Back to Journals » Journal of Inflammation Research » Volume 19
Astrocytes and Microglia Regulate Opioid Receptor-Driven Cancer Brain Metastasis and Neural Injury: Remodeling the Brain Microenvironment
Authors Cui D ![]() , Wang X
, Wang X ![]() , Shang X
, Shang X
Received 6 March 2026
Accepted for publication 15 May 2026
Published 27 May 2026 Volume 2026:19 606362
DOI https://doi.org/10.2147/JIR.S606362
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Junhao Wang
Di Cui, Xiang Wang, Xiaoling Shang
College of Basic Medical Sciences, Changchun University of Chinese Medicine, Changchun, Jilin, People’s Republic of China
Correspondence: Xiaoling Shang, Jingyue Economic Development Zone, No. 1035, Boshuo Road, Changchun City, Jilin Province, People’s Republic of China, Email [email protected]
Abstract: Brain metastasis is a common complication in the progression of multiple solid tumors and is associated with poor prognosis. Its occurrence is not determined solely by the intrinsic invasiveness of tumor cells. After tumor cells enter the central nervous system (CNS), their survival and expansion largely depend on the brain microenvironment. Circulating tumor cells (CTCs) must first cross the blood-brain barrier (BBB). They then encounter a defensive microenvironment centered on reactive astrocytes and disease-associated microglia. This “colonization bottleneck” prevents most disseminated cells from surviving. Under tumor-derived stimulation, neuroglial cells undergo functional reprogramming. They shift from a predominantly restrictive state to a tumor-supportive adaptive state. This shift involves amplified inflammation, enhanced immunosuppression, and altered metabolic coupling, which together reshape the metastatic niche in the brain. At the same time, disruption of microenvironmental homeostasis affects neuronal survival. This links brain metastasis with neuronal injury. Opioid receptors (ORs) are functionally expressed in astrocytes and microglia. Their activation regulates inflammatory responses, metabolic states, and immune signaling networks. They may serve as key signaling nodes that control functional shifts in neuroglial cells. Opioid receptor signaling does not directly drive tumorigenesis. Instead, it likely resets how neuroglial cells respond to external perturbations. This process amplifies brain microenvironmental dysregulation. As a result, it indirectly promotes brain metastatic colonization and aggravates neuronal injury. In summary, brain metastasis results from long-term interactions between tumor cells and the brain microenvironment. Elucidating glia-mediated selection mechanisms and their regulatory nodes will deepen the understanding of the biological nature of brain metastasis.
Keywords: brain metastasis, astrocytes and microglia, microenvironmental dysregulation, opioid receptor signaling, neural injury
Introduction
Brain metastasis from cancer typically occurs at late stages of malignant tumor progression and is one of the most destructive complications in current clinical practice, with extremely high difficulty of intervention.1 Its occurrence is often accompanied by progressive neurological dysfunction, a marked decline in quality of life, and poor long-term prognosis.2–5 Unlike common metastatic target organs such as the lung, liver, or bone, the formation of brain metastasis cannot be simply attributed to increased intrinsic invasiveness or enhanced circulatory dissemination of tumor cells. Instead, it largely depends on the selective constraints and remodeling capacity of the highly specialized microenvironment of the CNS.6 The structural integrity of the BBB, a glucose-centered metabolic pattern, and unique immune regulatory features together impose strong constraints on incoming tumor cells. These factors make the brain one of the most selective target organs in the metastatic cascade.7–9
For a long time, brain metastasis was considered the result of abnormal proliferation of tumor cells after crossing the BBB. However, accumulating basic and translational studies indicate that this process is not a single “tumor cell-driven event,” but rather a multicellular, coordinated process centered on dynamic remodeling of the brain microenvironment.9 In this context, glial cells, especially astrocytes and microglia, have been increasingly recognized as critical regulatory units during metastatic colonization.10 As key regulators of neural homeostasis, these cells initially restrict disseminated tumor cells through inflammation, phagocytosis, and barrier functions.11 Accumulating evidence indicates that this process is not solely tumor cell–driven. Instead, it is a coordinated multicellular process involving dynamic remodeling of the brain microenvironment.
Notably, this imbalance in the brain microenvironment does not only support tumor colonization. It also serves as an important pathological basis for neuronal injury and neurological dysfunction.12 Sustained amplification of neuroinflammation, disruption of metabolic homeostasis, and weakening of BBB function act in combination. They are considered key mechanisms underlying the progressive worsening of neurological symptoms in patients with brain metastasis.13 However, based on current research, the regulatory hub linking “brain metastasis-microenvironmental dysregulation-neuronal injury” has not been systematically integrated. The causal chain and key nodes remain insufficiently defined.
ORs are a class of G protein-coupled receptors that are widely expressed in the CNS. They have long been regarded as key regulators of pain perception.14,15 However, accumulating evidence indicates that ORs also participate in the regulation of inflammatory responses and the shaping of neuroglial cell functions. They exert multi-level roles in maintaining neural microenvironmental homeostasis.16 According to receptor subtypes, ORs are classified into μ (MOR), δ (DOR), and κ (KOR). MOR is most abundantly expressed in the CNS. DOR and KOR are functionally expressed in astrocytes and microglia and participate in processes such as inflammatory responses.17–19 Under physiological conditions, transient activation mediated by endogenous opioid peptides helps limit excessive inflammation and maintain neural homeostasis. However, under chronic stimulation or sustained exposure to exogenous opioid agonists, OR signaling becomes biased. This shift leads to amplified neuroglial inflammation, metabolic alterations, and immune imbalance.20,21 This subtype-dependent signaling is not simply “pro-inflammatory” or “anti-inflammatory.” Instead, it may reshape the functional spectrum of glial cells and alter brain microenvironmental homeostasis. In tumor research, OR signaling can influence the tumor microenvironment and indirectly regulate tumor progression and metastasis. Some studies suggest that MOR activation affects tumor-associated inflammation and immune cell function.22 However, in the context of brain metastasis, it remains unclear whether ORs regulate neuroglial cell states, amplify brain microenvironmental dysregulation, and promote metastatic colonization. Systematic integration is lacking. Based on this gap, an overlooked question emerges: in the specific context of brain metastasis, could ORs act through modulation of neuroglial functional states as intermediate factors, thereby amplifying brain microenvironmental dysregulation, exacerbating neuronal injury, and promoting metastatic colonization and progression?
Based on existing evidence, we propose that ORs serve as key signaling nodes linking neural homeostasis regulation and tumor microenvironment remodeling. They may bias the functional states of neuroglial cells, thereby amplifying the vicious cycle of “microenvironmental dysregulation-pro-tumor growth-neuronal injury” during brain metastasis. Under certain conditions, this process may further accelerate the initiation and progression of brain metastasis from cancer. This review is based on a comprehensive survey of relevant literature in the PubMed and Web of Science databases, focusing on the keywords “brain metastasis,” “neuroglial cells,” “ORs,” and “tumor microenvironment,” and integrates and summarizes research progress over the past 20 years (2006–2026). On this basis, we propose that this vicious cycle may involve several key components, including: (1) mechanisms of brain microenvironment remodeling mediated by neuroglial cells during metastatic colonization; (2) the roles of ORs in regulating neuroglial inflammation, metabolism, and immune functions; and (3) how opioid receptor imbalance links microenvironmental dysregulation with neuronal injury in the context of brain metastasis, thereby providing a theoretical basis for intervention strategies targeting the neuro-tumor microenvironment axis.
Brain Metastatic Colonization: A Selective Process Centered on the Brain Microenvironment
From the overall framework of the metastatic cascade, brain metastasis is not the passive endpoint of tumor cell dissemination but a highly selective colonization event. Even if CTCs successfully traverse the BBB, the vast majority are still eliminated at early stages within the brain and fail to form stable metastatic lesions23,24 (Figure 1). Definitions of relevant terms are provided in the Brief Glossary (Table 2).
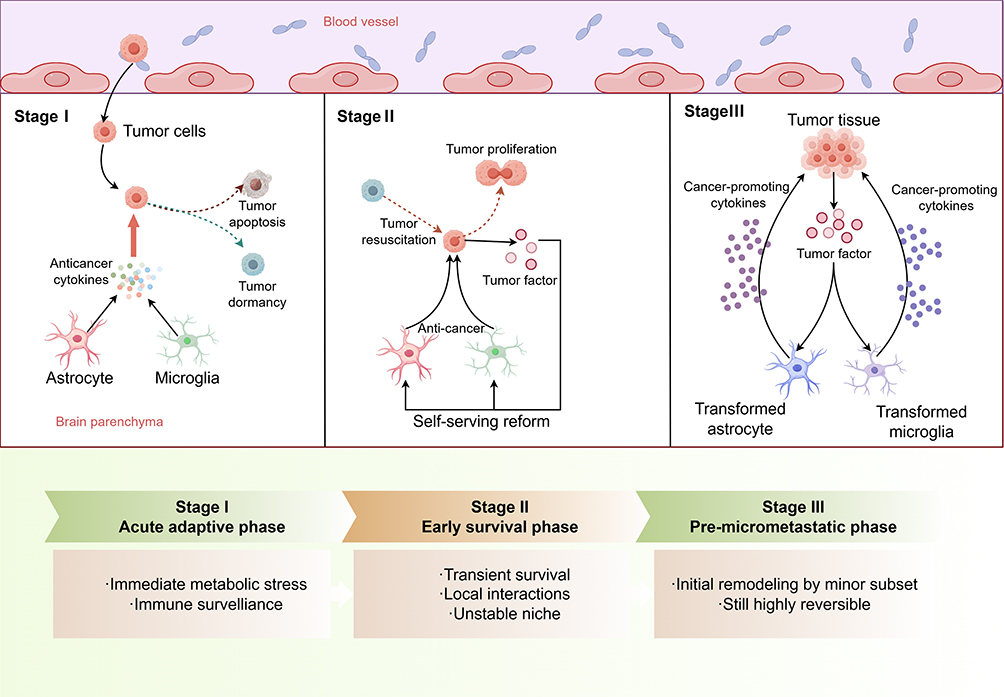 |
Figure 1 Brain Microenvironmental Dysregulation and the Formation of Tumor Metastasis (By Figdraw). Traversal of the BBB alone is insufficient to achieve brain metastasis, as disseminated tumor cells undergo multi-stage selection within the brain. In the initial stage (Stage I), metabolic and immune pressures create a colonization bottleneck. Subsequently (Stages II–III), surviving cells must acquire tolerance to the microenvironment or actively remodel it within an unstable niche. This process is inefficient and exhibits a degree of reversibility. Astrocytes and microglia continuously participate in regulating this selection process across different stages. |
During the immune clearance phase, that is, the “earliest stage” after CTCs enter the brain parenchyma, neuroglial cells predominantly exhibit a restrictive program. Astrocytes and microglia can rapidly sense the invasion of tumor cells and form the first line of defense against disseminated tumor cells through the release of pro-inflammatory factors, phagocytic activity, and enhancement of local barrier functions.25,26 For CTCs, this abruptly altered microenvironment imposes substantial survival pressure. They must rapidly adapt to this entirely new environment; otherwise, they face elimination. As a result, a large proportion of CTCs are cleared at this stage or are forced into a dormant state.27–29 From this perspective, the key determinant of brain metastatic colonization is not whether tumor cells enter the brain tissue, but whether they possess the capacity to remodel and “educate” the neuroglia-dominated brain microenvironment. This constitutes the immediate stress response and the first round of selection following CTCs traversal of the BBB.
CTCs that survive the initial stage can persist only transiently and have not yet acquired stable and sufficient survival support. Tumor cells then enter the early phase of immune equilibrium. At this stage, they have not established robust immune evasion mechanisms and cannot effectively resist recognition and attack by the host immune system.30 Accordingly, the surrounding microenvironment remains hostile and suppressive, with limited support for proliferation. The overall survival state is therefore highly unstable During this phase, tumor cells begin to influence the local environment through progressive interactions and initiate adaptive remodeling. Some early anti-tumor signals may undergo functional transition. For example, sustained type I interferon signaling can induce CCL2 and recruit monocyte-derived myeloid cells. This process gradually weakens immune clearance and lays the foundation for the subsequent establishment of an immunosuppressive environment.26
With continued remodeling of the local microenvironment, a small subset of tumor cells gradually acquires more favorable survival conditions and enters an active remodeling phase within immune equilibrium. For example, astrocytes in the brain can be induced to shift from a tumor-suppressive phenotype to a tumor-promoting phenotype, while the phagocytic and cytotoxic functions of microglia are concurrently suppressed. Tumor cells may also initiate metabolic reprogramming to acidify the brain microenvironment, thereby supporting their own survival, or promote the formation of neovascular networks to sustain colonization.9,31,32 At this stage, the microenvironment has undergone initial remodeling but remains reversible, thereby laying the foundation for subsequent tumor expansion.
This process also indicates that the brain microenvironment has features distinct from those of peripheral tissues, imposing additional selective pressure on tumor cells. Existing evidence suggests that brain tissue relies predominantly on glucose metabolism, and its tight metabolic coupling may limit the rapid adaptation of incoming cells, thereby constituting a form of metabolic “selection.” At the same time, immune surveillance in the brain is primarily mediated by microglia and astrocytes. Although these cells can exert clearance functions at early stages, some studies indicate that they may undergo functional transitions under sustained stimulation.33,34 In addition, the strict regulation of the BBB not only restricts the entry of most cells but also provides a relatively isolated growth environment for tumor cells that have already colonized, enabling further adaptation to the local niche.
Taken together, these factors indicate that brain metastasis is not a purely passive process, but a highly selective colonization event regulated by multiple microenvironmental factors. Although some evidence is derived from studies of central inflammation unrelated to brain metastasis or from peripheral tumor microenvironments, these models share common features in inflammatory states and patterns of glial activation, and thus provide informative references for understanding the functional consequences of inflammatory responses.
Remodeling of Neuroglial Functional States and Their Roles in Brain Metastasis-Related Processes
Under physiological conditions, glial cells maintain relatively stable functional states. They contribute to neural network homeostasis by coordinating inflammatory responses, energy metabolism, and support of synaptic structure and function.10,35–37 However, when the CNS is exposed to pathological stimuli, neuroglial responses are not simply “activation” or “suppression.” Their functional states exhibit marked heterogeneity and change dynamically with the nature and duration of the stimuli.38 Transient acute stimuli typically induce defensive and protective states. In contrast, when inflammatory signals persist, metabolic burden continues to increase, or tumor-associated factors exert sustained effects, the functional balance among different glial cell subsets is gradually disrupted, and cellular states shift in a directional manner.39 During this process, neuroglial cells transition from maintainers of homeostasis to key executors that drive pathological progression, thereby creating conditions that support intracranial tumor colonization and expansion.
Early Stage of Brain Metastasis: Reactive Gliosis and Neuroinflammation
During the micrometastatic stage of brain metastasis, neuroglial cells typically first exhibit a defensive activation state. Studies have shown that 2–4 weeks after resection of primary melanoma tumors, the characteristic marker of reactive astrocytes, GFAP, is upregulated.40 This proliferative response occurs well before the formation of metastatic lesions, indicating that astrocytes are early responders in the metastatic cascade. At this stage, focal neuroinflammatory responses can be induced, and by enhancing BBB integrity and restricting nutrient diffusion, an unfavorable survival environment for tumor cells is established.41 Disease-associated microglia participate in the early clearance of tumor cells through phagocytosis and antigen presentation. The coordinated actions of these two cell types maintain a predominantly anti-tumor microenvironment characterized by inflammation and immune defense.42 The glial response at this stage essentially serves the maintenance of neural homeostasis.
Under Sustained Stimulation:Changes in Glial Cell States and Characteristics of the Immune Microenvironment
Under such sustained stimulation, the functional states of neuroglial cells gradually shift and drive the reconstruction of the central microenvironment. Existing studies indicate that, in the context of chronic inflammation, metabolic stress, and prolonged intercellular signaling, astrocytes and microglia can transition from defensive phenotypes to states that support tumor survival and expansion. This transition is typically characterized by biased amplification of inflammatory responses, enhanced immunosuppressive signaling, and adaptive support for tumor metabolic demands.
For example, astrocytes can be recruited to brain metastases by Reelin secreted from lung cancer cells and can secrete pro-survival factors such as SERPINE1 to support the growth of small cell lung cancer. They can also activate the EGFR pathway through secretion of IL-11 to increase PD-L1 expression, while IL-11 binding to its intrinsic receptor further facilitates tumor immune evasion.43,44 A more conservative interpretation is that this “pro-tumor” shift does not represent a complete loss of glial function, but rather a redirected mode of homeostatic regulation, ultimately leading to the formation of a relatively permissive ecological niche for tumor colonization in the brain.
Intrinsic Link Between State Transition and Neuronal Injury
Functional reprogramming of neuroglial cells promotes tumor colonization and may also be accompanied by changes in the microenvironment surrounding neurons. Sustained activation of pro-inflammatory signaling, disruption of metabolic homeostasis, and weakening of BBB function are all associated with neuronal injury and synaptic dysfunction, although their causal relationships remain to be further validated. These observations suggest that brain metastasis-associated neuronal injury may involve multi-level links to alterations in glial cell function, rather than being solely attributable to direct tumor invasion of neurons (Figure 2).
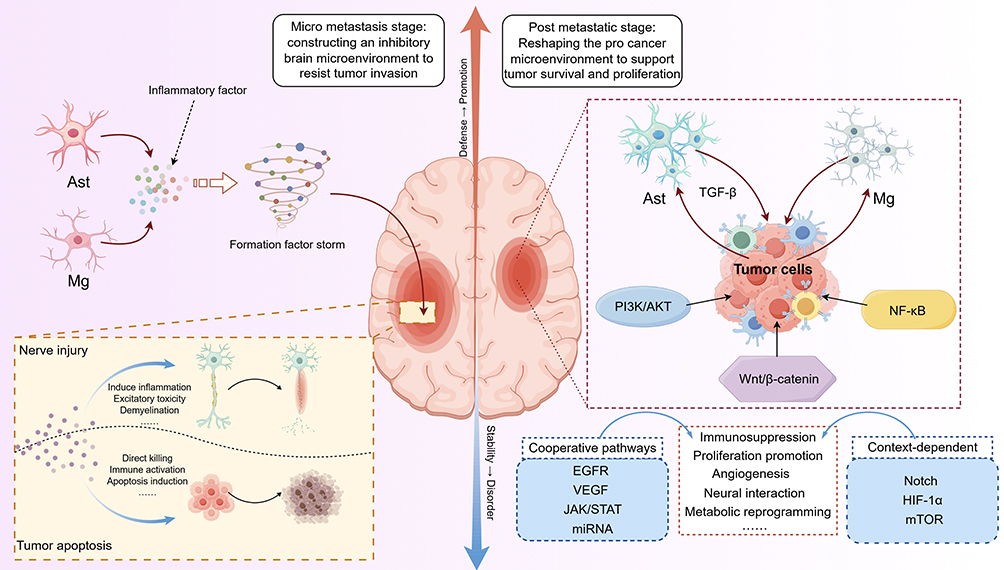 |
Figure 2 Schematic illustration of brain microenvironment remodeling centered on neuroglial cells (By Figdraw). During brain metastasis, astrocytes and microglia coordinately regulate inflammatory responses and immune surveillance through their interactions. Intercellular crosstalk engages multiple signaling pathways to remodel the brain microenvironment, thereby influencing tumor cell survival and adaptation.44–54 |
Amplified Inflammation and Neurotoxic Microenvironment
During the tumor-promoting stage, astrocytes and microglia are often in a persistently activated state. For example, in the context of melanoma and non-small cell lung cancer, the release of pro-inflammatory cytokines such as IL-6 and IL-8, together with the sustained involvement of signaling axes including NF-κB, STAT3, and CXCL10-CXCR3, establishes a relatively stable inflammatory background55–62 (Table 1). This response pattern may provide a relatively favorable microenvironment for tumor cell survival and is accompanied by reduced efficiency of immune clearance. From the perspective of the nervous system, this “persistently active” inflammatory state may be associated with certain long-term alterations in neuronal function. Some studies suggest that chronic inflammation may impair neuronal homeostasis over time. This effect may involve oxidative stress, excitotoxicity, and altered synaptic pruning.63–65 However, direct evidence for this sequence of events in the context of brain metastasis remains insufficient. Notably, such inflammatory responses often lack clear termination signals. Their effects may extend beyond the initial lesion site. Some studies indicate that these inflammatory signals can propagate along neural networks and, if sustained, may create a microenvironment with potentially adverse effects on overall neural circuitry. If confirmed, this process likely reflects gradual accumulation rather than abrupt change.
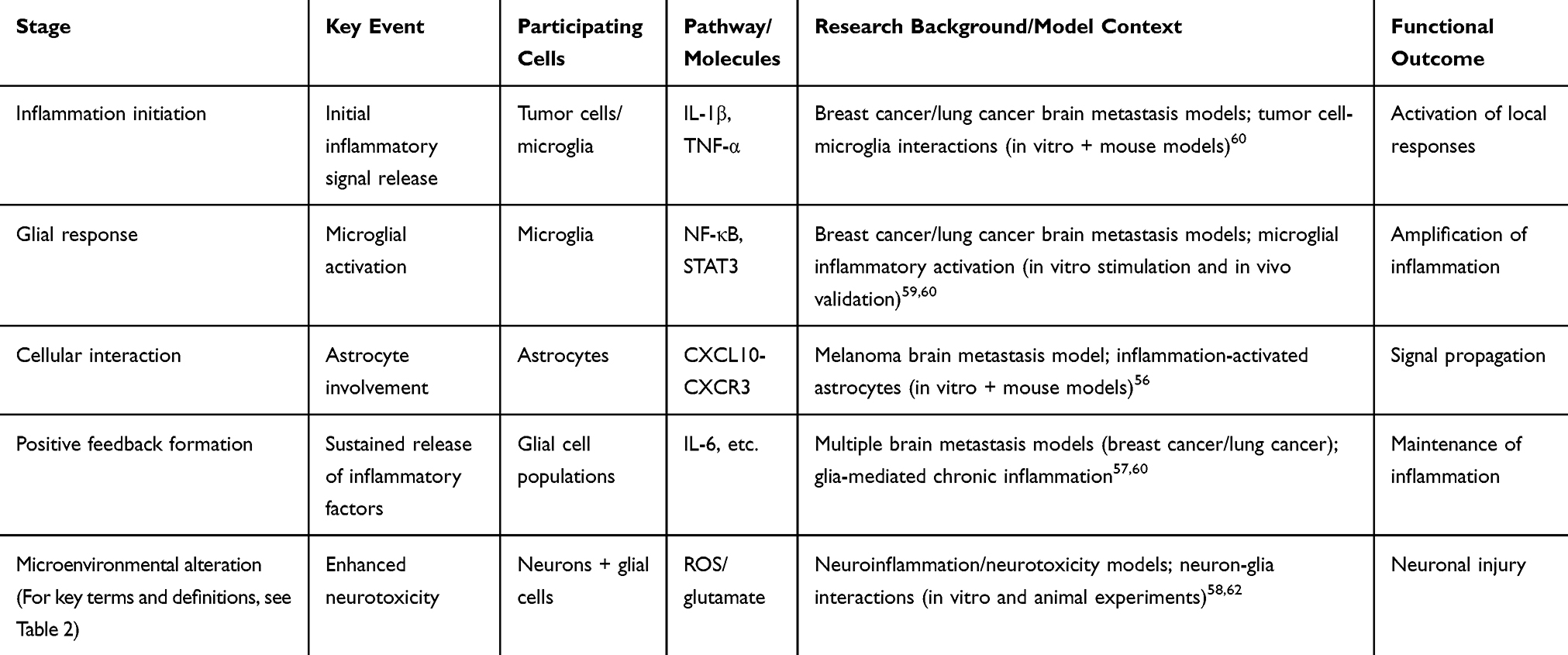 |
Table 1 Key Steps of the Inflammatory Cascade and Their Functional Consequences in Brain Metastasis |
 |
Table 2 Brief Glossary |
Disruption of Metabolic Coupling and Synaptic Dysfunction
Under physiological conditions, astrocytes maintain neuronal metabolic homeostasis and synaptic activity through multiple mechanisms, including the supply of energy substrates and the precise coordination of neurotransmitter cycling.66 However, under tumor-associated reprogramming states, this supportive role may shift, accompanied by changes in the allocation of metabolic resources and response priorities. This change does not necessarily manifest as overt “metabolic failure,” but may be associated with reduced efficiency of metabolic coupling, thereby affecting the buffering capacity of neurons in maintaining energy and neurotransmitter homeostasis.67 In this context, some studies have observed that synaptic plasticity shows decreased tolerance to minor metabolic disturbances, and the stability of neural networks may be correspondingly weakened. In other words, metabolic imbalance may be better understood as a persistently existing vulnerability rather than an acute injury triggered by a single event.
BBB Imbalance and Neural Network Vulnerability
Alongside inflammatory and metabolic alterations, shifts in neuroglial function may also affect the regulation of the perivascular microenvironment in the brain. Glial cells normally exert fine control over components such as vascular endothelial cells, pericytes, and the basement membrane. If this regulatory capacity is partially weakened, it may lead to dysregulation of the perivascular microenvironment, which is not necessarily equivalent to a simple structural disruption of the BBB. Such dysregulation may, on the one hand, facilitate the diffusion of tumor-associated factors within the brain, and on the other hand increase the exposure of neurons to abnormal inflammatory signals and metabolic stress.45,68 Its potential effects may extend beyond structural changes and could also involve alterations in the overall stability of neural networks. Therefore, neurological dysfunction associated with brain metastasis may not be fully attributable to neuronal death alone, but is more likely the result of the combined effects of multiple processes, including glial functional shifts, perivascular microenvironmental imbalance, and inflammatory and metabolic changes. The precise causal relationships among these factors remain to be further clarified.
ORs Influence Cancer Metastasis and Progression by Modulating the Tumor Microenvironment
Early clinical observations suggest that perioperative patterns of opioid use may be associated with postoperative recurrence risk in some cancer patients.69 This correlation has drawn attention to the potential role of ORs in tumor progression and has prompted subsequent mechanistic investigations at the molecular and cellular levels.
Subsequent basic and translational studies indicate that, across multiple solid tumors, changes in opioid receptor expression levels are associated with invasive tumor phenotypes and metastasis-related processes.19 For example, activation of DOR can influence signaling pathways such as JAK1/2-STAT3. This, in turn, alters the mode of communication between tumor cells and the surrounding microenvironment and, in specific contexts, is associated with enhanced migration and invasiveness of breast cancer cells. In addition, DOR signaling has been reported to be linked to processes such as epithelial-mesenchymal transition (EMT), suggesting that it may participate in tumor progression by modulating cellular plasticity and microenvironmental adaptability.18
Similarly, in certain experimental models, μ-opioid receptor (MOR)-related signaling can regulate tumor cell responses to microenvironmental cues through pathways such as PI3K/AKT and mTOR. This regulation affects cell migration, adhesion, and the formation of vascular-related structures.22,70–72 These changes may create a microenvironment that favors CTC survival and colonization. Current evidence suggests that ORs are not independent oncogenic drivers. Instead, they bias metastasis-related phenotypes by modulating the interactions between tumor cells and their surrounding microenvironment.
On this basis, a further question arises: whether these regulatory effects also operate in brain metastasis, a process that is highly dependent on microenvironmental adaptation, and whether they may have even greater significance in this context. The CNS represents a specialized niche with immune and metabolic characteristics that differ markedly from those of peripheral tissues. Its metastatic colonization process largely depends on remodeling of the local microenvironment, in which glial cells are considered key regulators.
Existing studies indicate that ORs in the CNS not only participate in classical pain regulation, but also contribute to maintaining the functional balance of the neural microenvironment by modulating inflammatory signaling pathways, glial cell activation states, and the expression of multiple cytokines. Moreover, the dynamic transitions of glial cell subsets not only reflect changes in cellular functional states, but may also be accompanied by reprogramming of opioid receptor expression and signaling responses.
It should be noted that the expression profiles and strength of evidence for ORs differ among glial cell types. In microglia, MOR and KOR show relatively consistent evidence of expression, whereas in astrocytes, receptor expression appears more heterogeneous and, to some extent, depends on specific inflammatory or experimental conditions.73–77
Based on these observations, it can be hypothesized that opioid receptor-related signaling may shape a brain microenvironment favorable for tumor cell survival and colonization by regulating glial cell activation states and their interactions with tumor cells, thereby exerting potential modulatory effects during the initiation and progression of brain metastasis. However, this inference is primarily based on the integration of cross-level evidence, and its precise mechanisms and applicability across different tumor types remain to be validated by further experimental studies.
Potential Regulatory Roles of ORs in Glial Cell-Cancer Cell Crosstalk Within the Brain Metastatic Microenvironment
Within the highly specialized microenvironment of brain metastasis, molecular systems that can alter glial response patterns may, in principle, also influence the formation of the metastatic niche. Based on this premise, we propose a “neuroglial sensitivity model,” in which the core role of ORs in the brain metastatic microenvironment may not lie in directly inducing inflammation or driving tumor-associated responses, but in resetting the effective sensing range and response slope of neuroglial cells to external perturbations.
This shift in the effective sensing range can be defined as a systematic change in the stimulus intensity required to trigger specific functional outputs in glial cells under the same external conditions. Such changes can be quantified by the following metrics: (i) the dose-response curves of inflammatory factor release; (ii) ratios associated with immune activation phenotypes in astrocytes and microglia (eg., the proportion of pro-inflammatory versus reparative features in microglia, and the relative proportions of reactive astrocyte subtypes); and (iii) metabolic shifts in astrocytes and microglia (eg., increases or decreases in the glycolysis/oxidative phosphorylation ratio). In terms of temporal scale, this process can be divided into short-term (minutes to hours, primarily reflecting receptor-mediated signaling transduction changes) and long-term (hours to days, involving transcriptional and epigenetic regulation) components. Different opioid receptor subtypes may play distinct roles in this process, thereby altering the response curves of glial cells to stimuli.
Based on the above definition, a falsifiable hypothesis can be further proposed: opioid receptor signaling, by modulating the effective sensing range of glial cells, may alter the distribution of their functional states under different stimulus intensities, thereby amplifying or suppressing inflammation-related cascade responses within a certain range. Specifically, ORs may be better understood as microenvironmental modulators. By regulating the functional states of neuroglial cells and their intrinsic signaling networks, they may indirectly shape the functional crosstalk between glial cells and tumor cells. In this way, inflammatory, stress, or chemotactic signals of a given intensity may be transformed into more propagative collective responses, thereby amplifying or attenuating their microenvironmental effects at the network level (Figure 3). However, whether they trigger a transition from homeostatic to pro-inflammatory or pro-tumor states within specific stimulus ranges, or instead suppress such transitions, remains to be determined by further experimental validation.
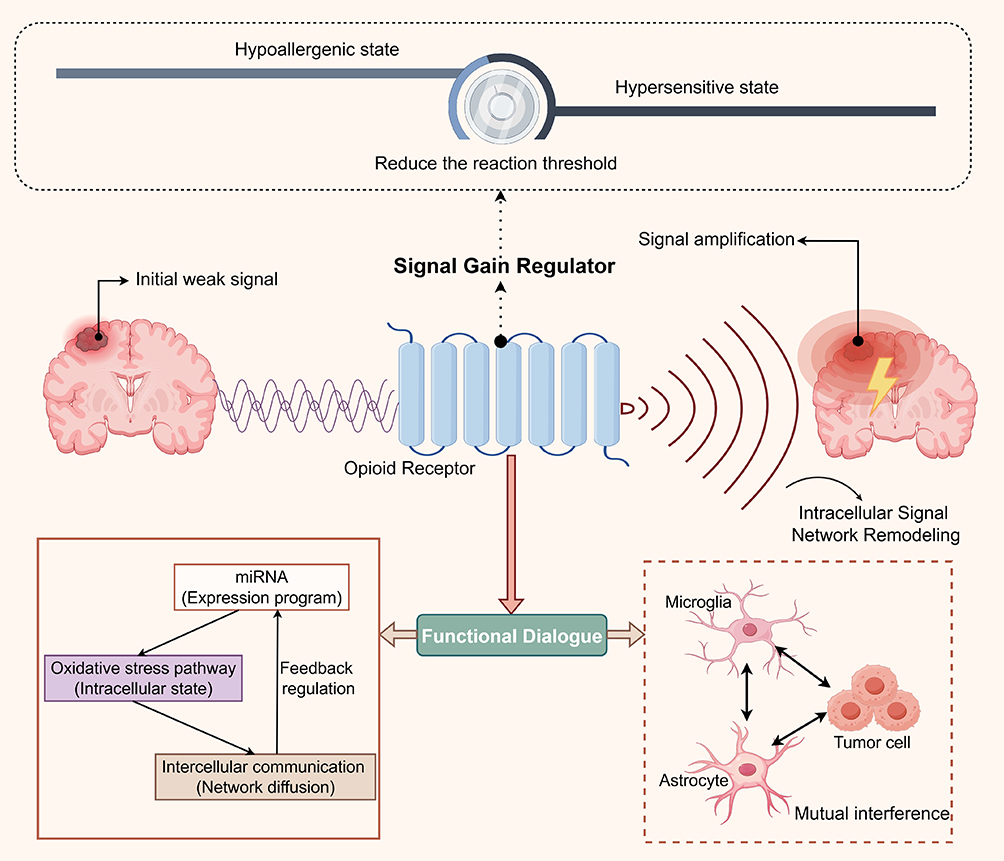 |
Figure 3 Schematic illustration of opioid receptor-mediated regulation of neuroglial sensitivity states (By Figdraw). Opioid receptor signaling may act as a “Signal Gain Modulator” by regulating the functional states of astrocytes and microglia and their intracellular signaling networks. In this process, local inflammatory, stress, or chemotactic signals are amplified and propagated, thereby influencing, in a context-dependent manner, glial cell-tumor cell interactions, metastatic colonization, and neuronal injury. |
Neuroglial Cells as the Critical Interface for Opioid Signaling Entry into the Brain Metastatic Niche
Within the framework of “opioid receptor-mediated resetting of the effective sensing range of glial cells,” neuroglial cells can be regarded as a key interface through which opioid signaling engages the brain metastatic microenvironment. As principal regulatory units of the brain microenvironment, astrocytes and microglia not only participate in the maintenance of inflammatory responses, but also largely determine the dynamic balance of local immune and metabolic states. Existing studies have shown that ORs are detectably expressed in these cell types and possess functional signaling capacity.78,79
In this context, ORs are more likely to influence the overall response trajectory of brain tissue to tumor-associated stimuli by modulating the response properties of glial cells, rather than by directly triggering inflammatory responses. For example, under specific stimulus conditions, MOR-related signaling can affect the release of inflammatory mediators from astrocytes and indirectly regulate the activation state of microglia through paracrine pathways. This process typically exhibits time-dependent characteristics, with more pronounced effects under conditions of sustained stimulation.19,80
Multilevel Signaling Networks Reset the Functional Phenotypes of Glial Cells
Further mechanistic studies suggest that the regulation of glial cells by opioid receptor signaling is not confined to single intracellular processes, but is achieved through amplification and integration within multi-level intercellular communication networks. This network includes classical signaling pathways (such as MAPK and JAK/STAT), microRNA-mediated post-transcriptional regulation, and extracellular vesicle (EV)-dependent long-range information transfer.14,81–84 In astrocytes, activation of KOR can engage stress-related pathways such as p38 MAPK, upregulate specific miRNA expression, and is accompanied by downregulation of homeostasis-related molecules such as xCT. These changes may, to some extent, disrupt glutamate metabolic balance and drive a shift toward a reactive state. At the same time, activation of μ-opioid receptor (MOR) can induce astrocytes to release extracellular vesicles enriched in miR-138. After uptake by microglia, these vesicles can influence their transcriptional programs and functional states through the TLR7 pathway.46,85 Overall, these mechanisms collectively form a non-linear regulatory network centered on microRNAs as key nodes, enabling opioid receptor signaling to coordinate the functional outputs of glial cell populations at both intracellular and intercellular levels.
Evidence Limitations and Context-Dependent Modes of Action in the Setting of Brain Metastasis
It should be noted that direct evidence is still lacking to demonstrate that opioid receptor signaling causally mediates specific interactions between glial cells and tumor cells during the development of brain metastasis. A more plausible interpretation is that ORs integrate signals related to inflammation, stress, and intercellular communication, and modulate the overall response pattern of the existing microenvironment, thereby influencing, to some extent, the conditions for tumor cell survival and colonization.
Within this framework, opioid receptor signaling is highly context-dependent, shaped by receptor subtype, ligand source, and exposure duration. Acute, physiological activation—typically driven by endogenous opioids—tends to restrain excessive inflammation, whereas sustained or chronic stimulation (for example, by exogenous agonists) may bias glial cells toward pro-inflammatory or dysregulated states. Distinct receptor subtypes are likely to contribute differentially. Thus, opioid receptors primarily modulate the threshold and gain of glial responses, enabling dynamic bidirectional regulation rather than fixed pro- or anti-inflammatory effects. This framework may reconcile divergent findings and underscores the need to consider stimulus intensity, duration, and microenvironmental context.
Future Perspectives
Although recent studies have increasingly highlighted the critical roles of neuroglial cells in brain metastasis and the potential involvement of ORs in mediating brain microenvironmental imbalance, several key scientific questions remain unresolved. Future research should systematically elucidate, at the mechanistic level, how ORs regulate the functional states of neuroglial cells in the context of brain metastasis, and further define their boundaries of action across different pathological conditions. Particular attention should be given to the molecular mechanisms by which ORs distinguish between endogenous endorphin signaling and exogenous opioid drug signaling, and whether this distinction determines the directional shift of neuroglial cells toward homeostatic support or inflammatory activation. At the same time, it remains to be determined whether inflammation-related signals, under sustained biased regulation by ORs, can act synergistically with tumor-derived signals to amplify responses and drive brain metastatic colonization and progression. These questions require validation using time-resolved and cell type-specific models.
Beyond the CNS, alterations in the peripheral immune system (such as T lymphocytes and B lymphocytes) can exacerbate inflammatory states within the CNS. At the same time, inflammatory factors released by activated astrocytes and microglia in the brain can enter the peripheral circulation via cerebrospinal fluid, further promoting activation of the peripheral immune system and amplifying inflammatory responses.86–88 Within this bidirectional interaction, it remains to be clarified whether the peripheral immune system participates in the expression of ORs and the regulation of their associated signaling pathways.
Overall, opioid receptor signaling pathways are more likely to represent a class of regulatable microenvironmental control nodes. Precise modulation of OR signaling may preserve analgesia and neuroprotection while limiting pro-inflammatory and pro-metastatic effects, thereby providing a new theoretical basis for achieving coordinated benefits of “analgesia-neuroprotection-metastasis control” in patients with brain metastasis from cancer.
Limitations
Some mechanistic inferences currently lack sufficient direct evidence, and the related conclusions rely, to some extent, on indirect or secondary evidence, requiring further experimental validation. In particular, in the context of brain metastasis, direct evidence for interactions between ORs and glial cells remains limited. Current understanding is largely derived from studies across different disease settings, and its applicability needs to be systematically confirmed in brain metastasis-specific models.
Abbreviations
CNS, Central nervous system; ORs, Opioid receptors; MOR, Mu opioid receptor; DOR, Delta opioid receptor; KOR, Kappa opioid receptor; CTCs, Circulating tumor cells; BBB, Blood-brain barrier; CCL2, C-C motif chemokine ligand 2; GFAP, Glial fibrillary acidic protein; SERPINE1, Serpin family E member 1; IL-11, Interleukin-11; EGFR, Epidermal growth factor receptor; PD-L1, Programmed death-ligand 1; IL-6, Interleukin-6; IL-8, Interleukin-8; NF-κB, Nuclear factor kappa B; STAT3, Signal transducer and activator of transcription 3; CXCL10, C-X-C motif chemokine ligand 10; CXCR3, C-X-C motif chemokine receptor 3; JAK1, Janus kinase 1; JAK2, Janus kinase 2; EMT, Epithelial-mesenchymal transition; PI3K, Phosphoinositide 3-kinase; AKT, Protein kinase B; mTOR, Mechanistic target of rapamycin; MAPK, Mitogen-Activated Protein Kinase; EV, extracellular vesicle; miRNA, MicroRNA; xCT, Cystine/glutamate antiporter; miR-138, MicroRNA-138; TLR7, Toll-like receptor 7.
Data Sharing Statement
Data sharing is not applicable to this article as no data were created or analysed in this study.
Acknowledgments
We appreciate all authors whose publications could be included in our review. We thank the FigDraw drawing platform for its support.
Author Contributions
All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. D.C. was mainly responsible for the conceptualization, investigation, writing - original draft and writing - review and editing of the manuscript. X.W. and X.L.S. contributed primarily to the conceptualization, supervision, and funding acquisition of the manuscript.
Funding
This research is supported by the National Natural Science Foundation of China (82505560) and the Jilin Provincial Science and Technology Development Program (YDZJ202501ZYTS758).
Disclosure
The author(s) report no conflicts of interest in this article.
References
1. Gerstberger S, Jiang Q, Ganesh K. Metastasis. Cell. 2023;186:1564–14. doi:10.1016/j.cell.2023.03.003
2. Maher EA, Mietz JL, Arteaga CL, et al. Brain metastasis: opportunities in basic and translational research. Cancer Res. 2009;69:6015–6020. doi:10.1158/0008-5472.CAN-08-4347
3. Johung KL, Yeh N, Desai NB, et al. Extended survival and prognostic factors for patients with ALK-rearranged non-small-cell lung cancer and brain metastasis. J Clin Oncol. 2016;34:123–129. doi:10.1200/JCO.2015.62.0138
4. Wang Y, Li X, Zhang H, et al. Development of graded prognostic assessment for breast cancer brain metastasis incorporating extracranial metastatic features: a retrospective analysis of 284 patients. BMC Cancer. 2024;24:1262. doi:10.1186/s12885-024-12983-3
5. Takeshita S, Takahashi Y, Saito T, et al. Prognostic factors for patients with brain metastasis from gynecological cancer: significance of treatment-free interval. Jpn J Clin Oncol. 2017;47:604–610. doi:10.1093/jjco/hyx060
6. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221:107753. doi:10.1016/j.pharmthera.2020.107753
7. Arvanitis CD, Ferraro GB, Jain RK. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat Rev Cancer. 2020;20:26–41. doi:10.1038/s41568-019-0205-x
8. De Leo A, Ugolini A, Vegliante MC, et al. Glucose-driven histone lactylation promotes the immunosuppressive activity of monocyte-derived macrophages in glioblastoma. Immunity. 2024;57:1105–1123.e8. doi:10.1016/j.immuni.2024.04.006
9. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31:326–341. doi:10.1016/j.ccell.2017.02.009
10. Ishibashi K, Hirata E. Multifaceted interactions between cancer cells and glial cells in brain metastasis. Cancer Sci. 2024;115:2871–2878. doi:10.1111/cas.16241
11. Allen NJ, Lyons DA. Glia as architects of central nervous system formation and function. Science. 2018;362:181–185. doi:10.1126/science.aat0473
12. Hou T, Ding H, Huang G, et al. Neural–tumor crosstalk and molecular targeting: mechanisms and therapeutic implications in cancer neuroscience. Cell Oncol. 2026;49:35. doi:10.1007/s13402-025-01151-9
13. Andersen BM, Faust Akl C, Wheeler MA, et al. Glial and myeloid heterogeneity in the brain tumour microenvironment. Nat Rev Cancer. 2021;21:786–802. doi:10.1038/s41568-021-00397-3
14. Che T, Roth BL. Molecular basis of opioid receptor signaling. Cell. 2023;186:5203–5219. doi:10.1016/j.cell.2023.10.029
15. Abdel Shaheed C, Maher CG, Williams KA, et al. Opioid analgesics for nociceptive cancer pain: a comprehensive review. CA. Cancer J Clin. 2024;74:286–313. doi:10.3322/caac.21823
16. Li Y, Wang X, Zhou Y, et al. Effects of opioid drugs on immune function in cancer patients. Biomed Pharmacother. 2024;175:116665. doi:10.1016/j.biopha.2024.116665
17. Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011;115:1363–1381. doi:10.1097/ALN.0b013e318238bba6
18. Tripolt S, Hofer D, Prantl L, et al. Opioids drive breast cancer metastasis through the δ-opioid receptor and oncogenic STAT3. Neoplasia. 2021;23:270–279. doi:10.1016/j.neo.2020.12.011
19. Zhang H, Sun J, Chen W, et al. Targeting the mu-opioid receptor for cancer treatment. Curr Oncol Rep. 2021;23:111. doi:10.1007/s11912-021-01107-w
20. Lacagnina MJ, Rivera PD, Bilbo SD. Glial and neuroimmune mechanisms as critical modulators of drug use and abuse. Neuropsychopharmacology. 2017;42:156–177. doi:10.1038/npp.2016.121
21. Schwartz EKC, De Aquino JP, Sofuoglu M. Glial modulators as novel therapeutics for comorbid pain and opioid use disorder. Br J Clin Pharmacol. 2024;90:3054–3066. doi:10.1111/bcp.16094
22. Wang X, Zhang Y, Li J, et al. μ-opioid receptor agonist facilitates circulating tumor cell formation in bladder cancer via the MOR/AKT/Slug pathway: a comprehensive study including randomized controlled trial. Cancer Commun. 2023;43:365–386. doi:10.1002/cac2.12408
23. Xu A, Liu Y, Wang Z, et al. Dynamic regulation of macrophage polarization in acute myocardial infarction and its therapeutic potential. J Inflamm Res. 2025;18:17363–17385. doi:10.2147/JIR.S543139
24. Izadi N, Saberi B, Kamali K, et al. Breaking boundaries: role of the brain barriers in metastatic process. Fluids Barriers CNS. 2025;22:3. doi:10.1186/s12987-025-00618-z
25. Evans KT, Ferguson SD, Lesniak MS, et al. Microglia promote anti-tumour immunity and suppress breast cancer brain metastasis. Nat Cell Biol. 2023;25:1848–1859. doi:10.1038/s41556-023-01273-y
26. Ma W, Kebir S, Kim SS, et al. Type I interferon response in astrocytes promotes brain metastasis by enhancing monocytic myeloid cell recruitment. Nat Commun. 2023;14:2632. doi:10.1038/s41467-023-38252-8
27. Neophytou CM, Kyriakou T-C, Papageorgis P. Mechanisms of metastatic tumor dormancy and implications for cancer therapy. Int J Mol Sci. 2019;20:6158. doi:10.3390/ijms20246158
28. Ghajar CM, Peinado H, Mori H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15:807–817. doi:10.1038/ncb2767
29. Dai J, Cimino PJ, Goubran HA, et al. Astrocytic laminin-211 drives disseminated breast tumor cell dormancy in brain. Nat Cancer. 2021;3:25–42. doi:10.1038/s43018-021-00297-3
30. Bates M, Jansson PJ, Richardson DR, et al. Circulating tumour cells: the good, the bad and the ugly. Biochim Biophys Acta Rev Cancer. 2023;1878:188863. doi:10.1016/j.bbcan.2023.188863
31. Klemm F, Maas RR, Bowman RL, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell. 2020;181:1643–1660.e17. doi:10.1016/j.cell.2020.05.007
32. Kloosterman DJ, Akkari L, Bowman RL, et al. Macrophage-mediated myelin recycling fuels brain cancer malignancy. Cell. 2024;187:5336–5356.e30. doi:10.1016/j.cell.2024.07.030
33. Sohrabi A, Lefebvre AEYT, Harrison MJ, et al. Microenvironmental stiffness induces metabolic reprogramming in glioblastoma. Cell Rep. 2023;42:113175. doi:10.1016/j.celrep.2023.113175
34. Bander ED, Rivera M, Cisse B, et al. The Benedict Arnold of the central nervous system tumor microenvironment? The role of microglia/macrophages in glioma. World Neurosurg. 2021;153:214–221. doi:10.1016/j.wneu.2021.06.121
35. Wright-Jin EC, Gutmann DH. Microglia as dynamic cellular mediators of brain function. Trends Mol Med. 2019;25:967–979. doi:10.1016/j.molmed.2019.08.013
36. Feng Y, Hu X, Zhang Y, et al. The role of microglia in brain metastases: mechanisms and strategies. Aging Dis. 2024;15:169–185. doi:10.14336/AD.2023.0514
37. Strickland MR, Alvarez-Breckenridge C, Gainor JF, et al. Tumor immune microenvironment of brain metastases: toward unlocking antitumor immunity. Cancer Discov. 2022;12:1199–1216. doi:10.1158/2159-8290.CD-21-0976
38. Escartin C, Galea E, Lakatos A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24:312–325. doi:10.1038/s41593-020-00783-4
39. Masuda T, Sankowski R, Staszewski O, et al. Microglia heterogeneity in the single-cell era. Cell Rep. 2020;30:1271–1281. doi:10.1016/j.celrep.2020.01.010
40. Schwartz H, Blacher E, Amer M, et al. Incipient melanoma brain metastases instigate astrogliosis and neuroinflammation. Cancer Res. 2016;76:4359–4371. doi:10.1158/0008-5472.CAN-16-0485
41. O’Brien ER, Kersemans V, Tredwell M, et al. Glial activation in the early stages of brain metastasis: TSPO as a diagnostic biomarker. J Nucl Med. 2014;55:275–280. doi:10.2967/jnumed.113.127449
42. Park ES, Kim SJ, Kim SW, et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proc Natl Acad Sci U S A. 2011;108:17456–17461.
43. Tang M, Chen Z, Wu X, et al. Brain metastasis from EGFR-mutated non-small-cell lung cancer: secretion of IL11 from astrocytes upregulates PD-L1 and promotes immune escape. Adv Sci. 2024;11:e2306348. doi:10.1002/advs.202306348
44. Qu F, Liu Z, Huang J, et al. Crosstalk between small-cell lung cancer cells and astrocytes mimics brain development to promote brain metastasis. Nat Cell Biol. 2023;25:1506–1519. doi:10.1038/s41556-023-01241-6
45. Michinaga S, Koyama Y. Dual roles of astrocyte-derived factors in regulation of blood–brain barrier function after brain damage. Int J Mol Sci. 2019;20:571. doi:10.3390/ijms20030571
46. Liao K, Niu F, Hu G, et al. Morphine-mediated release of miR-138 in astrocyte-derived extracellular vesicles promotes microglial activation. J Extracell Vesicles. 2020;10:e12027. doi:10.1002/jev2.12027
47. Liu L, Zhang Y, Li X, et al. Biological profile of breast cancer brain metastasis. Acta Neuropathol Commun. 2025;13:78. doi:10.1186/s40478-025-01983-4
48. Chen Q, Boire A, Jin X, et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493–498. doi:10.1038/nature18268
49. He W, He S, Wang Z, et al. Astrocyte elevated gene-1 (AEG-1) induces epithelial–mesenchymal transition in lung cancer through activating Wnt/β-catenin signaling. BMC Cancer. 2015;15:107. doi:10.1186/s12885-015-1124-1
50. Liu W, Zhang X, Li Y, et al. CD146⁺ pericyte-like lung cancer brain metastatic stem cells promote tumor angiogenesis through dual regulatory effects on the VEGF/VEGFR axis. Theranostics. 2026;16:1905–1924. doi:10.7150/thno.122241
51. Lahiri A, Biswas A, Dey S, et al. Endoplasmic reticulum stress amplifies cytokine responses in astrocytes via a PERK–EIF2A–JAK1 signaling axis. Glia. 2025;73:2273–2288. doi:10.1002/glia.70067
52. Guo R, Li X, Yang J, et al. Context-dependent regulation of Notch signaling in glial development and tumorigenesis. Sci Adv. 2023;9:eadi2167. doi:10.1126/sciadv.adi2167
53. Tamai S, Ichimura K, Saito N, et al. Tumor microenvironment in glioma invasion. Brain Sci. 2022;12:505. doi:10.3390/brainsci12040505
54. Jung B-K, Kim SY, Lee J, et al. Reduced secretion of lipocalin-2 from reactive astrocytes through autophagic and proteasomal regulation alleviates inflammatory stress and neuronal damage. Autophagy. 2023;19:2296–2317. doi:10.1080/15548627.2023.2180202
55. Rigg E, Mavroeidi P, Pardo OE, et al. Inhibition of extracellular vesicle-derived miR-146a-5p decreases progression of melanoma brain metastasis via Notch pathway dysregulation in astrocytes. J Extracell Vesicles. 2023;12:e12363. doi:10.1002/jev2.12363
56. Doron H, Amer M, Ershaid N, et al. Inflammatory activation of astrocytes facilitates melanoma brain tropism via the CXCL10–CXCR3 signaling axis. Cell Rep. 2019;28:1785–1798.e6. doi:10.1016/j.celrep.2019.07.033
57. Zou Y, Zheng S, Xie X, et al. Polyunsaturated fatty acids from astrocytes activate PPARγ signaling in cancer cells to promote brain metastasis. Cancer Discov. 2019;9:1720–1735. doi:10.1158/2159-8290.CD-19-0270
58. Priego N, Zhu L, Monteiro C, et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med. 2018;24:1024–1035. doi:10.1038/s41591-018-0044-4
59. Wang L, Cosset E, Kwon J, et al. Targeting the HSP47–collagen axis inhibits brain metastasis by reversing M2 microglial polarization and restoring antitumor immunity. Cell Rep Med. 2024;5:101533. doi:10.1016/j.xcrm.2024.101533
60. Jin Y, Xu Z, Yan Y, et al. Targeting polarized phenotype of microglia via IL6/JAK2/STAT3 signaling reduces non-small-cell lung cancer brain metastasis. Signal Transduct Target Ther. 2022;7:52. doi:10.1038/s41392-022-00872-9
61. Rodriguez-Baena FJ, Garcia-Romero N, Gonzalez-Tejedo C, et al. Microglial reprogramming enhances antitumor immunity and immunotherapy response in melanoma brain metastases. Cancer Cell. 2025;43:413–427.e9. doi:10.1016/j.ccell.2025.01.008
62. Pukrop T, Dehghani F, Chuang HN, et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia. 2010;58:1477–1489. doi:10.1002/glia.21022
63. Haroon E, Miller AH, Sanacora G. Inflammation, glutamate, and glia: a trio of trouble in mood disorders. Neuropsychopharmacology. 2017;42:193–215. doi:10.1038/npp.2016.199
64. Yang G, Li S, Wang Q, et al. Microglia-orchestrated neuroinflammation and synaptic remodeling: roles of pro-inflammatory cytokines and receptors in neurodegeneration. Front Cell Neurosci. 2025;19:1700692. doi:10.3389/fncel.2025.1700692
65. Gabrielli M, Battista N, Riganti L, et al. Microglial large extracellular vesicles propagate early synaptic dysfunction in Alzheimer’s disease. Brain. 2022;145:2849–2868. doi:10.1093/brain/awac083
66. Theparambil SM, Hosford PS, Ruminot I, et al. Adenosine signalling to astrocytes coordinates brain metabolism and function. Nature. 2024;632:139–146. doi:10.1038/s41586-024-07611-w
67. Ruan X, Li Y, Zhang H, et al. Breast cancer cell-secreted miR-199b-5p hijacks neurometabolic coupling to promote brain metastasis. Nat Commun. 2024;15:4549. doi:10.1038/s41467-024-48740-0
68. Chen T, Liu W, Zhang Y, et al. Cellular and molecular mechanisms of the blood–brain barrier dysfunction in neurodegenerative diseases. Fluids Barriers CNS. 2024;21:60. doi:10.1186/s12987-024-00557-1
69. Exadaktylos AK, Buggy DJ, Moriarty DC, et al. Can anesthetic technique for primary breast cancer surgery affect recurrence or metastasis? Anesthesiology. 2006;105:660–664. doi:10.1097/00000542-200610000-00026
70. Singleton PA, Mirzapoiazova T, Hasina R, et al. Increased μ-opioid receptor expression in metastatic lung cancer. Br J Anaesth. 2014;113:i103–i108. doi:10.1093/bja/aeu165
71. Lennon FE, Moss J, Singleton PA, et al. Overexpression of the μ-opioid receptor in human non-small cell lung cancer promotes Akt and mTOR activation, tumor growth, and metastasis. Anesthesiology. 2012;116:857–867. doi:10.1097/ALN.0b013e31824babe2
72. Scroope CA, Singleton Z, Hollmann MW, et al. Opioid receptor-mediated and non-opioid receptor-mediated roles of opioids in tumour growth and metastasis. Front Oncol. 2021;11:792290. doi:10.3389/fonc.2021.792290
73. Maduna T, Audouard E, Dembélé D, et al. Microglia express μ-opioid receptor: insights from transcriptomics and fluorescent reporter mice. Front Psychiatry. 2019;9:726. doi:10.3389/fpsyt.2018.00726
74. Pahan P, Xie JY. Microglial inflammation modulates opioid analgesic tolerance. J Neurosci Res. 2023;101:1383–1392. doi:10.1002/jnr.25199
75. Liu L, Xu Y, Dai H, et al. Dynorphin activation of kappa opioid receptor promotes microglial polarization toward M2 phenotype via the TLR4/NF-κB pathway. Cell Biosci. 2020;10:42. doi:10.1186/s13578-020-00387-2
76. Li Y, Liu J, Chen P, et al. Signaling mechanisms of μ-opioid receptor in the hippocampus: disinhibition versus astrocytic glutamate regulation. Cell Mol Life Sci. 2021;78:415–426. doi:10.1007/s00018-020-03595-8
77. Byrne LS, Peng J, Sarkar S, et al. Interleukin-1 beta-induced up-regulation of opioid receptors in the untreated and morphine-desensitized U87 MG human astrocytoma cells. J Neuroinflammation. 2012;9:252. doi:10.1186/1742-2094-9-252
78. Green JM, Sundman MH, Chou Y, et al. Opioid-induced microglia reactivity modulates opioid reward, analgesia, and behavior. Neurosci Biobehav Rev. 2022;135:104544. doi:10.1016/j.neubiorev.2022.104544
79. Toloff K, Woodcock E. Is the neuroimmune system a therapeutic target for opioid use disorder? A systematic review. Med Res Arch. 2022;10. doi:10.18103/mra.v10i8.2955.
80. Xie F, Liu Y, Chen Z, et al. Morphine induces inflammatory responses via both TLR4 and cGAS–STING signaling pathways. Cytokine. 2024;183:156737. doi:10.1016/j.cyto.2024.156737
81. Xu Y, Wang J, Zhang X, et al. δ-opioid receptor, microglia and neuroinflammation. Aging Dis. 2023;14:778–792. doi:10.14336/AD.2022.0912
82. Kao S-C, Zhao X, Lee CY, et al. Absence of μ-opioid receptor mRNA expression in astrocytes and microglia of rat spinal cord. NeuroReport. 2012;23:378–384. doi:10.1097/WNR.0b013e3283522e1b
83. Barbierato M, Zusso M, Skaper SD, et al. MicroRNAs: emerging role in the endogenous μ-opioid system. CNS Neurol Disord Drug Targets. 2015;14:239–250. doi:10.2174/1871527314666150116123932
84. Smith ACW, Kenny PJ. MicroRNAs regulate synaptic plasticity underlying drug addiction. Genes Brain Behav. 2018;17:e12424. doi:10.1111/gbb.12424
85. Chen Y, Guo L-B, Zan G-Y, et al. Astroglial kappa opioid receptor-mediated reduction of glutamate exchanger xCT in the prelimbic cortex underlies chronic stress-induced depressive-like behaviors. Mol Psychiatry. 2025. doi:10.1038/s41380-025-03441-y
86. Dantzer R. Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol Rev. 2018;98:477–504. doi:10.1152/physrev.00039.2016
87. Rogers TJ, Roy S. Editorial: the role of opioid receptors in immune system function. Front Immunol. 2022;12:832292. doi:10.3389/fimmu.2021.832292
88. Brejchova J, Holan V, Svoboda P. Expression of opioid receptors in cells of the immune system. Int J Mol Sci. 2020;22:315. doi:10.3390/ijms22010315
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.