurosci Ment Heal. 2014;1:1–11. | ||
Andrade DM, Tai P, Dalmau J, Wennberg R. Tonic seizures: a diagnostic clue of anti-LGI1 encephalitis? Neurology. 2011;77(24):2140. | ||
Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167–177. | ||
Amatoury M, Merheb V, Langer J, Wang XM, Dale RC, Brilot F. High-throughput flow cytometry cell-based assay to detect antibodies to N-methyl-D-aspartate receptor or dopamine-2 receptor in human serum. J Vis Exp. 2013;(81):e50935. | ||
Sinmaz N, Amatoury M, Merheb V, Ramanathan S, Dale RC, Brilot F. Autoantibodies in movement and psychiatric disorders: updated concepts in detection methods, pathogenicity, and CNS entry. Ann N Y Acad Sci. 2015;5:1–17. | ||
Pollak TA, McCormack R, Peakman M, Nicholson TR, David AS. Prevalence of anti-N-methyl-d-aspartate (NMDA) antibodies in patients with schizophrenia and related psychoses: a systematic review and meta-analysis. Psychol Med. 2013;46(1):1–13. | ||
Masopust J, Andrys C, Bazant J, Vysata O, Kuca K, Valis M. Anti-NMDA receptor antibodies in patients with a first episode of schizophrenia. Neuropsychiatr Dis Treat. 2015;11:619–623. | ||
Masdeu JC, González-Pinto A, Matute C, et al. Serum IgG antibodies against the NR1 subunit of the NMDA receptor not detected in schizophrenia. Am J Psychiatry. 2012;169(10):1120–1121. | ||
Rhoads J, Guirgis H, McKnight C, Duchemin AM. Lack of anti-NMDA receptor autoantibodies in the serum of subjects with schizophrenia. Schizophr Res. 2011;129(2–3):213–214. | ||
Pathmanandavel K, Starling J, Merheb V, et al. Antibodies to surface dopamine-2 receptor and N-methyl-D-aspartate receptor in the first episode of acute psychosis in children. Biol Psychiatry. 2015;77(6):537–547. | ||
Lancaster E, Leypoldt F, Titulaer MJ, et al. IgG antibodies to the NMDA receptor are distinct from IgA and IgM responses. Ann Neurol. 2014;1:822–832. | ||
Lennox BR, Coles AJ, Vincent A. Antibody-mediated encephalitis: a treatable cause of schizophrenia. Br J Psychiatry. 2012;200(2):92–94. | ||
Barry H, Hardiman O, Healy DG, et al. Anti-NMDA receptor encephalitis: an important differential diagnosis in psychosis. Br J Psychiatry. 2011;199(6):508–509. | ||
Kuppuswamy PS, Takala CR, Sola CL. Management of psychiatric symptoms in anti-NMDAR encephalitis: a case series, literature review and future directions. Gen Hosp Psychiatry. 2014;36(4):388–391. | ||
Chapman MR, Vause HE. Anti-NMDA receptor encephalitis: diagnosis, psychiatric presentation, and treatment. Am J Psychiatry. 2011;168(3):245–251. | ||
Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ, Dalmau J, Friedman D. Extreme delta brush; a unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurology. 2012;79(11):1094–1100. | ||
Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014;13(3):276–286. | ||
Ohkawa T, Satake S, Yokoi N, et al. Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis. J Neurosci. 2014;34(24):8151–8163. | ||
Pettingill P, Kramer HB, Coebergh JA, et al. Antibodies to GABAA receptor alpha1 and gamma2 subunits: Clinical and serologic characterization. Neurology. 2015;84(12):1233–1241. | ||
Lancaster E, Lai M, Peng X, et al. Antibodies to the GABAB receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010;9(1):67–76. | ||
Boronat A, Sabater L, Saiz A, Dalmau J, Graus F. GABA(B) receptor antibodies in limbic encephalitis and anti-GAD-associated neurologic disorders. Neurology. 2011;76(9):795–800. | ||
Höftberger R, Titulaer MJ, Sabater L, et al. Encephalitis and GABAB receptor antibodies: Novel findings in a new case series of 20 patients. Neurology. 2013;81(17):1500–1506. | ||
Lancaster E, Martinez-Hernandez E, Titulaer MJ, et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology. 2011;77(18):1698–1701. | ||
Mat A, Adler H, Merwick A, et al. Ophelia syndrome with metabotrophic glutamat receptor 5 antibodies in CSF. Neurology. 2013;2(C):1349–1350. | ||
Prüss H, Rothkirch M, Kopp U, et al. Limbic encephalitis with mGlur5 antibodies and immunotherapy-responsive prosopagnosia. Neurology. 2014;83(15):1384–1386. | ||
Dale RC, Merheb V, Pillai S, et al. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain. 2012;135(11):3453–3468. | ||
Hutchinson M, Waters P, McHugh J, et al. Progressive encephalomyelitis, rigidity and myoclonus: a novel glycine receptor antibody. Neurology. 2008:1289–1292. | ||
Piotrowicz A, Thümen A, Leite MI, Vincent A, Moser A. A case of glycine-receptor antibody-associated encephalomyelitis with rigidity and myoclonus (PERM): Clinical course, treatment and CSF findings. J Neurol. 2011;258(12):2268–2270. | ||
McKeon A, Martinez-Hernandez E, Lancaster E, et al. Glycine Receptor Autoimmune Spectrum With Stiff-Man Syndrome Phenotype. JAMA Neurol. 2013;70(1):44. | ||
Carvajal-González A, Leite MI, Waters P, et al. Glycine receptor antibodies in PERM and related syndromes: Characteristics, clinical features and outcomes. Brain. 2014;137(8):2178–2192. | ||
Vincent A, Irani SR. Caspr2 antibodies in patients with thymomas. J Thorac Oncol. 2010;5(10 Suppl 4):S277–S280. | ||
Boronat A, Gelfand JM, Gresa-Arribas N, et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol. 2013;73(1):120–128. | ||
Tobin WO, Lennon VA, Komorowski L, et al. DPPX potassium channel antibody: Frequency, clinical accompaniments, and outcomes in 20 patients. Neurology. 2014;83(20):1797–1803. | ||
Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol. 2010;67(4):470–478. | ||
Ali F, Rowley M, Jayakrishnan B, Teuber S, Gershwin ME, Mackay IR. Stiff-person syndrome (SPS) and anti-GAD-related CNS degenerations: Protean additions to the autoimmune central neuropathies. J Autoimmun. 2011;37(2):79–87. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.