Back to Journals » Drug Design, Development and Therapy » Volume 14
Atorvastatin Combined with Low-Dose Dexamethasone Treatment Protects Endothelial Function Impaired by Chronic Subdural Hematoma via the Transcription Factor KLF-2
Authors Fan Y, Wang D, Rao C ![]() , Li Y
, Li Y ![]() , Rong H, Wang Z
, Rong H, Wang Z ![]() , Zhang J
, Zhang J
Received 3 April 2020
Accepted for publication 22 July 2020
Published 12 August 2020 Volume 2020:14 Pages 3291—3299
DOI https://doi.org/10.2147/DDDT.S256050
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Yueshan Fan,1– 3,* Dong Wang1,2 ,* Chenxu Rao,1– 3,* Ying Li,1,2 Hongtao Rong,1,2 Zengguang Wang,1,2 Jianning Zhang1,2
1Department of Neurosurgery, Tianjin Medical University General Hospital, Tianjin 300052, People’s Republic of China; 2Tianjin Neurological Institute, Key Laboratory of Post-Neuroinjury Neuro-Repair and Regeneration in Central Nervous System, Ministry of Education and Tianjin City, Tianjin 300052, People’s Republic of China; 3Graduate School, Tianjin Medical University, Tianjin, 300000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jianning Zhang; Zengguang Wang Tel +86 22-60817448
Email [email protected]; [email protected]
Objective: Our previous study showed that the combination therapy with atorvastatin and low-dose dexamethasone protected endothelial cell function in chronic subdural hematoma (CSDH) injury. In this study, we aimed to investigate the mechanism underlying the effects of this combination therapy on CSDH-induced cell dysfunction.
Methods: Monocytes and endothelial cells were cocultured with CSDH patient hematoma samples to mimic the pathological microenvironment of CSDH. Monocytes (THP-1 cells) and endothelial cells (hCMEC/D3 cells) were cocultured in a transwell system for 24 h before stimulation with hematoma samples diluted in endothelial cell medium (ECM) at a 1:1 ratio. Tight junction markers were detected by Western blotting, PCR and immunofluorescence. hCMEC/D3 cells were collected for Western blot and PCR analyses to detect changes in the expression levels of vascular cell adhesion molecule (VCAM-1), intercellular adhesion molecule (ICAM-1), and Kruppel-like factor 2 (KLF-2). The IL-6, IL-10 and VEGF levels in the supernatant were measured by enzyme-linked immunosorbent assay (ELISA).
Results: KLF-2 expression in endothelial cells was decreased after stimulation with CSDH patient hematoma samples, but combination therapy with atorvastatin and low-dose dexamethasone reversed this trend. KLF-2 protected injured cells by increasing the expression of VE-cadherin and ZO-1; attenuating the expression of VCAM-1, ICAM-1, IL-6 and VEGF; and enhancing the expression of IL-10, all of which play pivotal roles in endothelial inflammation. Moreover, the effect of combination therapy with atorvastatin and low-dose dexamethasone was obviously reduced in endothelial cells with KLF-2 knockdown compared with normal cells.
Conclusion: Coculture with hematoma samples decreased KLF-2 expression in human cerebral endothelial cells. Combination therapy with atorvastatin and low-dose dexamethasone counteracted hematoma-induced KLF-2 suppression in human cerebral endothelial cells to attenuate robust endothelial inflammation and permeability. KLF-2 plays an important role in drug therapy for CSDH and may become the key factor in treatment and prognosis.
Keywords: chronic subdural hematoma, atorvastatin combined with low-dose dexamethasone treatment, endothelial inflammation and permeability, Kruppel-like factor 2
Introduction
Chronic subdural hematoma (CSDH) is one of the most common reasons for neurosurgical procedures in elderly individuals.1 Neurosurgery is the first-line treatment for this disease. Although the surgical cure rate can be as high as 80%, 20% of patients will recur and develop a CSDH requiring further surgery.2 In addition, because elderly individuals develop conditions associated with increasing age and commonly use anticoagulants, the prevalence of this neurological disease is obviously increasing.3,4 Thus, nonsurgical therapeutic approaches for CSDH must be identified. Emerging data have convincingly demonstrated that a multipathway physiological mechanism that involves inflammatory pathways, angiogenesis and growth factors, coagulopathy, hyperfibrinolysis, and exudation mediates the pathology of CSDH.5 Based on previous studies, several therapies, such as previously used a ntithrombotic drugs,6 angiotensin-converting enzyme inhibitors, atorvastatin and dexamethasone administered at presentation,7 have been proposed.
In our previous study, we demonstrated that atorvastatin is an effective and safe nonsurgical treatment for patients with CSDH. However, in the randomized clinical trial with 200 patients, up to 11.2% of the patients were not cured by atorvastatin and required further surgical treatment, which should be further discussed.8 Our recent study with four young patients indicated that atorvastatin combined with low-dose dexamethasone treatment completely cured three of the patients and obviously decreased the hematoma size in the remaining patient without any clinical complications.9 Additionally, with our further proof-of-concept Phase II clinical trial, we showed that atorvastatin combined with low-dose dexamethasone treatment is more effective than atorvastatin alone in reducing the hematoma size and improving neurological function in patients with CSDH.10 Moreover, the results of our basic research study were the same as those of our clinical study. We further screened a zinc finger transcription factor, Kruppel-like factor (KLF-2), by RNA sequencing and found that its expression was significantly increased in hematoma-injured cells after combination therapy. This study will be published in the near future.
KLF-2 is a member of a subfamily of zinc-finger transcription factors,11 whose members play critical roles in vascular function12 and are highly expressed in endothelial cells.13 Recent research has indicated that KLF-2 expression can be stimulated by shear stress,14 and inhibited by some injuries such as those caused by proinflammatory factors or ischemic stroke.13,15 Emerging data have convincingly demonstrated that KLF-2 is a “molecular switch” that regulates vessel function and disease.12 A recent study by Yusheng demonstrated that a high glucose level suppressed KLF-2 expression in human umbilical vein endothelial cells (HUVECs) and the suppression was counteracted by atorvastatin treatment.16 However, the effects of KLF-2 both on the process of CSDH and treatment with atorvastatin combined with low-dose dexamethasone remains unclear.
In this study, we hypothesized that KLF-2 plays important roles in both the process of CSDH and the treatment with atorvastatin combined with low-dose dexamethasone. Herein, in an attempt to better understand the mechanism underlying the efficacy of the combined treatment for the hematoma-induced injury, we mimicked the pathological microenvironment by coculturing monocytes and endothelial cells with CSDH patient hematoma samples to investigate the protective roles of KLF-2 in vascular inflammation and permeability.
Methods
Establishing a Cell Coculture and CSDH Patient Hematoma Sample Induced Injury Model
The human cerebral endothelial cell line, hCMEC/D3 was ordered from Sigma-Aldrich (Sigma-Aldrich, SCC075, St. Louis, MO, USA)), cultured with endothelial cell medium (ECM) (ScienCell Research Laboratories, California, USA), and grown on 0.1% collagen type I (Sigma-Aldrich, 08–115, St. Louis, MO, USA))-coated coverslips until a tight monolayer was formed. We obtained the human monocyte cell line Thp-1 from American Type Culture Collection (ATCC, TIB-202, Virginia, USA) and cultured it in RPMI 1640 medium (Gibco,11,875,093, Waltham, USA), supplemented with 10% FBS (Gibco, 26,140,079, Waltham, USA). We replaced the medium every other day.
Hematoma samples were collected from patients (n=10). The patient information is presented in Table 1. The supernatants of the hematoma samples were collected after centrifugation at 3000 rpm for 15 min and were stored at −80 °C. Before the hematoma sample was used, we diluted it with ECM at a 1:1 ratio, allowed it to stand for 6 h and then centrifuged it at 3000 rpm for 15 min. The supernatants were stored for further study.
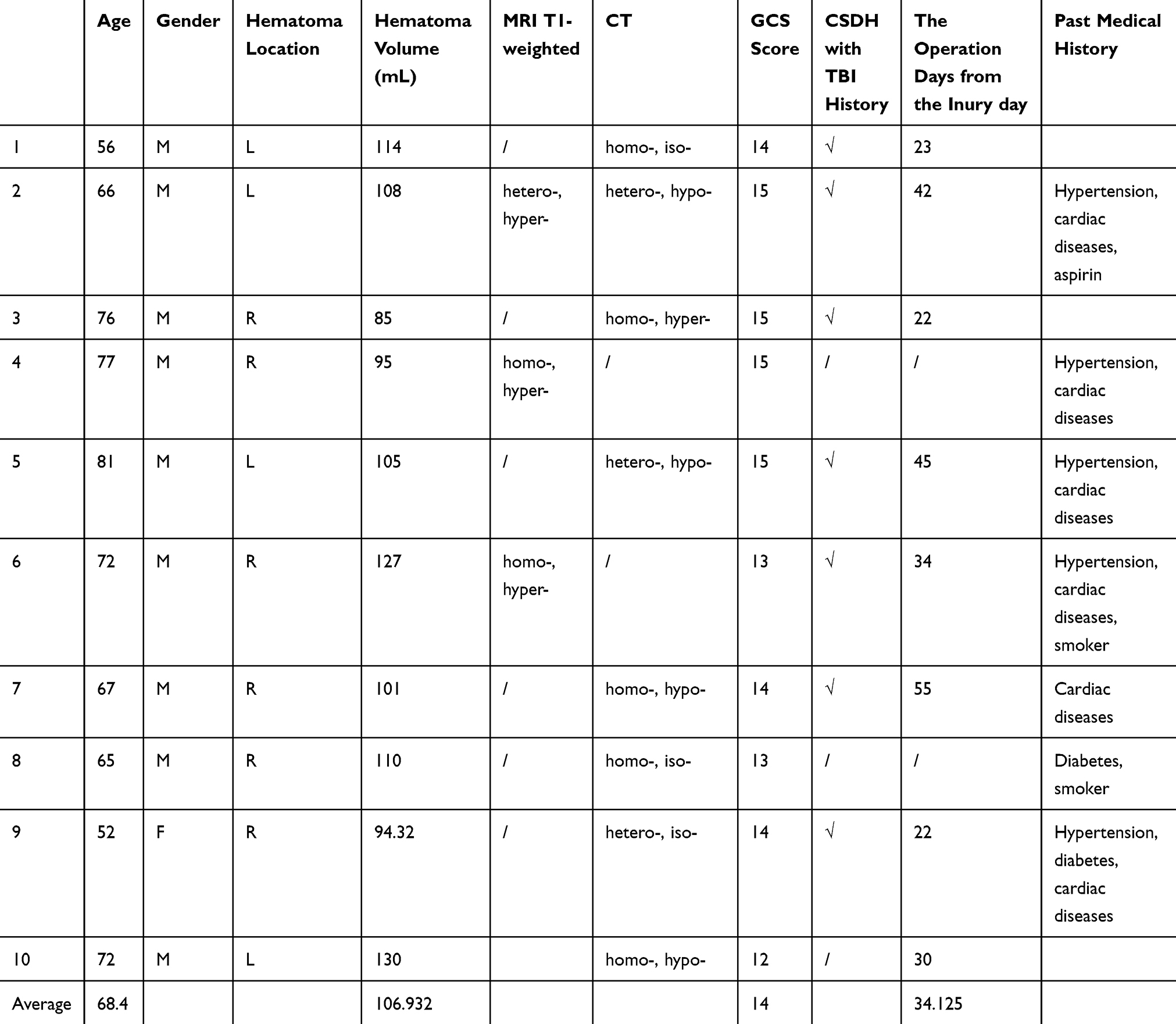 |
Table 1 The Information of the CSDH Patients |
Prior to cell stimulation, cells were cocultured in a transwell system to mimic the in vivo vascular microenvironment. Polycarbonate membranes with 0.4-µm pores were used to separate the upper and lower wells of a transwell chamber (Corning, 3412, New York, USA). We transplanted THP-1 cells into the upper well and hCMEC/D3 cells into the coated lower wells and incubated the cells for 24 h before hematoma simulation.
Subsequently, we added the diluted hematoma into the bottom wells. After 1h, we replaced the hematoma samples with ECM and RPMI 1640 medium, and PBS, atorvastatin (Sigma-Aldrich, PZ0001, St. Louis, MO, USA)), or dexamethasone (Sigma-Aldrich, D1756, St. Louis, MO, USA)), was added to the mixed medium, and the cells were cocultured with hCMEC/D3 cells for 1 h.
The experiment method and protocol were approved by Tianjin Medical University ethics committee of China on July 31, 2018 (approval No. IRB2018-088-01). This study was also in accordance with the Declaration of Helsinki. An written informed consent was obtained from each patient or a patient’s legal representative (When a patient’s consciousness is not good, the family member should sign for him).
Hopping Probe Ion Conductance Microscopy (HPICM) Scanning
As described previously,17,18 we detected changes in endothelial cell morphology at multiple timepoints after hematoma injury. The advantage of HPICM scanning is that it can scan living cells directly without causing any damage and has no effect on cell structure. We completed this experiment within 2 h to ensure that the cell status was stable.
Western Blot Analysis
The protocol was similar to that used in previous research.19 Membranes were incubated with anti-VE-cadherin (1:1000, Cell Signaling Technology, 2500, Danvers, Massachusetts, USA), anti-ZO-1 (1:1000, Cell Signaling Technology, 13,663, Danvers, Massachusetts, USA), anti-VCAM-1 (1:1000, Cell Signaling Technology, 13,662, Danvers, Massachusetts, USA), anti-ICAM-1 (1:1000, Abcam, ab18981) and anti-KLF-2 (1:250, Abcam, ab17008) antibodies. We then added horseradish peroxidase (HRP)-conjugated secondary IgG antibodies and incubated the membranes for 1 h at room temperature. Immunoreactions were visualized with an enhanced chemiluminescence (ECL) system (Millipore, Billerica, MA, United States), and protein band densities were quantified with ImageJ software. GAPDH was used as the normalization control.
Immunofluorescence Staining
We performed immunofluorescence staining as previously described.19 Cells were incubated overnight with a primary antibody specific for VE-cadherin (1:400, Cell Signaling Technology, 2500). Cells were then washed with PBS and incubated with an Alexa Fluor-conjugated anti-rabbit IgG antibody (1:500, Molecular Probes) for 1 h at room temperature. Nuclei were counterstained for 5 min with 4ʹ,6-diamidino-2-phenylindole (DAPI).
Pcr
PCR was performed as described previously.20 The sequences of the primers used for PCR are listed in Table 2.
 |
Table 2 The Sequences of Primers Used in This Study |
Enzyme-Linked Immunosorbent Assay (ELISA)
We detected the concentrations of IL-6, IL-10 and VEGF with ELISA kits (R&D) according to the manufacturer’s instructions.
SiRNA Transfection
SiRNA transfection was performed in vitro with Lipofectamine 3000 (Thermo Fisher Scientific). We obtained the siRNA-KLF-2 and siRNA control constructs from RiboBio. Then, 50 nmol siRNA-KLF-2 or siRNA control was added to Lipofectamine 3000, and the mixture was added to the cell culture medium and incubated for 6 h.We replaced the culture medium after 6 h. The cells were cultured for 24 h for further experiments.
Statistical Analysis
We analyzed the data with GraphPad Prism (IBM, USA) and presented them as the means ± SDs. One-way ANOVA was used to analyze the results. P-values of less than 0.05 were considered to indicate significant differences.
Results
The Effects of Combined Treatment on the Expression of KLF-2
Before we investigated the effect of combined low-dose atorvastatin and dexamethasone treatment on KLF-2 expression, we analyzed the changes in hCMEC/D3 cell morphology at multiple timepoints after hematoma injury by HPICM scanning. We found that the cell morphology significantly changed from 54 min to 78 min (Figure 1A). Based on these data, we chose 78 min as the injury timepoint. Likewise, we used the same time period to rescue injured cells with atorvastatin monotherapy or atorvastatin combined with low-dose dexamethasone treatment.
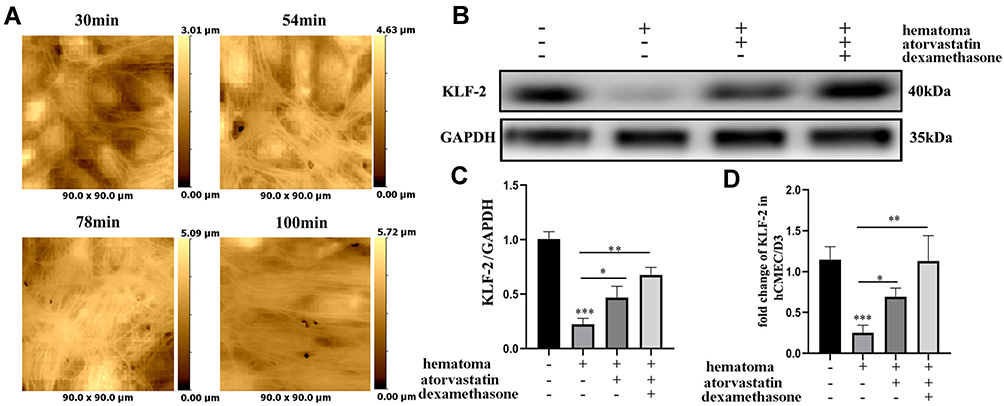 |
Figure 1 The expression of KLF-2 in endothelial cells was decreased after hematoma sample injury. (A) The morphological changes in cells at different time points after stimulation with hematoma supernatant observed by HPICM; (B) The changes in KLF-2 expression after treatment of hematoma supernatant-damaged cells with different interventions; (C) Gray value analysis of panel (B); (D) The changes in the KLF-2 mRNA level after treatment of hematoma-damaged cells with different interventions. *P < 0.05; **P < 0.01; ***P < 0.001. |
We measured the expression of KLF-2 (Figure 1B–D) by Western blotting and PCR. As shown in Figure 1B–D, the level of KLF-2 was decreased after hematoma injury, but this effect was reversed when we rescued injured cells with atorvastatin monotherapy or atorvastatin combined with low-dose dexamethasone treatment. KLF-2 expression was much higher in the combined treatment group than in the monotherapy group, which suggested that KLF-2 may play a crucial role in mediating the effect of the combined therapy on hematoma injury.
The Role of KLF-2 in the Vascular Permeability of hCMEC/D3 Cells After Hematoma Injury
Then, we further performed immunofluorescence staining and Western blotting to evaluate cell-cell junction protein markers in hematoma-stimulated hCEMC/D3 cells with KLF-2 knockdown that were cocultured with normal Thp-1 cells (Figure 2A–C). The vulnerability of the cells to hematoma injury increased after KLF-2 knockdown, as evidenced by the considerably decreased expression of VE-cadherin and ZO-1 in the knockdown cells compared with the normal cells.
 |
Figure 2 Hematoma sample stimulates cocultured cells and induces changes in the expression of tight junction proteins after knockdown of KLF-2 in endothelial cells. (A) The changes in tight junction proteins in endothelial cells after injury observed by immunofluorescence staining; (B) Western blot analysis of the changes in tight junction protein expression in endothelial cells with different genotypes after injury; (C). Gray value analysis of panel B. *P < 0.05. |
These data demonstrated that KLF-2 can effectively reduce the effects of hematoma injury on cells by decreasing vessel leakage, as indicated by the increased expression of tight junction proteins.
The Effect of KLF-2 on the Vascular Endothelial Inflammatory Reaction Between THP-1 and hCMEC/D3 Cells After Hematoma Injury
We detected changes in vascular cell adhesion molecule (VCAM-1) and intercellular adhesion molecule (ICAM-1) levels in hCMEC/D3 cells cocultured with THP-1 cells. After stimulation with hematoma samples, the expression of VCAM-1 and ICAM-1 was significantly increased a at both the protein (Figure 3A–B) and mRNA levels (Figure 3C) in KLF-2-ablated hCMEC/D3 cells compared with normal cells with hematoma injury.
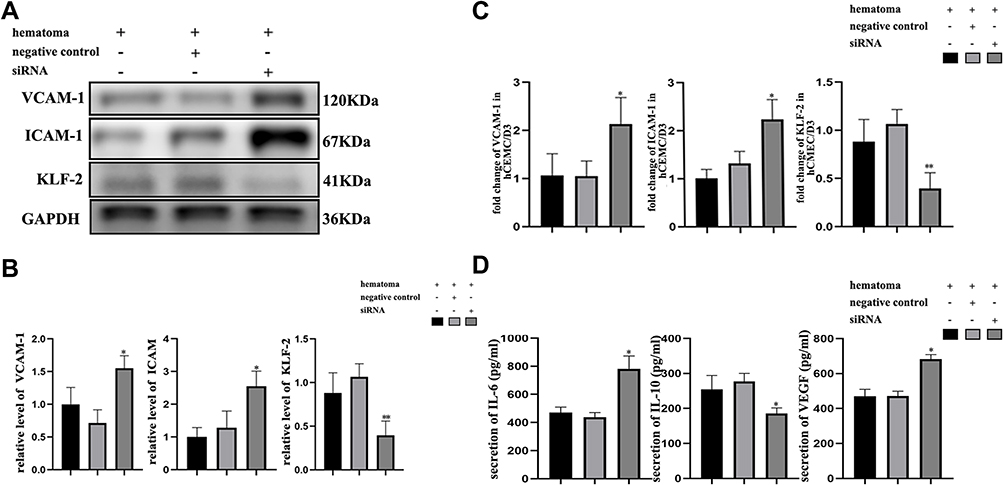 |
Figure 3 Hematoma sample stimulates cocultured cells and induces changes in the expression of vascular inflammation markers after ablation of KLF-2 in endothelial cells. (A) Western blot analysis of the changes in adhesion protein expression in endothelial cells with different genotypes after injury; (B) Gray value analysis of panel (A); (C) PCR analysis of the changes in adhesion protein mRNA levels in endothelial cells with different genotypes after injury; (D) ELISA analysis of the changes in inflammatory factor and VEGF expression in endothelial cells with different genotypes after injury. *P < 0.05, **P<0.01. |
Then, coculture supernatants were collected to observe the changes in the expression levels of inflammatory factors after hematoma injury (Figure 3D). The results suggested that knockdown cells secreted higher levels of the proinflammatory factor IL-6 and lower levels of the immunosuppressive factor IL-10 than normal cells. The concentration of VEGF in the sham group, which was approximately 40 pg/mL to 60 pg/mL (data not shown). However, the level of VEGF after hematoma stimulation was increased nearly tenfold compared with that in normal cell supernatants, the expression of VEGF in KLF-2-ablated cell supernatants was further increased compared with that in normal cell supernatants after hematoma injury.
As shown in Figure 3, KLF-2 protected cells from vascular inflammation and the proangiogenic microenvironment caused by hematoma injury by decreasing the expression of VCAM-1, ICAM-1, VEGF and IL-6 and inducing the expression of IL-10.
KLF-2 and Combined Low-Dose Atorvastatin and Dexamethasone Treatment Interact in Hematoma-Stimulated Injured Cells
Based on the abovementioned data, we further evaluated the functional differences between normal and KLF-2-ablated cells, which were rescued by monotherapy or combined treatment after hematoma injury. Endothelial inflammation and permeability function markers were evaluated by Western blotting, PCR and ELISA.
The results indicated that KLF-2 could influence the inhibitory effect of atorvastatin monotherapy or combined atorvastatin and low-dose dexamethasone treatment on the cellular damage. We focused on the lack of significant differences in VE-cadherin and ZO-1 expression at both the protein and mRNA levels (Figure 4A–C) between the injury group and the rescue group of KLF-2 knockdown cells, which demonstrated that vascular leakage was not obviously improved. We further performed Western blotting and PCR to detect changes in VCAM-1 and ICAM-1 levels and found no obvious differences between the injury group and the rescue group of KLF-2 knockdown cells (Figure 4A–C). In addition, the expression levels of cytokines did not differ significantly between the injury group and the rescue group of KLF-2 knockdown cells (Figure 4D). However, in treated normal cells, all phenomena were counteracted. Compared with the injury group, all treatment groups, especially the combined treatment group, exhibited greatly increased expression levels of VE-cadherin and ZO-1. The levels of VCAM-1 and ICAM-1 were lower in the treatment groups than in the injury group, consistent with the trends in the IL-6 and VEGF expression. However, the expression of IL-10 was induced in both treatment groups of normal cells compared with the injured cell group. We further compared the expression levels of markers of vascular leakage and markers of inflammation between the atorvastatin monotherapy and combined treatment groups. The data proved that compared with the monotherapy, the combined treatment could much more effectively protect cells from injury by attenuating the high levels of endothelial inflammation and permeability.
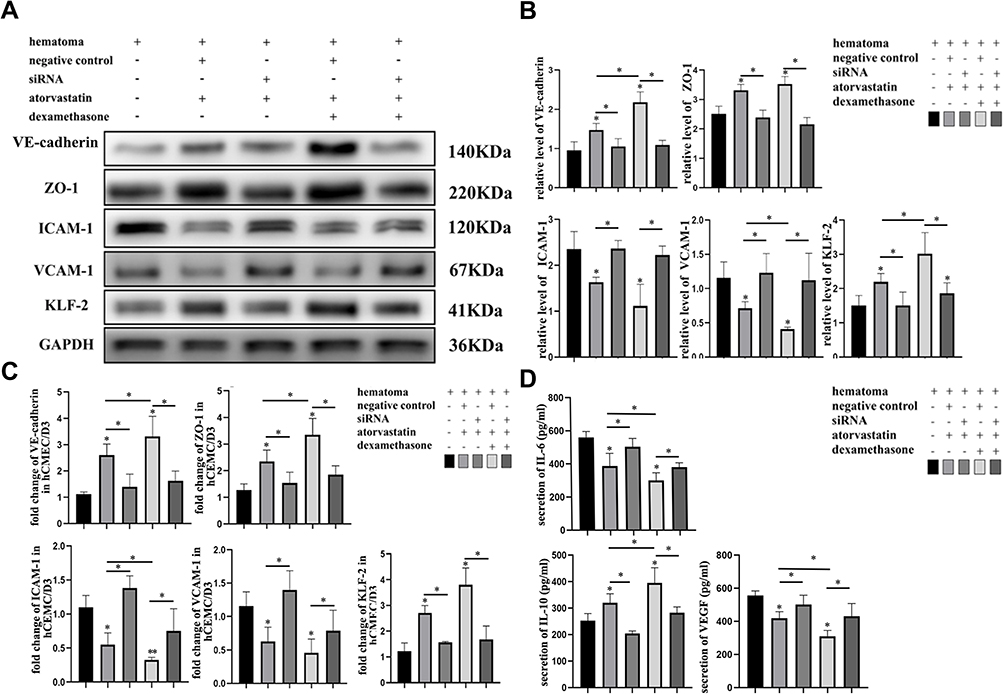 |
Figure 4 Hematoma sample stimulates cocultured cells and induces changes in the expression of vascular inflammation markers and tight junction proteins after ablation of KLF-2 in endothelial cells given different treatments. (A) Western blot analysis of the changes in tight junction and adhesion protein expression in endothelial cells with different genotypes given different treatments after injury; (B) Gray value analysis of panel (A); (C) PCR analysis of the changes in the mRNA levels of tight junction and adhesion proteins in endothelial cells with different genotypes given different treatments after injury; (D) ELISA analysis of the changes in inflammatory factor and VEGF expression in endothelial cells with different genotypes given different treatments after injury. *P < 0.05. |
Most importantly, as shown in Figure 4, KLF-2 played a pivotal role in mediating the effects of both monotherapy and combined treatment. The effects of these therapies were obviously reduced when KLF-2 expression was knocked down in the hCMEC/D3 cell line. We thus speculated that KLF-2 played a pivotal role in mediating the effects of both treatments, especially in the combined treatment.
Discussion
The involvement of complex processes such as angiogenesis, inflammation, and coagulopathy in the process of CSDH has gradually been established.21,22 Because of its encapsulated nature, the subdural cavity is closed and cerebrospinal fluid does not permeate the subdural space containing blood after hematoma formation.23 Therefore, the sources of mediators that drive inflammation and angiogenesis include cells such as endothelial cells, inflammatory cells and fibroblasts, which reside in membranes.24
The results of our previous clinical and basic research showed that the effect of combined treatment was better than that of atorvastatin monotherapy in both clinical and basic research. The expression of KLF-2 was significantly increased after treatment of cells with hematoma-stimulated injury. Interestingly, KLF-2 expression was much higher in the combined treatment group than in the monotherapy group. These results suggested that KLF-2 plays an important role in the process of rescuing injured cells, especially in the context of combined treatment.
To further investigate the specific mechanism underlying the efficacy of the combination therapy in CSDH, we used an endothelial cell-monocyte-hematoma sample coculture system to mimic the microenvironment of the CSDH subdural cavity. The levels of both inflammatory and angiogenic factors were assessed to evaluate the extent of injury. Emerging data have demonstrated that the expression of adhesion proteins, such as VCAM-1 and ICAM-1, plays pivotal roles in the inflammatory response as these proteins can induce leukocyte attachment and rolling on the endothelial surface.25,26 A study demonstrated that KLF-2 can reduce the expression of VCAM-1 and ICAM-1 in a high-glucose environment,16 but no studies have addressed the effects of KLF-2 on CSDHs. In this study, we identified that hematoma-induced injury could reduce the expression of KLF-2, which causes robust vascular inflammation and permeability, mediated mainly by increased VCAM-1 and ICAM-1 expression and diminished VE-cadherin and ZO-1 expression. In addition, KLF-2 ablation exacerbated hematoma-induced injury in cocultured cells. In contrast to the KLF-2 suppression observed in hematoma injury, strongly increased KLF-2 expression was observed in cells treated with monotherapy and especially in cells treated with combined treatment, and this increase in expression counteracted the alterations caused by hematoma injury. Furthermore, KLF-2 ablation diminished the therapeutic effects of both the monotherapy and the combined therapy on rescuing injured cells. Compared with atorvastatin monotherapy, the combined treatment was significantly more effective in both inducing KLF-2 expression and protecting cells from hematoma injury. This result suggests that KLF-2 is a critical factor regulating the effectiveness of the combined treatment against hematoma injury in endothelial cells.
However, this study has some limitations. The specific mechanism by which KLF-2 mediates CSDH needs to be determined. In addition, the change in the KLF-2 level and its influence on the effect of the combined treatment should be evaluated in vivo. The level of KLF-2 in hematoma samples should be measured to investigate whether it differs significantly between hematoma tissue and CSDH patient serum.
Overall, our results indicate that atorvastatin combined with low-dose dexamethasone can counteract hematoma-induced KLF-2 suppression in human cerebral endothelial cells, attenuating the robust endothelial inflammation and permeability. KLF-2 plays an important role in drug therapy for CSDH and may become the key factor in treatment and prognosis.
Acknowledgments
We thank Liu Li, Weiyun Cui, Fanglian Chen Shu Zhang, Ying Li and Liqun He from Tianjin Neurological Institute for their technical support.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest and report no conflicts of interest for this work.
References
1. Chen JC, Levy ML. Causes, epidemiology, and risk factors of chronic subdural hematoma. Neurosurg Clin N Am. 2000;11(3):399–406. doi:10.1016/S1042-3680(18)30101-3
2. Allison A, Edlmann E, Kolias AG, et al. Statistical analysis plan for the Dex-CSDH trial: a randomised, double-blind, placebo-controlled trial of a 2-week course of dexamethasone for adult patients with a symptomatic chronic subdural haematoma. Trials. 2019;20(1):698. doi:10.1186/s13063-019-3866-6
3. Almenawer SA, Farrokhyar F, Hong C, et al. Chronic subdural hematoma management: a systematic review and meta-analysis of 34,829 patients. Ann Surg. 2014;259(3):449–457. doi:10.1097/SLA.0000000000000255
4. Miah IP, Holl DC, Peul WC, et al. Dexamethasone therapy versus surgery for chronic subdural haematoma (DECSA trial): study protocol for a randomised controlled trial. Trials. 2018;19(1):575. doi:10.1186/s13063-018-2945-4
5. Holl DC, Volovici V, Dirven CMF, et al. Pathophysiology and Nonsurgical Treatment of Chronic Subdural Hematoma: from Past to Present to Future. World Neurosurg. 2018;116(402–411):e402. doi:10.1016/j.wneu.2018.05.037
6. Adelborg K, Grove EL, Sundboll J, Laursen M, Schmidt M. Sixteen-year nationwide trends in antithrombotic drug use in Denmark and its correlation with landmark studies. Heart. 2016;102(23):1883–1889. doi:10.1136/heartjnl-2016-309402
7. Poon MTC, Al-Shahi Salman R. Association between antithrombotic drug use before chronic subdural haematoma and outcome after drainage: a systematic review and meta-analysis. Neurosurg Rev. 2018;41(2):439–445. doi:10.1007/s10143-017-0860-x
8. Jiang R, Zhao S, Wang R, et al. Safety and efficacy of atorvastatin for chronic subdural hematoma in Chinese patients: A randomized clinical trial. JAMA Neurol. 2018;75(11):1338–1346. doi:10.1001/jamaneurol.2018.2030
9. Huang J, Li L, Zhang J, et al. Treatment of relapsed chronic subdural hematoma in four young children with atorvastatin and low-dose dexamethasone. Pharmacotherapy. 2019;39(7):783–789. doi:10.1002/phar.2276
10. Wang D, Gao C, Xu X, et al. Treatment of chronic subdural hematoma with atorvastatin combined with low-dose dexamethasone: phase II randomized proof-of-concept clinical trial. J Neurosurg. 2020;1–9.
11. Anderson KP, Kern CB, Crable SC, Lingrel JB. Isolation of a gene encoding a functional zinc finger protein homologous to erythroid Kruppel-like factor: identification of a new multigene family. Mol Cell Biol. 1995;15(11):5957–5965. doi:10.1128/MCB.15.11.5957
12. Atkins GB, Jain MK. Role of Kruppel-like transcription factors in endothelial biology. Circ Res. 2007;100(12):1686–1695. doi:10.1161/01.RES.0000267856.00713.0a
13. SenBanerjee S, Lin Z, Atkins GB, et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199(10):1305–1315. doi:10.1084/jem.20031132
14. Zhong F, Lee K, He JC. Role of Kruppel-like factor-2 in kidney disease. Nephrology. 2018;23(Suppl 4):53–56. doi:10.1111/nep.13456
15. Shi H, Sheng B, Zhang F, et al. Kruppel-like factor 2 protects against ischemic stroke by regulating endothelial blood brain barrier function. Am J Physiol Heart Circ Physiol. 2013;304(6):H796–H805. doi:10.1152/ajpheart.00712.2012
16. Liu YS, Xu DL, Huang ZW, Hao L, Wang X, Lu QH. Atorvastatin counteracts high glucose-induced Kruppel-like factor 2 suppression in human umbilical vein endothelial cells. Postgrad Med. 2015;127(5):446–454. doi:10.1080/00325481.2015.1039451
17. Novak P, Li C, Shevchuk AI, et al. Nanoscale live-cell imaging using hopping probe ion conductance microscopy. Nat Methods. 2009;6(4):279–281. doi:10.1038/nmeth.1306
18. Liu X, Yang X, Zhang B, et al. High-resolution morphological identification and characterization of living neuroblastoma SK-N-SH cells by hopping probe ion conductance microscopy. Brain Res. 2011;1386:35–40. doi:10.1016/j.brainres.2011.02.053
19. Rong H, Fan Y, Yang M, et al. Brain-derived microparticles activate microglia/macrophages and induce neuroinflammation. Brain Res. 2018;1694:104–110. doi:10.1016/j.brainres.2018.05.015
20. Wang D, Li T, Tian Y, et al. Effects of atorvastatin on chronic subdural hematoma: a preliminary report from three medical centers. J Neurol Sci. 2014;336(1–2):237–242. doi:10.1016/j.jns.2013.11.005
21. Edlmann E, Giorgi-Coll S, Whitfield PC, Carpenter KLH, Hutchinson PJ. Pathophysiology of chronic subdural haematoma: inflammation, angiogenesis and implications for pharmacotherapy. J Neuroinflammation. 2017;14(1):108. doi:10.1186/s12974-017-0881-y
22. Fu S, Li F, Bie L. Drug therapy for chronic subdural hematoma: bench to bedside. J Clin Neurosci. 2018;56:16–20. doi:10.1016/j.jocn.2017.07.034
23. Suzuki M, Endo S, Inada K, et al. Inflammatory cytokines locally elevated in chronic subdural haematoma. Acta Neurochir (Wien). 1998;140(1):51–55. doi:10.1007/s007010050057
24. Hohenstein A, Erber R, Schilling L, Weigel R. Increased mRNA expression of VEGF within the hematoma and imbalance of angiopoietin-1 and −2 mRNA within the neomembranes of chronic subdural hematoma. J Neurotrauma. 2005;22(5):518–528. doi:10.1089/neu.2005.22.518
25. DiChiara MR, Kiely JM, Gimbrone MA, Lee ME, Perrella MA, Topper JN. Inhibition of E-selectin gene expression by transforming growth factor beta in endothelial cells involves coactivator integration of Smad and nuclear factor kappaB-mediated signals. J Exp Med. 2000;192(5):695–704. doi:10.1084/jem.192.5.695
26. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76(2):301–314. doi:10.1016/0092-8674(94)90337-9
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.