")
Back to Journals » Journal of Blood Medicine » Volume 12
Asymptomatic Joint Bleeding and Joint Health in Hemophilia: A Review of Variables, Methods, and Biomarkers
Authors Gooding R, Thachil J, Alamelu J, Motwani J, Chowdary P
Received 30 January 2021
Accepted for publication 19 March 2021
Published 1 April 2021 Volume 2021:12 Pages 209—220
DOI https://doi.org/10.2147/JBM.S304597
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Martin H Bluth
Richard Gooding,1 Jecko Thachil,2 Jayanthi Alamelu,3 Jayashree Motwani,4 Pratima Chowdary5
1Leicester Haemophilia Centre, Haemostasis & Thrombosis Unit, Leicester Royal Infirmary, Leicester, UK; 2Haematology Department, Manchester Royal Infirmary, Manchester, UK; 3Haemophilia Department, Evelina Children’s Hospital – St Thomas’ Hospital, London, UK; 4Haemophilia Comprehensive Care Centre, Birmingham Children’s Hospital, Birmingham, UK; 5Katharine Dormandy Haemophilia Centre, Royal Free Hospital, London, UK
Correspondence: Richard Gooding
Leicester Haemophilia Centre, Haemostasis & Thrombosis Unit, Level 2, Osborne Building, The Leicester Royal Infirmary, Infirmary Square, Leicester, LE1 5WW, UK
Tel +44 116 258 6500
Fax +44 116 258 6120
Email [email protected]
Abstract: Joint health is a key contributor to quality of life in patients with hemophilia. However, variables that impact long-term joint outcomes have not been comprehensively defined. A systematic literature search identified publications relating to joint health in patients with hemophilia. Studies clearly show that early, sustained prophylaxis with factor replacements improves long-term joint outcomes. However, a subset of patients appear to develop arthropathy despite maintaining excellent bleeding outcomes, which suggests possible recurrent asymptomatic bleeding into the joints in these patients. Furthermore, limited data are available on how long-acting factor VIII and factor IX replacement therapies could impact long-term joint outcomes. Many variables were identified as potential indicators that a patient may develop hemophilic arthropathy, including genetic mutations, endogenous factor VIII and IX levels, bone health, and physical activity levels. Tools for the diagnosis and monitoring of hemophilic arthropathy are critical to detect early joint damage, so that management can be adjusted accordingly. Imaging techniques, particularly magnetic resonance imaging, can detect synovial changes, a strong predictor for the future development of hemophilic arthropathy. In addition, several biomarkers associated with cartilage and bone formation, vascularization, and angiogenesis could potentially identify the onset and progression of early joint damage. Since the development of hemophilic arthropathy is complex, a comprehensive therapeutic approach is necessary for the effective prevention of arthropathy in patients with hemophilia.
Keywords: hemophilia, asymptomatic bleeding, joint disease, hemarthrosis, hemophilic arthropathy, hemarthropathy
Introduction
Joint bleeds can lead to inflammation and destruction of the joint, which ultimately results in arthropathy, a significant morbidity in patients with hemophilia. Recurrent hemarthrosis results in the accumulation of hemosiderin, synovial inflammatory changes, cartilage degradation and, in late stages, joint destruction.1,2 Hemophilic arthropathy is associated with increased pain, loss of employment, school absenteeism, and a reduction in physical wellbeing and quality of life.3–7
Effective prevention of joint deterioration is limited by the relatively limited knowledge of the pathogenesis of hemophilic arthropathy.1 In addition, although commonly associated with severe disease, it is not currently possible to accurately predict which patients will develop hemophilic arthropathy as patients may develop arthropathy with few or no joint bleeds.8–12 In patients with established joint damage, joint deterioration often progresses even if few or no further joint bleeds occur.13 These observations suggest the occurrence of bleeding that is not detected by the patients, and this recurrent asymptomatic joint bleeding might contribute to the development of hemophilic arthropathy.9,11,13
Early prophylaxis with factor replacement concentrates can prevent joint bleeding, and thus the development of arthropathy.12,14 Furthermore, identifying the variables that influence the progression of hemophilic arthropathy, including asymptomatic joint bleeding, may highlight modifiable aspects that could aid patients, or help to identify patients who may benefit from more intensive therapy in the future. Here, we firstly present the findings from a systematic literature review to identify variables that influence joint deterioration in patients with hemophilia. Secondly, we discuss methods to help identify patients with hemophilia who are at risk of joint deterioration and we consider the impact of long-acting factor replacement therapies on joint outcomes. We also consider which biomarkers may be associated with bone health in these patients. Lastly, we consider future research and practice directions for the prevention of arthropathy in patients with hemophilia.
Joint Outcomes in Hemophilia: A Systematic Literature Review
Methods
A systematic literature search was conducted in both EMBASE and PubMed, according to PRISMA guidelines,15 on November 2018 and updated in July 2020 to identify variables that could influence the development of arthropathy, as assessed by joint scores, in patients with hemophilia. Search terms were designed to select publications according to the patient population, treatment administered, and outcomes reported. The search was limited to articles published in English, with no specified date range for PubMed and 1947 to present for EMBASE.
The following search term was used: Hemophilia AND (Magnetic Resonance Imaging OR Diagnostic imaging OR Ultrasonography OR Radiography OR Arthroscopy OR Synovectomy OR Arthralgia OR Hemarthrosis OR synovitis OR Synovial Fluid OR Synovial Membrane OR Hyperalgesia OR Brain Infarction OR Cerebral Hemorrhage OR Subclinical OR Silent OR Cartilage OR Hemochromatosis OR Iron OR Joint OR Articular OR Hemosiderin). The search terms were derived from an exploded MeSH term analysis that identified related terms.
All publications retrieved by this search strategy were individually assessed against pre-defined inclusion and exclusion criteria (Table 1). The aim was to identify original articles that looked at joint health outcomes assessed by joint scores. Publications underwent an initial screen based on the title and abstract using these inclusion and exclusion criteria. Potentially relevant publications then underwent a second screen based on the full text of the article. The relevant data from all eligible publications were collected and aggregated to allow further analysis.
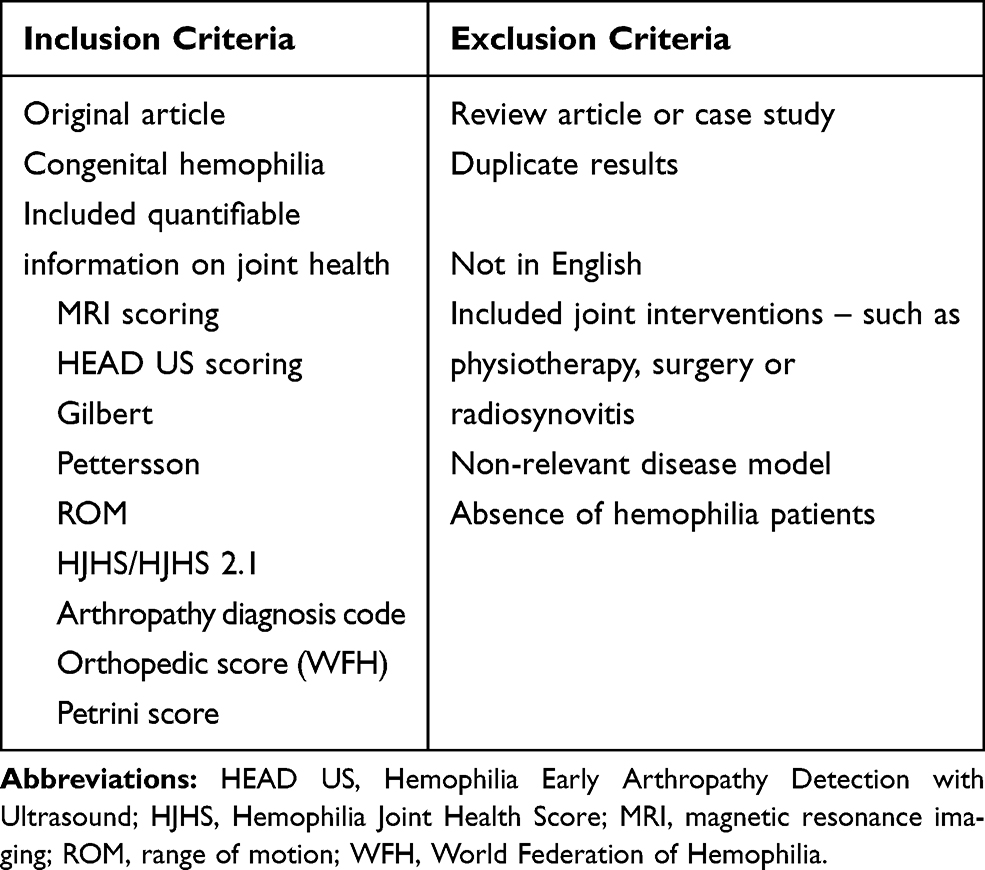 |
Table 1 Systematic Literature Review Inclusion and Exclusion Criteria |
Results
The initial search identified 5,380 results. Following the initial abstract and title screen, 861 articles were identified that met the initial screening criteria. The full-text screen identified 256 publications that measured arthropathy and recorded additional data on patients (Figure 1). Due to the wide variability in the outcomes considered, formal study quality assessment and meta-analytic evaluation were not performed.
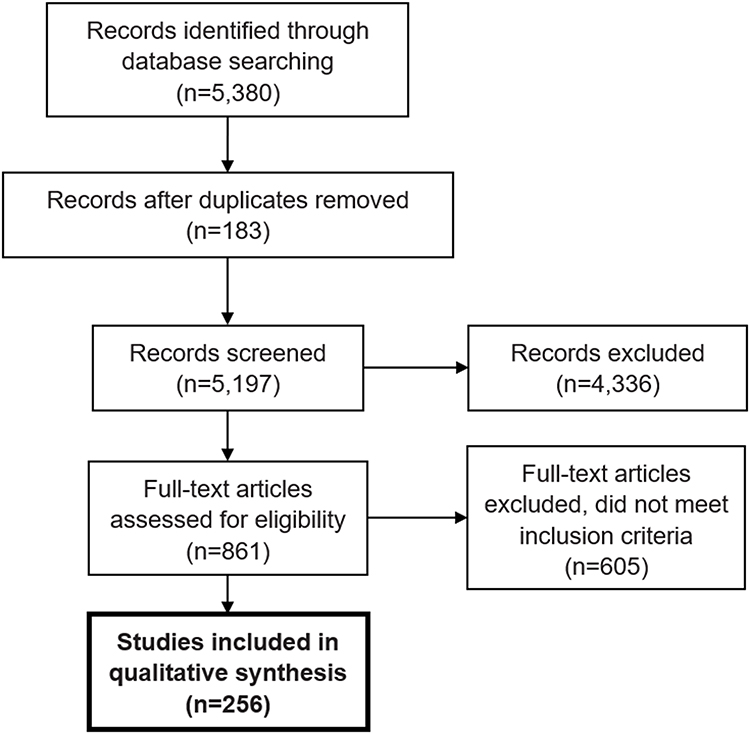 |
Figure 1 PRISMA systematic literature review flow chart. |
Variables Influencing Joint Outcomes
The key variables that may influence joint outcomes highlighted by the literature search results were disease severity, gene mutations, bone mineral density (BMD), physical activity, and prophylaxis with factor replacement concentrates.
Disease Severity
Low endogenous FVIII/FIX activity levels (~1 IU/dl, 1% of normal) are known to be associated with higher rates of arthropathy due to the increased susceptibility to bleeding.16–21 Patients with severe hemophilia (<1 IU/dl, <1% of normal) are more prone to joint damage than patients with mild or moderate hemophilia.17 However, patients with moderate (1–5 IU/dl, 1–5% of normal) and mild (5–40 IU/dl, 5–40% of normal) hemophilia are still susceptible to developing arthropathy.16,20,22,23 Factor levels of 1–3 IU/dl are now considered insufficient to fully prevent bleeding in all patients with hemophilia.12 Even though the majority of patients with moderate hemophilia have fewer joint bleeds than patients with severe hemophilia, a subset of these patients (~30% of patients) experience bleeding and long-term joint damage.16,22 Patients with mild hemophilia have a higher likelihood of developing chronic arthropathy than the general population and asymptomatic bleeding has been suggested as its potential cause.23 It is also notable that hemophilia carriers with factor activity levels >40 IU/dl (40% of normal) can experience decreases in joint range of motion (ROM) and structural joint changes.18,19,21
Gene Mutations
Several studies have correlated gene mutations with joint outcomes in patients with hemophilia (Table 2).24–30 An investigation conducted in 14 hemophilia treatment centers in the United States found that a total of 613 single nucleotide polymorphism (SNP) markers were significantly correlated with ROM scores on at least two joint pairs.24 This study also identified multiple inflammatory- or immune-related genes that were associated with arthropathy; SNPs in the NOD2 (associated with musculoskeletal manifestations in inflammatory bowel disease) and TLR10 (role in pathogen recognition and activation of innate immunity) genes were potentially strongly associated with arthropathy, but only a small number of patients carried the variant alleles.24 In another study, the underlying factor VIII (FVIII) mutation type (missense, nonsense, frameshift, intron 22 inversion) was not found to be significantly associated with joint deterioration in a cohort of hemophilia A patients with minimal access to hemostatic treatment.25 In contrast, a study from India found that severe molecular defects of the FVIII or factor IX (FIX) gene were associated with disease severity, including poor joint outcomes, as patients with inversions, deletions, insertions and nonsense mutations had an approximate four-fold increased risk of having a severe disease phenotype.26 Additionally, functional polymorphisms in the FVIII gene also affect the phenotype; for example, patients with an FVII353Q allele had an increased risk of a severe phenotype.26 However, this study did not identify any significant association with inflammatory or immunoregulatory cytokines. Two publications noted that HLA B27 mutations (associated with seronegative spondyloarthritis) were correlated with synovitis and arthropathy27,28 and another reported that HFE mutations (associated with hemochromatosis), resulting in reduced iron absorption, contributed to the development of arthropathy.29 A small study reported that patients with the MTHFR variants MTHFR 677TT or MTHFR 1298AC had a higher number of affected joints or a higher degree of effusion, respectively, compared to patients with other genotypes.30 Furthermore, this study also found a positive association between genetic biomarkers related to inflammation and number of subchondral cysts.
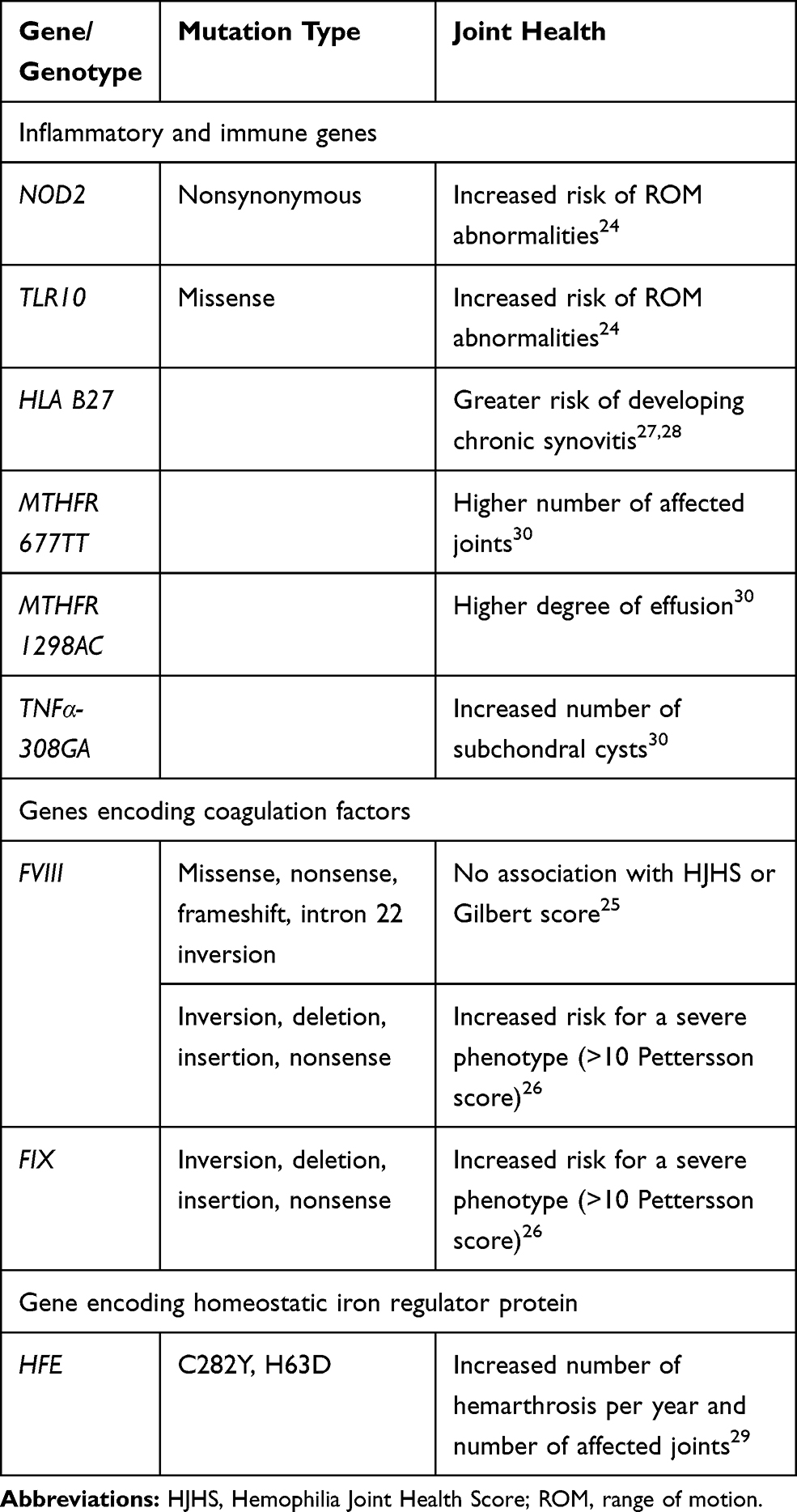 |
Table 2 Gene Mutations and Genotypes Associated with Joint Health in Patients with Hemophilia |
Prophylaxis
Publications largely agreed that prophylaxis with factor replacement therapy reduces the rate of arthropathy development compared to episodic treatment.8,14,31–38 Several publications showed that an early start to prophylaxis (before 2–3 years of age) reduces the risk of developing arthropathy.33,35,36,39–42 However, it has been reported that early prophylaxis is not sufficient to completely prevent joint damage.8–10,42 Prophylaxis initiated later in a patient’s life has been shown to still provide benefits over episodic treatment with lower bleeding rates and improved joint health over time.14,34,35 However, no reduction in structural arthropathy progression was observed, suggesting that pre-existing joint arthropathy may be irreversible,14 further emphasizing the importance of primary prophylaxis. Continued prophylaxis also appears to be important; patients who stopped prophylaxis in adulthood showed similar bleeding rates to those on prophylaxis but had significantly higher rates of arthropathy over time.43,44
Bone Mineral Density and Physical Activity
Several studies found that low BMD correlated with arthropathy.45–55 It has been suggested that the chronic pain associated with arthropathy leads to reduced mobility and an avoidance of weight-bearing activity, ultimately resulting in decreased BMD.45–53 Overall, studies indicated that physical activity did not negatively affect joint health and may even improve joint health in patients with hemophilia.56–59 This may be due to improved muscle strength, balance and co-ordination seen in patients who maintain physical activity.
Identification of Patients with Hemophilia at Risk of Joint Deterioration
Imaging
Magnetic resonance imaging (MRI) is considered the most sensitive measure for joint assessment.13 MRI provides detailed joint images and can detect early damage (ie, synovial hypertrophy) and soft tissue changes in joints as well as bleeds (Table 3).13,60–62 MRI can also identify whether bleeds have occurred in a joint by detecting hemosiderin in the joint.13,60,63,64 However, MRI can be expensive, time-consuming and difficult to access, and often requires sedation in children.13,60 Ultrasound is less sensitive to early joint changes than MRI and is not able to discriminate between soft tissue types,60,64 but is a cheaper imaging modality, more widely available, and easy to access.13,64 Ultrasound can be used to detect synovial hypertrophy, and cartilage and bone surface abnormalities (Table 3).13,60,61,63,65 In addition, it can also be used to rapidly assess multiple joints at once.61 Patients only need to remain still for a short timeframe, which can be valuable when studying children’s joints, and ultrasound can be incorporated into routine check-ups.60,61,63,64 X-rays can also be used to assess joint deterioration and have demonstrated a good correlation with ultrasound and physical assessments, but low correlation with MRI findings.66–68 X-rays show only late (ie, osteochondral) joint changes,13 and when radiographic changes are detected, arthropathy is frequently already advanced.13,62,65
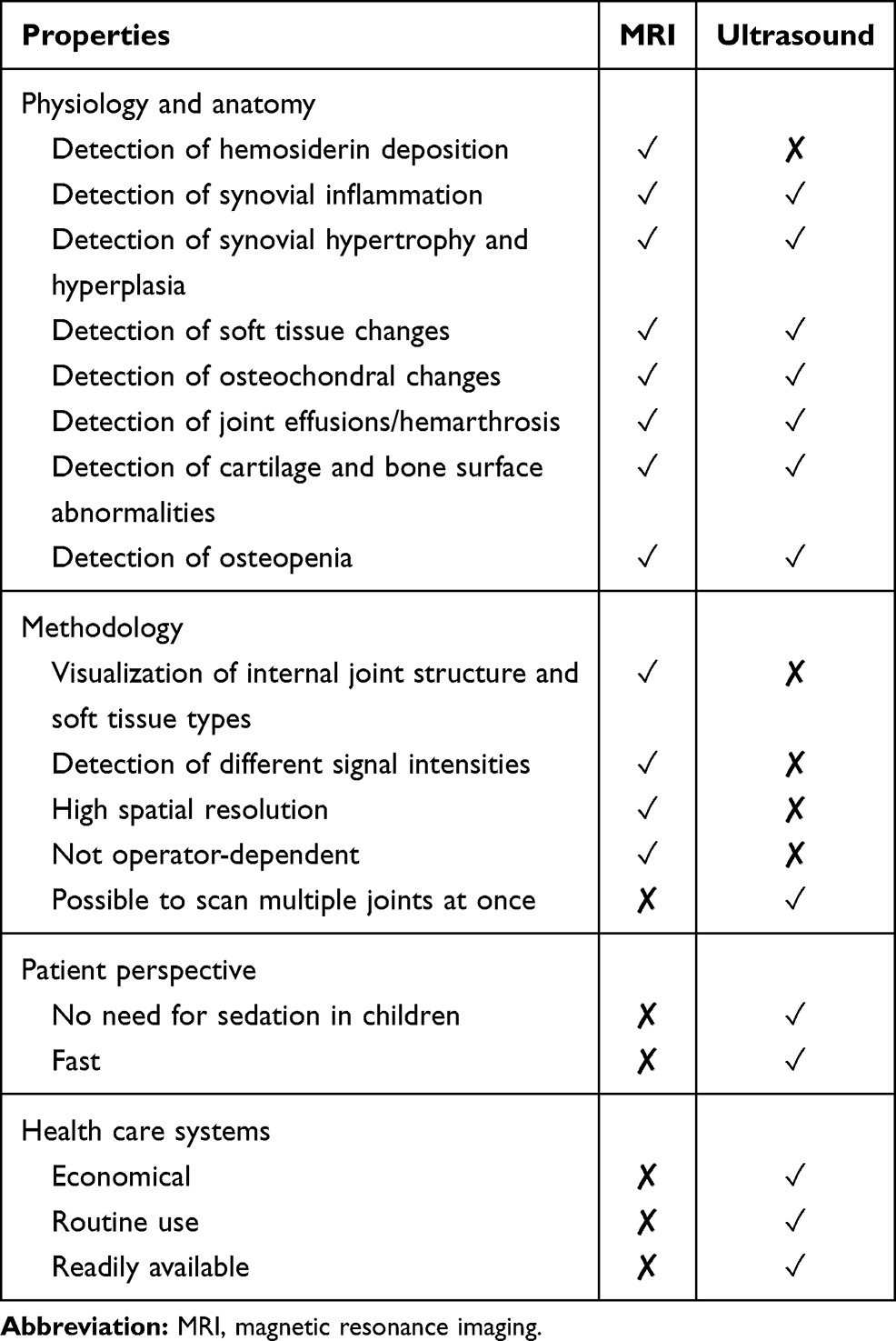 |
Table 3 Properties of Magnetic Resonance Imaging (MRI) and Ultrasound |
Clinical Joint Scores
Physical examination scales such as the Hemophilia Joint Health Score (HJHS) have been successfully used to assess joint outcomes, without requiring imaging techniques.69–72 However, studies have found that the HJHS is not as sensitive as MRI or ultrasound, and thus assessment methods should be combined to generate a more detailed analysis of joints, until more accurate methods of assessment and scoring are available.65,67 Smaller studies have also looked at using thermal imaging for inflammation, surface electromyography for muscle function and balance analysis for the lower limbs to detect early signs of joint deterioration.73–75 These methods are not well characterized and are not routinely used for assessing joint outcomes.
Importantly, several studies have reported that bleeding rates do not or only weakly correlate with other outcomes such as MRI or HJHS,8,67,71,76,77 and that joints deemed as “normal” according to physical examination or x-ray show abnormalities on MRI.65,68,71 Therefore, joints that appear normal may be affected by asymptomatic bleeding. MRI is highly sensitive and can detect early joint damage, even before clinical symptoms manifest, and these MRI changes have been shown to strongly predict future development of arthropathy.62 However, because MRI is not easily accessible, ultrasound may be a good affordable alternative.13 Imaging techniques used in combination with thorough physical examination could provide accurate joint outcomes assessment tools for early diagnosis and subsequent monitoring of joint damage. For instance, MRI can be used to monitor normal joints in patients with hemophilia, including patients with mild hemophilia, in order to detect early arthropathic changes, which could allow for treatment adjustment and potentially prevent further joint deterioration, irreversible functional impairment, and future need for orthopedic surgery. Similarly, patients with mild damage could be monitored by ultrasound and patients with established disease could be monitored by joint scores and targeted imaging.
Impact of Long-Acting Factor Replacement Therapy on Joint Outcomes
Prophylaxis with long-acting factor replacement therapies in both hemophilia A and B has been associated with a reduced number of joint bleeds and resolution of target joints during clinical trials.78 Recently, Zanon et al studied whether adherence to prophylaxis impacted on joint outcomes (HJHS) and involvement in physical activity.79 The study reported a decrease in HJHS and the number of total target joints and an increase in physical activity levels in patients who had high adherence to prophylaxis compared to patients with no or low adherence.79 Since poor adherence to prophylaxis might result in worsened joint outcomes, the use of long-acting products may help to avoid the low trough levels of FVIII and FIX that put patients at higher risk of bleeds (and then lead to joint problems).
The majority of the studies discussed in the systematic literature review that specifically investigated joint health and prophylaxis used standard-acting products. Recently, Malec et al compared joint health (HJHS) in patients with severe hemophilia A and B receiving standard-acting and long-acting products.80 The study reported no differences in mean HJHS for either group of patients receiving standard-acting compared to those receiving long-acting products. However, joint health data were collected at a single time point, and thus the study did not assess the time of switch to long-acting products nor the status of joints over time after switching to long-acting products. Further data on arthropathy development in patients on long-acting products, as well as on patients switching from standard-acting to long-acting products, are needed.
Biomarkers and Bone Health in Patients with Hemophilia
Recurrent bleeding into the joints causes the synovium to hypertrophy in order to clear blood from the joint space, the synovium is overwhelmed and iron (hemosiderin) accumulates leading to synovial angiogenesis and inflammation.1 The synovium produces pro-inflammatory cytokines and proteases, and this chronic proliferation of inflammatory cells causes the cartilage to breakdown. Furthermore, the constant presence of blood in the joint leads to bone changes, bone remodeling, loss of BMD and osteoporosis.1 Consequently, biomarkers of inflammation, and cartilage, bone and synovium changes could provide a prospective method to detect and monitor asymptomatic joint bleeding as well as early joint damage.
Several studies have identified biomarkers in the blood or urine that could be indicative of joint deterioration. Table 4 provides an overview of these potential biomarkers of bone health. Proteins associated with inflammation (CRPM, hsCRP), cartilage destruction (C2M, CTX-II, COMP, ADAMTS5) and bone turnover (PINP, CTX-I) have been detected in hemophilia patients with joint disease.81 Even though not all these biomarkers were correlated with radiological or physical joint assessments, the combination of C2M, CRPM and ADAMTS5 was able to distinguish hemophilia patients with joint disease from healthy controls with high accuracy.81 Another study reported that several biomarkers of cartilage deterioration (CTX-II, C1,2C, CS-846 and COMP) correlated with radiographic joint damage in patients with hemophilic arthropathy, but bone biomarkers (CTX-I, C1,2C) did not.82 One study found that serum levels of the key bone turnover markers sRANKL and OPG were significantly lower in patients with hemophilia than in healthy controls (p<0.05 and p<0.001, respectively), and these had an inverse correlation with joint outcomes (MRI and ultrasound).83 The levels of serum sclerostin, another key regulator of bone formation, were significantly elevated in children with hemophilia versus healthy controls (p=0.028) and had a positive correlation with joint damage (HJHS); elevated levels of serum sclerostin in patients with hemophilia might be indicative of a high risk for developing osteoporosis.52
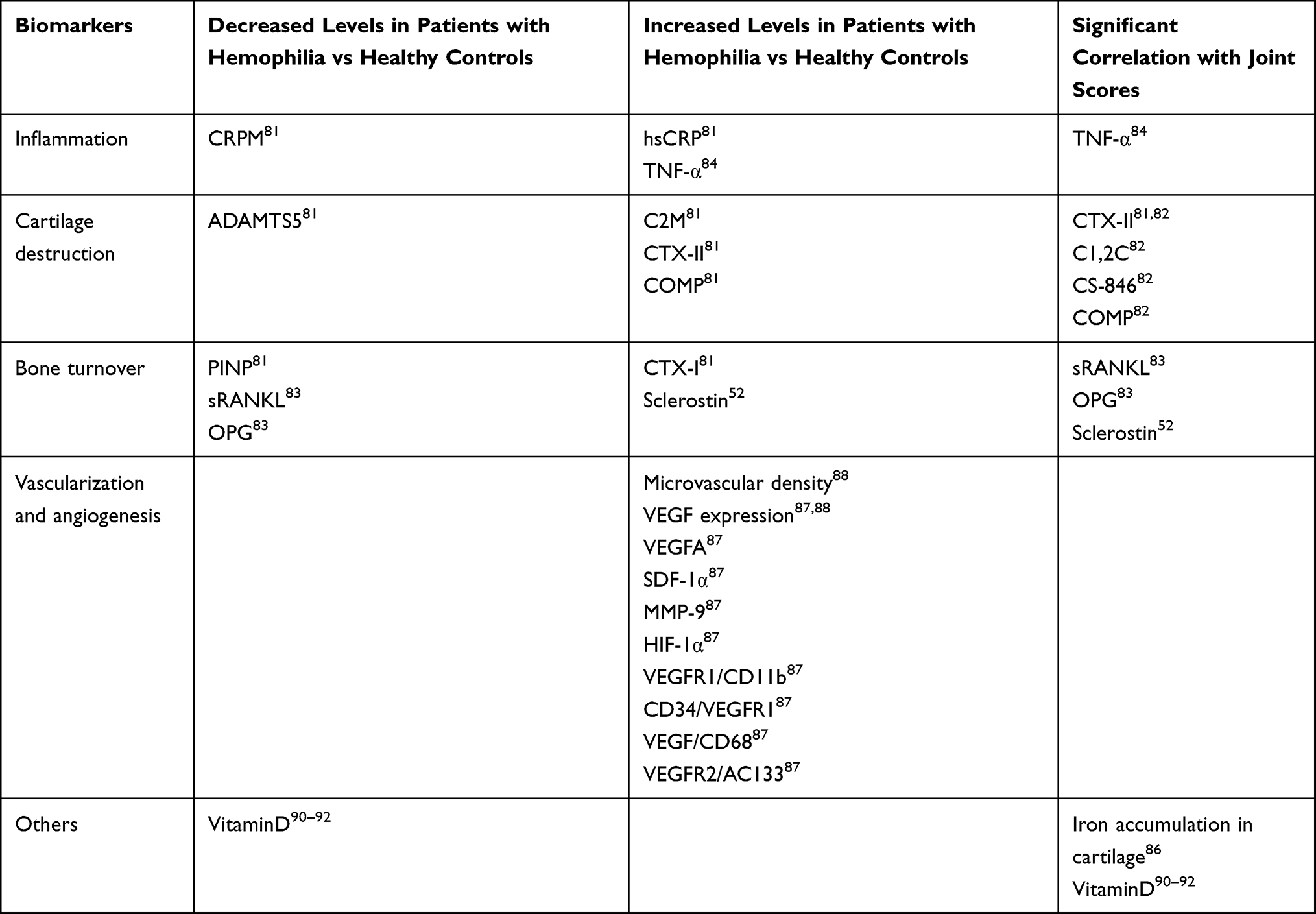 |
Table 4 Potential Biomarkers of Bone Health |
In another study, serum levels of the proinflammatory cytokine TNF-α were significantly elevated in patients with hemophilic arthropathy compared to healthy controls (p<0.0001).84 Moreover, TNF-α levels were positively correlated with the number of joint bleeds, degree of synovial hypertrophy, and clinical and ultrasound joint outcomes. The authors suggest that TNF-α is involved in the progression of hemophilic arthropathy and could be a suitable biomarker to detect joint deterioration in patients with hemophilia.84 However, TNF-α is an acute phase inflammatory marker that is elevated in a range of conditions, including rheumatoid arthritis, and as such is not specific to hemophilic arthropathy.85 Iron accumulation in cartilage is another biomarker that has been shown to correlate with joint damage and progression of hemophilic arthropathy, further it has the advantage of being easily detected with MRI T2* sequences.86 The development and validation of iron quantification MRI methods could assist in detecting asymptomatic joint bleeding, and provide a tool to evaluate and adjust treatment in patients with hemophilia.86
Vascularization and angiogenesis have been found to be increased in patients with joint damage. One study found that proangiogenic factors and proangiogenic macrophage/monocyte cells were up-regulated in patients with joint disease and expression of VEGFR2/AC133 endothelial progenitor cells and CD34/VEGFR1 hematopoietic progenitor cells were increased.87 Sera from patients with joint damage induced an angiogenic response in endothelial cells, while peripheral blood mononuclear cells from these subjects induced synovial cell proliferation.87 In another study, microvascular density and VEGF expression were significantly increased (p<0.005 and p=0.02, respectively) in synovial tissue from patients with hemophilic arthropathy compared to healthy controls.88
As mentioned above, there is evidence of an association between hemophilic arthropathy and low BMD. Furthermore, it has been shown that FVIII or FIX deficiency results in reduced BMD, with a quarter of patients with hemophilia having osteoporosis.89 Low vitamin D levels have also been associated with decreased BMD, physical activity, quality of life and poor joint health.90–92 These studies suggest that routine assessment of serum levels of vitamin D could guide early diagnosis of joint damage and treatment in patients with hemophilia, particularly in children. In patients with hemophilia, prevention of poor bone health should include primary prophylaxis, a diet rich in calcium and vitamin D, physiotherapy, and weight-bearing physical activities.52,89–91
Prevention of Arthropathy in Patients with Hemophilia: Future Directions in Research and Practice
There is robust evidence that prophylaxis with factor replacement therapies reduces the number of joint bleeds and rate of arthropathy development; however, the optimal trough level to protect patients from joint damage has not been determined yet. Furthermore, factor replacement alone may not be sufficient to prevent joint damage. A recent study by Zhou et al reported that joint bleeding events were not associated with time spent below certain clotting factor thresholds, and that vascularity changes also played a role in joint bleeding.93 Currently, an ongoing clinical trial (NCT03358836) is studying whether a long-acting FIX product with an intended trough level of >10% could provide better joint protection in patients with severe hemophilia B than the standard trough of 1% FIX. This study will also evaluate the early diagnosis of joint damage using ultrasound. The results from this study will be important, particularly since recent World Federation of Hemophilia guidelines have deemed factor trough levels of 1–3% insufficient to fully prevent bleeding in all patients with hemophilia.12 However, results are not expected until 2027.
As previously discussed, several studies have identified potential targets for the prevention and treatment of arthropathy in patients with hemophilia, including cartilage iron, inflammation, bone remodeling, cartilage regeneration and vascular remodeling. Accordingly, a pilot study of the safety and efficacy of local anti-VEGF therapy with intra-articular bevacizumab (Avastin®) for prevention of recurrent hemarthroses at target joints is currently ongoing in Taiwan in patients with chronic hemophilic synovitis and was due for completion in late 2020 (NCT02060305).
Furthermore, it is not yet known what level of joint protection emergent non-factor replacement therapies and gene therapy may provide. Some of these products are still in development and it will be some time until their effects on joint health, and prevention and treatment of hemophilic arthropathy can be analyzed.
Finally, physiotherapists play a valuable role in assessing patients’ joint status and in aiding recovery of function after a joint bleed, but many centers lack a dedicated hemophilia physiotherapist. Dedicated hemophilia ‘joint assessors’ could play an important role in providing dedicated time for thorough physical examination and joint scores, and in monitoring the progress of joint deterioration. Currently, home-based physiotherapy programs are being evaluated in people with hemophilia as an inexpensive accessible intervention with the potential to improve joint health.94,95 Individualized home-based exercise programs, with access to online/virtual tools and support from physiotherapists, have been positively received with encouraging results.94–96 Future randomized controlled clinical trials will be able to provide evidence of the clinical efficacy of such programs.
Conclusions
Currently, the best treatment option for patients with hemophilia is prophylaxis, particularly in young patients for whom primary prophylaxis is vital for the prevention of bleeding and to promote healthy joints. However, maintaining factor levels over a certain threshold does not fully prevent joint bleeds. Joint abnormalities and reduction of ROM have been reported even in patients who started prophylaxis early, in patients with mild hemophilia and in hemophilia carriers. In these cases, the occurrence of asymptomatic joint bleeding might be undetected for a significant amount of time before it is identified, and intervention can be provided. Therefore, early detection of changes in “normal” joints becomes crucial. To this end, sensitive diagnostic methods such as MRI or ultrasound are critical for early diagnosis of joint damage, monitoring of deterioration and treatment guidance, particularly in patients with normal physical and x-ray assessments. Although specific biomarkers of joint damage are still lacking, a few candidates such as cartilage iron, and inflammatory, cartilage destruction and bone formation factors have the potential to become novel targets for early diagnosis, prevention and treatment of joint damage in patients with hemophilia. The future of treatment of patients with hemophilia may be a therapeutic approach consisting of primary prophylaxis with factor replacement, calcium and vitamin D supplementation, physiotherapy and weight-bearing exercise programs, together with routine monitoring of cartilage iron, vitamin D, and key inflammatory and bone formation biomarker levels, as well as joint status with sensitive imaging and physical assessment tools.
Abbreviations
BMD, bone mineral density; FVIII, factor VIII; FIX, factor IX; HJHS, Hemophilia Joint Health Score; MRI, magnetic resonance imaging; ROM, range of motion; SNP, single nucleotide polymorphism.
Acknowledgments
Medical writing assistance was provided by Anna Mestres-Missé of Meridian HealthComms Ltd (Plumley, UK) in accordance with Good Publication Practice (GPP3) guidelines, funded by CSL Behring.
Funding
The authors received no specific funding for this work.
Disclosure
Dr Richard Gooding reports personal fees from CSL Behring, during the conduct of the study. Dr Jecko Thachil reports personal fees from CSL Behring, Takeda, Shire, Roche Chugai, and Sobi, during the conduct of the study. Professor Pratima Chowdary reports non-financial support from CSL Behring, during the conduct of the study; grants from and advisory committee for Pfizer, Bayer, CSL Behring, Freeline, Novo Nordisk, Sobi, Chugai, Roche, Takeda, Sanofi, and Spark; personal fees from BioMarin and UniQure, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. van Vulpen LFD, Holstein K, Martinoli C. Joint disease in haemophilia: pathophysiology, pain and imaging. Haemophilia. 2018;24(S6):44–49. doi:10.1111/hae.13449
2. Rodriguez-Merchan EC. Musculo-skeletal manifestations of haemophilia. Blood Rev. 2016;30(5):401–409. doi:10.1016/j.blre.2016.04.008
3. Chen CM, Huang KC, Chen CC, et al. The impact of joint range of motion limitations on health-related quality of life in patients with haemophilia A: a prospective study. Haemophilia. 2015;21(3):e176–e184. doi:10.1111/hae.12644
4. Holstein K, von Mackensen S, Bokemeyer C, Langer F. The impact of social factors on outcomes in patients with bleeding disorders. Haemophilia. 2016;22(1):46–53. doi:10.1111/hae.12760
5. Khair K, Holland M, Bladen M, Griffioen A, McLaughlin P, von Mackensen S. Study of physical function in adolescents with haemophilia: the SO-FIT study. Haemophilia. 2017;23(6):918–925. doi:10.1111/hae.13323
6. O’Hara J, Walsh S, Camp C, et al. The impact of severe haemophilia and the presence of target joints on health-related quality-of-life. Health Qual Life Outcomes. 2018;16(1):84. doi:10.1186/s12955-018-0908-9
7. Rodriguez-Merchan EC. Treatment of musculo-skeletal pain in haemophilia. Blood Rev. 2018;32(2):116–121. doi:10.1016/j.blre.2017.09.004
8. Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. doi:10.1056/NEJMoa067659
9. Olivieri M, Kurnik K, Pfluger T, Bidlingmaier C. Identification and long-term observation of early joint damage by magnetic resonance imaging in clinically asymptomatic joints in patients with haemophilia A or B despite prophylaxis. Haemophilia. 2012;18(3):369–374. doi:10.1111/j.1365-2516.2011.02682.x
10. Kraft J, Blanchette V, Babyn P, et al. Magnetic resonance imaging and joint outcomes in boys with severe hemophilia A treated with tailored primary prophylaxis in Canada. J Thromb Haemost. 2012;10(12):2494–2502. doi:10.1111/jth.12025
11. Di Minno MND, Iervolino S, Soscia E, et al. Magnetic resonance imaging and ultrasound evaluation of “healthy” joints in young subjects with severe haemophilia A. Haemophilia. 2013;19(3):e167–e173. doi:10.1111/hae.12107
12. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1–158. doi:10.1111/hae.14046
13. Rodriguez-Merchan EC, Jimenez-Yuste V, Aznar JA, et al. Joint protection in haemophilia. Haemophilia. 2011;17(S2):1–23. doi:10.1111/j.1365-2516.2011.02615.x
14. Manco-Johnson MJ, Lundin B, Funk S, et al. Effect of late prophylaxis in hemophilia on joint status: a randomized trial. J Thromb Haemost. 2017;15(11):2115–2124. doi:10.1111/jth.13811
15. Moher D, Liberati A, Tetzlaff J, Altman DG; Prisma Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. doi:10.1371/journal.pmed.1000097
16. Fischer K, van der Bom JG, Mauser-Bunschoten EP, et al. Endogenous clotting factor activity and long-term outcome in patients with moderate haemophilia. Thromb Haemost. 2000;84(6):977–980. doi:10.1055/s-0037-1614159
17. den Uijl IEM, Biesma D, Grobbee D, Fischer K. Turning severe into moderate haemophilia by prophylaxis: are we reaching our goal? Blood Transfus. 2013;11(3):364–369. doi:10.2450/2012.0092-12
18. Gilbert L, Rollins L, Hilmes M, et al. Haemophilia A carriers demonstrate pathological and radiological evidence of structural joint changes. Haemophilia. 2014;20(6):e426–e429. doi:10.1111/hae.12535
19. Sidonio RF, Mili FD, Li T, et al. Females with FVIII and FIX deficiency have reduced joint range of motion. Am J Hematol. 2014;89(8):831–836. doi:10.1002/ajh.23754
20. Soucie JM, Monahan PE, Kulkarni R, Konkle BA, Mazepa MA. The frequency of joint hemorrhages and procedures in nonsevere hemophilia A vs B. Blood Adv. 2018;2(16):2136–2144. doi:10.1182/bloodadvances.2018020552
21. Osooli M, Donfield SM, Carlsson KS, et al. Joint comorbidities among Swedish carriers of haemophilia: a register-based cohort study over 22 years. Haemophilia. 2019;25(5):845–850. doi:10.1111/hae.13831
22. den Uijl IEM, Biesma D, Grobbee D, Fischer K. Outcome in moderate haemophilia. Blood Transfus. 2014;12(Suppl 1):s330–s336. doi:10.2450/2012.0091-12
23. Osooli M, Lovdahl S, Steen Carlsson K, et al. Comparative burden of arthropathy in mild haemophilia: a register-based study in Sweden. Haemophilia. 2017;23(2):e79–e86. doi:10.1111/hae.13166
24. Gomperts ED, Schwarz J, Donfield SM, et al. The importance of genetic factors for the development of arthropathy: a longitudinal study of children and adolescents with haemophilia A. Thromb Haemost. 2017;117(2):277–285. doi:10.1160/TH16-06-0440
25. Khanum F, Bowen DJ, Kerr BC, Collins PW. Joint health scores in a haemophilia A cohort from Pakistan with minimal or no access to factor VIII concentrate: correlation with thrombin generation and underlying mutation. Haemophilia. 2014;20(3):426–434. doi:10.1111/hae.12326
26. Jayandharan GR, Nair SC, Poonnoose PM, et al. Polymorphism in factor VII gene modifies phenotype of severe haemophilia. Haemophilia. 2009;15(6):1228–1236. doi:10.1111/j.1365-2516.2009.02080.x
27. Shankarkumar U, Ghosh K, Mohanty D. Novel HLA B*2714 and B*2708 allele associations in seronegative spondarthritis patients and haemophilia patients with chronic synovitis in India. Tissue Antigens. 2003;62(2):175–178. doi:10.1034/j.1399-0039.2003.00074.x
28. Ghosh K, Shankarkumar U, Shetty S, Mohanty D. Chronic synovitis and HLA B27 in patients with severe haemophilia. Lancet. 2003;361(9361):933–934. doi:10.1016/S0140-6736(03)12763-8
29. Cruz E, Porto G, Morais S, Campos M, de Sousa M. HFE mutations in the pathobiology of hemophilic arthropathy. Blood. 2005;105(8):3381–3382. doi:10.1182/blood-2004-11-4342
30. López-Jiménez JJ, Ortega-Cervantes R, Luna-Záizar H, et al. Genetic biomarkers related to hemarthrosis, inflammation, and cartilage structure in pediatric patients with hemophilic arthropathy. Mol Genet Genomic Med. 2019;7(11):e979. doi:10.1002/mgg3.979
31. Fischer K, van der Bom JG, Molho P, et al. Prophylactic versus on-demand treatment strategies for severe haemophilia: a comparison of costs and long-term outcome. Haemophilia. 2002;8(6):745–752. doi:10.1046/j.1365-2516.2002.00695.x
32. Collins P, Faradji A, Morfini M, Enriquez MM, Schwartz L. Efficacy and safety of secondary prophylactic vs. on-demand sucrose-formulated recombinant factor VIII treatment in adults with severe hemophilia A: results from a 13-month crossover study. J Thromb Haemost. 2010;8(1):83–89. doi:10.1111/j.1538-7836.2009.03650.x
33. Gringeri A, Lundin B, von Mackensen S, Mantovani L, Mannucci PM; Esprit Study Group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT study). J Thromb Haemost. 2011;9(4):700–710. doi:10.1111/j.1538-7836.2011.04214.x
34. Tagliaferri A, Feola G, Molinari AC, et al. Benefits of prophylaxis versus on-demand treatment in adolescents and adults with severe haemophilia A: the POTTER study. Thromb Haemost. 2015;114(1):35–45. doi:10.1160/TH14-05-0407
35. Oldenburg J, Zimmermann R, Katsarou O, et al. Controlled, cross-sectional MRI evaluation of joint status in severe haemophilia A patients treated with prophylaxis vs. on demand. Haemophilia. 2015;21(2):171–179. doi:10.1111/hae.12539
36. Manco-Johnson MJ, Soucie JM, Gill JC. Prophylaxis usage, bleeding rates, and joint outcomes of hemophilia, 1999 to 2010: a surveillance project. Blood. 2017;129(17):2368–2374. doi:10.1182/blood-2016-02-683169
37. Oldenburg J, Kulkarni R, Srivastava A, et al. Improved joint health in subjects with severe haemophilia A treated prophylactically with recombinant factor VIII Fc fusion protein. Haemophilia. 2018;24(1):77–84. doi:10.1111/hae.13353
38. Warren BB, Jacobson L, Kempton C, et al. Factor VIII prophylaxis effects outweigh other hemostasis contributors in predicting severe haemophilia A joint outcomes. Haemophilia. 2019;25(5):867–875. doi:10.1111/hae.13778
39. Fischer K, van der Bom JG, Mauser-Bunschoten EP, et al. The effects of postponing prophylactic treatment on long-term outcome in patients with severe hemophilia. Blood. 2002;99(7):2337–2341. doi:10.1182/blood.V99.7.2337
40. Bladen M, Main E, Hubert N, Koutoumanou E, Liesner R, Khair K. Factors affecting the haemophilia joint health score in children with severe haemophilia. Haemophilia. 2013;19(4):626–631. doi:10.1111/hae.12108
41. Nijdam A, Foppen W, van der Schouw YT, Mauser-Bunschoten EP, Schutgens REG, Fischer K. Long-term effects of joint bleeding before starting prophylaxis in severe haemophilia. Haemophilia. 2016;22(6):852–858. doi:10.1111/hae.12959
42. Warren BB, Thornhill D, Stein J, et al. Young adult outcomes of childhood prophylaxis for severe hemophilia A: results of the Joint Outcome Continuation Study. Blood Adv. 2020;4(11):2451–2459. doi:10.1182/bloodadvances.2019001311
43. Nijdam A, Foppen W, De Kleijn P, et al. Discontinuing early prophylaxis in severe haemophilia leads to deterioration of joint status despite low bleeding rates. Thromb Haemost. 2016;115(5):931–938. doi:10.1160/TH15-08-0637
44. Fischer K, Van Der Bom JG, Prejs R, et al. Discontinuation of prophylactic therapy in severe haemophilia: incidence and effects on outcome. Haemophilia. 2001;7(6):544–550. doi:10.1046/j.1365-2516.2001.00560.x
45. Wallny TA, Scholz DT, Oldenburg J, et al. Osteoporosis in haemophilia - an underestimated comorbidity? Haemophilia. 2007;13(1):79–84. doi:10.1111/j.1365-2516.2006.01405.x
46. Nair AP, Jijina F, Ghosh K, Madkaikar M, Shrikhande M, Nema M. Osteoporosis in young haemophiliacs from western India. Am J Hematol. 2007;82(6):453–457. doi:10.1002/ajh.20877
47. Abdelrazik N, Reda M, El-Ziny M, Rabea H. Evaluation of bone mineral density in children with hemophilia: mansoura University children hospital (MUCH) experience, Mansoura, Egypt. Hematology. 2007;12(5):431–437. doi:10.1080/10245330701383700
48. Alioglu B, Selver B, Ozsoy H, Koca G, Ozdemir M, Dallar Y. Evaluation of bone mineral density in Turkish children with severe haemophilia A: Ankara hospital experience. Haemophilia. 2012;18(1):69–74. doi:10.1111/j.1365-2516.2011.02587.x
49. Naderi A, Nikvarz M, Arasteh M, Shokoohi M. Osteoporosis/osteopenia and hemophilic arthropathy in severe hemophilic patients. Arch Iran Med. 2012;15(2):82–84. doi:1012152/AIM.006
50. Anagnostis P, Vakalopoulou S, Slavakis A, et al. Reduced bone mineral density in patients with haemophilia A and B in Northern Greece. Thromb Haemost. 2012;107(3):545–551. doi:10.1160/TH11-08-05563
51. Anagnostis P, Vakalopoulou S, Vyzantiadis TA, et al. The clinical utility of bone turnover markers in the evaluation of bone disease in patients with haemophilia A and B. Haemophilia. 2014;20(2):268–275. doi:10.1111/hae.12271
52. El-Mikkawy DME, Elbadawy MA, Abd El-Ghany SM, Samaha D. Serum sclerostin level and bone mineral density in pediatric hemophilic arthropathy. Indian J Pediatr. 2019;86(6):515–519. doi:10.1007/s12098-019-02855-1
53. Wells AJ, McLaughlin P, Simmonds JV, et al. A case-control study assessing bone mineral density in severe haemophilia A in the UK. Haemophilia. 2015;21(1):109–115. doi:10.1111/hae.12565
54. Kempton CL, Antun A, Antoniucci DM, et al. Bone density in haemophilia: a single institutional cross-sectional study. Haemophilia. 2014;20(1):121–128. doi:10.1111/hae.12240
55. Kiper Unal HD, Comert Ozkan M, Atilla FD, et al. Evaluation of bone mineral density and related parameters in patients with haemophilia: a single center cross-sectional study. Am J Blood Res. 2017;7(5):59–66.
56. Harris S, Boggio LN. Exercise may decrease further destruction in the adult haemophilic joint. Haemophilia. 2006;12(3):237–240. doi:10.1111/j.1365-2516.2006.01214.x
57. Groen WG, Takken T, van der Net J, Helders PJ, Fischer K. Habitual physical activity in Dutch children and adolescents with haemophilia. Haemophilia. 2011;17(5):e906–e912. doi:10.1111/j.1365-2516.2011.02555.x
58. Cuesta-Barriuso R, Torres-Ortuno A, Pérez-Alenda S, Jose Carrasco J, Querol F, Nieto-Munuera J. Sporting activities and quality of life in children with hemophilia: an observational study. Pediatr Phys Ther. 2016;28(4):453–459. doi:10.1097/PEP.0000000000000296
59. Timmer MA, Veenhof C, de Kleijn P, de Bie RA, Schutgens REG, Pisters MF. Movement behaviour patterns in adults with haemophilia. Ther Adv Hematol. 2020;11:2040620719896959. doi:10.1177/2040620719896959
60. Doria AS, Keshava SN, Mohanta A, et al. Diagnostic accuracy of ultrasound for assessment of hemophilic arthropathy: MRI correlation. Am J Roentgenol. 2015;204(3):W336–W347. doi:10.2214/AJR.14.12501
61. De la Corte-rodriguez H, Rodriguez-Merchan EC, Alvarez-Roman MT, Martin-Salces M, Martinoli C, Jimenez-Yuste V. The value of HEAD-US system in detecting subclinical abnormalities in joints of patients with hemophilia. Expert Rev Hematol. 2018;11(3):253–261. doi:10.1080/17474086.2018.1435269
62. Foppen W, van der Schaaf IC, Beek FJA, Mali WPTM, Fischer K. MRI predicts 5-year joint bleeding and development of arthropathy on radiographs in hemophilia. Blood Adv. 2020;4(1):113–121. doi:10.1182/bloodadvances.2019001238
63. Foppen W, van der Schaaf IC, Beek FJA, Mali WPTM, Fischer K. Diagnostic accuracy of point-of-care ultrasound for evaluation of early blood-induced joint changes: comparison with MRI. Haemophilia. 2018;24(6):971–979. doi:10.1111/hae.13524
64. von Drygalski A, Moore RE, Nguyen S, et al. Advanced hemophilic arthropathy: sensitivity of soft tissue discrimination with musculoskeletal ultrasound. J Ultrasound Med. 2018;37(8):1945–1956. doi:10.1002/jum.14541
65. Poonnoose PM, Hilliard P, Doria AS, et al. Correlating clinical and radiological assessment of joints in haemophilia: results of a cross sectional study. Haemophilia. 2016;22(6):925–933. doi:10.1111/hae.13023
66. Klukowska A, Czyrny Z, Laguna P, Brzewski M, Serafin-Krol MA, Rokicka-Milewska R. Correlation between clinical, radiological and ultrasonographical image of knee joints in children with haemophilia. Haemophilia. 2001;7(3):286–292. doi:10.1046/j.1365-2516.2001.00509.x
67. Fischer K, Nijdam A, Holmstrom M, et al. Evaluating outcome of prophylaxis in haemophilia: objective and self-reported instruments should be combined. Haemophilia. 2016;22(2):e80–e86. doi:10.1111/hae.12901
68. Pergantou H, Matsinos G, Papadopoulos A, Platokouki H, Aronis S. Comparative study of validity of clinical, X-ray and magnetic resonance imaging scores in evaluation and management of haemophilic arthropathy in children. Haemophilia. 2006;12(3):241–247. doi:10.1111/j.1365-2516.2006.01208.x
69. Feldman BM, Funk SM, Bergstrom BM, et al. Validation of a new pediatric joint scoring system from the International Hemophilia Prophylaxis Study Group: validity of the hemophilia joint health score. Arthritis Care Res. 2011;63(2):223–230. doi:10.1002/acr.20353
70. Fischer K, de Kleijn P. Using the Haemophilia Joint Health Score for assessment of teenagers and young adults: exploring reliability and validity. Haemophilia. 2013;19(6):944–950. doi:10.1111/hae.12197
71. Oymak Y, Yildirim AT, Yaman Y, et al. The effectiveness of tools for monitoring hemophilic arthropathy. J Pediatr Hematol Oncol. 2015;37(2):e80–e85. doi:10.1097/MPH.0000000000000210
72. Timmer MA, Foppen W, Schutgens RE, Pisters MF, Fischer K. Comparing findings of routine haemophilia joint health score and haemophlia early arthropathy detection with ultrasound assessments in adults with haemophilia. Haemophilia. 2017;23(2):e141–e143. doi:10.1111/hae.13147
73. Seuser A, Kurnik K, Mahlein AK. Infrared thermography as a non-invasive tool to explore differences in the musculoskeletal system of children with hemophilia compared to an age-matched healthy group. Sensors. 2018;18(2):518. doi:10.3390/s18020518
74. Seuser A, Navarrete-Duran M, Auerswald G, Mancuso ME. Muscle function deterioration in patients with haemophilia: prospective experience from Costa Rica. Haemophilia. 2018;24(4):e230–e241. doi:10.1111/hae.13455
75. García-Massó X, Ye-Lin Y, Garcia-Casado J, Querol F, Gonzalez LM. Assessment of haemophilic arthropathy through balance analysis: a promising tool. Comput Methods Biomech Biomed Engin. 2019;22(4):418–425. doi:10.1080/10255842.2018.1561877
76. Iem DU, De Schepper AMA, Camerlinck M, Grobbee DE, Fischer K. Magnetic resonance imaging in teenagers and young adults with limited haemophilic arthropathy: baseline results from a prospective study. Haemophilia. 2011;17(6):926–930. doi:10.1111/j.1365-2516.2011.02513.x
77. Lundin B, Ljung R, Pettersson H. MRI scores of ankle joints in children with haemophilia–comparison with clinical data. Haemophilia. 2005;11(2):116–122. doi:10.1111/j.1365-2516.2005.01061.x
78. Chowdary P. Extended half-life recombinant products in haemophilia clinical practice – expectations, opportunities and challenges. Thromb Res. 2020;196:609–617. doi:10.1016/j.thromres.2019.12.012
79. Zanon E, Tagliaferri A, Pasca S, et al. Physical activity improved by adherence to prophylaxis in an Italian population of children, adolescents and adults with severe haemophilia A: the SHAPE Study. Blood Transfus. 2020;18(2):152–158. doi:10.2450/2019.0040-19
80. Malec LM, Cheng D, Witmer CM, et al. The impact of extended half-life factor concentrates on prophylaxis for severe hemophilia in the United States. Am J Hematol. 2020;95(8):960–965. doi:10.1002/ajh.25844
81. Hua B, Olsen EHN, Sun S, et al. Serological biomarkers detect active joint destruction and inflammation in patients with haemophilic arthropathy. Haemophilia. 2017;23(4):e294–e300. doi:10.1111/hae.13196
82. Jansen NWD, Roosendaal G, Lundin B, et al. The combination of the biomarkers urinary C-terminal telopeptide of type II collagen, serum cartilage oligomeric matrix protein, and serum chondroitin sulfate 846 reflects cartilage damage in hemophilic arthropathy. Arthritis Rheum. 2009;60(1):290–298. doi:10.1002/art.24184
83. Melchiorre D, Milia AF, Linari S, et al. RANK-RANKL-OPG in hemophilic arthropathy: from clinical and imaging diagnosis to histopathology. J Rheumatol. 2012;39(8):1678–1686. doi:10.3899/jrheum.120370
84. Manetti M, Linari S, Romano E, et al. TNF-alpha/TNF-R system may represent a crucial mediator of proliferative synovitis in hemophilia A. J Clin Med. 2019;8(7):939. doi:10.3390/jcm8070939
85. Parameswaran N, Patial S. Tumor necrosis factor-alpha signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20(2):87–103. doi:10.1615/CritRevEukarGeneExpr.v20.i2.10
86. von Drygalski A, Barnes RFW, Jang H, et al. Advanced magnetic resonance imaging of cartilage components in haemophilic joints reveals that cartilage hemosiderin correlates with joint deterioration. Haemophilia. 2019;25(5):851–858. doi:10.1111/hae.13802
87. Acharya SS, Kaplan RN, Macdonald D, Fabiyi OT, DiMichele D, Lyden D. Neoangiogenesis contributes to the development of hemophilic synovitis. Blood. 2011;117(8):2484–2493. doi:10.1182/blood-2010-05-284653
88. Zetterberg E, Palmblad J, Wallensten R, Morfini M, Melchiorre D, Holmström M. Angiogenesis is increased in advanced haemophilic joint disease and characterised by normal pericyte coverage. Eur J Haematol. 2014;92(3):256–262. doi:10.1111/ejh.12227
89. Rodriguez-Merchan EC, Valentino LA. Increased bone resorption in hemophilia. Blood Rev. 2019;33:6–10. doi:10.1016/j.blre.2018.05.002
90. Eldash HH, Atwa ZT, Saad MA. Vitamin D deficiency and osteoporosis in hemophilic children: an intermingled comorbidity. Blood Coagul Fibrinolysis. 2017;28(1):14–18. doi:10.1097/MBC.0000000000000519
91. Gamal Andrawes N, Hashem Fayek M, Salah El-Din N, Atef Mostafa R. Effect of low-dose factor VIII prophylaxis therapy on bone mineral density and 25(OH) vitamin D level in children with severe haemophilia A. Haemophilia. 2020;26(2):325–332. doi:10.1111/hae.13917
92. Abbasnezhad A, Habibi M, Abdolkarimi B, Zare S, Fazeli Moghadam E, Choghakhori R. Serum concentrations of vitamin D, calcium, phosphorus and trace minerals in adults and children with haemophilia A: association with disease severity, quality of life, joint health and functional status. Int J Hematol Oncol Stem Cell Res. 2020;14(1):56–71.
93. Zhou JY, Barnes RFW, Foster G, Iorio A, Cramer TJ, von Drygalski A. Joint bleeding tendencies in adult patients with hemophilia: it’s not all pharmacokinetics. Clin Appl Thromb Hemost. 2019;25:1076029619862052. doi:10.1177/1076029619862052
94. Lobet S, Meite N, Koninckx MI, et al. Implementation and assessment of a self- and community-based rehabilitation programme in patients with haemophilia from Cote d’Ivoire. Haemophilia. 2019;25(5):859–866. doi:10.1111/hae.13833
95. Timmer MA, Kloek CJJ, de Kleijn P, et al. A blended physiotherapy intervention for persons with hemophilic arthropathy: development study. J Med Internet Res. 2020;22(6):e16631. doi:10.2196/16631
96. Hashem F, Stephensen D, Drechsler WI, et al. Muscle strengthening intervention for boys with haemophilia: developing and evaluating a best-practice exercise programme with boys, families and health-care professionals. Health Expectations. 2020;23:1350–1361.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.