Back to Journals » Cancer Management and Research » Volume 11
ATR activated by EB virus facilitates chemotherapy resistance to cisplatin or 5-fluorouracil in human nasopharyngeal carcinoma
Authors Zhang B, Cui B, Du J, Shen X, Wang K ![]() , Chen J, Xiao L, Sun C, Li Y
, Chen J, Xiao L, Sun C, Li Y
Received 10 September 2018
Accepted for publication 19 November 2018
Published 9 January 2019 Volume 2019:11 Pages 573—585
DOI https://doi.org/10.2147/CMAR.S187099
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Kenan Onel
Bo Zhang,1,* Bomiao Cui,1,* Jintao Du,2 Xin Shen,1 Kun Wang,1 Jiao Chen,1 Liying Xiao,1 Chongkui Sun,1 Yan Li1
1State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu 610041, Sichuan, China; 2Department of Otorhinolaryngology–Head and Neck Surgery, West China Hospital, Chengdu 610041, Sichuan, China
*These authors contributed equally to this work
Purpose: Epstein–Barr virus (EBV) infection is closely associated with nasopharyngeal carcinoma (NPC) and increases the chemotherapy resistance of tumor cells. Although the mechanism by which EBV manipulates ataxia telangiectasia mutation (ATM)-mediated DNA damage response in NPC has been extensively studied, the relationship between ATR (ATM and Rad-3 related) and EBV infection is largely unexplored, and also the role of ATR in chemotherapy resistance in EBV-positive NPC has not been specifically reported.
Materials and methods: Levels of γ-H2AX, latent membrane protein 1 (LMP1), and EBV-encoded RNA in clinical NPC and nasopharyngeal inflammation (NPI) specimens were examined using immunohistochemistry and in situ hybridization. The effects of EBV infection, chemotherapy drugs cisplatin (CDDP) and 5-fluorouracil (5-FU) treatment, and ATR silencing were assessed in NPC cells in vitro using immunofluorescence, Western blot, and flow cytometry.
Results: A notable increase of γ-H2AX expression was examined in the EBV-positive NPC clinical specimens. Additionally, we observed that the phosphorylation of ATR/checkpoint kinase 1 (CHK1) pathway protein was gradually activated along with the duration of EBV exposure in NPC cell lines, which was obviously inhibited after ATR depletion. Moreover, EBV infection promoted the resistance of NPC cells to CDDP and 5-FU, whereas the chemosensitivity of cells was significantly enhanced following ATR knockdown. Furthermore, ATR depletion caused both S-phase cell arrest and apoptosis, enhanced p53 phosphorylation, and impaired the formation of Rad51.
Conclusion: Our data suggest that EBV activation of ATR-mediated DNA damage response might result in chemotherapy resistance to CDDP and 5-FU in NPC. Accordingly, ATR knockdown may serve as an effective treatment strategy for chemotherapy-resistant, EBV-positive NPC.
Keywords: ATR, EBV, NPC, chemotherapy resistance, cisplatin, 5-fluorouracil, ATRi, CNE1, Rad51, p53
Introduction
During the lifespan of cells, many internal and external factors (such as virus infection, oxygen free radical, ultraviolet radiation) can destroy the chemical structure of DNA and affect the integrity and stability of cell genome. To ensure genome integrity after DNA damage, the cells usually initiate a checkpoint mechanism to inhibit cell cycle progression, with this response dependent upon two major protein kinases, ataxia telangiectasia-mutated (ATM) and ATR (ATM and Rad-3-related),1 which function under the action of a positive feedback mechanism that leads to the binding of multiple downstream response molecules to the DNA strand breaks. Through excitation, amplification, and multi-pathway coordination, p53 and the downstream checkpoint kinase (CHK)1/CHK2 are activated, thereby allowing signal transmission to DNA strand breaks.2 The cell cycle checkpoint regulation is an important mechanism related to host cell response to genomic damage caused by certain viruses, such as the Epstein–Barr virus (EBV). Therefore, the DNA damage response (DDR) can be dysregulated by EBV infection, with EBV-encoded viral proteins.3 Former studies have shown that EBV infection activates DNA damage checkpoints by promoting the phosphorylation of ATM and CHK2 pathway and the formation of 53BP1 foci.4,5 Conversely, an ATM/Chk2-mediated DDR pathway suppresses EBV transformation of primary human B cells.6 The manipulation of ATM-mediated DDR by EBV has been extensively studied, but much less is known about ATR-mediated DDR by EBV infection.
EBV is an oncogenic herpesvirus causally implicated in nasopharyngeal carcinoma (NPC) and African endemic Burkitt’s lymphoma. EBV in the plasma levels of NPC patients was associated with the degree of lymph node metastasis, tumor burden, and poor prognosis.7,8 Currently, chemotherapy, together with radiotherapy, is the most common and important clinical treatment of NPC, which is essential for controlling the local recurrence and distant metastasis of tumors. Cisplatin (CDDP) and 5-fluorouracil (5-FU) are the main effective chemotherapeutic drugs with the mechanism of initiation of DNA damage in order to kill tumor cells.4,9,10 However, chemoresistance has become a major obstacle to NPC treatment and represents the main cause of treatment failure. Notably, numerous studies report that DDR is closely correlated with tumor cell chemoresistance.11–13 Activation of DDR allows the cells to self-repair and resist external damage by activating downstream cyclins and apoptotic proteins, thereby achieving resistance to chemotherapy. Therefore, DDR inhibition is currently considered the cause of tumor cell sensitivity to chemotherapy by inducing cell death or aging without initiating checkpoints and effective DNA repair.14–18 In particular, some studies have investigated antitumor treatment via ATR inhibition combined with chemotherapy drugs.7,19 However, the relationship among EBV infection, ATR-mediated DDR pathway, and chemoresistance in NPC remains unknown. After all, how to enhance the initial responses and/or to counter the emergence of resistance in NPC is intense interest.
In the present study, we have evaluated the relationship among EBV infection, ATR-CHK1 activity, and the chemosensitivity of NPC. A key implication of our work is that it provides the clinical rationale that evaluates ATR regulation in combination with CDDP or 5-FU in EBV-positive cells.
Materials and methods
Collection of NPC and nasopharyngeal inflammation (NPI) clinical specimens
In total, we collected 50 paraformaldehyde-fixed paraffin-embedded NPC tissues with histopathology reports. The average age of the patients was 48±11 years, with 20 patients with NPI (average age: 44±17 years) selected as controls. All 70 tissue specimens were gathered from 2010 to 2012. The study was carried out under the approval and supervision of the Ethics Committee of Sichuan University (WCSHIRB2011100), and written informed consents were obtained from each patient.
Immunohistochemistry (IHC) and in situ hybridization
EBV latent membrane protein 1 (LMP1) was shown to act as an oncogene and was expressed in NPC. EBV-encoded RNA (EBER) in situ hybridization can be a gold standard for detecting EBV.20 Combined LMP1 and EBER assay is an economical and specific method to detect EBV infection. Meanwhile, phosphorylation of histone H2AX (γ-H2AX) is a well-characterized marker of DNA breaks and activation of DNA repair.21 Therefore, we used IHC and in situ hybridization to analyze whether EBV infection is associated with DDR according to γ-H2AX level. The method in detail is as follows. An IHC streptavidin–peroxidase three-step method was used to assess pathological specimens. Slides were incubated with a primary antibody against γ-H2AX (1:100; Santa Cruz Biotechnology, Dallas, TX, USA), which is the histone variant H2AX phosphorylated at Ser139 in the C-terminal region or LMP1 (1:100; Santa Cruz Biotechnology), and detected using the ChemMate EnVision kit (Dako, Carpinteria, CA, USA). Immune reactivity was analyzed and quantified using ImageScope software (Aperio Technology, Vista, CA, USA). For LMP1-negative samples, EBER was detected using in situ hybridization according to the manufacturer’s protocol (Pan Path, Amsterdam, The Netherlands).
Cell lines, EBV infection, and chemotherapy
The following cell lines were used in this study: CNE1 (an EBV-negative, low-differentiated NPC cell line) and B95-8 (a marmoset EBV-immortalized B cell line). CNE1 and B95-8 cells were obtained from the State Key Laboratory of Oral Diseases (Sichuan, China) and cultured in Roswell Park Memorial Institute-1640 (HyClone, Logan, UT, USA) with 10% FBS (Gibco, Grand Island, NY, USA) and 1% penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA USA). All cells were cultured at 37°C in a humidified incubator with 5% CO2.
B95-8 cells were cultured in a P2 laboratory according to a previously reported method.22 EBV was isolated from B95-8 cells by multigelation and ultrasonication of cells in the late log phase, followed by filtration through a 0.22 µm filter and further purification using PEG6000 (P8250; Solarbio, Beijing, China). The viral titer was calculated at 1.0×109 copies/mL using real-time PCR according to the EBV nucleic acid quantitative detection kit (Sansure Biotech, Changsha, China).
EBV infection was performed according to the method reported by Bejarano et al.23 Briefly, the CNE1 cells were adjusted to 5×105/well in six-well plates and 5×103 cells/well in 96-well plates using Roswell Park Memorial Institute-1640 medium with 10% FBS and 1% penicillin–streptomycin, inoculated in plates, then infected with EBV suspension at a multiplicity of infection (MOI) of 100, and cultured at 37°C. The cells were passaged every 2 days and reinoculated with freshly EBV suspension after each passage. Stably infected CNE1 cells were used in subsequent experiments.
The EBV-positive (EBV+) CNE1 or EBV-negative (EBV−) CNE1 cells were treated with CDDP (A8321; APExBIO, Boston, MA, USA) at different concentrations (0, 5, 10, 20, 40 µM) or 5-FU (A4071; APExBIO) at different concentrations (0, 12.5, 25, 50, 100 µM) for 24, 48, and 72 hours.
CDDP at a concentration of 10 µM or 5-FU at 25 µM (lower than IC50) for 48 hours was used for the following chemotherapy experiments.
Immunofluorescence
CNE1 cells with a cell count of 5×105 were inoculated in six-well plate preset with sterilized coverslips. After growing overnight, 100 MOI EBVs were used to infect the cells for 24 hours. Then the coverslips were taken out and the cells on them were fixed with 4% paraformaldehyde for 15 minutes at room temperature, after which the cells were permeabilized with 0.2% Triton X-100 for 5 minutes, followed by 30 minutes of blocking in 4% FBS. The cells were incubated with green fluorescence antibodies against γ-H2AX (1:200; Signalway Antibody, College Park, MD, USA) for 2 hours and then with an Alexa Fluor 594 dye-conjugated secondary antibody (1:500; Thermo Fisher Scientific) for 1 hour before counterstaining of the nuclei with DAPI (1:1,000; Sigma-Aldrich, St Louis, MO, USA) at room temperature. The slides were covered with fluorescence mounting medium (Dako, Glostrup, Denmark) and photographed under a fluorescence microscope (Olympus, Tokyo, Japan).
Western blot
We incubated the CNE1 cells with 100 MOI EBV for an additional 6, 12, or 24 hours and detected the phosphorylation level of ATR pathway proteins.
Cells were trypsinized, harvested, washed, and lysed in ice-cold buffer (Beyotime, Jiangsu, China). Western blot was performed according to standard procedures. Primary antibodies against ATR (1:1,000; Abcam, Cambridge, UK), phosphorylated (p)-ATR (1:1,000; Abcam), γ-H2AX (1:500; Santa Cruz Biotechnology), p-replication protein A (RPA)32 (1:1,000; Santa Cruz Biotechnology), CHK1 (1:500; Santa Cruz Biotechnology), p-CHK1 (1:500; Santa Cruz Biotechnology), Rad51 (1:10,000; Abcam), and p-p53 (1:1,000; Abcam) were diluted in 5% bovine serum albumin. GAPDH (1:1,000; Santa Cruz Technology) was used as an internal control.
Plasmid transfection
EBV+ CNE1 cells were transfected with an ATR-targeting shRNA plasmid (shATR; sc-29763-SH; Santa Cruz Biotechnology), called ATR interference (ATRi). Negative control (shNC; Santa Cruz Biotechnology) vectors were constructed using scrambled shRNA sequences. Stable transfectants were obtained within 2–3 weeks by treating with puromycin (Thermo Fisher Scientific) at 2 µg/mL. Then, we chose the maintaining concentration of 1 µg/mL puromycin.
Cell proliferation assay
We examined the effect of the chemotherapy drugs CDDP and 5-FU on the viability and proliferation of CNE1 cells using a Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan). Briefly, NPC cells were seeded in 96-well plates at a density of 103 cells per well and duplicates of four wells each were set for each time point. After treatment by the indicated materials for 48 hours, CCK-8 was added and the absorbance at 450 nm was measured using a Varioskan Flash microplate reader (Thermo Fisher Scientific). The cell viability was calculated as: cell viability=[A(experimental) −A(blank)]/[A(control)−A(blank)]. Cell inhibition=1−cell viability.
Cell cycle and apoptosis assessment
EBV+ CNE1 cells transfected with shNC and shATR were exposed to 10 µM CDDP or 25 µM 5-Fu for 48 hours and the following cell cycle and apoptosis assays were performed.
The cells were fixed and permeabilized overnight with 70% ethanol precooled at 4°C. The cells were then incubated with RNase A at 37°C for 30 minutes and stained with propidium iodide (PI) at 4°C for 30 minutes (cell cycle detection kit; KeyGEN, Jiangsu, China). Cell cycle status was assayed using a flow cytometer (Beckman Coulter, Brea, CA, USA).
To evaluate apoptosis, an apoptosis detection kit (KeyGEN) was used according to the manufacturer’s protocol. Cells were digested with trypsin without EDTA, 500 µL of binding buffer was added to the cell suspension, and Annexin-fluorescein isothiocyanate and PI were used for staining. Apoptosis was assayed using a flow cytometer (Beckman Coulter).
Statistical analysis
SPSS software (version 19.0; IBM Corporation, Armonk, NY, USA) and GraphPad Prism software (version 6; GraphPad Software, San Diego, CA, USA) were used for statistical analysis. Data are presented as the mean±SD, and one-way analysis of variance was used to compare means. Student’s t-test was used for pairwise comparisons between groups. Chi-squared test was used to analyze clinical samples, and P<0.05 was considered statistically significant.
Ethics approval and informed consent
This study was approved by the local Ethics Committee of the West China Hospital of Stomatology, Sichuan University (WCSHIRB2011100) and conducted in accordance with the Declaration of Helsinki. The experiments were performed in accordance with approved guidelines.
Results
EBV increased γ-H2AX expression in NPC specimens
To determine the presence of EBV infection in 50 NPC and 20 NPI specimens, we examined the expression level of EBV LMP1 using IHC. The results indicated positivity rates of 76% in NPC (38/50; Figure 1A and B) and 25% in NPI (5/20; Figure 1C and D). In LMP1-negative samples, we evaluated EBER levels by in situ hybridization, which demonstrated that 10 of the 27 LMP1-negative samples were EBER positive, 9 of which were NPC (Figure 1E and F) and 1 was NPI. Therefore, EBV positivity was found in 47 of 50 NPC samples, which was significantly higher than the positivity in the NPI group (P<0.05; Figure 1G).
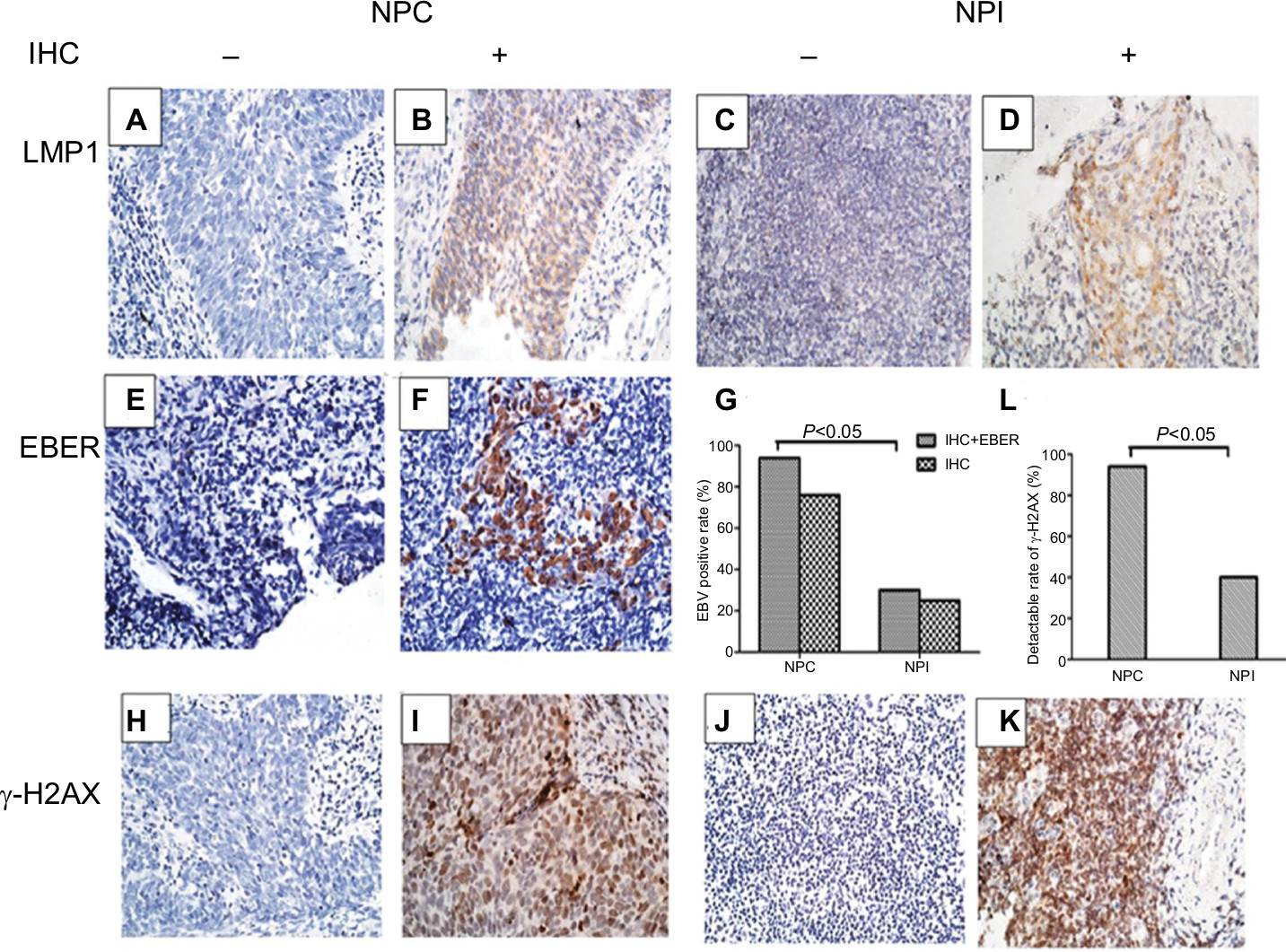 | Figure 1 LMP1, EBER, and γ-H2AX expression in the clinical samples of NPC and NPI. Notes: (A–F) Representative IHC results for LMP1 (A: negative; B: positive) in human NPC samples or (C: negative; D: positive) in human NPI samples. Representative in situ hybridization results for EBER (E: negative; F: positive) in human NPC samples. (G) EBV-positive rate detection by IHC and in situ hybridization in NPC and NPI samples. (H–K) Representative IHC results for γ-H2AX (H: negative; I: positive) in human NPC samples or (J: negative; K: positive) in human NPI samples. (L) Percentage of γ-H2AX-positive specimens in NPC and NPI samples. Magnification 400×. Abbreviations: EBER, EBV encoded RNA; EBV, Epstein–Barr virus; IHC, immunohistochemistry; NPC, nasopharyngeal carcinoma; NPI, nasopharyngeal inflammation. |
We then determined the levels of γ-H2AX in all NPC and NPI specimens (Figure 1H–K). Results showed that 94% (47/50) of the NPC samples displayed γ-H2AX positivity, whereas only 40% (8/20) of the NPI samples were positive, indicating that there were significantly more DDR in NPC than in NPI specimens (P<0.05; Figure 1L). Additionally, we assessed the relationship between the γ-H2AX level and EBV infection in the NPC specimens. Elevated γ-H2AX levels were observed in 97.9% of EBV-positive NPC biopsies (46/47), whereas this was observed in only one-third of the EBV-negative NPC samples (P<0.05; Table 1).
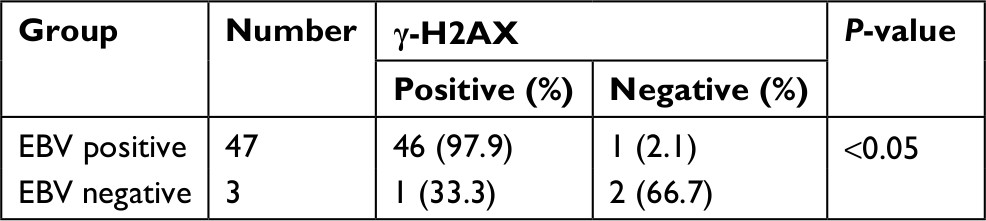 | Table 1 Correlation analysis of EBV and γ-H2AX detection rates in NPC clinical specimens Abbreviations: EBV, Epstein–Barr virus; NPC, nasopharyngeal carcinoma. |
EBV mediated activation of the ATR/CHK1 pathway in NPC cells
Figure 2A shows that the number of γ-H2AX foci in EBV-positive cells was significantly higher than that in EBV-negative cells, indicating that EBV infection resulted in an accumulation of DNA damage in NPC cells. We then incubated the CNE1 cells with EBV for different time periods. As shown in Figure 2B, phosphorylation of ATR, H2AX, CHK1, and RPA32 increased over a short period upon prolonged EBV exposure. By contrast, after ATR underwent stable knockdown, the amounts of p-RPA32 and γ-H2AX detected in EBV+ CNE1 cells decreased obviously. As expected, phosphorylation of the kinase CHK1 and ATR was also attenuated upon ATR depletion (Figure 2C). These results indicate that ATR is the key factor involved in EBV-mediated activation of the ATR/CHK1 pathway in NPC cells.
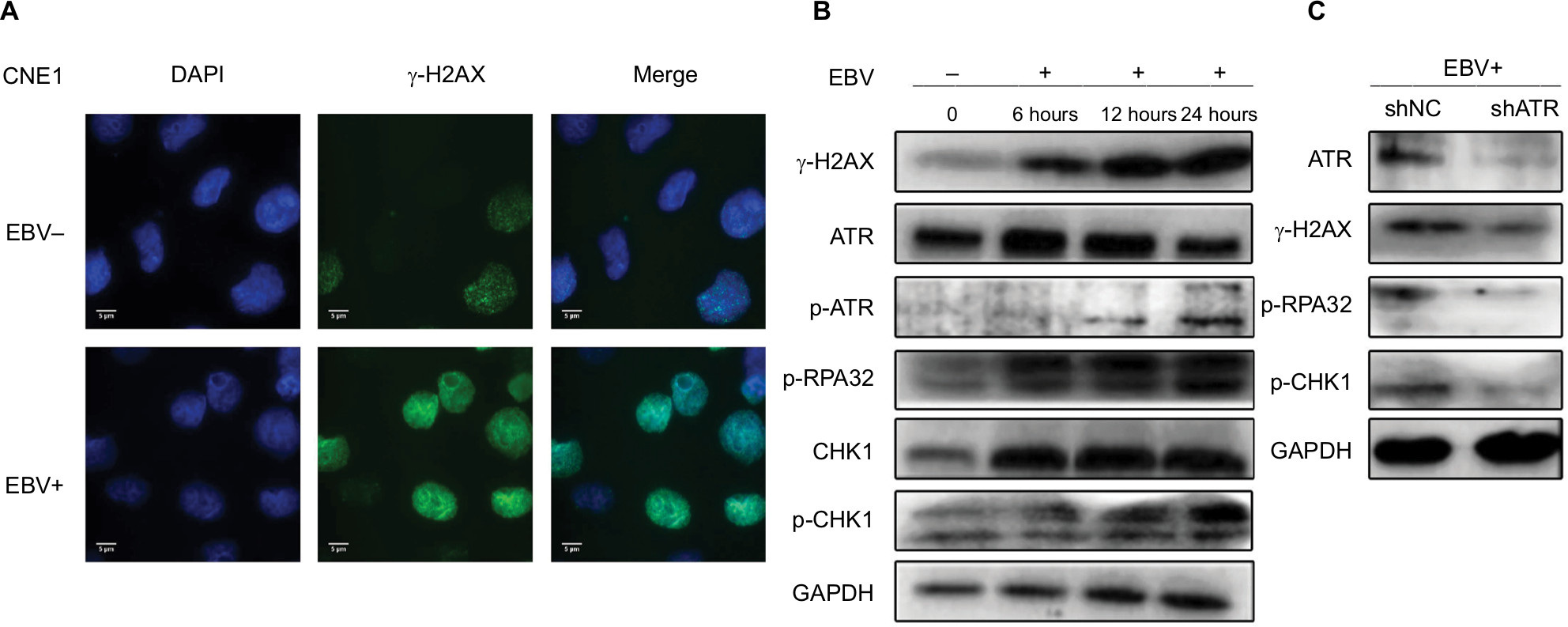 | Figure 2 EBV upregulates ATR-signaling pathway proteins in CNE1 cells. Notes: (A) Immunostaining for γ-H2AX after 24 hours of EBV treatment. (B) CNE1 cells infected with EBV for 6, 12, or 24 hours were subjected to Western blot using the indicated antibodies. (C) ATR depletion inhibits phosphorylation of ATR pathway proteins. Plasmids expressing shATR and shNC were transfected into EBV-positive CNE1 cells. Abbreviations: ATM, ataxia telangiectasia mutation; ATR, ATM and Rad-3 related; EBV, Epstein–Barr virus. |
EBV mediated chemoresistance to CDDP or 5-FU, which was reversed by ATRi
Two main chemotherapeutic drugs (CDDP and 5-FU) used for NPC treatment were employed to examine the cellular responses. With CDDP taken as an example, the 50% inhibitory concentrations (IC50) of CDDP in EBV+ group and EBV− group were 22.28 and 12.16 µM, respectively (Figure 3A). Following ATR-shRNA transfection, the IC50 of CDDP in EBV+ group decreased from 22.68 μM to 12.13 µM, which was similar to that of EBV− group (Figure 3B). What is more, in EBV+ group, the inhibition rate of 40 µM CDDP for 48 hours increased from 54.4% to 73.7% by adding additional shATR. Meanwhile, the cells’ viability was the lowest, about 34.8% (Figure 3A–C). The same situation was observed in 5-FU-treated EBV+ group and shATR group. Taken together, EBV infection significantly enhanced the viability of CNE1 cells and promoted the resistance to CDDP or 5-FU, whereas ATR depletion increased the chemosensitivity of the EBV+ cells in a dose- and time-dependent manner.
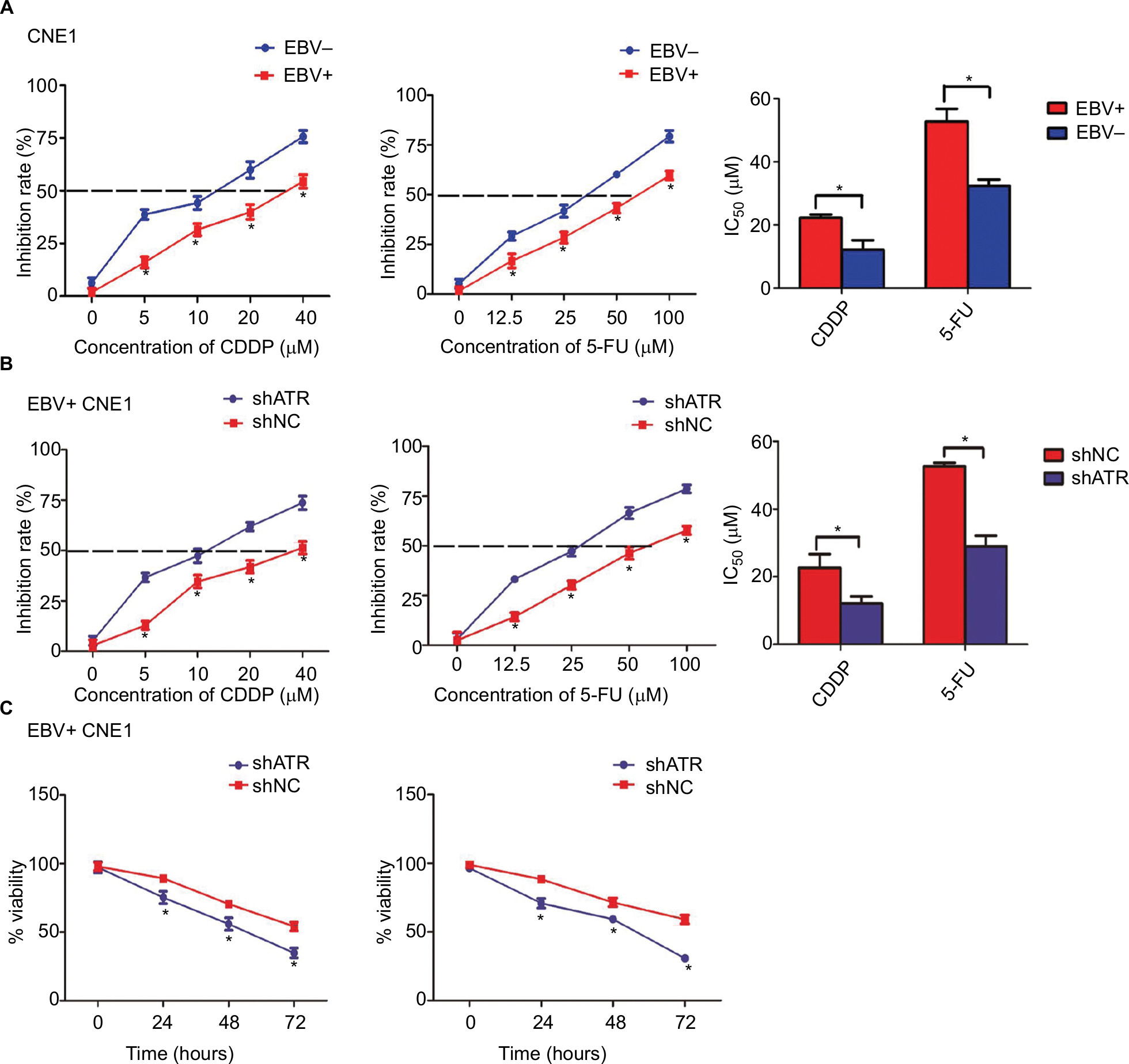 | Figure 3 ATR interference enhances the chemosensitivity of EBV-positive CNE1 cells to CDDP or 5-FU in vitro. Notes: (A) EBV increased the resistance of CNE1 cells to CDDP or 5-FU. (B, C) ATR knockdown enhanced CDDP and 5-FU cytotoxicity in a dose- and time-dependent manner. EBV-positive CNE1 cell proliferation and viability were measured using CCK-8 assays. Data represent the mean±SD of quadruplicate determinations. *P<0.05. Abbreviations: 5-FU, 5-fluorouracil; ATM, ataxia telangiectasia mutation; ATR, ATM and Rad-3 related; CCK-8, Cell Counting Kit-8; CDDP, cisplatin; EBV, Epstein–Barr virus. |
ATRi induced apoptosis in EBV-positive chemotherapy cells
To determine whether ATR knockdown enhanced the cells’ apoptosis in EBV+ chemotherapy group, the cells were stained with Annexin V-fluorescein isothiocyanate and PI, following which flow cytometry (FCM) analysis was performed. As shown in Figure 4, with CDDP and 5-FU treatment, the apoptosis rate in the ATRi group was significantly enhanced to 22.93% and 22.98% respectively, as compared with that in the control group (5.76% and 6.44%, respectively). These data indicate that ATR knockdown-enhanced sensitivity against CDDP or 5-FU in EBV+ CNE1 cells was through promotion of cell apoptosis.
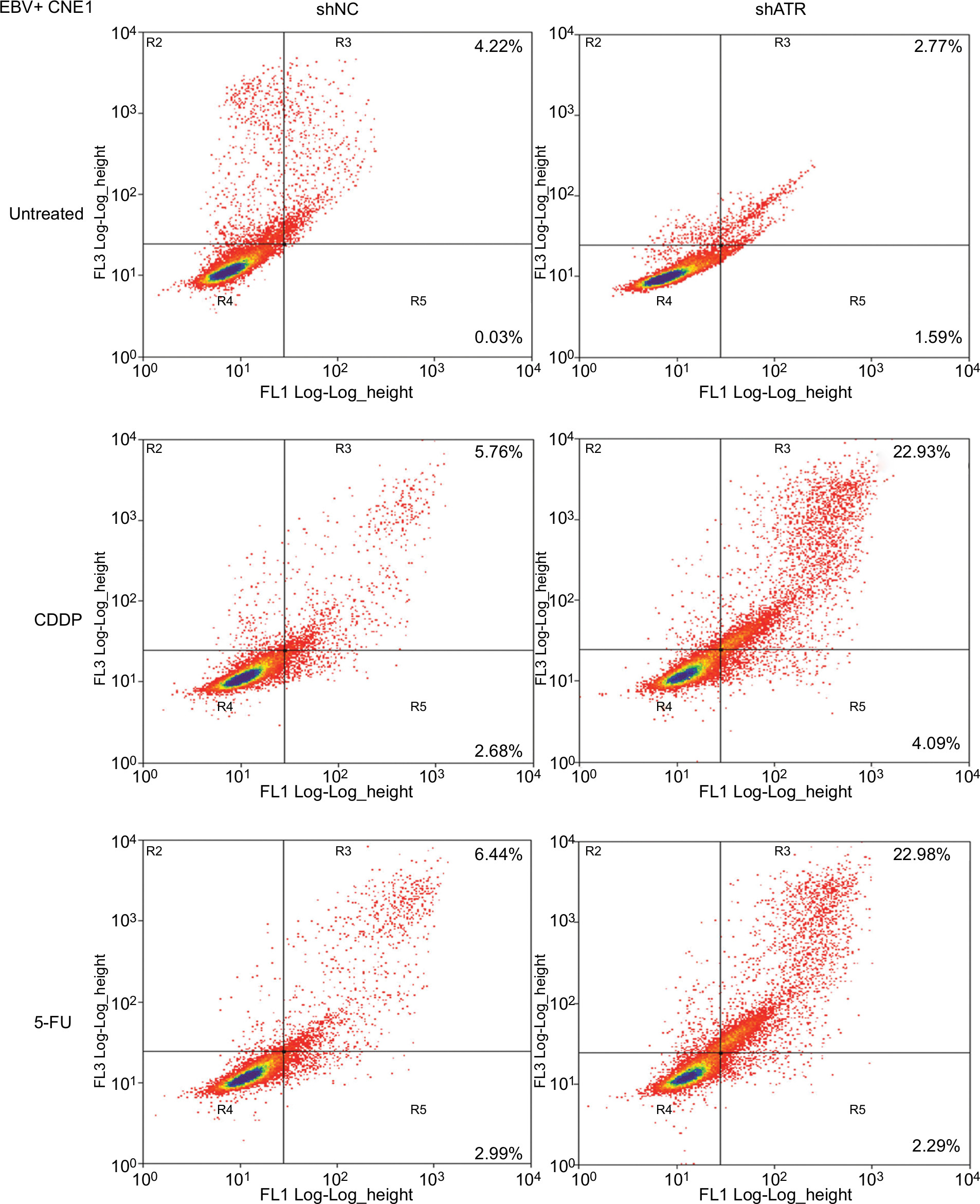 | Figure 4 ATRi enhances EBV-positive CNE1 cell apoptosis after CDDP or 5-FU treatment. Notes: Apoptotic cells were evaluated by FCM using Annexin V-FITC and PI staining (early apoptosis + late apoptosis). Abbreviations: 5-FU, 5-fluorouracil; ATM, ataxia telangiectasia mutation; ATR, ATM and Rad-3 related; ATRi, ATR interference; CDDP, cisplatin; EBV, Epstein–Barr virus; FCM, flow cytometry; FITC, fluorescein isothiocyanate; PI, propidium iodide. |
ATRi resulted in S-phase arrest in EBV-positive chemotherapy cells
We then used FCM to assess whether ATR knockdown altered the cell cycle in EBV+ chemotherapy group. After treatment with CDDP or 5-FU, the ATRi cells displayed a significantly larger fraction in the S phase (31.07% or 34.71%, respectively) relative to shNC-transfected cells (15.3% or 13.5%, respectively), as shown in Figure 5A. These findings suggest that ATR silencing promoted cell cycle arrest in EBV-infected NPC cells following CDDP or 5-FU treatment (Figure 5B).
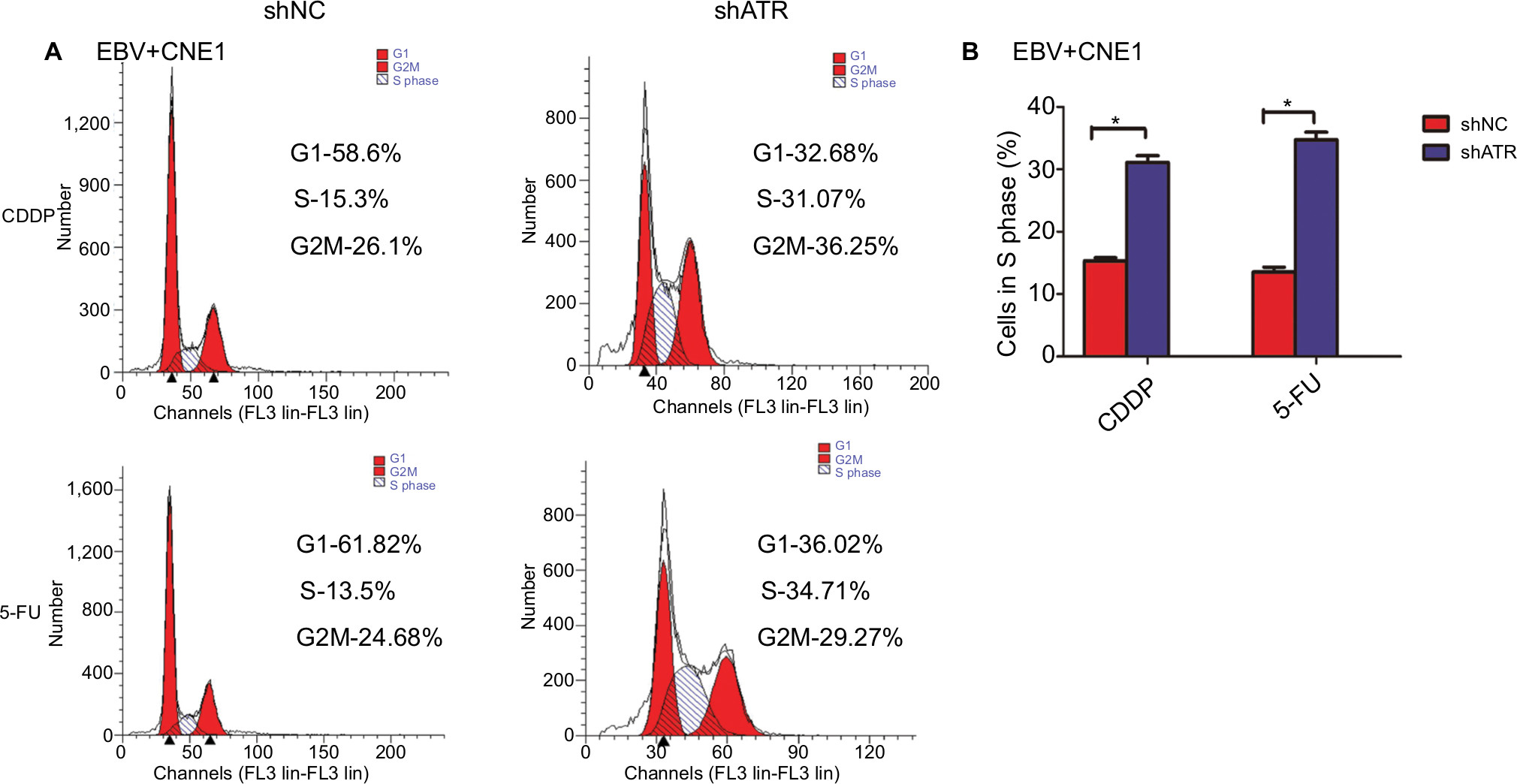 | Figure 5 ATRi promotes cell cycle arrest. Notes: (A) shNC- and shATR-transfected cells were exposed to 10 µM CDDP or 25 µM 5-FU for 48 hours, followed by treatment with RNase A+PI and cell cycle evaluation by FCM. (B) Statistics assay for cells in S phase. *P<0.05. Abbreviations: 5-FU, 5-fluorouracil; ATM, ataxia telangiectasia mutation; ATR, ATM and Rad-3 related; ATRi, ATR interference; CDDP, cisplatin; EBV, Epstein–Barr virus; FCM, flow cytometry; PI, propidium iodide. |
ATRi enhanced p53 phosphorylation and impaired the formation of Rad51
p53 is a key effector in DDR and can be phosphorylated at Ser15 by ATR, thereby inducing cell cycle arrest and/or apoptosis through transcriptional and non-transcriptional mechanisms.24 Rad51 is frequently detected in multiple discrete subnuclear structures (nuclear foci), which participate in the DNA repair process and might represent sites of ongoing homologous recombination (HR).25 To determine the mechanism of the cell cycle arrest and apoptosis, as well DNA repair situation, we investigated p53 and Rad51 levels following ATRi in EBV+ chemotherapy group. Western blot results showed that ATRi decreased Rad51 levels and increased p53 phosphorylation (Figure 6).
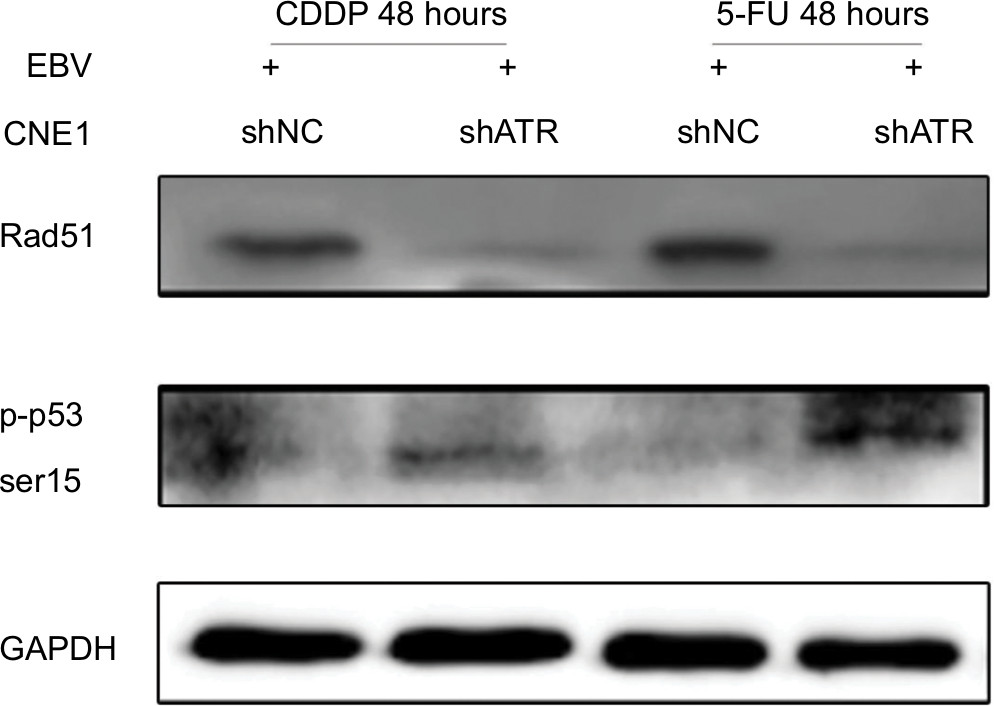 | Figure 6 ATRi increases p53 phosphorylation and impairs Rad51 formation. Notes: shATR- and shNC-transfected EBV-positive CNE1 cells were treated with 10 µM CDDP or 25 µM 5-FU for 48 hours, and cell lysates were immunoblotted with the indicated antibodies. Abbreviations: 5-FU, 5-fluorouracil; ATM, ataxia telangiectasia mutation; ATR, ATM and Rad-3 related; ATRi, ATR interference; CDDP, cisplatin; EBV, Epstein–Barr virus. |
Our findings indicate that EBV activated the ATR-mediated DDR pathway characterized by enhanced phosphorylation of the pathway proteins RPA32, CHK1, and H2AX. Additionally, EBV combined ATR increased the chemoresistance to CDDP or 5-FU. In EBV+ chemotherapy group, ATR silencing resulted in cell cycle arrest and apoptosis, which might be influenced by increased p53 phosphorylation and impaired Rad51 formation, resulting in apoptosis and ineffective HR repair. These results support a model in which EBV enhances ATR pathway protein expression and plays an important role in inducing chemotherapy resistance in NPC. Therefore, ATR regulation might represent a promising strategy for EBV-positive NPC (Figure 7).
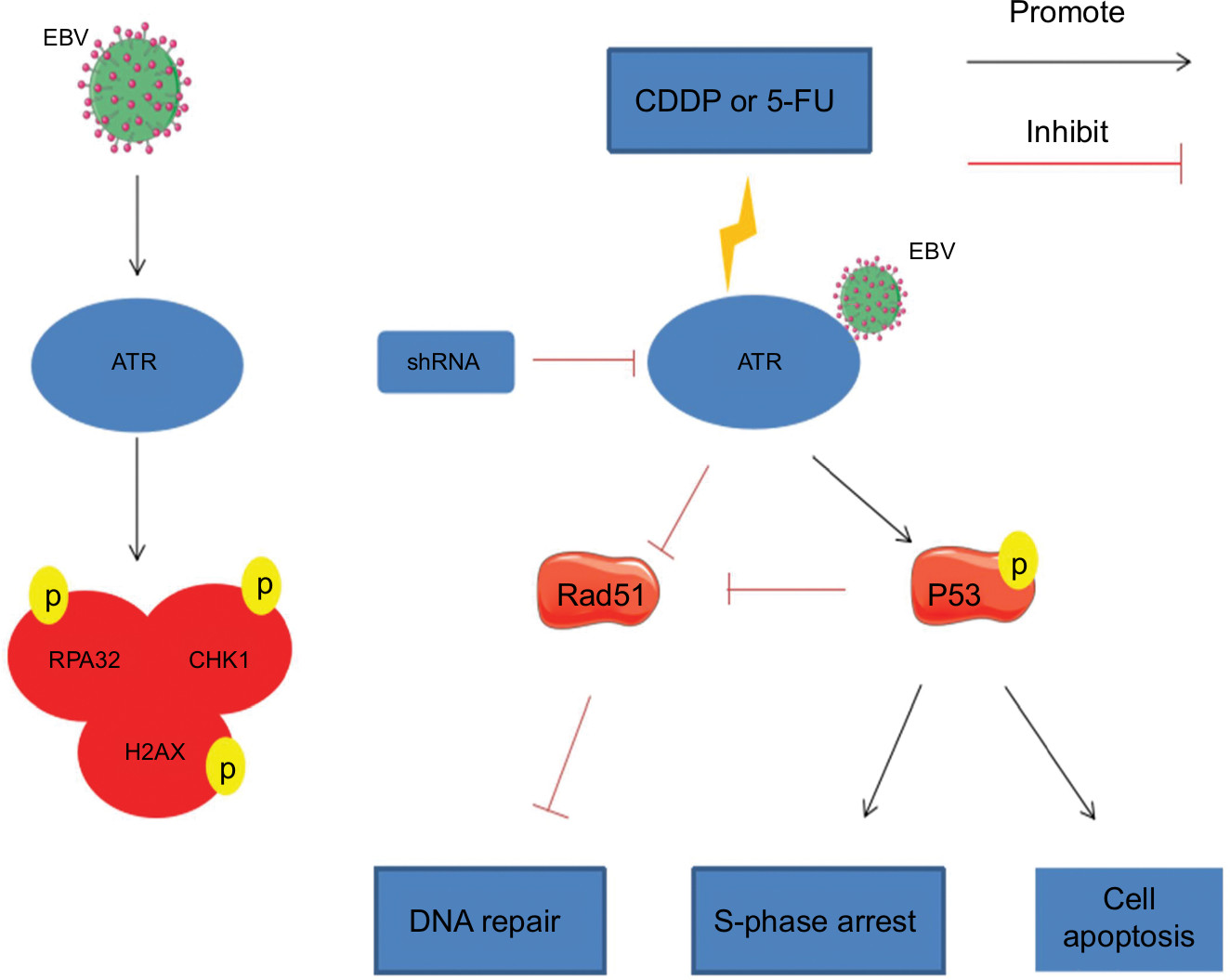 | Figure 7 Model for EBV related chemoresistance. Notes: EBV activates the ATR-mediated DDR pathway, which is characterized by enhanced phosphorylation of the pathway proteins RPA32, CHK1, and H2AX. During CDDP- or 5-FU-induced DDR in EBV-positive CNE1, ATRi leads to enhanced p53 phosphorylation, resulting in S-phase arrest and initiation of a p53-mediated apoptosis pathway. Meanwhile, ATRi also impairs Rad51 formation, leading to ineffective HR repair. Abbreviations: 5-FU, 5-fluorouracil; ATM, ataxia telangiectasia mutation; ATR, ATM and Rad-3 related; ATRi, ATR interference; CDDP, cisplatin; DDR, DNA damage response; EBV, Epstein–Barr virus; HR, homologous recombination. |
Discussion
EBV infection is generally considered the main cause of NPC, and EBV LMP1 is considered to play an important role in NPC development and progression based on its high detection rate, thereby making it an ideal marker of EBV infection.26 Moreover, in situ hybridization using single-stranded DNA probes for EBER, a small-molecule RNA that constitutes an EBV-encoded early product, exhibits extremely high specificity and sensitivity in detecting the presence of EBV in paraffin sections. The fact that EBER can be clearly localized renders it as the gold standard for detecting EBV.20,27 Therefore, in the present study, we detected EBV using these two methods, with detection rates as high as 94%, which was higher than the detection rate by IHC (76%) and suggested that this protocol could significantly increase the EBV detection rate. Additionally, in the collected NPC specimens, we found that the phosphorylation ratio of H2AX determined using γ-H2AX, a sensitive biomarker for DDR,21,28,29strongly correlated with the EBV infection rate, which is consistent with previous findings.4,30 Despite the limited number of EBV-negative clinical samples, this finding suggests that EBV infection during NPC development promoted DDR. Further experiments in the culture cells revealed similar results, with levels of γ-H2AX in CNE1 cells continuing to increase along with the duration of EBV infection. This was consistent with the results obtained from clinical specimens, as well as with the findings of Hau et al.31
EBV infection activates ATM signal transduction to stimulate DNA damage checkpoints;4,32,33 however, activation of the ATR replication checkpoint pathway in NPC has not been extensively studied. The present study revealed the effect of EBV infection on the ATR pathway in NPC cells. We found that phosphorylation of ATR, H2AX, RPA32, and CHK1 rapidly increased with prolonged EBV exposure, with a similar effect of EBV exposure detected in B lymphocytes.34,35 The ATR-CHK1 pathway primarily mediates DNA replication stress via a biochemically complex process, with γ-H2AX serving as a hallmark event for ATR pathway activation. Typically, RPA is recruited to single-stranded DNA fragments to cope with replication stress, which leads to ATR recruitment and activation33 and triggers the phosphorylation of downstream molecules, including CHK1.36 Furthermore, recent evidence37 suggests that an increase in RPA might function as an EBV-mediated strategy to stabilize replication forks during replication stress. Additionally, phosphorylation of RPA32 represents a reliable marker for ATR activation.38 Therefore, we verified that EBV infection in NPC cells led to an increase in replication stress and, consequently, RPA32 recruitment to chromatin and ATR and CHK1 activation. However, a recent study showed that EBV activated only ATR and RPA32, but decreased CHK1 expression, whereas inhibiting signal transducer and activator of transcription 3 expression enhanced CHK1 phosphorylation.39 These discrepancies might be due to signal transducer and activator of transcription 3 levels in EBV-positive NPC cells and lymphoma cells or due to specific types of EBV used. Furthermore, to investigate whether ATR functions as a key protein in the pathway, we performed shRNA-mediated knockdown, which attenuated the levels of its downstream molecules. This was consistent with the findings of Mordasini et al,34 who used EBV to infect B cells and found that ATR downregulation decreased CHK1 phosphorylation and EBV transformation efficiency, whereas CHK1 downregulation did not affect B-cell transformation.
CDDP and 5-FU are antitumor drugs that induce DNA double- or single-strand breaks, activate the cell cycle checkpoint-signaling network, trigger cell cycle arrest, and cause DNA damage, resulting in apoptosis.4,7,8,40,41 However, such DNA damage can be repaired, thereby protecting the genetic and physiological activities of the cells and allowing their survival.42 Therefore, the DDR (a genomic stability mechanism) provides a theoretical basis for the design and improved efficacy of DNA metabolism drugs. ATR plays an important role in the DDR through a complex network of signaling pathways that regulate cell cycle checkpoints, DNA repair, and cell death.43,46 Under conditions where ATR kinase is inhibited, the cells exhibit a high sensitivity to DNA-metabolizing toxicants (eg, chemotherapeutic drugs).47 Because our experiments demonstrated that ATR functions as a key factor in pathway protein phosphorylation following EBV-induced DNA damage in NPC cells, we determined the effect of ATR on drug resistance in EBV-positive CNE1 cells. Using the CCK-8 assay, we found that EBV infection increased the resistance to CDDP or 5-FU in CNE1 cells, whereas ATRi reversed the sensitivity to chemotherapy in EBV-positive CNE1 cells in a dose- and time-dependent manner.
Given these findings, we used FCM to detect the cell cycle changes, with the results showing that ATRi induced S-phase arrest in the EBV-positive chemotherapy group. A previous report showed that LMP1 can prevent chemotherapy-induced cytotoxicity and S-phase arrest, thereby causing chemotherapy resistance.44 In our experiments, ATR silencing enhanced the sensitivity of EBV-positive NPC cells to CDDP or 5-FU, possibly due to the relief of the EBV-induced prevention of S-phase arrest. ATR plays an important role in the implementation of S-phase cell cycle checkpoints during normal S-phase progression and in response to DNA damage. Moreover, ATR and p53 are closely associated; p53 can be significantly phosphorylated on Ser15 by ATR, which impairs the ability of MDM2 to bind to p53, promoting p53 accumulation and functional activation in response to DNA damage.45 Additionally, previous studies revealed that p53 plays an important role in the activity of S-phase DNA damage checkpoints and transcriptionally activates p21, resulting in decreased cyclin A (cyclin-dependent kinase) activity and causing S-phase arrest.46 Our results showed that p53 phosphorylation was enhanced in ATR-silenced EBV-positive chemotherapy group and was consistent with results reported by Wang et al.47 Therefore, we consider that S-phase cell cycle arrest might be associated with increased p53 phosphorylation.
DNA repair procedure is a primary cause of chemotherapy drug resistance.48 Notably, ATR kinase is a key regulator of HR,49–51 with ATR-CHK1 reportedly promoting HR by regulating HR factors.52 ATR-CHK1 enhances the phosphorylation of Rad51,53 Fanconi anemia complementation group E,54 and Fanconi anemia complementation group D255 to recruit HR proteins to DNA lesions for repair. Since the elevated Rad51 level is associated with radiation and chemotherapy resistance in tumor cells,56,57 our results showed ATR silencing increased the sensibility and reduced Rad51 levels in EBV-positive CNE1 cells, which would inhibit the establishment of effective HR repair, then increase the accumulation of DNA damage, and lead to tumor cell death. These findings are in agreement with those ATR inhibition studies.58,59
Moreover, p53 can inhibit the HR pathway in a non-transcriptionally dependent manner, mainly through directly binding to Rad51, which inhibits Rad51-mediated strand invasion.60 Furthermore, the role of p53/Rad51 repair complexes is dependent upon p53 phosphorylation at Ser15.61 Arias-Lopez et al62 found that p53 binds to the promoter region of Rad51, thereby downregulating Rad51 mRNA and protein levels and inhibiting the formation of Rad51 foci.Therefore, we consider that Rad51 impairment is not only directly related to the inhibition of ATR expression, but might also result from increased p53 phosphorylation, although this requires experimental verification.
Conclusion
Our results demonstrated that the ATR-CHK1 pathway was rapidly activated by EBV infection, and that ATR is a key factor for the protein phosphorylation of this pathway in NPC cells. Furthermore, EBV could result in poor sensitivity to chemotherapy. In EBV-positive CDDP or 5-FU chemosensitive NPC cells, ATR knockdown could induce apoptosis and cell cycle arrest, which was related to the inhibition of Rad51-mediated DNA repair and the promotion of p53-mediated death. Our results thus revealed a novel role of EBV in the coordination of ATR signaling pathway to chemoresistance.
Data sharing statement
All data generated or analyzed during this study are included in this published article.
Acknowledgments
We would like to thank Prof Hong Li for helping with virus culture and isolation at the Key Laboratory of Birth Defects and Related Diseases of Women and Children, Ministry of Education, West China Second University Hospital, Sichuan University. This study was supported by the National Natural Science Foundation of China (81172579 and 81502351).
Author contributions
BZ, BC, and XS designed and performed the experiments and analyzed the results. BZ, BC, JD, XS, CS, and YL conceived and designed the project, interpreted the data, and wrote the manuscript. JD and JC were involved in data acquisition, analysis, and interpretation. KW and LX were involved in data interpretation and discussions. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297(5581):547–551. | ||
Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. | ||
Hau PM, Tsao SW. Epstein–Barr virus hijacks DNA damage response transducers to orchestrate its life cycle. Viruses. 2017;9(11):341. | ||
Poon RY. DNA damage checkpoints in nasopharyngeal carcinoma. Oral Oncol. 2014;50(5):339–344. | ||
Choudhuri T, Verma SC, Lan K, Murakami M, Robertson ES. The ATM/ATR signaling effector Chk2 is targeted by Epstein–Barr virus nuclear antigen 3C to release the G2/M cell cycle block. J Virol. 2007;81(12):6718–6730. | ||
Nikitin PA, Yan CM, Forte E, et al. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein–Barr virus transformation of primary human B cells. Cell Host Microbe. 2010;8(6):510–522. | ||
Chen WH, Tang LQ, Guo SS, et al. Prognostic value of plasma Epstein–Barr virus DNA for local and regionally advanced nasopharyngeal carcinoma treated with cisplatin-based concurrent chemoradiotherapy in intensity-modulated radiotherapy era. Medicine. 2016;95(5):e2642. | ||
Chen M, Yin L, Wu J, et al. Impact of plasma Epstein–Barr virus–DNA and tumor volume on prognosis of locally advanced nasopharyngeal carcinoma. Biomed Res Int. 2015;2015:1–5. | ||
Wagner JM, Karnitz LM. Cisplatin-induced DNA damage activates replication checkpoint signaling components that differentially affect tumor cell survival. Mol Pharmacol. 2009;76(1):208–214. | ||
Srinivas US, Dyczkowski J, Beißbarth T, et al. 5-Fluorouracil sensitizes colorectal tumor cells towards double stranded DNA breaks by interfering with homologous recombination repair. Oncotarget. 2015;6(14):12574–12586. | ||
Kwok M, Davies N, Agathanggelou A, et al. Synthetic lethality in chronic lymphocytic leukaemia with DNA damage response defects by targeting the ATR pathway. Lancet. 2015;385(1):S58. | ||
Carroll BL, Pulkoski-Gross MJ, Hannun YA, Obeid LM. CHK1 regulates NF-κB signaling upon DNA damage in p53-deficient cells and associated tumor-derived microvesicles. Oncotarget. 2016;7(14):18159–18170. | ||
Wang L, Mosel AJ, Oakley GG, Peng A. Deficient DNA damage signaling leads to chemoresistance to cisplatin in oral cancer. Mol Cancer Ther. 2012;11(11):2401–2409. | ||
Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3(5):421–429. | ||
Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci U S A. 1999;96(24):13777–13782. | ||
Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 2000;14(3):278–288. | ||
Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol. 2003;5(3):255–260. | ||
Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. | ||
Caporali S, Levati L, Starace G, et al. AKT is activated in an ataxia-telangiectasia and Rad3-related-dependent manner in response to temozolomide and confers protection against drug-induced cell growth inhibition. Mol Pharmacol. 2008;74(1):173–183. | ||
Danielsson K, Nylander E, Sjöström M, Ebrahimi M. Epstein–Barr virus is not detected in mucosal lichen planus. Med Oral Patol Oral Cir Bucal. 2018;23(5):e560–e563. | ||
Gruhne B, Sompallae R, Masucci MG. Three Epstein–Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene. 2009;28(45):3997–4008. | ||
Glaser R, Zhang HY, Yao KT, et al. Two epithelial tumor cell lines (HNE-1 and HONE-1) latently infected with Epstein–Barr virus that were derived from nasopharyngeal carcinomas. Proc Natl Acad Sci U S A. 1989;86(23):9524–9528. | ||
Bejarano MT, Masucci MG, Klein G, Klein E. T-cell-mediated inhibition of EBV-induced B-cell transformation: recognition of virus particles. Int J Cancer. 1988;42(3):359–364. | ||
Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. | ||
Zeng Q, Wang Z, Liu C, et al. Knockdown of NFBD1/MDC1 enhances chemosensitivity to cisplatin or 5-fluorouracil in nasopharyngeal carcinoma CNE1 cells. Mol Cell Biochem. 2016;418(1–2):137–146. | ||
vera-Sempere FJ, Burgos JS, Botella MS, Cordoba J, Gobernado M. Immunohistochemical expression of Epstein–Barr virus-encoded latent membrane protein (LMP-1) in paraffin sections of EBV-associated nasopharyngeal carcinoma in Spanish patients. Eur J Cancer B Oral Oncol. 1996;32B(3):163–168. | ||
Gulley ML, Tang W. Laboratory assays for Epstein–Barr virus-related disease. J Mol Diagn. 2008;10(4):279–292. | ||
Cha H, Lowe JM, Li H, et al. Wip1 directly dephosphorylates gamma-H2AX and attenuates the DNA damage response. Cancer Res. 2010;70(10):4112–4122. | ||
Bonner WM, Redon CE, Dickey JS, et al. γH2AX and cancer. Nat Rev Cancer. 2008;8(12):957–967. | ||
Fang CY, Lee CH, Wu CC, et al. Recurrent chemical reactivations of EBV promotes genome instability and enhances tumor progression of nasopharyngeal carcinoma cells. Int J Cancer. 2009;124(9):2016–2025. | ||
Hau PM, Deng W, Jia L, et al. Role of ATM in the formation of the replication compartment during lytic replication of Epstein–Barr virus in nasopharyngeal epithelial cells. J Virol. 2015;89(1):652–668. | ||
Choudhuri T, Verma SC, Lan K, Murakami M, Robertson ES. The ATM/ATR signaling effector Chk2 is targeted by Epstein–Barr virus nuclear antigen 3C to release the G2/M cell cycle block. J Virol. 2007;81(12):6718–6730. | ||
Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. | ||
Mordasini V, Ueda S, Aslandogmus R. Activation of ATR-Chk1 pathway facilitates EBV-mediated transformation of primary tonsillar B-cells. Oncotarget. 2016;8:6461. | ||
Hafez AY, Messinger JE, Mcfadden K, et al. Limited nucleotide pools restrict Epstein–Barr virus-mediated B-cell immortalization. Oncogenesis. 2017;6(6):e349. | ||
Burrows AE, Elledge SJ. How ATR turns on: TopBP1 goes on ATRIP with ATR. Genes Dev. 2008;22(11):1416–1421. | ||
Toledo LI, Altmeyer M, Rask MB, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155(5):1088–1103. | ||
Anantha RW, Vassin VM, Borowiec JA. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J Biol Chem. 2007;282(49):35910–35923. | ||
Koganti S, Hui-Yuen J, Mcallister S, et al. STAT3 interrupts ATR-Chk1 signaling to allow oncovirus-mediated cell proliferation. Proc Natl Acad Sci U S A. 2014;111(13):4946–4951. | ||
Adamsen BL, Kravik KL, de Angelis PM. DNA damage signaling in response to 5-fluorouracil in three colorectal cancer cell lines with different mismatch repair and TP53 status. Int J Oncol. 2011;39(3):673–682. | ||
Rezaee M, Sanche L, Hunting DJ. Cisplatin enhances the formation of DNA single- and double-strand breaks by hydrated electrons and hydroxyl radicals. Radiat Res. 2013;179(3):323–331. | ||
Jordan P, Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci. 2000;57(8–9):1229–1235. | ||
Kidiyoor GR, Kumar A, Foiani M. ATR-mediated regulation of nuclear and cellular plasticity. DNA Repair (Amst). 2016;44:143–150. | ||
Mei YP, Zhou JM, Wang Y, et al. Silencing of LMP1 induces cell cycle arrest and enhances chemosensitivity through inhibition of AKT signaling pathway in EBV-positive nasopharyngeal carcinoma cells. Cell Cycle. 2007;6(11):1379–1385. | ||
Tibbetts RS, Brumbaugh KM, Williams JM, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13(2):152–157. | ||
Rohaly G, Chemnitz J, Dehde S, et al. A novel human p53 isoform is an essential element of the ATR-intra-S phase checkpoint. Cell. 2005;122(1):21–32. | ||
Wang G, Bhoopalan V, Wang D, Wang L, Xu X. The effect of caffeine on cisplatin-induced apoptosis of lung cancer cells. Exp Hematol Oncol. 2015;4:5. | ||
Rosell R, Lord RV, Taron M, Reguart N. DNA repair and cisplatin resistance in non-small-cell lung cancer. Lung Cancer. 2002;38(3):217–227. | ||
Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9(8):616–627. | ||
Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5(9):a012716. | ||
Buisson R, Niraj J, Rodrigue A, et al. Coupling of homologous recombination and the checkpoint by ATR. Mol Cell. 2017;65(2):336–346. | ||
Qiu Z, Oleinick NL, Zhang J. ATR/CHK1 inhibitors and cancer therapy. Radiother Oncol. 2018;126(3):450–464. | ||
Sørensen CS, Hansen LT, Dziegielewski J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7(2):195–201. | ||
Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D’Andrea AD. Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2007;27(8):3098–3108. | ||
Zhi G, Wilson JB, Chen X, et al. Fanconi anemia complementation group FANCD2 protein serine 331 phosphorylation is important for Fanconi anemia pathway function and BRCA2 interaction. Cancer Res. 2009;69(22):8775–8783. | ||
Maacke H, Jost K, Opitz S, et al. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19(23):2791–2795. | ||
Nakanoko T, Saeki H, Morita M, et al. Rad51 expression is a useful predictive factor for the efficacy of neoadjuvant chemoradiotherapy in squamous cell carcinoma of the esophagus. Ann Surg Oncol. 2014;21(2):597–604. | ||
Yazinski SA, Comaills V, Buisson R, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017;31(3):318–332. | ||
Peasland A, Wang LZ, Rowling E, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer. 2011;105(3):372–381. | ||
Bertrand P, Saintigny Y, Lopez BS. p53’s double life: transactivation-independent repression of homologous recombination. Trends Genet. 2004;20(6):235–243. | ||
Restle A, Janz C, Wiesmüller L. Differences in the association of p53 phosphorylated on serine 15 and key enzymes of homologous recombination. Oncogene. 2005;24(27):4380–4387. | ||
Arias-Lopez C, Lazaro-Trueba I, Kerr P, et al. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006;7(2):219–224. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.