Back to Journals » Journal of Blood Medicine » Volume 12
Autoimmune Acquired Factor XIII Deficiency: A Case Report
Received 29 October 2020
Accepted for publication 29 December 2020
Published 9 February 2021 Volume 2021:12 Pages 63—68
DOI https://doi.org/10.2147/JBM.S288634
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Martin H Bluth
Ana Marco,1,2 Pascual Marco1– 3
1Thrombosis and Hemostasis Department, Hematology Service, University General Hospital in Alicante, Alicante, Spain; 2Biomedical Health Research Institute, University General Hospital in Alicante, Alicante, Spain; 3Department of Clinical Medicine, Miguel Hernández University, Alicante, Spain
Correspondence: Ana Marco
University General Hospital in Alicante, Pintor Baeza 12, Alicante, 03010, Spain
Tel +34 965913863
Fax +34 965913869
Email [email protected]
Abstract: Autoimmune acquired factor XIII (FXIII) deficiency is a rare disorder characterized by severe spontaneous hematomas and autoantibodies against FXIII. High mortality rates have been reported (18% within a year of diagnosis). We present a 70-year-old patient with recurrent muscular hematomas. The basic hemostasis study and the coagulation factors were within normal ranges. The aggregation platelet study was also normal and von Willebrand disease was excluded. Bearing in mind the recurrent bleeding history and the described laboratory results, we considered a FXIII deficiency, that was confirmed (FXIII< 10%). In addition, we suspected an acquired FXIII deficiency since the patient did not report a personal or family history of bleeding and FXIII gene sequencing study was normal. Non-immune causes were ruled out, and plasma autoantibodies against FXIII were detected. Immunosuppression was rapidly initiated to eradicate inhibitor as was hemostatic treatment to obtain bleeding control. Currently, the patient is asymptomatic, but a low level of FXIII inhibitor remains.
Keywords: acquired disease, autoantibodies, factor XIII deficiency, hematoma, spontaneous
Introduction
Coagulation factor XIII (FXIII) is a hetero-tetrameric zymogen that plays a key role in clot stabilization. FXIII is comprised of two A and two B subunits and is activated by thrombin and calcium.1 However, other additional functions have been described. Although FXIII-B is generated in the liver, FXIII-A is produced by hematopoietic cells. Therefore, FXIII contributes to pregnancy maintenance, bone and cartilage growth and wound healing.1
FXIII deficiency is characterized by variable bleeding episodes depending on FXIII levels. Congenital FXIII deficiency is an autosomal recessive rare disease with an incidence of 1.5 cases per 2 million people. It is characterized by severe and spontaneous bleeding, including umbilical and intracranial hemorrhage at birth, usually in case of severe deficiency (FXIII<1%). Other symptoms include soft tissue bleeding, recurrent miscarriages, bruising and hemarthroses.2 Inhibitor development against FXIII in inherited FXIII deficiency in patients receiving FXIII replacement therapy is also infrequent but may induce severe bleeding.3 However, acquired FXIII deficiency is more common and can be classified as immune and non-immune disease. Acquired immune-mediated FXIII deficiency implies the presence of an autoantibody targeting FXIII epitopes which may lead to severe bleeding. The clinical presentation is variable from mucocutaneous and intramuscular bleedings to life-threatening hemorrhages such as intracranial, intra-thoracic or intra-peritoneal.4 In addition, clinical bleeding in any acquired immune factor deficiency does not correlate with factor level or inhibitor titer.5 Ichinose et al described the largest series in the literature, including 93 patients with a large proportion of patients from Japan (59).6 Around 20% died within the first year of diagnosis. Non-immune causes are more common but rarely cause bleeding and are related to excessive consumption or decreased production of FXIII.7
An early diagnosis is essential, as acquired immune FXIII deficiency can be fatal if proper treatment is not rapidly provided.
Case Report
A 70-year-old male was admitted to our clinic for assessment of a spontaneous hematoma in the lower right extremity. Antiplatelet and anticoagulant treatment was not reported. The ultrasound revealed a 14 cm quadriceps hematoma. The patient also referred the presence of provoked hematomas in upper and lower extremities in the last 6 months, which evolved adequately with tranexamic acid. The initial blood test including a basic hemostasis study and a complete blood count was within normal ranges. The platelet function study and the von Willebrand profile were also normal. All coagulation factors were normal, except for the FXIII antigen of 9% confirmed in 2 different samples. Fibrinogen and alfa2-antiplasmin were in the range of normality. Factor activity assays (extrinsic and intrinsic pathways) were performed with a one-stage coagulant assay using factor-deficient plasma in automated coagulometer (ACL-Top, IL, Bedford, USA). Von Willebrand factor antigen and ristocetin cofactor activity were performed by chemiluminescence (HemosIL AcuStar, IL, Bedford, USA) in automated coagulometer (ACL-AcuStar, IL, Bedford, USA) in poor platelet plasma. Fibrinogen was measured using the coagulative Clauss method (STA-Liquid-Fib, Diagnostica Stago, Paris, France) in poor platelet plasma (STA-Rac, Diagnostica Stago, Paris, France) in automated coagulometer and alfa2-antiplasmin was determined by chromogenic assay (ACL-Top, IL, Bedford, USA) in poor platelet plasma, following the manufacturer´s recommendations. FXIII antigen was determined using the automated latex immunoassay (FXIII antigen latex reagent, IL, Bedford, USA) in automated coagulometer (ACL-Top, IL, Bedford, USA), following the manufacturer´s instructions.
We considered an acquired immune FXIII deficiency, confirmed by Bethesda assay (inhibitor titer of 12 BU) and absence of mutation in FXIII gene sequencing study. The Bethesda assay quantifies the amount of inhibitor that neutralizes 50% residual factor in an equal mixture of normal plasma and patient plasma in 2 hours at 37º.8
We also excluded any underlying disease and immune dysregulation. Initial weight-adjusted treatment (weight of 70 Kg) with prednisone 1 mg/kg/day (70 mg daily) for 15 days and immunoglobulins 1g/kg/day (70 g daily) for 2 days was prescribed. The patient presented an appropriate clinical and analytical improvement, with a remarkable decrease in the autoantibody titer. We monitored FXIII levels and performed the thrombin generation test before starting steroids, 1 week and 2 weeks after. The patient had an increase in FXIII levels throughout the scheduled visits associated with an enlargement in the endogenous thrombin potential and a higher thrombin peak (Figure 1). This improvement was confirmed by the thromboelastometry, which showed an increase in clot firmness and a shortened clot formation time (Figure 2). Thrombin generation was determined by the fluorometric method described by Hemker,9 modified and automated (STA Genesia, Diagnostica Stago, Paris, France) in poor platelet plasma. We used the STG-Bleedscreen reagent (Diagnostica Stago, Paris, France) that contains a mixture of phospholipids and a low recombinant tissue factor concentration. Thrombin generation assay provides information on the lag time of thrombin generation, the endogenous thrombin potential, the peak of thrombin concentration, the time to reach the maximum peak of thrombin and the time to thrombin neutralization. Rotational thromboelastometry (ROTEM®, IL, Bedford, USA) provides global information about the dynamics of clot development, stabilization and dissolution using citrated whole blood. The following ROTEM® parameters were measured in EXTEM (provides similar information to prothrombin time) and FIBTEM (analyzes the functional fibrinogen component): clotting time, clot formation time and maximum clot firmness. EXTEM and FIBTEM are fibrinogen and FXIII dependent.
 |
Figure 1 Changes in thrombin generation depending on FXIII antigen. (A) Before starting steroids. Baseline FXIII of 5%. (B) The patient received steroids for 1 week. Baseline FXIII of 34%. (C) The patient received steroids for 2 weeks. Baseline FXIII of 45%. Reference within normal parameters is shown in green and patient´s sample in blue. |
 |
Figure 2 Changes in thromboelastography. (A) The patient received corticosteroids for 1 week. (B) The patient received steroids for 2 weeks. Abbreviations: CT, clotting time; CFT, clot formation time; MCF, maximum clot firmness. |
However, the patient had poor tolerance to corticosteroids with hypertension and anxiety, so we decided their discontinuation. No new bleeding episodes were reported. Due to persistence of the inhibitor, rituximab was then proposed, but since the patient refused, immunosuppressive therapy was changed to azathioprine 50 mg daily. One week later the patient referred vomiting and diarrhea, so azathioprine was discontinued. The patient remained clinically stable. One month after discontinuation of immunosuppression, the patient was admitted to the emergency department with thoracic oppressive pain and minimal effort dyspnea for 24 hours. The blood count revealed mild leukocytosis with neutrophilia (12.27 x103/µL leukocytes and 10.53 x103/µL neutrophils), hemoglobin of 14 gr/dl (previous was 16 gr/dl) and platelets of 248 x103/µL. The basic hemostatic study was normal, the baseline FXIII level was 20% and the inhibitor titer was 3 BU. The chest x-ray and CT scan objectified the presence of a mediastinal hematoma. Hemostatic treatment with fresh frozen plasma (15 mL/kg) and tranexamic acid (500 mg/8 hours) as well as immunosuppressive therapy with rituximab (375 mg/m2 weekly for 4 weeks, 678 mg weekly) were administered (body surface of 1.81 m2). Factor XIII concentrate was not necessary, due to clinical stability. No further bleedings were reported. The patient is currently receiving corticosteroids at low doses (20 mg daily), with an excellent clinical evolution. The patient is asymptomatic with no relevant side effects. The last blood sample (5 months after initial diagnosis) was taken 1 month after corticosteroids 20 mg daily were initiated and revealed a FXIII of 55% and an antibody titer of 1 BU. Changes in hemostatic parameters are shown in Table 1.
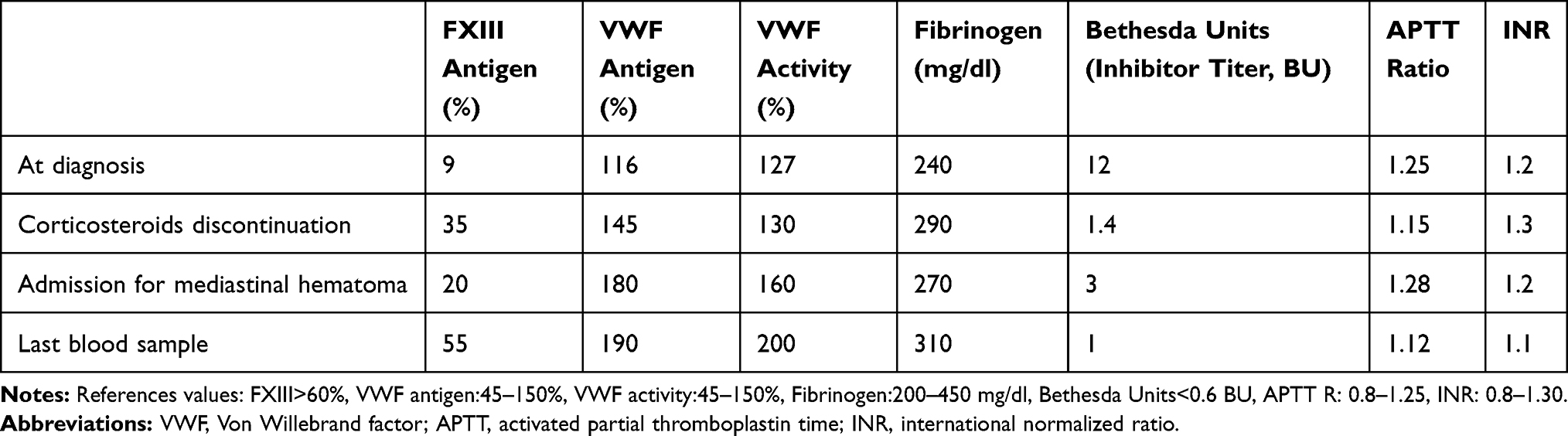 |
Table 1 Changes in Hemostatic Parameters Throughout Time |
Discussion
Acquired immune FXIII deficiency is a rare disease caused by the presence of an autoantibody which targets FXIII epitopes and may cause severe and life-threatening bleeding. About 50% of the cases are idiopathic. Autoimmune diseases and malignancies are the most prevalent underlying conditions. Intramuscular and subcutaneous bleeding events are the most frequently reported.4
Evaluating differential diagnosis, congenital FXIII deficiency was ruled out, as the patient referred a negative personal and familiar bleeding tendency and no mutation in FXIII gene sequencing study was detected. Non-immune-mediated causes of acquired FXIII deficiency including increased consumption or decreased production like recent surgery, liver disease, cancer, sepsis or interfering medication were also discarded. In these cases, life-threatening bleeding events rarely appear, FXIII levels normally range between 20% and 70% and antibodies against FXIII are not detected. Here we report a major bleeding with FXIII level below 10% and presence of antibodies against FXIII. Finally, other autoimmune coagulation factor deficiencies such as acquired hemophilia or acquired Von Willebrand disease were not considered because all coagulation factors and von Willebrand factor (antigen and activity) were within normal range except for FXIII.
Our patient met the diagnostic criteria for immune-mediated acquired FXIII deficiency proposed by the ISTH.4 He referred no previous personal or family bleeding history and was not receiving anticoagulant or antiplatelet treatment. In our laboratory, the identification of neutralizing autoantibodies was confirmed using a 1:1 mixing test of patient plasma and normal control plasma, with quantification of the inhibitor by the Bethesda assay. Most inhibitors against FXIII are directed against A subunit.4,10 In our patient, it was not possible to identify the inhibitor specificity although the reagent to determine FXIII antigen is based on an antibody highly specific for A subunit.
The sequencing study of FXIII gene detected no mutation, supporting the final diagnosis of acquired immune FXIII deficiency.
Thrombin generation and thromboelastometry could be useful global hemostasis tests for monitoring clinical and biological evolution. To our knowledge, these global tests in acquired immune FXIII deficiency have not been previously reported. Our patient presented a significant decrease in thrombin generation compared with normal reference plasma at diagnosis that improved as FXIII increased and inhibitor titer decreased (Figure 1). In agreement with Gosh et al, we described a reduction in the peak height of thrombin and endogenous thrombin potential at diagnosis although they only determine basal Factor XIII levels in patients with inherited FXIII deficiencies.11 We suggest that as FXIII levels increase, more thrombin is generated that binds to FXIII, promoting a positive feedback and activation of the coagulation cascade.12 These results were endorsed by the thromboelastography that showed an increase in maximum clot firmness and a shortened clot formation time (Figure 2A and B). Theusinger et al obtained similar results in critical patients in the postoperative period of a major surgery (acquired non-immune FXIII deficiencies).13
Regarding the management of these patients, hemostatic and antibody eradication therapy should be rapidly initiated. However, given the rarity of this disease, evidence-based recommendations are lacking.14 High-dose FXIII concentrates (50–100 IU/kg) or FXIII-containing blood products like fresh frozen plasma or cryoprecipitate could be an optimal approach to achieve hemostatic control. However, they could show no effectiveness in the presence of high titer inhibitor.14 Other hemostatic treatments including antifibrinolytics like tranexamic acid together with FXIII administration have been associated with favorable effects.14 Recombinant factor VII has been rarely reported probably due to effectiveness of FXIII concentrates or fresh frozen plasma. However, it should be considered in severe bleedings with no response to FXIII-containing products.15 Immunosuppressive treatments include corticosteroids alone or in combination with cyclophosphamide. Rituximab, ciclosporin or immunoglobulins have also presented successful results.15,16 Our patient received first-line immunosuppressive treatment with corticosteroids and immunoglobulins and hemostatic treatment with tranexamic acid. Although immunosuppression was initially effective, steroids were discontinued due to side effects. Although no new bleeding events appeared, a second-line immunosuppression treatment with azathioprine was initiated, that was also discontinued due to side effects. Four weeks later, the patient developed a mediastinal hematoma that evolved favorably with rituximab and hemostatic treatment with fresh frozen plasma and tranexamic acid.
A fatal intracranial hemorrhage in an adult patient with comorbidities diagnosed of an acquired FXIII deficiency has been recently published. This patient received rituximab as immunosuppression and hemostatic treatment with fresh frozen plasma and rFVIIa.17
Our patient developed a major bleeding episode. Neither a triggering disease nor an underlying drug was identified. However, the patient evolved favorably with an appropriate response to immunosuppression and hemostatic treatment. Generally speaking, even with this atypical favorable presentation, we suggest, in agreement with the literature, an early diagnosis and management to prevent further morbidity and mortality.
Long-term outcomes are variable. Around 50–68% achieved antibody eradication without bleeding recurrence.14,18 If no bleeding episodes are reported, there is no evidence to support the administration of FXIII prophylaxis in patients with recurrent or persistent antibodies. A long-term follow-up is necessary, as this is a chronic disease and relapse may occur.7,14 Our patient currently receives corticosteroids 20 mg daily and remains asymptomatic with a low inhibitor titer.
Ethics and Consent
The study participant has given written informed consent to participate and publish the data. Institutional approval was not required for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Komaromi I, Bagoly Z, Muszbeck L. Factor XIII: novel structural and functional aspects. J Thromb Haemost. 2011;9:9–20. doi:10.1111/j.1538-7836.2010.04070.x
2. Levy JH, Greenberg C. Biology of factor XIII and clinical manifestations of factor XIII deficiencies. Transfusion. 2013;53:1120–1131. doi:10.1111/j.1537-2995.2012.03865.x
3. Muszbek L, Penzes K, Katona E. Auto and alloantibodies against factor XIII: laboratory diagnosis and clinical consequences. J Thromb Haemost. 2018;16:822–832. doi:10.1111/jth.13982
4. Ichinose A, Kohler HP, Philippou H; on behalf of the Factor XIII and Fibrinogen SSC Subcommittee of the ISTH. Recommendations for ISTH/SSC criterion 2015 for autoimmune acquired factor XIII/13 deficiency. Thromb Haemost. 2016;116:772–774. doi:10.1160/TH-16-05-0362
5. Baudo F, Colllins P, Huth-kuhne A, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) registry. Blood. 2012;120:39–46. doi:10.1182/blood-2012-02-408930
6. Ichinose A; on behalf of the Japanese Collaborative Research Group on AH13. Autoimmune acquired factor XIII deficiency due to anti-factor XIII/13 antibodies: a summary of 93 patients. Blood Rev. 2017;31:37–45. doi:10.1016/j.blre.2016.08.002
7. Muszbek L, Katona E. Diagnosis and management of congenital and acquired FXIII deficiencies. Semin Thromb Hemost. 2016;42:429–439. doi:10.1055/s-0036-1572326
8. Duncan E, Collecutt M, Street A. Nijmegen-bethesda assay to measure factor VIII inhibitors. Methods Mol Biol. 2013;992:321–333.
9. Hemker H, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33:4–15. doi:10.1159/000071636
10. Kohler HP, Ichinose A, Seitz R, Ariens AR, Muszbek L. on behalf of the factor XIII and fibrinogen SSC subcommittee of the ISTH. J Thromb Haemost. 2011;9:1404–1406. doi:10.1111/j.1538-7836.2011.04315.x
11. Ghosh K, Mota L, Shetty S, Kulkarni B. Spectrum of changes in endogenous thrombin potential due to heritable disorders of coagulation. Blood Coagul Fibrinolysis. 2008;19:577–580. doi:10.1097/MBC.0b013e328307089d
12. Colman RW, Hirsh J, Marder VJ, Clowes AW, George JN. Hemostasis and Thrombosis: Basic Principles and Clinical Practice.Lippincott Williams & Wilkins.2006. Vol. 319. 1078–1079.
13. Theusinger OM, Baulig W, Asmis LM, Seifert B, Spahn DR. In vitro factor XIII supplementation increases clot firmness in Rotation Thromboelastometry (ROTEM). Thromb Haemost. 2010;104:385–391. doi:10.1160/TH09-12-0858
14. Sheng Yan MT, Rydz N, Goodyear D, Sholzberg M. Acquired factor XIII deficiency: a review. Transfus Apher Sci. 2018;57:724–730. doi:10.1016/j.transci.2018.10.013
15. Boehlen F, Casini A, Chizzolini C, et al. Acquired factor XIII deficiency: a therapeutic challenge. Thromb Haemost. 2013;109:479–487. doi:10.1160/TH12-08-0604
16. Franchini M, Frattini F, Crestani S, Bonfanti C. Acquired FXIII inhibitors: a systematic review. J Thromb Thrombolysis. 2013;36:109–114. doi:10.1007/s11239-012-0818-3
17. Di Micco P, Gussoni G, Pieralli F, Campanini M, Dentali F, Fontanella A. Acquired factor XIII deficiency inducing recurrent and fatal bleeding, description of a case. J Blood Med. 2020;11:43–45. doi:10.2147/JBM.S232115
18. Kessel R, Hu C, Shore-Lesserson L, Rand J, Manwani D. A child with acquired factor XIII deficiency: case report and literature review. Haemophilia. 2013;19:814–826. doi:10.1111/hae.12145
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.