Back to Journals » Cancer Management and Research » Volume 7
Atypical teratoid rhabdoid tumors: a population-based clinical outcomes study involving 174 patients from the Surveillance, Epidemiology, and End Results database (1973–2010)
Authors Lau C, Mahendraraj K, Chamberlain R
Received 14 May 2015
Accepted for publication 8 July 2015
Published 18 September 2015 Volume 2015:7 Pages 301—309
DOI https://doi.org/10.2147/CMAR.S88561
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Kenan Onel
Christine SM Lau,1,2 Krishnaraj Mahendraraj,1 Ronald S Chamberlain1–3
1Department of Surgery, Saint Barnabas Medical Center, Livingston, NJ, USA; 2Saint George's University School of Medicine, Grenada, West Indies; 3Department of Surgery, New Jersey Medical School, Rutgers University, Newark, NJ, USA
Introduction: Atypical teratoid rhabdoid tumors (ATRTs) are rare, highly malignant embryonal tumors of the central nervous system (CNS) accounting for 20% of CNS tumors in children under the age of 3. This study examines a large cohort of ATRT patients to determine demographic, clinical, and pathologic factors which impact prognosis and survival.
Methods: Demographic and clinical data were abstracted on 174 ATRT patients (171 pediatric patients age <20 and 3 adult patients age ≥20) from the Surveillance, Epidemiology, and End Results database (1973–2010). Standard statistical methodology was used.
Results: A total of 174 ATRT cases (mean age of 2.84 years) were identified. ATRT had a higher incidence in males (56.3%), Caucasians (59.1%), and children <3 years of age (80.5%), P<0.001. The most common primary sites were the cerebellum (17.8%), ventricles (16.1%), and frontal lobe (12.6%). Mean overall survival was 3.2±0.4 years, while overall and cancer-specific mortality were 63.2% and 56.3%, respectively, P=0.005. Most ATRT cases were treated with surgery alone (58.0%), followed by a combination of surgery and radiation (34.3%), no treatment (6.5%), and radiation alone (1.2%). The use of combination therapy has increased significantly (16.1%) since 2005 (P<0.001), while primary surgical resection and radiation therapy rates remain relatively unchanged. The longest survival was observed among ATRT patients receiving combination therapy (5.9±0.7 years), followed by radiation alone (2.8±1.2 years), and surgery alone (1.9±0.4 years), P<0.001. Multivariable analysis identified only distant metastases (OR =4.6) as independently associated with increased mortality, whereas combination therapy (OR =0.4) was associated with reduced mortality, P<0.005.
Conclusion: ATRT is a rare and highly aggressive embryonal malignancy of the CNS that presents more often as locoregional tumors >4 cm in male Caucasian children of age <3 years, involving the cerebellum, ventricles, or frontal lobe. Combination therapy significantly improves survival, and its use has been increasing since 2005.
Keywords: childhood brain cancer, pediatric cancer, central nervous system, rare tumors, SEER database
Introduction
Central nervous system (CNS) tumors constitute the second most common malignancy in the pediatric population, following leukemia.1 Atypical teratoid rhabdoid tumors (ATRTs) are a rare, aggressive, and highly malignant embryonal tumor of the CNS, comprising approximately 3% of pediatric brain tumors, and 20% of CNS tumors in children under the age of 3.2–4 First described in the late 1980s, ATRT is associated with an extremely poor prognosis, with mean overall survival ranging from 6 to 18 months.2–6 Diagnosing ATRT has been difficult, and it is often misdiagnosed as primitive neuroendocrine tumor (PNET), medulloblastoma, or other CNS tumors.7,8 Historically, ATRT has been treated with surgical resection alone; however, more recent studies have demonstrated similar benefits to adjunct radiation.9,10
Current knowledge regarding ATRT is limited, and only rare case reports or small retrospective observational studies exist.3,9 The demographic, pathologic, and clinical factors influencing prognosis and survival in ATRT patients are not well understood. The current study examines a large cohort of ATRT patients from the Surveillance, Epidemiology, and End Results (SEER) database in an effort to identify demographic, clinical, and treatment strategies that may impact the clinical outcomes in a modern ATRT cohort.
Methods
Data for the current study were extracted from the SEER database provided by the National Cancer Institute between 1973 and 2010. SEER Stat software version 8.0.4 (National Cancer Institute, Bethesda, MD, USA) was utilized to extract data from 18 SEER registries (Alaska Native Tumor Registry, Arizona Indians, Cherokee Nation, Connecticut, Detroit, Georgia Center for Cancer Statistics, Greater Bay Area Cancer Registry, Greater California, Hawaii, Iowa, Kentucky, Los Angeles, Louisiana, New Jersey, New Mexico, Seattle-Puget Sound, and Utah). A total of 174 patients with histologically confirmed ATRT were identified and exported to IBM SPSS®v20.2 (IBM Corporation, Armonk, NY, USA). A total of 174 patients with a primary diagnosis of ATRT were identified to form the final study cohort, using the SEER International Classification of Disease for Oncology (ICD-O-3) codes 9508/3. Demographic and clinical data extracted included age, sex, race, tumor stage, tumor size, primary tumor site, and type of treatment received (surgery, radiation, both or unknown/no treatment). The term “no treatment” refers to no reported treatment. Patients with in situ cancers, those with nonspecific site of tumor origin, and those in whom histologic confirmation of their cancer was not available were excluded from the final study cohort. Endpoints examined included overall survival, mortality, and cancer-specific mortality. Categorical variables were compared using the chi-square test, and continuous variables were compared using Student’s t-test and analysis of variance. Multivariable analysis using the “backward Wald” method was performed to calculate odds ratios (ORs) and determine independent factors affecting survival. Missing and unknown data were excluded from the multivariable analysis. Kaplan–Meier analysis was used to compare long-term actuarial survival between groups. Statistical significance was accepted at the level of P<0.05.
Results
Demographic data
A total of 174 ATRT cases were reported in the SEER database over the 38-year study period (1973–2010). Among these, 171 ATRT patients (98.3%) were aged 19 years or younger, and the median age at diagnosis was 1.00 year (mean age was 2.84 years), P<0.001 (Table 1). Specifically, 140 patients (80.5%) were ≤3 years, while 31 patients (17.8%) were between 4 and 19 years old, and 3 patients (1.7%) were ≥20 years old, P<0.001. There were 98 male patients (56.3%) and 76 female patients (43.7%), with a male to female ratio of 1.3:1, P<0.001.
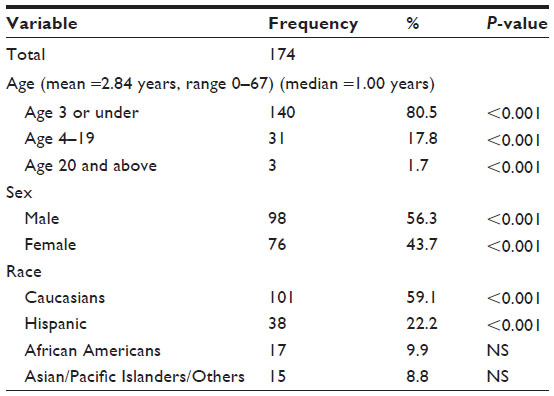 | Table 1 Demographic profiles of 174 patients with ATRT from the SEER database, 1973–2010 |
The majority of ATRT cases occurred among Caucasians (101 cases; 59.1%, P<0.001), followed by Hispanics (38 patients; 22.2%, P<0.001), African–Americans (17 patients; 9.9%, P=not significant [NS]), and Asian/Pacific Islanders (15 patients; 8.8%, P>0.05).
Tumor characteristics
The most common primary site for ATRT was the cerebellum (17.8%; 31 cases), followed by the ventricles (16.1%, 28 cases), frontal lobe (12.6%, 22 cases), brain stem (12.1%, 21 cases), temporal lobe (5.7%, 10 cases), parietal lobe (4.0%, 7 cases), pineal gland (3.4%, 6 cases), cerebrum (2.9%, 5 cases), occipital lobe (1.7%, 3 cases), overlapping lesions of the brain (6.9%, 12 cases), or other parts of the brain (16.7%, 29 cases), P=NS (Table 2). Eighty-seven percent (120 cases) of ATRT cancer were locoregional at presentation, while 13.0% (18 cases) had distant metastasis, P<0.005. Around 60.2% (80 cases) of ATRT tumors were greater than 4 cm in size, 34.6% (46 cases) were between 2 and 4 cm, and 5.3% (7 cases) were less than 2 cm.
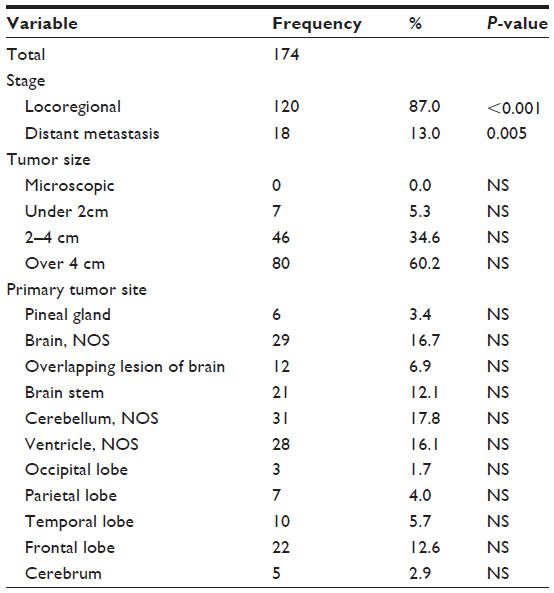 | Table 2 Tumor characteristics of 174 patients with ATRT from the SEER database, 1973–2010 |
Treatment
Fifty-eight percent of the ATRT patients (98 patients, P=0.02) were treated initially with surgery, while 1.2% (2 patients, P=NS) were initially treated with primary external beam radiation therapy (Tables 3 and 4). Around 34.3% (58 patients, P=0.005) received both surgery and radiation, while 6.5% (11 patients, P=NS) had neither surgery nor radiation treatment. ATRT cases managed with a combination of surgery and radiation increased by 16.1% over the study period, from 22.3% between 1973 and 2004 to 38.4% after 2005, P<0.001. The number of ATRT cases treated with combination surgery and radiation showed a 6.9% increase between 2004 and 2005 (3.4%–10.3%), a 10.3% increase between 2006 and 2007 (5.2%–15.5%), and a 3.4% increase between 2009 and 2010 (13.8%–17.2%) was observed, P<0.001 (Figure 1). The use of primary radiation therapy and surgical resection alone remained approximately the same (0.0% in the 1973–2004 period increasing to 1.7% in the post-2005 period for radiation treatment alone and 56.3% in the 1973–2004 period increasing to 58.7% in the post-2005 period for surgical resection alone), P<0.001. There was an 11.3% decrease in the number of ATRT patients receiving no treatment, from 14.6% in the 1973–2004 period to 3.3% in the post-2005 period, P<0.001. Mean survival for patients treated with primary radiation therapy was significantly improved compared to those treated with surgery alone (15.4±0.4 years vs 13.6±0.3 years, P<0.001). The longest survival was seen among ATRT patients receiving both surgery and radiation (5.9±0.7 years), followed by radiation alone (2.8±1.2 years), surgery alone (1.9±0.4 years), and finally those receiving neither surgery nor radiation (0.3±0.2 years), P<0.001.
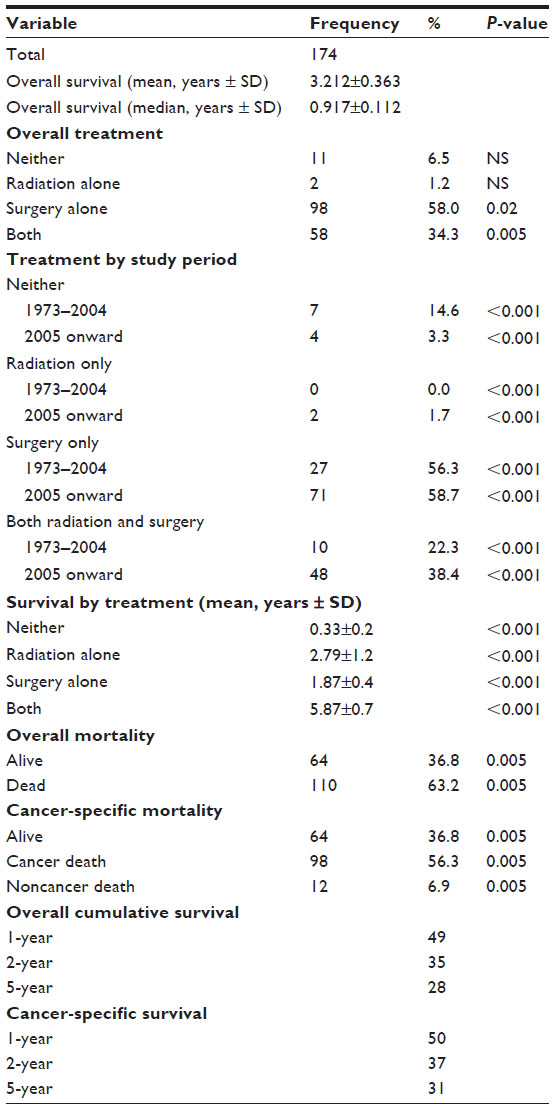 | Table 3 Treatment and survival outcomes of 174 patients with ATRT from the SEER database, 1973–2010 |
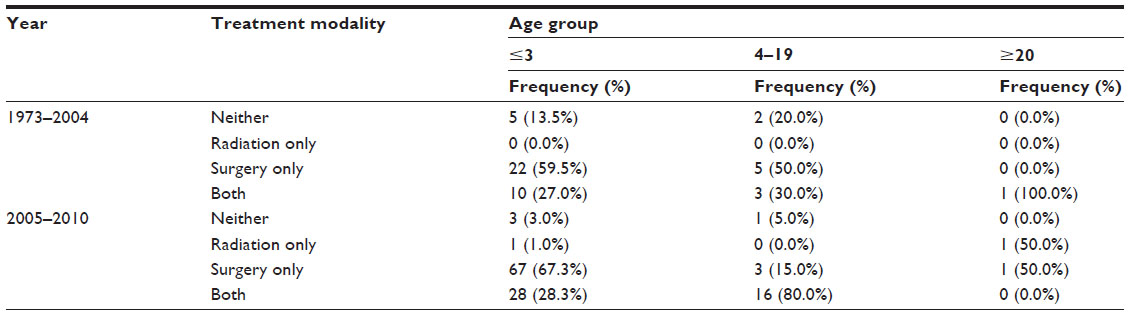 | Table 4 Comparing treatment profiles for different age groups between 1973–2004 and 2005–2010 for 174 patients with ATRT from the SEER database, 1973–2010 |
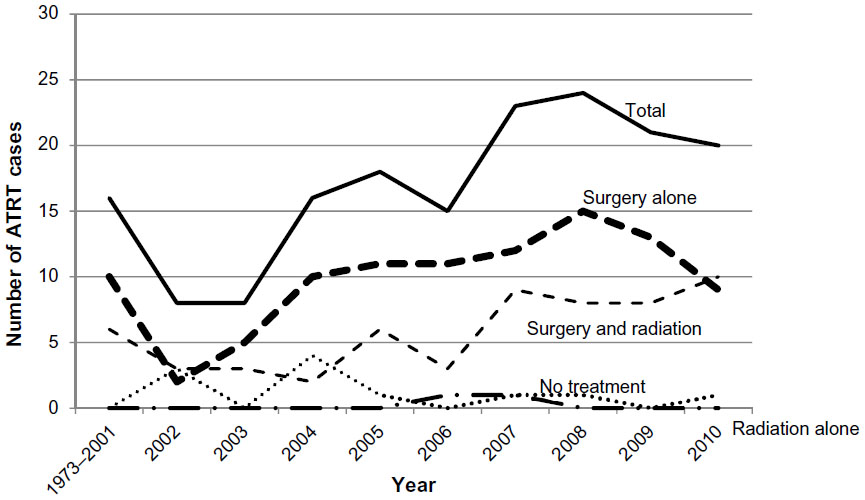 | Figure 1 Trends in incidence rate and treatment modality for ATRT from the SEER database, 1973–2010. |
Outcomes
Overall and cancer-specific mortality were 63.2% and 56.3%, respectively, for the entire cohort, P<0.005 (Figures 2 and 3). Mean overall survival was 3.212±0.363 years. ATRT patients undergoing surgery and radiation therapy experienced significant survival benefit (mean survival 5.87±0.7 years), compared to those treated with primary surgical therapy (1.87±0.4 years) or radiation alone (2.79±1.2 years), P<0.001. Patients receiving no therapy had the lowest survival of 0.33±0.2 years, P<0.001.
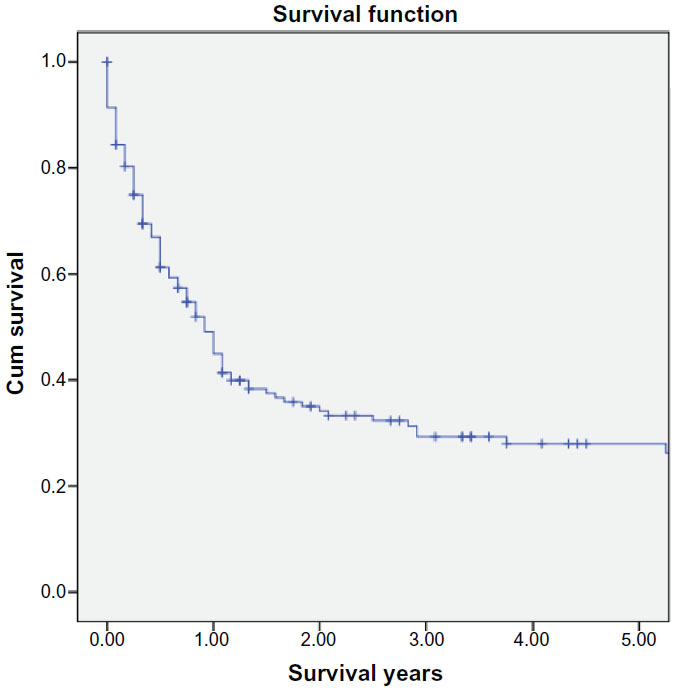 | Figure 2 Kaplan–Meier curves illustrating actuarial survival for patients with ATRT from the SEER database, 1973–2010. |
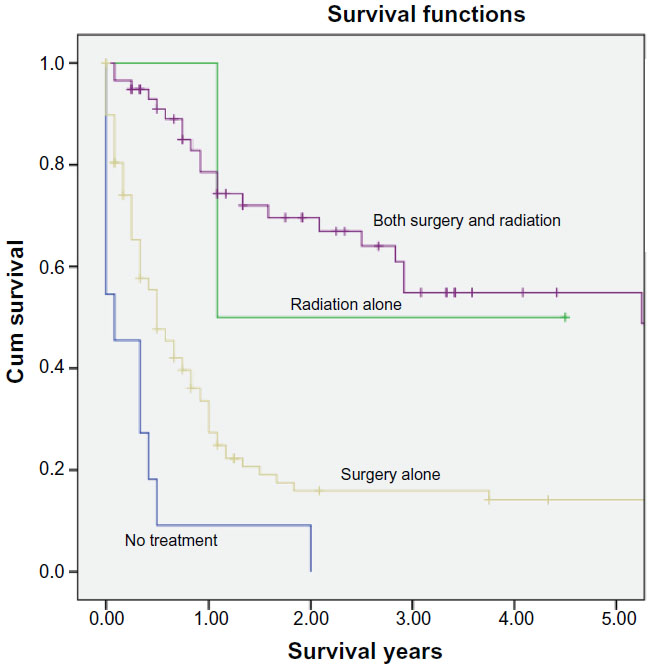 | Figure 3 Kaplan–Meier curves illustrating actuarial survival by treatment modality for patients with ATRT from the SEER database, 1973–2010. |
When stratified by ethnicity, Caucasians had the longest survival (4.17±0.5 years), followed by Hispanics (2.48±0.7 years) and Asian/Pacific Islanders/other races (1.84±1.0 years). African–Americans had the shortest survival (1.21±0.4 years). Males had a longer survival than females (3.52±0.5 years vs 2.90±0.5 years). There were no significant survival differences between different ethnicities (P=0.634) or sexes (P=0.443).
Multivariable analysis
Multivariable analysis identified only distant metastases (OR =4.6, CI =2.7–6.9) as independently associated with increased mortality, P<0.005. Conversely, combination surgery and radiation treatment (OR =0.4, CI =0.1–2.0) was independently associated with reduced mortality, P<0.005.
Discussion
ATRT is a rare and highly malignant embryonal tumor of the CNS. Based on the current study, ATRT is more prevalent among male Caucasian children under 3 years of age, which is consistent with prior studies.2–4 ATRT most commonly presents in the cerebellum, ventricles, and frontal lobe, with a locoregional distribution and tumor sizes greater than 4 cm. Distant metastasis occurred in 13% of patients, which is consistent with a prior report by Athale et al,11 in which 14% of the patients had metastasis. Most metastatic lesions remain in the CNS, 58% of which involved the spinal cord and subarachnoid space with possible leptomeningeal spread.11 Pulmonary and peritoneal metastases have also been reported, but are rare.11 The clinical presentation of ATRT depends on the location of the primary tumor at diagnosis and is typically associated with nonspecific symptoms, including lethargy, vomiting, and/or failure to thrive.3,7,8
Histologically, ATRT tumors are highly cellular neoplasms that are composed of sheets or nests of rhabdoid cells, which contain an eccentric round nucleus with prominent nucleolus and a plump cell body.6–8 Mitotic figures are typically present, as well as necrosis with calcifications.8,12 PNET components, which are present in almost two-thirds of ATRT, are typically made up of small basophilic cells, with high nuclear-to-cytoplasmic ratios.8 Mesenchymal components, found in approximately one third of ATRT cases, consist of loosely arrayed spindle-shaped cells separated by pale ground substance.8 The least common histological feature of ATRT is the epithelial component, such as glandular or squamous epithelium.7,8
Advancements in immunohistochemistry (IHC) have allowed for more accurate diagnosis of ATRT, making it possible to initiate earlier appropriate therapy.8 There are no IHC stains which are pathognomonic for ATRT; however, these tumors almost always express epithelial membrane antigen, vimentin, smooth-muscle actin, and glial fibrillary acid protein. In contrast, germ-cell tumor markers, including alpha-fetoprotein, placental alkaline phosphatase, and human chorionic gonadotropin, are always negative.6–8,10 Chromosome 22 abnormalities can also be detected, ranging from partial deletions to monosomy.6–8,10,13 One of the most common abnormalities is a deletion of region 11.2 of the long arm of chromosome 22 (22q11.2), causing a frameshift or nonsense mutation in the hSNF5/INI1 gene, and subsequently leading to loss-of-function of the tumor suppressor gene.14 IHC showing the absence of nuclear INI1 staining in tumor cells can aid in the diagnosis of ATRT.15
Advances in radiological imaging has permitted better delineation and improved sensitivity in detecting ATRT lesions. Historically, ATRTs were diagnosed only after surgical excision of the tumor; and until recently, these were often indistinguishable from PNET.7,8 ATRT appears similar to PNET radiologically, with increased density on unenhanced computerized tomography (CT) images and inhomogenous contrast enhancement, often associated with cysts and hemorrhages.8,10 Warmuth-Metz et al16 studied 9 CT and 32 magnetic resonance imaging (MRI) images from a total of 33 children with ATRT in Germany and observed that all 9 CT scans showed hyperdensity, and 3 of the images (25%) had evidence of calcifications. MRI was more varied. On T1-weighted images, 12 of the 27 scans showed homogenous hypointensity while the other 15 showed inhomogenous intensities.16 On T2-weighted images, 17 of the 30 scans showed mixed intensities with predominantly hypointense values.16 Of the 13 predominantly homogenous tumors, 5 were homogenous hypointense, 4 were homogenous isointense, and 4 were homogenous hyperintense.16 Almost half of the patients showed hemorrhage, and 63% of scans showed strong enhancement after intravenous administration of contrast medium.16
Until recently, treatment of ATRT has been by surgical resection alone. In the current study, more than half of all ATRT patients were treated with surgery, and survival was poor (1.87±0.4 years). Complete surgical resection is the most important factor in predicting the prognosis of ATRT patients; however, total or near-total resection is difficult to achieve and is accomplished in less than one-third of all patients.2,17 As part of the Canadian Pediatric Brain Tumor Consortium, Lafay-Cousin et al18 studied 50 children ≤18 years of age with CNS ATRT. Fifteen of the patients underwent gross total resection and had a 2-year overall survival of 60%, compared to ten patients who underwent subtotal resection and achieved a 2-year overall survival of only 21% (P=0.03).18
The use of radiation therapy in addition to surgical resection as a treatment for ATRT has increased from 22.3% of cases between 1973 and 2004, to 38.4% of cases since 2005. This change in treatment paradigm has occurred as a result of several small cohort studies which have reported significant benefits to adjunct radiation.3,19 Chen et al19 reported a significant relationship between overall survival and the interval between surgery and the initiation of radiation therapy as well as the interval between surgery and completion of radiation therapy. These authors also demonstrated that the total dose of radiation therapy (>50 Gy) was the only significant predictive factor for improved failure-free survival (P=0.037). Although there was no significant difference in overall survival rate, this was attributed to the limited sample population. Tekautz et al3 reported that radiation was associated with prolonged survival in older children and adult ATRT patients, while younger children benefitted only if they received radiation therapy early in the course of treatment. In this study, combination of surgery and radiation conferred the longest mean survival of 5.87±0.7 years, which is consistent with prior reports.5,20 Buscariollo et al20 (2012) reported on 144 ATRT patients in a retrospective study and noted that those who received initial radiation therapy had a better overall survival when compared to those who did not (34 months to not yet reached vs 6 months, P<0.001).
Radiation therapy in ATRT pediatric patients has typically been delayed, given the young age of the patients and the vulnerability to adverse neurocognitive effects on the developing brain.20,21 Evidence from early studies suggest that patients who receive radiation at a very young age often suffer severe long-term neurological, cognitive, and developmental effects, including a decline in intelligence quotient.22,23 Radcliffe et al24 demonstrated more severe cognitive impairment among children <3 years of age, compared to older children receiving brain radiation. Mulhern et al25 in the Pediatric Oncology Group studied 22 children between the ages of 1.7 and 14.8 years with medulloblastomas, randomized to receive either standard dose cranial irradiation of 36 Gy (n=13) or reduced-dose cranial irradiation of 23.4 Gy (n=9). Children <9 years of age, experienced a mean decline of 22.50 points on intelligence quotient scores with standard radiation dose, compared to children >9 years of age, who experienced a mean decline of 7.00 points. Younger children <9 years of age receiving standard radiation dose had lower scores than those receiving reduced radiation dose (P=0.030).25
Various theories about the underlying mechanism for cognitive dysfunction have been proposed. This includes the direct, irradiation-induced gradual loss of glial cells in demyelination and inhibition of white matter development.22 Oligodendrocytes and their progenitor cells, responsible for producing myelin, are the most radiosensitive glial cells.26 Death of this cell type as a result of radiation initiates release of oxygen species from astrocytes, causing an inflammatory response and impairing neurogenesis.26 Another proposed explanation is that the indirect effect of irradiation on white matter, through damage to the microvasculature, leads to hypoxia and ischemia.22,26,27
Recent pharmacological approaches to managing learning and cognitive problems induced by cancer treatment have shown significant potential.22 Methylphenidate has been used in children with attention-deficit disorder following radiation therapy and has been shown to improve teacher and parent report of attention.26,28 Modafinil and donepezil have also been shown to improve attention and global functioning in adults after radiation therapy; however, its use has not yet been studied in children.26 Further development of innovative strategies to preserve neurocognitive function in children undergoing brain cancer treatment is required.
Since ATRT is most prevalent in young children, at a time when their brains are most vulnerable to the neurocognitive impairments caused by radiation, conformal focal irradiation methods have recently been utilized in hope of limiting or eliminating the amount of neurodevelopmental impairment that occurs. Conformal focal irradiation and proton radiotherapy have been successfully studied in young children with other forms of CNS embryonal tumors, including ependymoma and medulloblastomas.20,29,30 Its use in the treatment of ATRT is relatively new and limited. De Amorim et al4 conducted the first clinical series reporting the use of proton therapy in pediatric ATRT, involving ten pediatric patients (median age 1.8 years, range 15 days–19.3 years). At a median follow-up of 27.3 months from the initiation of radiation therapy, nine patients had no evidence of disease, and one patient died.4
Chemotherapy has been well studied for the management of ATRT. Many patients receive chemotherapy in addition to surgical resection, with the aim of postponing or avoiding radiation therapy, especially in younger children under the age of 3. In an early study, Burger et al6 reported that patients treated with chemotherapy had a dismal prognosis, with most dying within 12 months. Poor response to conventional chemotherapy methods used to treat medulloblastomas has led to the evaluation of a variety of alternative chemotherapy regimens. The Canadian Pediatric Brain Tumor Consortium,18 reported on 18 patients who received high-dose chemotherapy (HDC) regimens and noted longer 2-year overall survival compared to those who received conventional chemotherapy (47.9% vs 27.3%, P=0.036). Although different chemotherapy regimens were used in this study, most patients received a methotrexate-based induction, followed by high-dose carboplatin and thiotepa.18 Despite some benefit associated with chemotherapy treatment, tumor progression is relatively common with survival rates remaining low, often requiring repeat surgery and radiation.5,20,21,31 Athale et al11 conducted a review of 79 pediatric ATRT patients treated with chemotherapy and reported a median survival of only 17.3 months (range: 1.5–93 months) and an overall mortality of 67%. It was also reported that disease progression was the major cause of death, with craniospinal spread occurring in 58% of the patients.11
There are several limitations to this study which should be considered. First, the SEER database does not accurately code for all important clinical factors such as socioeconomic status, geography, tumor depth, and method of diagnostic confirmation, which may have had an influence on survival. Second, information on diagnostic imaging and follow-up are lacking. Data on surgical and radiation therapy were available in the SEER database; however, information on chemotherapy received was not, which limited this study’s ability to evaluate the impact of adjuvant or neoadjuvant therapy. There may also be an element of selection bias, since SEER registries are more likely to sample from urban than from rural areas. Despite these limitations, the SEER database has data obtained from 14% of the US population, and these findings can be generalized to the overall population.
Summary
ATRT is a rare and highly malignant tumor of the CNS, associated with a poor survival and high mortality. The current study represents the largest ATRT study to date, establishing trends in demographics, management, and clinical outcomes in a large US patient cohort. ATRT presents more often among Caucasian males under the age of 3 and tends to occur in the cerebellum, ventricles, or frontal lobes of the brain with locoregional tumors greater than 4 cm in size, with up to 13% of the patients developing distant metastasis. Advances in diagnostic imaging have allowed for better diagnosis of ATRT. While surgery remains the main method of treatment for ATRT, radiation has emerged as a promising adjunct or primary modality of treatment, nearly doubling overall survival. Despite prolonged survival, overall mortality exceeds 60%, and most affected children suffer long-term neurocognitive and developmental impairments. Future studies optimizing radiation regimens to limit or avoid neurocognitive impairments are needed to permit more precise multimodality management of these patients. The role of chemotherapy in ATRT treatment remains controversial, and despite anecdotal success, the optimal chemotherapy regimen remains unclear. All ATRT patients should be enrolled into clinical trials or registries to allow for more defined multimodality management to optimize clinical outcomes for these patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Patel S, Bhatnagar A, Wear C, et al. Are pediatric brain tumors on the rise in the USA? Significant incidence and survival findings from the SEER database analysis. Childs Nerv Syst. 2014;30:147–154. | |
Packer RJ, Biegel JA, Blaney S, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol. 2002;24:337–342. | |
Tekautz TM, Fuller CE, Blaney S, et al. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol. 2005;23:1491–1499. | |
De Amorim BK, Sethi R, Trofimov A, et al. Early clinical outcomes using proton radiation for children with central nervous system atypical teratoid rhabdoid tumors. Int J Radiat Oncol Biol Phys. 2013;86:114–120. | |
Hilden JM, Meerbaum S, Burger P, et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. 2004;22:2877–2884. | |
Burger PC, Yu IT, Tihan T, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol. 1998;22:1083–1092. | |
Rorke LB, Packer R, Biegel J. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol. 1995;24:21–28. | |
Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996;85:56–65. | |
Chen ML, McComb JG, Krieger MD. Atypical teratoid/rhabdoid tumors of the central nervous system: management and outcomes. Neurosurg Focus. 2005;18:E8. | |
Strother D. Atypical teratoid rhabdoid tumors of childhood: diagnosis, treatment and challenges. Expert Rev Anticancer Ther. 2005;5:907–915. | |
Athale UH, Duckworth J, Odame I, Barr R. Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies. J Pediatr Hematol Oncol. 2009;31:651–663. | |
Parwani AV, Stelow EB, Pambuccian SE, Burger PC, Ali SZ. Atypical teratoid/rhabdoid tumor of the brain: cytopathologic characteristics and differential diagnosis. Cancer. 2005;105:65–70. | |
Biegel JA, Allen CS, Kawasaki K, Shimizu N, Budarf ML, Bell CJ. Narrowing the critical region for a rhabdoid tumor locus in 22q11. Genes Chromosomes Cancer. 1996;16:94–105. | |
Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. | |
Schittenhelm J, Nagel C, Meyermann R, Beschorner R. Atypical teratoid/rhabdoid tumors may show morphological and immunohistochemical features seen in choroid plexus tumors. Neuropathology. 2011;31:461–467. | |
Warmuth-Metz M, Bison B, Dannemann-Stern E, Kortmann R, Rutkowski S, Pietsch T. CT and MR imaging in atypical teratoid/rhabdoid tumors of the central nervous system. Neuroradiology. 2008;50:447–452. | |
Zimmerman MA, Goumnerova LC, Proctor M, et al. Continuous remission of newly diagnosed and relapsed central nervous system atypical teratoid/rhabdoid tumor. J Neurooncol. 2005;72:77–84. | |
Lafay-Cousin L, Hawkins C, Carret AS, et al. Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer. 2012;48:353–359. | |
Chen YW, Wong TT, Ho DM, et al. Impact of radiotherapy for pediatric CNS atypical teratoid/rhabdoid tumor (single institute experience). Int J Radiat Oncol Biol Phys. 2006;64:1038–1043. | |
Buscariollo DL, Park HS, Roberts KB, Yu JB. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a surveillance, epidemiology, and end results analysis. Cancer. 2012;118:4212–4219. | |
Squire SE, Chan MD, Marcus KJ. Atypical teratoid/rhabdoid tumor: the controversy behind radiation therapy. J Neurooncol. 2007;81:97–111. | |
Mulhern RK, Merchant TE, Gajjar A, Reddick WE, Kun LE. Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol. 2004;5:399–408. | |
Duffner PK, Cohen ME, Thomas PR, Lansky SB. The long-term effects of cranial irradiation on the central nervous system. Cancer. 1985;56:1841–1846. | |
Radcliffe J, Bunin GR, Sutton LN, Goldwein JW, Phillips PC. Cognitive deficits in long-term survivors of childhood medulloblastoma and other noncortical tumors: age-dependent effects of whole brain radiation. Int J Dev Neurosci. 1994;12:327–334. | |
Mulhern RK, Kepner JL, Thomas PR, Armstrong FD, Friedman HS, Kun LE. Neuropsychologic functioning of survivors of childhood medulloblastoma randomized to receive conventional or reduced-dose craniospinal irradiation: a Pediatric Oncology Group study. J Clin Oncol. 1998;16:1723–1728. | |
Castellino SM, Ullrich NJ, Whelen MJ, Lange BJ. Developing interventions for cancer-related cognitive dysfunction in childhood cancer survivors. J Natl Cancer Inst. 2014;106(8). pii:dju186. | |
Hopewell JW, van der Kogel AJ. Pathophysiological mechanisms leading to the development of late radiation-induced damage to the central nervous system. Front Radiat Ther Oncol. 1999;33:265–275. | |
Conklin HM, Lawford J, Jasper BW, et al. Side effects of methylphenidate in childhood cancer survivors: a randomized placebo-controlled trial. Pediatrics. 2009;124:226–233. | |
Merchant TE, Hua CH, Shukla H, Ying X, Nill S, Oelfke U. Proton versus photon radiotherapy for common pediatric brain tumors: comparison of models of dose characteristics and their relationship to cognitive function. Pediatr Blood Cancer. 2008;51:110–117. | |
Merchant TE, Li C, Xiong X, Kun LE, Boop FA, Sanford RA. Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Oncol. 2009;10:258–266. | |
Ginn KF, Gajjar A. Atypical teratoid rhabdoid tumor: current therapy and future directions. Front Oncol. 2012;2:114. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.