Back to Journals » Clinical and Experimental Gastroenterology » Volume 10
Autoimmune atrophic gastritis: current perspectives
Authors Minalyan A, Benhammou JN, Artashesyan A, Lewis MS, Pisegna JR
Received 14 October 2016
Accepted for publication 17 January 2017
Published 7 February 2017 Volume 2017:10 Pages 19—27
DOI https://doi.org/10.2147/CEG.S109123
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Andreas M. Kaiser
Artem Minalyan,1 Jihane N Benhammou,1 Aida Artashesyan,1 Michael S Lewis,2 Joseph R Pisegna1
1Division of Gastroenterology, Hepatology and Parenteral Nutrition, 2Department of Pathology and Laboratory Medicine, VA Greater Los Angeles Healthcare System, Los Angeles, CA, USA
Abstract: At present there is no universally accepted classification for gastritis. The first successful classification (The Sydney System) that is still commonly used by medical professionals was first introduced by Misiewicz et al in Sydney in 1990. In fact, it was the first detailed classification after the discovery of Helicobacter pylori by Warren and Marshall in 1982. In 1994, the Updated Sydney System was proposed during the International Workshop on the Histopathology of Gastritis followed by the publication in The American Journal of Surgical Pathology by Dixon et al. Using the new classification, distinction between atrophic and nonatrophic gastritis was revised, and the visual scale grading was incorporated. According to the Updated Sydney System Classification, atrophic gastritis is categorized into multifocal (H. pylori, environmental factors, specific diet) and corpus-predominant (autoimmune). Since metaplasia is a key histological characteristic in patients with atrophic gastritis, it has been recommended to use the word “metaplastic” in both variants of atrophic gastritis: autoimmune metaplastic atrophic gastritis (AMAG) and environmental metaplastic atrophic gastritis. Although there are many overlaps in the course of the disease and distinction between those two entities may be challenging, the aim of this review article was to describe the etiology, epidemiology, pathogenesis, diagnosis, clinical manifestations and treatment in patients with AMAG. However, it is important to mention that H. pylori is the most common etiologic factor for the development of gastritis in the world.
Keywords: autoimmune gastritis, pernicious anemia, gastric carcinoid
Epidemiology
The prevalence of pernicious anemia (PA; one of the distinctive manifestations of autoimmune metaplastic atrophic gastritis [AMAG]) is ~0.1% in the general population and ~2% in those older than 60 years.1 For many years, it was suggested that PA is the disease of elderly females of Northern European ethnicity. Subsequently, it was shown that the prevalence of PA is similar among all populations (white, African American and non-white Hispanic).2 Based on the analysis of the data from 156 patients affected by PA, in 1978, Carmel and Johnson3 concluded that black women develop PA at younger age than women of all other races (53±16 years, p<0.001). Similar to other autoimmune conditions, AMAG is more common in females than in males (3:1 ratio). Strong association between AMAG and other autoimmune disorders has been well documented.4 It is believed that AMAG and PA might be underdiagnosed in all parts of the world. One of the explanations could be the fact that almost all forms of micro- and macrocytic anemia are treated with iron, folic acid and cobalamin without further identifying the etiology of the anemia. Moreover, in many cases, biopsy samples of the gastric mucosa are inadequate or taken from the wrong location.5
Pathogenesis
The understanding of the pathogenesis of AMAG is somewhat challenging for several reasons: 1) the prevalence of AMAG is relatively low, 2) in many cases there is a concurrent Helicobacter pylori-induced gastritis and 3) no or minimal manifestations are present in early stages of the disease. Nevertheless, we know that both genetic and environmental factors play a role in the development of autoimmune gastritis. Using murine models, it was possible to discover autoimmune gastritis susceptibility genes (Gasa1, 2, 3 and 4) on chromosomes 4 and 6 and H2 region. Interestingly, three of these genes are located on the same locus as non-obese diabetic mouse diabetes mellitus (DM) susceptibility genes, which could explain the strong association between AMAG and DM type 1 (T1DM).6,7
Despite a lack of understanding of the complex stages of AMAG pathogenesis, histological changes have been well studied (Figures 1 and 2). The initial changes include infiltration of oxyntic mucosa by lymphocytes and plasma cells. The uneven destruction of parietal cells with preserved islands of relatively normal oxyntic mucosa leads to the appearance of “islands in the sea”. This phenomenon, also known as gastric pseudopolyposis, is similar to colonic lesions in patients with ulcerative colitis.8,9 Hypochlorhydria, or the decrease/loss of hydrochloric acid secretion, then develops as a result of the combination of two factors: 1) loss of oxyntic mucosa and 2) disruption of maturation of parietal cells.10 Absence of negative feedback from parietal cells induces G-cell hyperplasia and increased gastric secretion in the tantrum, which, in turn, leads to parietal cell pseudo hypertrophy resembling proton pump inhibitor-induced effects on parietal cells.11 It is worth mentioning that this phenomenon cannot develop in atrophic glands, since anti-parietal cell antibodies also bind selectively to H+/K+ ATPase.12 Another important effect of elevated gastric secretion is its direct stimulation and proliferation of enterochromaffin-like (ECL) cells, which depends on severity and can be hyperplastic, dysplastic and neoplastic.13 Progression of ECL cell hyperplasia to neoplastic subtype can result in carcinoid tumor formation, which will be described subsequently. Areas of metaplasia develop within gastric body and fundus: pseudopyloric, intestinal metaplasia (IM) and pancreatic. Pseudopyloric metaplasia (“oxyntic antralization”) develops in oxyntic glands from mucus-secreting cells phenotypically resembling antral mucous cells. Although histologically pseudopyloric metaplasia may be indistinctive from antral gland cells, pepsinogen (PG) immunostaining can be helpful in differentiating: pepsinogen I (PGI) is characteristic of cells of oxyntic mucosa and is absent in the antrum.14 Recently, a new term to describe pseudopyloric metaplasia has been widely used: spasmolytic polypeptide-expressing metaplasia (SPEM). The etiology of SPEM is not clear. Several theories have been proposed: 1) longstanding inflammation of oxyntic mucosa, 2) acute direct effect of toxic substances on parietal cells (DMP-777)15,16 and 3) transdifferentiation of chief cells.17 Chief cells and SPEM express MAL2. This protein is involved in vesicle trafficking and considered the first trafficking protein, which is upregulated in SPEM.18 It has been shown that SPEM is a preneoplastic gastric lesion19 that can also transform into IM.20 IM is the replacement of glandular, foveolar and surface epithelium of gastric mucosa by intestinal epithelium. Depending on the histological features, two subtypes have been described: small intestinal and colonic. They, in turn, can be grouped into three types, type 1, or small intestinal, with brush border, “complete”, which are replaced fully by small intestinal epithelium with all types of cells (goblet, Paneth, etc), expresses syalomucins. Type 3, or colonic type, without brush border, “incomplete”, are metaplastic cells that are formed in the neighborhood of normal gastric cells, expresses sulfomucins. Type 2, or colonic type, without brush border, “incomplete”, where metaplasia is similar to Type 3, expresses both gastric and intestinal mucins.21 Apart from the abovementioned pathological changes, inflammatory and hyperplastic polyps develop in the later stages of AMAG.22 Oxyntic mucosa can be totally absent in advanced disease.
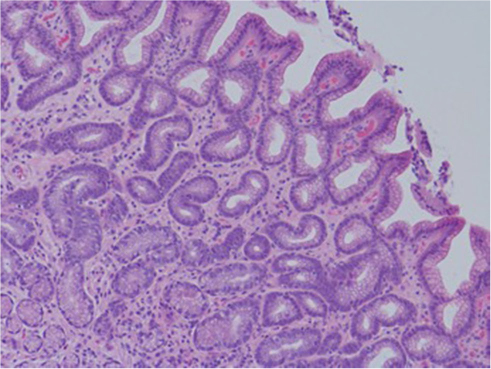 | Figure 1 Human gastric biopsy showing gastric gland atrophy. |
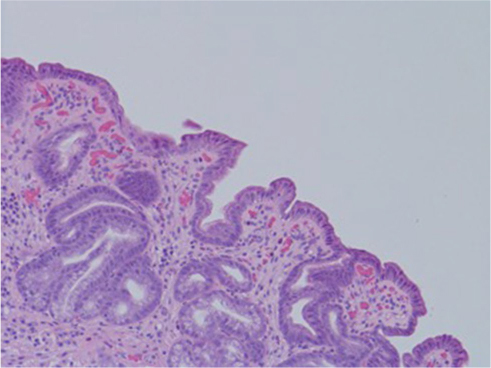 | Figure 2 Human gastric biopsy showing lymphocytic infiltrate. |
Association with H. pylori
It is a well-known fact that H. pylori can cause atrophic gastritis on its own. On the other hand, it is important to remember that in many cases patients with AMAG may have concurrent H. pylori infection.23 A third possible scenario is the progression of H. pylori-induced gastritis to AMAG. It is thought that etiology of this process is antigenic mimicry or cross-reactivity.24 Many patients with H. pylori develop a wide spectrum of antibodies, which includes antifoveolar, anticanalicular and the classic APCA. The most frequently detected antibodies are anticanalicular antibodies that, similar to APCA, are directed against H+/K+ ATPase (proton pump).25 Despite many overlaps in the course of the disease, it is important to differentiate AMAG from H. pylori-induced gastritis. AMAG affects only the body and fundus of the stomach, while H. pylori-induced lesions are known to have a multifocal pattern (Table 1).26 Other important factors that help differentiate between AMAG and H. pylori-induced gastritis are 1) ECL hyperplasia (more common in AMAG), 2) parietal cell pseudohypertrophy (in AMAG; can be seen in H. pylori infection secondary to proton pump inhibitor use) and 3) oxyntic gland involvement (more common in AMAG).
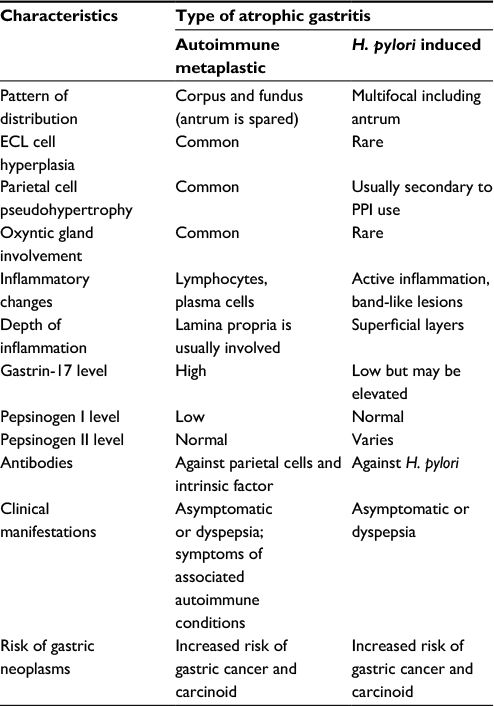 | Table 1 Comparison of AMAG with H. pylori-induced atrophic gastritis Abbreviations: AMAG, autoimmune metaplastic atrophic gastritis; H. pylori, Helicobacter pylori; ECL, enterochromaffin like; PPI, proton pump inhibitor. |
Additionally, inflammatory changes are quite distinctive in both of these conditions: lymphocytes and plasma cells infiltrating lamina propria with involvement of deep layers (AMAG), superficial band-like lesions with active inflammation (H. pylori-induced atrophic gastritis).27 Gastrin-17 (high in AMAG, low although may be elevated in H. pylori infection) and PGI levels (low in AMAG, normal in H. pylori infection) may also be utilized in patients at risk for gastric cancer (GC).28
Association with other autoimmune conditions
It has been well recognized that AMAG has tendency to occur more often in patients with other autoimmune conditions.29 Up to one-third of patients with autoimmune thyroid disease (AITD) and 6–10% of patients with T1DM have concurrent AMAG.30,31 Patients with polyglandular autoimmune (PGA) syndromes (especially PGA type 3B, which always includes AITD) are known to have a high prevalence of PA.32 The other autoimmune conditions that have been known to co-occur with AMAG (or its advanced form PA) are vitiligo, Addison’s disease, myasthenia gravis and perioral cutaneous autoimmune conditions (especially, erosive oral lichen planus).33–35
Familial cases of pernicious anemia (FPA) have been described including those in twins, suggesting genetic component of the disease.36 However, more studies need to be conducted to shed more light on how genetic variations predispose to the development of AMAG.37 Very rare cases of coexistence of two inherited disorders (FPA and hereditary hemochromatosis) have also been described in the literature.38
Diagnosis
In this section, we discuss the most common diagnostic tests used for screening and confirming a diagnosis of AMAG: serologic tests, endoscopy and histopathology.
Serological tests
The detection of target-specific antibodies has been well recognized as an effective method for screening and confirmation purposes. Among antibodies used for the diagnosis of AMAG, the following group is most widely used: APCA, anti-intrinsic factor antibodies (AIFAs) and anti-H. pylori antibodies (anti-HP-IgM and anti-HP-IgG). The significance of the measurement of anti-HP-antibodies is due to the frequent coexistence of H. pylori-induced gastritis and AMAG. APCA are known to have high sensitivity (~80%) but low specificity for PA.39 In contrast, AIFAs have been shown to be less sensitive (~50%) but more specific for PA.40 In patients with PA and negative AIFA, seroconversion may occur later in the course of the disease.41 Measurement of gastrin level (mainly Gastrin-17) is another diagnostic test that can be abnormal in patients with AMAG due to oxyntic gland atrophy, leading to elevated gastrin production by G-cells in the antrum. The combined assay of APCA, AIFA and anti-HP-antibodies along with the measurement of serum gastrin (Gastrin-17) are also known as “serological gastric biopsy”. It has been recognized to be effective for diagnostic purposes and highly correlates with the histological findings.42
The hypergastrinemia observed may be clinically difficult to differentiate from the other causes of hypergastrinemia. One method to identify the cause of hypergastrinemia is to measure the level of gastric acid output. With atrophic gastritis, the level of gastric acid output is low (<5 mEq/h), whereas with the hypergastrinemia associated with Zollinger-Ellison syndrome, the gastric acid output is >10 mEq/h. Furthermore, the secretin test is negative in >80% of patients.43 Another commonly overlooked cause of hypergastrinemia is gastric outlet obstruction and gastric dysmotility, which may require an upper endoscopy and radiological studies to be evaluated. With gastric dysmotility, the parietal cell complement will be either normal or increased.44 The measurement of PGI and PGII as well as PGI/PGII ratio can also be used to suspect AMAG. Specifically, PGI is secreted by the chief cells of oxyntic mucosa of gastric corpus and fundus, while PGII is produced by the chief cells and mucous neck cells of the entire gastric mucosa. Therefore, oxyntic atrophy is characteristic of AMAG, resulting in a decrease in PGI level along with low PGI/PGII ratio (<3).45 PGII level is mainly unaffected by atrophy of oxyntic mucosa.
Patients with laboratory confirmation of macrocytic anemia require serum vitamin B12 measurement. When high suspicion of PA is present, normal vitamin B12 level should be followed by homocysteine (HCys) and methylmalonic acid (MMA) measurements.46 However, it has also been shown that the serum levels of all three tests (vitamin B12, HCys and MMA) can fluctuate over time and even be normal in cobalamin-responsive disorders.47
The measurement of the plasma chromogranin A (CgA) level has been suggested for the diagnosis of AMAG, ECL hyperplasia and gastric carcinoids. CgA levels have been found to correlate well with the degree of ECL hyperplasia in patients with AMAG. In one study, the elevated CgA is thought to be associated with the development of nonalcoholic fatty liver disease (NAFLD) and is undergoing additional investigation.48 At the same time, the specificity of CgA level is too low (23%) to reveal gastric carcinoids in that group of patients.49
In other conditions, CgA can lead to false-positive results, including in inflammatory bowel diseases, hepatocellular carcinomas50 or other conditions such as with the chronic use of proton pump inhibitors or in renal insufficiency.
Endoscopy
In Western countries, endoscopic techniques for the diagnosis of AMAG have been used mainly for biopsy sampling purposes. The limitations related to endoscopy are the following: 1) low sensitivity, 2) low specificity and 3) interobserver variability.51 However, with the advent of novel endoscopic modalities (magnifying endoscopy, narrow-band imaging [NBI] and autofluorescence imaging [AFI]), it has become possible to detect minimal atrophic changes of the gastric mucosa.52 For instance, it has been shown that normal gastric corpus and fundus mucosal microvessels are characterized by the presence of 1) subepithelial capillary network (SECN) resembling honeycomb and 2) collecting venules in regular shape and appearance. In patients with atrophic gastritis, these normal findings are not seen.53 It is worth noting that none of the endoscopic findings should be used alone to make a diagnosis of atrophic gastritis. In fact, the combination of them (for example, vascular pattern and swelling of areae gastrica) can significantly improve diagnostic accuracy.54 As previously mentioned, in patients with high suspicion of atrophic gastritis, endoscopy has been used to get biopsy samples. According to the updated Sydney system recommendations, five biopsy samples should be obtained: two from the corpus, two from the antrum and one from the incisura angularis.55
Histopathology
Biopsy with subsequent histology is considered the most reliable method for the diagnosis of metaplastic atrophic gastritis. Changes described in the “Pathogenesis” section may be seen. In some cases, when severe inflammation is present, it is hard to reliably evaluate oxyntic atrophy. Another limitation can happen when interpreting histological results from patients with early stages of AMAG. Several diagnostic hints have been suggested that may be characteristic of AMAG before profound loss of oxyntic mucosa: 1) infiltrates of lymphocytes and plasma cells in lamina propria, 2) focal atrophy of oxyntic mucosa along with SPEM or IM, 3) pseudohypertrophy of parietal cells and 4) ECL hyperplasia (at least, linear level).
Since ECL hyperplasia is the precursor of gastric carcinoid tumors, it is important to stain corpus and fundus samples with CgA and synaptophysin.56 Additionally, staining for gastrin (negative in case of corpus/fundus biopsy) can help in localizing the site of a biopsy sample.57
Clinical manifestations
The symptoms related to atrophic gastritis develop slowly and, in fact, may have a long asymptomatic period. In many cases, it is challenging to diagnose AMAG or its advanced form PA based only on clinical manifestations. The affected patients may present with a variety of signs and symptoms ranging from weakness to severe neurological manifestations such as paranoia (megaloblastic madness).58 That is why among medical professionals, PA is also known as a “great pretender”. In this article, we would like to categorize clinical manifestations into three major groups: hematological, neurological and gastrointestinal.
Hematological manifestations
Iron-deficiency anemia (IDA) is one of the earliest presentations of AMAG that develops due to achlorhydria from oxyntic atrophy. Several studies have shown that refractory or unexplained IDA should increase clinicians’ suspicion of AMAG not only in adults but also in pediatric population.59–61 PA is the most common cause of cobalamin deficiency.62 Although the underlying mechanisms of how cobalamin deficiency causes megaloblastosis have not been fully elucidated, it is believed that it is due to cobalamin’s essential role in DNA synthesis.63 The asymptomatic increase in mean corpuscular volume and hypersegmented neutrophils are usually the initial laboratory findings in PA. With progression of the disease patients may complain of the weakness, light-headedness and palpitations. In advanced stages, symptoms of angina and congestive heart failure (peripheral edema and shortness of breath) may manifest.64 Other very significant laboratory findings include thrombocytopenia, increased levels of LDH and bilirubin (signs of hemolysis). In rare cases, schistocytes in the peripheral smear may mimic other more serious conditions, such as disseminated intravascular coagulopathy (DIC) or thrombotic thrombocytopenic purpura/hemolytic uremic syndrome (TTP/HUS).65 Cobalamin deficiencies have been associated with increased risk of thrombosis likely as a result of hyperhomocysteinemia. Elevated levels of HCys cause endovascular dysfunction. This, in turn, activates the coagulation system, increases platelet aggregation and constricts vessels. Acute myocardial infarction and pulmonary embolism have been described in young patients with severe hyperhomocysteinemia secondary to PA.66,67
Neurological manifestations
Neurological manifestations of cobalamin deficiency have long been associated with demyelination followed by axonal damage and eventually neuronal death.68 The pathophysiological changes are not fully understood, however, MMA accumulation has been recognized to have direct impact on myelin formation and attributes to the myriad of neurological problems in patients with cobalamin deficiency.69 Interestingly, neurological symptoms associated with cobalamin deficiency may be present in the absence of hematological changes.70,71 Subacute combined degeneration (SCD) is one of the most prominent neurological manifestations of PA. Posterior and lateral columns of the cervical and upper thoracic segments of the spinal cord are involved. This results in sensory abnormalities (usually initial symptoms) such as loss of vibratory and position sensations along with distal paresthesias. In advanced stages, corticospinal tract can also be affected.72 Although in many situations, the neurological damage is irreversible, it is crucial to timely treat these patients with parenteral vitamin B12 in order to stop progression and improve neurological deficits.73 Peripheral neuropathy is another common neurological manifestation of PA. The initial symptoms include paresthesia and numbness of the lower extremities. In this regard, it can be very difficult to distinguish clinically between peripheral neuropathy and SCD.74 Both asymptomatic and overt optic neuropathy cases have been described in the medical literature.75
Neuropsychiatric conditions associated with PA include mania, depression, obsessive-compulsive disorder (OCD), psychosis and dementia.76 It is worth mentioning that the use of high dose of vitamins B12, B6 and folic acid has been shown to slow the shrinkage of the whole brain as well as reduce the cerebral atrophy in patients with Alzheimer’s disease (AD).77
Gastrointestinal manifestations
Counterintuitively, AMAG rarely causes gastrointestinal symptoms. It has been suggested that this is due to the fact that most of the upper gastrointestinal-related complaints are secondary to increased level of hydrochloric acid. Hence, AMAG resulting in hypo- or even achlorhydria usually presents only with vague dyspeptic symptoms such as bloating, early satiety and epigastric discomfort.78 Atrophic glossitis, which usually presents as burning sensation of the tongue, has also been known as an early clinical manifestation of cobalamin deficiency.79
Neoplastic complications
The incidence of gastric neoplasms is higher in patients with AMAG compared to the general population.80,81 Overall, AMAG has been associated with the development of two types of gastric neoplasms: intestinal type GC and type I gastric carcinoid (TIGC).82
Intestinal-type gastric cancer
Two main factors that have been known to explain the occurrence of GC in AMAG patients are IM and concurrent H. pylori infection, which per se is the most common trigger of IM of the gastric mucosa.83 It should be pointed out that eradication of H. pylori in patients with known precancerous lesions (gastric atrophy, IM or gastric dysplasia) does not significantly lower the incidence of GC.84 The pathogenesis of the development of GC has been described by Correa and Piazuelo85 and is better known as “Correa cascade”. A simplified model of the cascade can be divided into following steps: 1) normal gastric mucosa, 2) nonatrophic gastritis, 3) multiple atrophic gastritis without (initially) and with (later) IM, 4) dysplasia progressing from low grade to high grade and 5) GC.86 It should be noted that AMAG without concurrent H. pylori infection is not considered to be part of the abovementioned cascade. Not all patients with H. pylori-induced gastritis develop GC. The chances are higher when certain virulence factors are present. For instance, CagA-positive strains of H. pylori have been shown to have significantly higher risk of the development of peptic ulcer and GC compared to CagA-negative strains.87 Another well-known virulence factor known to be associated with GC is VacA gene. Variations in this gene can be explained by the presence of signal (s) and middle (m) regions.88 Several studies have demonstrated the increased incidence of peptic ulcer and GC in patients infected with s1m1 compared to those infected with s2m2.89
Type I gastric carcinoid
Although it has been recognized that AMAG is associated with increased risk of the development of TIGC, the exact incidence is not known. There are three known types of gastric carcinoids: type I is associated with AMAG, type II can be present in patients with multiple endocrine neoplasia (MEN) I and Zollinger-Ellison syndrome and type III, the most aggressive variant, usually occurs sporadically. Serum gastrin levels vary depending on the type of gastric carcinoid: types I and II – high, and type III – normal (Table 2).90 The pathologic changes in T1GC are initiated by the loss of negative feedback by parietal cells on gastrin secretion. Hypo-/achlorhydria subsequently results in hypergastrinemia which, in turn, has trophic effects on ECL cells. ECL hyperplasia followed by ECL dysplasia is considered precancerous lesions and over time can progress to T1GC.91 The detection of body polyps during endoscopy in patients with AMAG is strongly associated with the presence of TIGC. Patients with TIGC are usually asymptomatic, although symptoms of dyspepsia and IDA can be present. That is why the diagnosis is usually made during gastroscopy.92
 | Table 2 Comparison of different types of gastric carcinoids Abbreviations: AMAG, autoimmune metaplastic atrophic gastritis; MEN1, multiple endocrine neoplasia type 1; ZES, Zollinger-Ellison syndrome; ECL, enterochromaffin like; N, normal. |
Treatment
The management strategies for AMAG and its advanced form PA depend on clinical manifestations, laboratory and imaging results. The therapy with the supplementation of iron, folic acid and cobalamin is commonly recommended in the early stages prior to progression to PA. Oral vitamin B12 supplement is used since it has been shown 1% of free vitamin B12 can be absorbed in the small intestine via passive diffusion.93 However, in patients with neurological manifestations, parenteral administration of vitamin B12 is currently recommended.
Given the fact that AMAG is highly associated with other autoimmune conditions, appropriate diagnostic investigation should be conducted especially for disorders such as autoimmune thyroiditis and T1DM.
Detection and subsequent treatment for H. pylori infection is another very important therapeutic strategy due to associated risks related to H. pylori coinfection. Since it has been confirmed by several studies that H. pylori infection can induce autoimmune process in the gastric lining including oxyntic mucosa, eradication of H. pylori can decrease the levels of antibodies associated with AMAG and has been proven effective to cure early stages of autoimmune gastritis.94,95
T1GC has an excellent prognosis, therefore, different approaches have been suggested: surveillance, polypectomy and antrectomy (removal of gastrin-producing part of the stomach).96 One of the potential treatments for TIGC has been recently introduced. Netazepide, a gastrin receptor antagonist, has been shown to decrease the plasma level of CgA along with tumor number and size.97–99 Another treatment option is to consider using somatostatin analogs. In one prospective study in a population of 107 with chronic atrophic gastritis, the authors showed that the use of somatostatin analogs resulted in a reduction in median gastrin and CgA levels and thus should be considered as effective therapy.100 A new and emerging form of therapy for the treatment of neuroendocrine tumors is the use of peptide receptor radiotherapy (PRRT), although these newer therapies have not been prospectively studied to date for autoimmune atrophic gastritis.101
Acknowledgments
This work received grant support from Department of Veterans Affairs RR&D Merit Review (JRP) I01 RX000194; Human Studies CORE through CURE: Digestive Diseases Research Center supported by NIH grant P30DK41301 and NIH NIDDK T32 NIH5T32DK07180 (JNB).
Disclosure
The authors report no conflicts of interest in this work.
References
Andres E, Serraj K. Optimal management of pernicious anemia. J Blood Med. 2012;3:97–103. | ||
Park JY, Lam-Himlin D, Vemulapalli R. Review of autoimmune metaplastic atrophic gastritis. Gastrointest Endosc. 2013;77(2):284–292. | ||
Carmel R, Johnson CS. Racial patterns in pernicious anemia. Early age at onset and increased frequency of intrinsic-factor antibody in black women. N Engl J Med. 1978;298(12):647–650. | ||
Lam-Tse WK, Batstra MR, Koeleman BP, et al. The association between autoimmune thyroiditis, autoimmune gastritis and type 1 diabetes. Pediatr Endocrinol Rev. 2003;1(1):22–37. | ||
Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis – pathogenesis, pathology and management. Nat Rev Gastroenterol Hepatol. 2013;10(9):529–541. | ||
Silveira PA, Wilson WE, Esteban LM, et al. Identification of the Gasa3 and Gasa4 autoimmune gastritis susceptibility genes using congenic mice and partitioned, segregative and interaction analyses. Immunogenetics. 2001;53(9):741–750. | ||
Baxter AG, Jordan MA, Silveira PA, Wilson WE, Van Driel IR. Genetic control of susceptibility to autoimmune gastritis. Int Rev Immunol. 2005;24(1–2):55–62. | ||
Ikeda T, Senoue I, Hara M, Tsutsumi Y, Harasawa S, Miwa T. Gastric pseudopolyposis: a new clinical manifestation of type A gastritis. Am J Gastroenterol. 1985;80(2):82–90. | ||
Krasinskas AM, Abraham SC, Metz DC, Furth EE. Oxyntic mucosa pseudopolyps: a presentation of atrophic autoimmune gastritis. Am J Surg Pathol. 2003;27(2):236–241. | ||
Judd LM, Gleeson PA, Toh BH, van Driel IR. Autoimmune gastritis results in disruption of gastric epithelial cell development. Am J Physiol. 1999;277(1 pt 1):G209–G218. | ||
Chlumska A, Boudova L, Benes Z, Zamecnik M. Autoimmune gastritis. A clinicopathologic study of 25 cases. Cesk Patol. 2005;41(4):137–142. | ||
Stolte M, Bethke B, Ruhl G, Ritter M. Omeprazole-induced pseudohypertrophy of gastric parietal cells. Z Gastroenterol. 1992;30(2):134–138. | ||
Solcia E, Fiocca R, Villani L, Luinetti O, Capella C. Hyperplastic, dysplastic, and neoplastic enterochromaffin-like-cell proliferations of the gastric mucosa. Classification and histogenesis. Am J Surg Pathol. 1995;19(Suppl 1):S1–S7. | ||
Li P, He C, Sun L, Dong N, Yuan Y. Pepsinogen I and II expressions in situ and their correlations with serum pesignogen levels in gastric cancer and its precancerous disease. BMC Clin Pathol. 2013;13(1):1–10. | ||
Goldenring JR, Ray GS, Coffey RJ Jr, et al. Reversible drug-induced oxyntic atrophy in rats. Gastroenterology. 2000;118(6):1080–1093. | ||
Nomura S, Yamaguchi H, Ogawa M, Wang TC, Lee JR, Goldenring JR. Alterations in gastric mucosal lineages induced by acute oxyntic atrophy in wild-type and gastrin-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2005;288(2):G362–G375. | ||
Nozaki K, Ogawa M, Williams JA, et al. A molecular signature of gastric metaplasia arising in response to acute parietal cell loss. Gastroenterology. 2008;134(2):511–522. | ||
Weis VG, Petersen CP, Mills JC, Tuma PL, Whitehead RH, Goldenring JR. Establishment of novel in vitro mouse chief cell and SPEM cultures identifies MAL2 as a marker of metaplasia in the stomach. Am J Physiol Gastrointest Liver Physiol. 2014;307(8):G777–G792. | ||
Halldorsdottir AM, Sigurdardottrir M, Jonasson JG, et al. Spasmolytic polypeptide-expressing metaplasia (SPEM) associated with gastric cancer in Iceland. Dig Dis Sci. 2003;48(3):431–441. | ||
Goldenring JR, Nam KT, Wang TC, Mills JC, Wright NA. Spasmolytic polypeptide-expressing metaplasia and intestinal metaplasia: time for reevaluation of metaplasias and the origins of gastric cancer. Gastroenterology. 2010;138(7):2207–2210, 2210.e1. | ||
Correa P, Piazuelo MB, Wilson KT. Pathology of gastric intestinal metaplasia: clinical implications. Am J Gastroenterol. 2010;105(3):493–498. | ||
Abraham SC, Singh VK, Yardley JH, Wu TT. Hyperplastic polyps of the stomach: associations with histologic patterns of gastritis and gastric atrophy. Am J Surg Pathol. 2001;25(4):500–507. | ||
Veijola LI, Oksanen AM, Sipponen PI, Rautelin HIK. Association of autoimmune type atrophic corpus gastritis with Helicobacter pylori infection. World J Gastroenterol. 2010;16(1):83–88. | ||
Negrini R, Savio A, Poiesi C, et al. Antigenic mimicry between Helicobacter pylori and gastric mucosa in the pathogenesis of body atrophic gastritis. Gastroenterology. 1996;111(3):655–665. | ||
Faller G, Steininger H, Eck M, Hensen J, Hann EG, Kirchner T. Antigastric autoantibodies in Helicobacter pylori gastritis: prevalence, in-situ binding sites and clues for clinical relevance. Virchows Arch. 1996;427(5):483–486. | ||
Genta RM. Helicobacter pylori, inflammation, mucosal damage, and apoptosis: pathogenesis and definition of gastric atrophy. Gastroenterology. 1997;113(6 suppl):S51–S55. | ||
Torbenson M, Abraham SC, Boitnott J, Yardley JH, Wu TT. Autoimmune gastritis: distinct histological and immunohistochemical findings before complete loss of oxyntic glands. Mod Pathol. 2002;15(2):102–109. | ||
De Re V, Orzes E, Canzonieri V, et al. Pepsinogens to distinguish patients with gastric intestinal metaplasia and Helicobacter pylori infection among populations at risk for gastric cancer. Clin Transl Gastroenterol. 2016;7(7):e183. | ||
Castoro C, Le Moli R, Arpi ML, et al. Association of autoimmune thyroid diseases, chronic atrophic gastritis and gastric carcinoid: experience from a single institution. J Endocrinol Invest. 2016;39(7):779–784. | ||
Centanni M, Marignani M, Gargano L, et al. Atrophic body gastritis in patients with autoimmune thyroid disease: an under diagnosed association. Arch Intern Med. 1999;159(15):1726–1730. | ||
De Block CE, De Leeuw IH, Van Gaal LF. Autoimmune gastritis in type 1 diabetes: a clinically oriented review. J Clin Endocrinol Metab. 2008;93(2):363–371. | ||
Ness-Abramof R, Nabriski DA, Braverman LE, et al. Prevalence and evaluation of B12 deficiency in patients with autoimmune thyroid disease. Am J Med Sci. 2006;332(3):119–122. | ||
Amerio P, Tracanna M, De Remigis P, et al. Vitiligo associated with other autoimmune diseases: polyglandular autoimmune syndrome types 3B+C and 4. Clin Exp Dermatol. 2006;31(5):746–749. | ||
Zelissen PM, Bast EJ, Croughs RJ. Associated autoimmunity in Addison’s disease. J Autoimmun. 1995;8(1):121–130. | ||
Chang KH, Lyu RK, Ro LS, Wu YR, Chen CM. Coexistence of pernicious anemia and myasthenia gravis – a rare combination of autoimmune diseases in Taiwan. J Formos Med Assoc. 2006;105(11):946–949. | ||
Delva PL, MacDonell JE, MacIntosh OC. Megaloblastic anemia occurring simultaneously in white female monozygotic twins. Can Med Assoc J. 1965;92(21):1129–1131. | ||
Banka S, Ryan K, Thomson W, Newman WG. Pernicious anemia – genetic insights. Autoimmun Rev. 2011;10(8):455–459. | ||
Bonafoux B, Henry L, Delfour C, et al. Association of familial pernicious anaemia and hereditary haemochromatosis. Acta Haematol. 2008;119(1):12–14. | ||
Rusak E, Chobot A, Krzywicka A, Wenzlau J. Anti-parietal cell antibodies – diagnostic significance. Adv Med Sci. 2016;61(2):175–179. | ||
Khan S, Del-Duca C, Fenton E, et al. Limited value of testing for intrinsic factor antibodies with negative gastric parietal cell antibodies in pernicious anaemia. J Clin Pathol. 2009;62(5):439–441. | ||
Ottesen M, Feldt-Rasmussen UF, Andersen J, Hippe E, Schouboe A. [Pernicious anemia. A study of initial forms of the disease and diagnostic significance of determination of the intrinsic factor antibody and parietal cell antibody]. Ugeskr Laeger. 1992;154(52):3758–3762. | ||
Antico A, Tampoia M, Villalta D, Tonutti E, Tozzoli R, Bizzaro N. Clinical usefulness of the serological gastric biopsy for the diagnosis of chronic autoimmune gastritis. Clin Dev Immunol. 2012;2012:520970. | ||
Shah P, Singh MH, Yang YX, Metz DC. Hypochlorhydria and achlorhydria are associated with false-positive secretin stimulation testing for Zollinger-Ellison syndrome. Pancreas. 2013;42(6):932. | ||
Phan J, Benhammou JN, Pisegna JR. Gastric hypersecretory states: investigation and management. Curr Treat Options Gastroenterol. 2015;13(4):386–397. | ||
Carmel R. Pepsinogens and other serum markers in pernicious anemia. Am J Clin Pathol. 1988;90(4):442–445. | ||
Wainwright P, Narayanan S, Cook P. False-normal vitamin B12 results in a patient with pernicious anaemia. Clin Biochem. 2015;48(18):1366–1367. | ||
Solomon LR. Cobalamin-responsive disorders in the ambulatory care setting: unreliability of cobalamin, methylmalonic acid, and homocysteine testing. Blood. 2005;105(3):978–985. | ||
Wu PB, Deng YZ, Shu YX, Tan SY, Li M, Fang G. Increased plasma CgA levels associated with nonalcoholic fatty liver disease. Turk J Gastroenterol. 2015;26(5):404–407. | ||
Peracchi M, Gebbia C, Basilisco G, et al. Plasma chromogranin A in patients with autoimmune chronic atrophic gastritis, enterochromaffin-like cell lesions and gastric carcinoids. Eur J Endocrinol. 2005;152(3):443–448. | ||
Massironi S, Fraquelli M, Paggi S, et al. Chromogranin A levels in chronic liver disease and hepatocellular carcinoma. Dig Liver Dis. 2009;41(1):31–35. | ||
Park YH, Kim N. Review of atrophic gastritis and intestinal metaplasia as a premalignant lesion of gastric cancer. J Cancer Prev. 2015;20(1):25–40. | ||
Dai Y-C, Tang Z-P, Zhang Y-L. How to assess the severity of atrophic gastritis. World J Gastroenterol. 2011;17(13):1690–1693. | ||
Anagnostopoulos GK, Ragunath K, Shonde A, Hawkey CJ, Yao K. Diagnosis of autoimmune gastritis by high resolution magnification endoscopy. World J Gastroenterol. 2006;12(28):4586–4587. | ||
Nomura S, Ida K, Terao S, et al. Endoscopic diagnosis of gastric mucosal atrophy: multicenter prospective study. Dig Endosc. 2014;26(6):709–719. | ||
Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International workshop on the histopathology of gastritis, Houston 1994. Am J Surg Pathol. 1996;20(10):1161–1181. | ||
Wong HH, Chu P. Immunohistochemical features of the gastrointestinal tract tumors. J Gastrointest Oncol. 2012;3(3):262–284. | ||
Sepulveda AR, Patil M. Practical approach to the pathologic diagnosis of gastritis. Arch Pathol Lab Med. 2008;132(10):1586–1593. | ||
Smith ADM. Megaloblastic madness. Br Med J. 1960;2(5216):1840–1845. | ||
Hershko C, Hoffbrand A, Keret D, et al. Role of autoimmune gastritis, Helicobacter pylori and celiac disease in refractory or unexplained iron deficiency anemia. Haematologica. 2005;90(5):585–595. | ||
Gonçalves C, Oliveira ME, Palha AM, Ferrão A, Morais A, Lopes AI. Autoimmune gastritis presenting as iron deficiency anemia in childhood. World J Gastroenterol. 2014;20(42):15780–15786. | ||
Miguel N, Costa E, Santalha M Jr, et al. Refractory iron-deficiency anemia and autoimmune atrophic gastritis in pediatric age group: analysis of 8 clinical cases. J Pediatr Hematol Oncol. 2014;36(2):134–139. | ||
Stabler SP. Clinical practice. Vitamin B12 deficiency. N Engl J Med. 2013;368(2):149–160. | ||
Tefferi A, Pruthi RK. The biochemical basis of cobalamin deficiency. Mayo Clin Proceed. 1994;69(2):181–186. | ||
Asimacopoulos PJ, Groves MD, Fischer DK, et al. Pernicious anemia manifesting as angina pectoris. South Med J. 1994;87(6):671–672. | ||
Tadakamalla AK, Talluri SK, Besur S. Pseudo-thrombotic thrombocytopenic purpura: a rare presentation of pernicious anemia. N Am J Med Sci. 2011;3(10):472–474. | ||
Melhem A, Desai A, Hofmann MA. Acute myocardial infarction and pulmonary embolism in a young man with pernicious anemia-induced severe hyperhomocysteinemia. Thromb J. 2009;7(1):1–5. | ||
Shamkani WA, Jafar NS, Narayanan SR, Rajappan AK. Acute myocardial infarction in a young lady due to vitamin B12 deficiency induced hyperhomocysteinemia. Heart Views. 2015;16(1):25–29. | ||
Metz J. Cobalamin deficiency and the pathogenesis of nervous system disease. Annu Rev Nutr. 1992;12:59–79. | ||
Francis G, Hohol K, Jawad Z, Ayer A, Toth C. Methylmalonic acid accumulation and the development of peripheral neuropathy (IN1-1.010). Neurology. 2012;78(1 suppl):IN1–IN1. | ||
Ralapanawa DM, Jayawickreme KP, Ekanayake EM, Jayalath WA. B(12) deficiency with neurological manifestations in the absence of anaemia. BMC Res Notes. 2015;8:458. | ||
Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720–1728. | ||
Hemmer B, Glocker FX, Schumacher M, Deuschl G, Lücking CH. Subacute combined degeneration: clinical, electrophysiological, and magnetic resonance imaging findings. J Neurol Neurosurg Psychiatry. 1998;65(6):822–827. | ||
Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZMN. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063–1068. | ||
Saperstein DS, Barohn RJ. Peripheral neuropathy due to cobalamin deficiency. Curr Treat Options Neurol. 2002;4(3):197–201. | ||
Chu C, Scanlon P. Vitamin B12 deficiency optic neuropathy detected by asymptomatic screening. BMJ Case Rep. 2011;2011:bcr0220113823. | ||
Metzler D, Miller WH, Stephen Edwards C. Psychiatric manifestation of vitamin B-12 deficiency: an update. Jefferson J Psychiatry. 1991;9(2). | ||
Douaud G, Refsum H, de Jager CA, et al. Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc Natl Acad Sci U S A. 2013;110(23):9523–9528. | ||
Lahner E, Annibale B. Pernicious anemia: new insights from a gastroenterological point of view. World J Gastroenterol. 2009;15(41):5121–5128. | ||
Zhu JC, Wang YF, Sheng J, Chen FX, Tang GY. [Atrophic glossitis is attributed to cobalamin deficiency]. Shanghai J Stomatol. 2013;22(1):58–62. | ||
Vannella L, Lahner E, Osborn J, Annibale B. Systematic review: gastric cancer incidence in pernicious anaemia. Aliment Pharmacol Ther. 2013;37(4):375–382. | ||
Vannella L, Sbrozzi-Vanni A, Lahner E, et al. Development of type I gastric carcinoid in patients with chronic atrophic gastritis. Aliment Pharmacol Ther. 2011;33(12):1361–1369. | ||
Lahner E, Esposito G, Galli G, Annibale B. Atrophic gastritis and pre-malignant gastric lesions. Transl Gastrointest Cancer. 2015;4(4):272–281. | ||
Schneller J, Gupta R, Mustafa J, Villanueva R, Straus EW, Raffaniello RD. Helicobacter pylori infection is associated with a high incidence of intestinal metaplasia in the gastric mucosa of patients at inner-city hospitals in New York. Dig Dis Sci. 2006;51(10):1801–1809. | ||
Wong BC, Lam SK, Wong WM, et al; China Gastric Cancer Study Group. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291(2):187–194. | ||
Correa P, Piazuelo MB. The gastric precancerous cascade. J Dig Dis. 2012;13(1):2–9. | ||
Yong X, Tang B, Li B-S, et al. Helicobacter pylori virulence factor CagA promotes tumorigenesis of gastric cancer via multiple signaling pathways. Cell Commun Signal. 2015;13(1):1–13. | ||
Yamaoka Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat Rev Gastroenterol Hepatol. 2010;7(11):629–641. | ||
van Doorn LJ, Figueiredo C, Sanna R, et al. Clinical relevance of the cagA, vacA, and iceA status of Helicobacter pylori. Gastroenterology. 1998;115(1):58–66. | ||
Wroblewski LE, Peek RM, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23(4):713–739. | ||
Zhou K, Ho W. Gastric carcinoids: classification and Diagnosis. In: Pisegna RJ, editor. Management of Pancreatic Neuroendocrine Tumors. New York, NY: Springer New York; 2015:83–93. | ||
Burkitt MD, Pritchard DM. Review article: pathogenesis and management of gastric carcinoid tumours. Aliment Pharmacol Ther. 2006;24(9):1305–1320. | ||
Li T-T, Qiu F, Qian ZR, Wan J, Qi X-K, Wu B-Y. Classification, clinicopathologic features and treatment of gastric neuroendocrine tumors. World J Gastroenterol. 2014;20(1):118–125. | ||
Andres E, Fothergill H, Mecili M. Efficacy of oral cobalamin (vitamin B12) therapy. Expert Opin Pharmacother. 2010;11(2):249–256. | ||
Stolte M, Meier E, Meining A. Cure of autoimmune gastritis by Helicobacter pylori eradication in a 21-year-old male. Z Gastroenterol. 1998;36(8):641–643. | ||
Faller G, Winter M, Steininger H, et al. Decrease of antigastric autoantibodies in Helicobacter pylori gastritis after cure of infection. Pathol Res Pract. 1999;195(4):243–246. | ||
Nikou GC, Angelopoulos TP. Current concepts on gastric carcinoid tumors. Gastroenterol Res Pract. 2012;2012:287825. | ||
Dockray GJ. Clinical endocrinology and metabolism. Gastrin. Best Pract Res Clin Endocrinol Metab. 2004;18(4):555–568. | ||
Moore AR, Boyce M, Steele IA, Campbell F, Varro A, Pritchard DM. Netazepide, a gastrin receptor antagonist, normalises tumour biomarkers and causes regression of type 1 gastric neuroendocrine tumours in a nonrandomised trial of patients with chronic atrophic gastritis. PLoS One. 2013;8(10):e76462. | ||
Fossmark R, Sordal O, Jianu CS, et al. Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide (YF476) results in regression of tumours and normalisation of serum chromogranin A. Aliment Pharmacol Ther. 2012;36(11–12):1067–1075. | ||
Massironi S, Zilli A, Fanetti I, Ciafardini C, Conte D, Peracchi M. Intermittent treatment of recurrent type-1 gastric carcinoids with somatostatin analogues in patients with chronic autoimmune atrophic gastritis. Dig Liver Dis. 2015;47(11):978–983. | ||
Kwekkeboom DJ, Krenning EP. Peptide receptor radionuclide therapy in the treatment of neuroendocrine tumors. Hematol Oncol Clin North Am. 2016;30(1):179–191. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.