Back to Archived Journals » Hypoxia » Volume 6
Astrocyte HIF-2α supports learning in a passive avoidance paradigm under hypoxic stress
Authors Leiton CV ![]() , Chen E, Cutrone A
, Chen E, Cutrone A ![]() , Conn K, Mellanson K
, Conn K, Mellanson K ![]() , Malik DM, Klingener M, Lamm R, Cutrone M, Petrie IV J
, Malik DM, Klingener M, Lamm R, Cutrone M, Petrie IV J ![]() , Sheikh J
, Sheikh J ![]() , DiBua A
, DiBua A ![]() , Cohen B, Floyd TF
, Cohen B, Floyd TF ![]()
Received 11 May 2018
Accepted for publication 23 June 2018
Published 8 November 2018 Volume 2018:6 Pages 35—56
DOI https://doi.org/10.2147/HP.S173589
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Cindy V Leiton,1,2 Elyssa Chen,2 Alissa Cutrone,3 Kristy Conn,2 Kennelia Mellanson,4 Dania M Malik,5 Michael Klingener,6 Ryan Lamm,7 Michael Cutrone,8 John Petrie IV,9 Joher Sheikh,10 Adriana DiBua,11 Betsy Cohen,12 Thomas F Floyd13–15
1Department of Pathology, Stony Brook University, Stony Brook, NY, USA; 2Department of Anesthesiology, Stony Brook University, Stony Brook, NY, USA; 3Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, USA; 4Department of Pharmacological Sciences, Stony Brook University, Stony Brook, NY, USA; 5Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Philadelphia, PA, USA; 6Department of Genetics, Stony Brook University, Stony Brook, NY, USA; 7Department of General Surgery, Thomas Jefferson University Hospital, Philadelphia, PA, USA; 8Department of Anesthesiology, Duke University Medical Center, Durham, NC, USA; 9Department of Biochemistry & Molecular Biology, Bloomberg School of Public health, Johns Hopkins University, Baltimore, MD, USA; 10Department of Physiology and Biophysics, Georgetown University, Washington, DC, USA; 11Department of Chemistry, Hofstra University, Hempstead, NY, USA; 12Computer Science Department, Swarthmore College, Swarthmore, PA, USA; 13Department of Anesthesiology and Pain Management, University of Texas Southwestern, Dallas, TX, USA; 14Department of Cardiothoracic Surgery, University of Texas Southwestern, Dallas, TX, USA; 15Department of Radiology, University of Texas Southwestern, Dallas, TX, USA
Background: The brain is extensively vascularized, uses ~20% of the body’s oxygen, and is highly sensitive to changes in oxygen. While synaptic plasticity and memory are impaired in healthy individuals by exposure to mild hypoxia, aged individuals appear to be even more sensitive. Aging is associated with progressive failure in pulmonary and cardiovascular systems, exposing the aged to both chronic and superimposed acute hypoxia. The HIF proteins, the “master regulators” of the cellular response to hypoxia, are robustly expressed in neurons and astrocytes. Astrocytes support neurons and synaptic plasticity via complex metabolic and trophic mechanisms. The activity of HIF proteins in the brain is diminished with aging, and the increased exposure to chronic and acute hypoxia with aging combined with diminished HIF activity may impair synaptic plasticity.
Purpose: Herein, we test the hypothesis that astrocyte HIF supports synaptic plasticity and learning upon hypoxia.
Materials and Methods: An Astrocyte-specific HIF loss-of-function model was employed, where knock-out of HIF-1α or HIF-2α in GFAP expressing cells was accomplished by cre-mediated recombination. Animals were tested for behavioral (open field and rotarod), learning (passive avoidance paradigm), and electrophysiological (long term potentiation) responses to mild hypoxic challenge.
Results: In an astrocyte-specific HIF loss-of-function model followed by mild hypoxia, we identified that the depletion of HIF-2α resulted in an impaired passive avoidance learning performance. This was accompanied by an attenuated response to induction in long-term potentiation (LTP), suggesting that the hippocampal circuitry was perturbed upon hypoxic exposure following HIF-2α loss in astrocytes, and not due to hippocampal cell death. We investigated HIF-regulated trophic and metabolic target genes and found that they were not regulated by HIF-2α, suggesting that these specific targets may not be involved in mediating the phenotypes observed.
Conclusion: Together, these results point to a role for HIF-2α in the astrocyte’s regulatory role in synaptic plasticity and learning under hypoxia and suggest that even mild, acute hypoxic challenges can impair cognitive performance in the aged population who harbor impaired HIF function.
Keywords: astrocyte, hypoxia, HIF, learning, LTP, tripartite synapse, memory, glia, cognitive function
Introduction
Aging can be accompanied by a spectrum of progressive organ system dysfunctions, including heart failure, COPD, anemia, cerebrovascular disease, and others, all of which result in chronic systemic hypoxia.1 These chronic disease states are subject to periods of acute exacerbation that further increase the level of hypoxic stress. Each of these diseases has also been associated with cognitive impairment.2–14 Even in healthy individuals,15,16 exposure to mild–moderate hypoxia or anemic hypoxia17–20 is known to cause cognitive impairment. Evidence suggests that even modest levels of hypoxia may impair brain-derived neurotrophic factor (BDNF) synthesis and synaptic plasticity.21,22 With aging in humans and in rodent models, memory appears to be more greatly impacted by hypoxia.23–28
The HIF transcription system29 is the “master” regulator of the cellular response to hypoxia.30,31 Under normoxia, HIF-α subunits are hydroxylated and degraded. Under hypoxic conditions, HIF degradation is suppressed, resulting in HIF stabilization and accumulation.32 Effective HIF function is required for hypoxic sensing,33 adaptation to hypoxia, and cellular function in all organs including the brain34–36 and in regions such as the hippocampus.30,37 HIF transcriptionally regulates hundreds of hypoxia-responsive genes,38,39 the products of which balance oxygen availability, delivery, and changes in metabolism to adjust for its decrease, all to ultimately result in cell survival.40
Aging is characterized by diminished HIF activity in many organs, including the brain.41–46 Several reports describe altered HIF function with aging that include increased cytoplasmic localization,47 reduced DNA binding,48 and decreased HIF target gene expression.49 Aging coincides with diminished HIF-1α in the rodent brain,42,46,48,50–52 but HIF-2α has received considerably less attention. In the aged murine brain, HIF-1α stabilization is impaired and HIF-2α stabilization is maximally elevated at baseline, unable to increase in response to hypoxic stress.46 In addition, the expression of prolyl hydroxylases critical in regulating HIF-α degradation is abnormal46 and the level of erythropoietin (EPO), a canonical HIF target gene, is markedly diminished at baseline and does not increase under hypoxic stress.53 Finally, vascular endothelial growth factor (VEGF), a major HIF-1α target gene, is induced under hypoxic stress in aged animals to a significantly reduced degree than in young animals.51,53 All of these features highlight molecular signatures of impaired hypoxic response associated with aging. An insidious loss of HIF activity therefore appears to collide with an ever-increasing exposure to chronic and acute hypoxia with aging. While HIF transcriptionally controls thousands of genes involved in the hypoxic response, the role of HIF in learning and synaptic plasticity has received relatively little attention.54–60
Astrocytes are key integrators of synaptic plasticity, performing and regulating processes that support synaptic function and hippocampal-related behavior61 to include modulating ions, signaling proteins and neurotransmitters,62–68 providing metabolic53,56,69 and trophic53,65,70–72 supports, and modulating neurovascular coupling.73–75 Similar processes have been shown to be regulated by HIF proteins in other systems, raising the question of whether HIF modulates these processes required for synaptic functions upon hypoxia in the brain.
Here, we test the hypothesis that astrocyte HIF supports learning under mild–moderate hypoxia, as is frequently seen with chronic and acute exacerbations of chronic disease states that accompany aging.
Materials and methods
Generation of knock-out (KO) mice
All experimental procedures described were approved by the Institutional Animal Care and Use Committee (IACUC) at Stony Brook University and are in accordance with the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health. The generation of astrocyte-selective HIF-1α and HIF-2α KO mice was previously described.71 Mice expressing Cre recombinase directed by the glial fibrillary acidic protein (GFAP) promoter (B6.Cg-Tg(Gfap-Cre)73.12Mvs/J, stock no: 012886)76 and mice homozygous of loxP-flanked HIF-1α (B6.129-Hif1αtm3Rsjo/J, stock no: 007561)77 and HIF-2α (also known as endothelial PAS domain-containing protein 1 [EPAS1]) (STOCK Epas1tm1Mcs/J, stock no: 008407)70 alleles were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). To generate astrocyte-selective HIF KOs, GFAP-Cre Transgenic (Tg) mice were first bred to HIFFl/Fl mice. The obtained GFAP-Cre Tg/HIFFl/WT mice were further crossed with mice carrying the same floxed HIF alleles (HIF-1αFl/Fl or HIF-2αFl/Fl) to finally achieve the complete deletion of both HIF alleles, resulting in GFAP-Cre Tg/HIF-1αFl/Fl (referred to as GFAP-Cre+/HIF-1αFl/Fl) and GFAP-Cre Tg/HIF-2αFl/Fl (referred to as GFAP-Cre+/HIF-2αFl/Fl). Wild type (WT) (C57BL6J, stock no: 000664) was also purchased from the Jackson Laboratory and bred as control and is referred to as C57.
Genotyping
Genomic DNA was isolated from tail biopsies collected at 21 days of age using the DNeasy genomic DNA isolation kit (Qiagen NV, Venlo, the Netherlands) following the procedure described by the manufacturer. The following primers were used in PCRs to detect the Cre transgene, 5′-GCG GTCTGGCAGTAAAAACTATC-3′ and 5′-GTGAAA CAGCATTGCTGTCACTT-3′; internal positive control, 5′-CTAGGCCACAGAATTGAAAGATCT-3′ and 5′-GTAG GTGGAAATTCTAGCATCATCC-3′; HIF-1α wild-type or floxed allele, 5′-TGCATGTGTATGGGTGTTTTG-3′ and 5′-GAAAACTGTCTGTAACTTCATTTCC-3′; and HIF-2α wild-type or floxed allele, 5′-GAGAGCAGCTTCTCCTG GAA-3′ and 5′-TGTAGGCAAGGAAACCAAGG-3′. The PCR products were visualized on a 3% agarose gel containing 0.01% ethidium bromide. The expected PCR product sizes are as follows: Cre Tg, 100 bp; internal positive control, 324 bp; HIF-1α wild type, 100 bp; HIF-1α-floxed mutant, 120 bp; HIF-2α wild type, 182 bp; and HIF-2α-floxed mutant, 220 bp.
Hypoxic exposure
The exposure to 12% O2 was carefully chosen. In adult rats, 12% O2 results in a pO2 ≈ 62 Torr.78 Assuming a normal alveolar-arterial (A-a) gradient, using the alveolar gas equation (PaO2 = FIO2 [PB-47] -1.2[PaCO2]), 12% O2 should yield an arterial pO2 ≈ 40 mmHg, which is equivalent to hemoglogin saturation (Hgb-SaO2) in the range of 80–85%. Bell et al,79 in healthy volunteers, demonstrated that exposure to 12% O2 yields Hgb-SaO2 in the range of 85–93%. Individuals with systemic diseases accompanied by hypoxia frequently experience this level of hypoxia chronically, intermittently, and acutely.80–83 This mild–moderate level of hypoxia elicits cognitive failure even in healthy young individuals.17,84 Finally, exposure to 12% O2, for a period of 6 hours, produces a robust increase in HIF expression in brain tissue.85–87
Morphology and hematocrit measurements
Body and wet organ weights were measured in male animals at 3–6 months of age immediately after euthanasia. Hematocrit was measured in males of the same age from blood samples taken immediately following euthanasia. Blood was collected in EDTA vacutainers, where blood was drawn into a standard hematocrit capillary tube, sealed with clay, and centrifuged.
Behavioral phenotyping
All protocols were performed in accordance with University Laboratory Animal Resources guidelines and were approved by the IACUC. Mice were housed in a 12-hour light–dark cycled, temperature-controlled room, with freely available food and water. Experiments were conducted during the light phase between 8 am and 6 pm. Male mice, aged 3–4 months, in five groups, such as C57B6J, GFAP-Cre-/HIF-1αFl/Fl, GFAP-Cre+/HIF-1αFl/Fl, GFAP-Cre-/HIF-2αFl/Fl, and GFAP-Cre+/HIF-2αFl/Fl, underwent the following behavioral analyses. Evaluators were blinded to group during testing.
Open-field (OF) testing
OF measures baseline exploratory behavior and general activity. The OF apparatus is constructed of gray plastic (45 cm ×45 cm ×40 cm; Coulbourn Instruments, Whitehall, PA, USA). A personal compurter-linked overhead video camera (LifeCam Studio™ Webcam; Microsoft Corporation, Redmond, WA, USA) positioned above the apparatus recorded each 10-minute trial. Total path distance, average speed, center, middle, and outer time were analyzed using the HVS Image software (Mountain View, CA, USA).
Rotarod (RR) testing
The RR apparatus tests motor function and coordination and may offer insight into motor activity/coordination and learning, as animals can improve their ability to stay on the rotating drum with repeated trials. The apparatus comprises a rotating drum (Harvard Apparatus, Holliston, MA, USA), which is accelerated from 4 to 40 revolutions per minute over the course of 10 minutes. Each mouse was placed individually on a drum, and the time to falling from the drum was recorded as latency (seconds). Three trials were conducted per day, for 3 consecutive days.
Passive avoidance (PA) learning paradigm
Learning was examined using a PA fear motivated paradigm, which requires activation of the hippocampus, as well as the amygdala and perirhinal cortex.88 The apparatus used for this test (Ugo Basile SRL, Varese, Italy) employs a standard light and dark chamber. Because mice prefer the dark, they move into the dark box when placed in the light box. When the animal moves into the dark side of the box, a foot shock (0.3 mA × 3 seconds) was delivered. Latency is recorded automatically.
The PA paradigm tests contextual fear and learning that associates with place and the shock. This paradigm creates a robust memory trace in a single training evolution. Animals were moved to laboratory 30 minutes prior to experimentation. On day 1, animals underwent a habituation trial in the PA apparatus during which they were allowed to freely move between the light and dark sides of the box. On day 2, they were again placed in the light side of the box and the time it took for them to enter the dark side was measured. On entry into the dark side, animals received a 3 second, 0.3 mA foot shock. Immediately following the foot shock, animals were returned to their cages and placed within either a room air (21% O2) or a 12% O2 environment in the BioSpherix (Redfield, NY, USA) chamber, for a period of 6 hours. After exposure to air or hypoxia, all animals recovered at 21% O2 until testing on day 3. On day 3 (24 hours after the foot shock or 18 hours after recovery from the air or hypoxia exposures), animals were tested for recall. Animals were again placed in the light side of the box, and the time to enter the dark side of the box was measured. A limit upon time to enter the dark side was placed at 300 seconds.
Field recordings for long-term potentiation (LTP)
Hippocampal slice preparation
Acute hippocampal slices were prepared from 3-month-old male mice. To activate HIF and the expression of related proteins, C57B6J, GFAP-Cre+/HIF-1αFl/Fl, and GFAP-Cre+/HIF-2αFl/Fl animals were exposed to 6 hours of 12% O2 or air (21% O2). Mice were then deeply anesthetized with isoflurane before decapitation. The brain was quickly removed and immersed in ice-cold artificial cerebrospinal fluid (ACSF). Transverse hippocampal slices (400 µm) were incubated at 32°C and continually perfused with oxygenated ACSF containing 124 mM NaCl, 2.5 mM KCl, 2.0 mM MgSO4, 2.0 mM CaCl2, 26.0 mM NaHCO3, 1.25 mM NaH2PO4, and 10 mM glucose. Slices were equilibrated for 1 hour before recording.
Data acquisition
Postsynaptic potentials were evoked by stimulating the Schaffer collateral pathway and recorded from the CA1 region of the hippocampus. Monopolar electrodes were used for stimulation. Single 0.1 ms square wave pulses were delivered through constant current isolation units (A385; WPI, Sarasota, FL, USA) at a frequency of 0.05 Hz. Field excitatory postsynaptic potentials (fEPSPs) in hippocampal CA1 stratum radiatum were recorded with ACSF-filled glass electrodes. Postsynaptic responses were amplified by an amplifier (Axoclamp 900A; Molecular Devices LLC, Sunnyvale, CA, USA). Theta-burst stimulation of three trains (10 seconds inter-train interval), each consisting of 15 (200 ms interstimulus interval [ISI]) ×4 pulses (100 Hz), was used to induce LTP. Data were digitized by a Digidata 1440 module (Molecular Devices LLC).
Data analysis
Initial analysis of LTP data was conducted in Clampfit 10.4 (Molecular Devices LLC). The slope of fEPSP (20–80% level of the falling phase) was measured. Experiments were excluded from the experimental data set if: 1) the baseline was unstable (variability >±20%), 2) the stimulation did not result in ≥ 20% increase in fEPSP slope, lasting for at least 60 minutes, and free of drift ≥ 20%, or 3) the fEPSP amplitude was <1 mV.
Primary mouse astrocyte cultures
Neonatal mice (postnatal days 1–3) were decapitated, cortices were dissected, and meninges were removed. Cortices were then minced and dissociated in papain and plated on poly-d-lysine (PDL)-coated flasks in DMEM supplemented with 10% FBS and penicillin–streptomycin (regular growth media). After three media changes every 3 days, oligodendrocytes and microglia were eliminated via mechanical agitation as previously described.89 Astrocytes were then trypsinized and plated in PDL-coated tissue culture dishes for experiments in regular growth media. Cells were maintained at 37°C in a 7.5% CO2, 95% air incubator (20% O2). Hypoxia was controlled using the ProOx 110 tissue culture system (BioSpherix).
Protein lysate preparation and immunoblotting
Immediately after exposure, cells were quickly washed in ice-cold PBS and lysed in boil buffer (20 mM Tris, pH 7.4, 1% sodium dodecyl sulfate, protease and phosphatase inhibitors; EMD Millipore, Billerica, MA, USA). Boil buffer was heated to 95°C and added immediately to cells. Scraped cells in buffer were placed in a 1.5 mL microcentrifuge tube and heated for 15 minutes at 95°C. A sample aliquot was isolated for protein concentration measurement using the Bradford protein assay (Bio-Rad Laboratories Inc., Hercules, CA, USA). Eight percent SDS-PAGE gels were used to resolve HIF-1α and HIF-2α proteins, and membranes were incubated with either anti-HIF-1α or anti-HIF-2α antibodies (AF1935 or AF2997; R&D Systems, Inc., Minneapolis, MN, USA), at 1:500, overnight at 4°C. The loading control antibody, anti-p115 (cat no: 612261 at 1:1000; BD Biosciences, San Jose, CA, USA), was coincubated with anti-HIF antibody. Antibodies detecting Cre recombinase were purchased (cat no: 69050; EMD Millipore) and used at 1:1000, while GFAP (cat no: Z0334; Dako Denmark A/S, Glostrup, Denmark) was also used at 1:1000. Enhanced chemiluminescence reagents (PN: 826-13460; LI-COR Biosciences, Lincoln, NE, USA) were used as a detection system. The blot was visualized using a C-DiGit imaging system (LI-COR Biosciences). Densitometry was measured using the Image Studio™ software (LI-COR Biosciences).
qRT-PCR
Immediately after normoxic or hypoxic exposure, cells were lysed with 350 mL of the PureLink RNA Mini Kit lysis buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with β-mercaptoethanol and scraped and collected into 1.5 mL tubes. Samples were frozen at -80°C until they were processed for RNA extraction using the manufacturer’s protocol, in addition to DNase I treatment for the removal of genomic DNA contamination. The SuperScript III First-Strand cDNA Synthesis Kit was then used to synthesize cDNA, of which 15 ng was loaded into reactions using TaqMan assay probes and Fast TaqMan Master Mix. All targets were normalized to 18S rRNA. TaqMan probes were as follows: glucose transporter 1 (Glut-1), Mm00441480_m1; pyruvate dehydrogenase kinase-1 (PDK-1), Mm00554300_m1; lactate dehydrogenase A (LDHA), Mm01612132_g1; monocarboxylate transporter 1 (MCT-4), Mm00525195_m1; EPO, Mm00433126_m1; and 18S, Mm03928990_g1.
DNA recombination
DNA from brain, liver, and kidney from C57B6J, GFAP-Cre-negative and -positive HIF-1α, and HIF-2α-floxed animals was extracted with the Qiagen DNeasy kit according to the manufacturer’s protocol. A total of 25 ng of genomic DNA was amplified by multiplex PCR with primers to distinguish the WT-, 2-loxP-, and Cre-mediated recombination-generated 1-loxP alleles. Primer sequences are as follows: HIF-1α F1, 5′-TTGGGGATGAAAACATCTGC-3′; F2, 5′-GCAGTTAAGAGCACTAGTTG-3′; and R, 5′-GGAGC TATCTCTCTAGACC-3′. The product sizes are as follows: HIF-1α 1-lox allele, 270 bp; 2-lox allele, 260 bp; and WT allele, 240 bp. Primer sequences are as follows: HIF-2α F1, 5′-CAGGCAGTATGCCGCTAATTCCAGTT-3′; F2, 5′-CTTCTTCCATCATGGGATCTGGGACT-3′; and R, 5′-GCTAACACTGTACAAAGAGTAGC-3′. The product sizes are as follows: HIF-2α 1-lox allele, 360 bp; 2-lox allele, 460 bp; and WT allele, 430 bp.
Immunofluorescence
Mice were transcardially perfused with PBS followed by 10% PBS-buffered formalin (Sigma-Aldrich Co., St Louis, MO, USA), and the brain was embedded in paraffin following standard procedure. For immunofluorescent staining, 5 µm sections were rehydrated in an ethanol series and subjected to microwave antigen retrieval with citrate buffer (HK087-20K; BioGenex, Fremont, CA, USA). Sections were blocked for nonspecific binding with 10% normal goat serum (Thermo Fisher Scientific) at room temperature (RT) for 1 hour before undergoing incubation with primary antibodies. Rabbit anti-Cre (1:100 in PBS with 3% BSA; cat no: PRB-106P; Covance, Princeton, NJ, USA) and mouse anti-GFAP monoclonal (1:800, cat no: MAB360; EMD Millipore) antibodies were used to detect Cre recombinase and endogenous GFAP, respectively. Primary antibody incubation was carried out overnight at 4°C. After washing with Tris Buffered Saline-Tween 20 (TBST), sections were incubated with secondary Alexa Fluor 488-conjugated goat antirabbit and Alexa Fluor 555 goat antimouse antibodies (both from Thermo Fisher Scientific) for 1 hour at RT in the dark. Nuclei were counterstained with DAPI (Sigma-Aldrich Co.). Confocal fluorescent images were obtained by a Zeiss LSM 510 NLO confocal scan head mounted on a Zeiss Axiovert 200M on an inverted-based microscope.
Apoptosis analysis
After the 6-hour hypoxic exposure, a subset of n=3 animals were either immediately perfused or returned to room air for 18 hours before undergoing perfusion. Animals were perfused with PBS followed by 10% buffered formalin. After the brains were removed, they were postfixed for 24 hours in 10% buffered formalin before being switched to 70% ethanol. Brains were then grossly prepped, processed, and paraffin-embedded. Five-micron thick sections were cut upon identification of the hippocampus and then affixed onto slides. The slides were then processed for immunohistochemistry where the slides were de-paraffinized in xylene and rehydrated in ethanol. The tissues were then immersed in a Coplin jar with citrate buffer and pressure cooked at 120°C for 10 minutes for antigen retrieval. The tissues were then washed with water and treated with 1% hydrogen peroxide for 5 minutes to block endogenous peroxidase signal. Subsequently, the tissues were blocked with 5% horse serum for 5 minutes and then overlaid with anti-CC3 antibody (#9661; Cell Signaling, Danvers, MA, USA) at 1:3000 in blocking solution (Dako Denmark A/S) overnight in a humid chamber at 4°C. The next day, the slides were washed in TBST and incubated with biotinylated antirabbit secondary at 1:3000 (Vector Labs, Burlingame, CA, USA) for 30 minutes at RT in blocking solution, followed with avidin–biotin complex for 30 minutes at RT. The tissues were then stained with light hematoxylin for 10 minutes and washed and cover slipped with mounting media. Images from the slides were taken at 5× using a true color camera on a Leica microscope. Representative images are insets cropped from original 5× images from one of the three animals analyzed per condition. Positive control tissue was derived from a postnatal day 8 pup injected with 10% ethanol and treated for 8 hours as per Olney et al.90
Statistical analysis
Sample sizes were estimated in the following manner. Although considered an excellent screening test for learning performance, PA testing is reported to be associated with a considerable variance in latency.91 Early trials in our laboratory indeed confirmed this variability. We therefore established our sample size for this measure, based on having sufficient power to detect a difference (D) between air and hypoxic exposures in any one genotype, where the standard deviation (s) was 1.5 (D). Using α=0.05, 80% power and a two-way ANOVA, we would require n = 3 mice/group. Lacking empirical data for the LTP studies, we estimated the range of sample sizes required to detect a difference (D), where s=0.5–1.0 (D), using α=0.05, and a two-way ANOVA. Sample sizes 6–17/group would be required to obtain the power of 0.80. We therefore settled on sample sizes 8–12/group for these studies. For all other supportive studies, sample sizes 6–8/group were empirically chosen, assuming s=0.5 (D), offering a power of 0.8–1.0, with P=0.05.
Parametric data were analyzed using the unpaired, two-tailed Student’s t-test. Nonparametric data were analyzed by Wilcoxon rank-sum test. Analysis of PA testing requires special treatment, as in this test, a ceiling of 300 seconds was used. For data with a ceiling or cutoff such as this, the most appropriate approach employs multivariable Cox proportional hazard modeling.92 A multivariable Cox proportional hazard model was constructed to test the impact of exposure (21% O2 and 12% O2) within each genotype. Within genotype, hazard ratios were calculated, with P<0.05 considered statistically significant. Finally, multivariable regression was employed to test group differences in the rate of learning on the RR apparatus (ie, dependency of speed and latency upon the variable groups [C57B6J, GFAP-Cre-/HIF-1αFl/Fl or GFAP-Cre+/HIF-1αFl/Fl, and GFAP-Cre-/HIF-2αFl/Fl or GFAP-Cre+/HIF-2αFl/Fl], days 1–3, and the group–day interaction). All analyses were performed using JMP® 9.02 (SAS Institute Inc., Cary, NC, USA).
Results
Generation of KOs
We generated astrocyte HIF-1α- and HIF-2α-floxed or KO mice using a GFAP promoter-driven Cre recombination from commercially available mice (maintained on a C57B6/J background; used as a background control and referred to as C57). Mice expressing GFAP promoter-driven Cre recombinase were crossed with loxP-flanked HIF-1α or HIF-2α allele carrying animals, yielding HIF-1α or HIF-2α deletion to generate either GFAP-Cre-/HIF-1αFl/Fl or GFAP-Cre+/HIF-1αFl/Fl and GFAP-Cre-/HIF-2αFl/Fl or GFAP-Cre+/HIF-2αFl/Fl mice, of which the GFAP-Cre- animals constituted the littermate, nonrecombined, floxed controls and the GFAP-Cre+ constituted the KO animals in GFAP-expressing cells. DNA recombination analysis confirmed the brain-specific excision of the HIF-1α and HIF-2α alleles (Figure 1A). We further detected Cre recombinase expression in GFAP-Cre+/HIF-1αFl/Fl and GFAP-Cre+/HIF-2αFl/Fl but not in GFAP-Cre-/HIF-1αFl/Fl and GFAP-Cre-/HIF-2αFl/Fl littermate controls (Figure 1B). Acute chronic hypoxic challenge (0.1% O2 for 6 hours) of primary cultured cortical astrocytes from each genotype demonstrated near complete HIF reduction in astrocytes isolated from both GFAP-Cre+/HIF-1αFl/Fl and GFAP-Cre+/HIF-2αFl/Fl, as little to no HIF-1α or HIF-2α protein was observed upon hypoxic exposure, respectively, and no compensation or off-target effects on the isoform not targeted for deletion was observed (Figure 1C). These results provide evidence that the HIF-1α and HIF-2α are adequately depleted from GFAP-expressing cells in the brain.
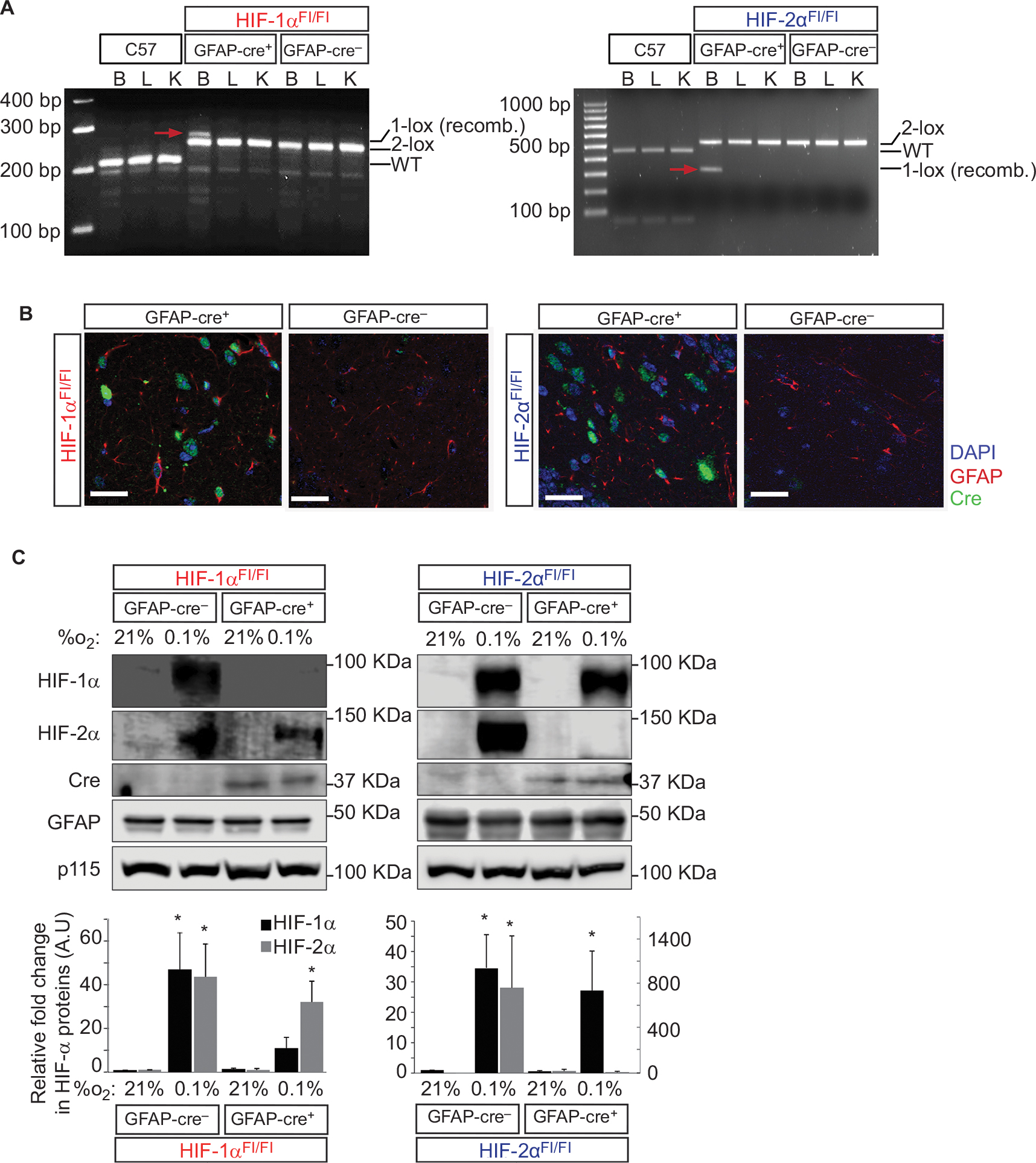 | Figure 1 Characterization of HIF-1α and HIF-2α knock-out in GFAP-expressing cells. Notes: (A) DNA recombination assays from brain, liver, and kidney tissues show organ specific, HIF-allele recombination of each genotype. HIF-1α Cre-mediated recombination generates 1-loxP allele of 270 bp. The nonrecombined 2-lox allele is 260 bp, and the WT allele is 240 bp. Product size for the HIF-2α-recombined 1-lox allele is 360 bp, that for the nonrecombined 2-lox allele is 460 bp, and that for the WT allele is 430 bp. Red arrows indicate the recombined bands that are only observed in B (brain) tissues from Cre-expressing animals. (B) Cre-recombinase (green) expression is visible only in Cre-expressing knock-out animals in tissue samples of the hippocampus. Scale bar =20 µm. GFAP = red, DAPI = blue. (C) Characterization of primary mouse cortical astrocytes isolated from neonatal cerebral cortices from either GFAP-Cre-negative or GFAP-Cre-positive HIF-1αFl/Fl or HIF-2αFl/Fl animals. Astrocytes were exposed to 21% or 0.1% O2 for 6 hours and lysed and assessed for the expression of HIF-1α, HIF-2α, Cre recombinase (Cre), GFAP, and p115 proteins. GFAP was detected as a marker for astrocytes, and p115, an endoplasmic reticulum-expressing protein, was used as a loading control. Quantitative analysis of HIF-1α and HIF-2α proteins was normalized to p115-loading control and plotted as a fold change relative to the respective Cre-negative control at 21% O2 (mean ± SE, n=6–8/group). *P<0.05. All statistical analyses were carried out in comparison to the respective GFAP-Cre- control at 21% O2. Abbreviations: AU, arbitrary units; B, brain; bp, base pairs; GFAP, glial fibrillary acidic protein; K, kidney; L, liver; WT, wild type. |
Morphologic phenotyping
Morphologic phenotyping for all strains revealed that gross appearance was similar (Figure 2A). Moreover, body (n=29–43/group) and organ (n=8–24/group) weights were comparable (Figure 2B). The single exception is the increased spleen weight in GFAP-Cre-/HIF-2αFl/Fl animals, when compared with the GFAP-Cre+/HIF-2αFl/Fl littermates and C57. There was no significant difference in spleen weight between GFAP-Cre+/HIF-2αFl/Fl and C57. Hematocrit was modestly lower in GFAP-Cre+/HIF-2αFl/Fl and GFAP-Cre-/HIF-2αFl/Fl than C57. Hematocrit also showed a modest but significant difference between GFAP-Cre+/HIF-2αFl/Fl and GFAP-Cre-/HIF-2αFl/Fl mice. These results are supported by previous work where no changes were found between C57 and GFAP-Cre-/HIF-1αFl/Fl and GFAP-Cre-/HIF-2αFl/Fl animals.71
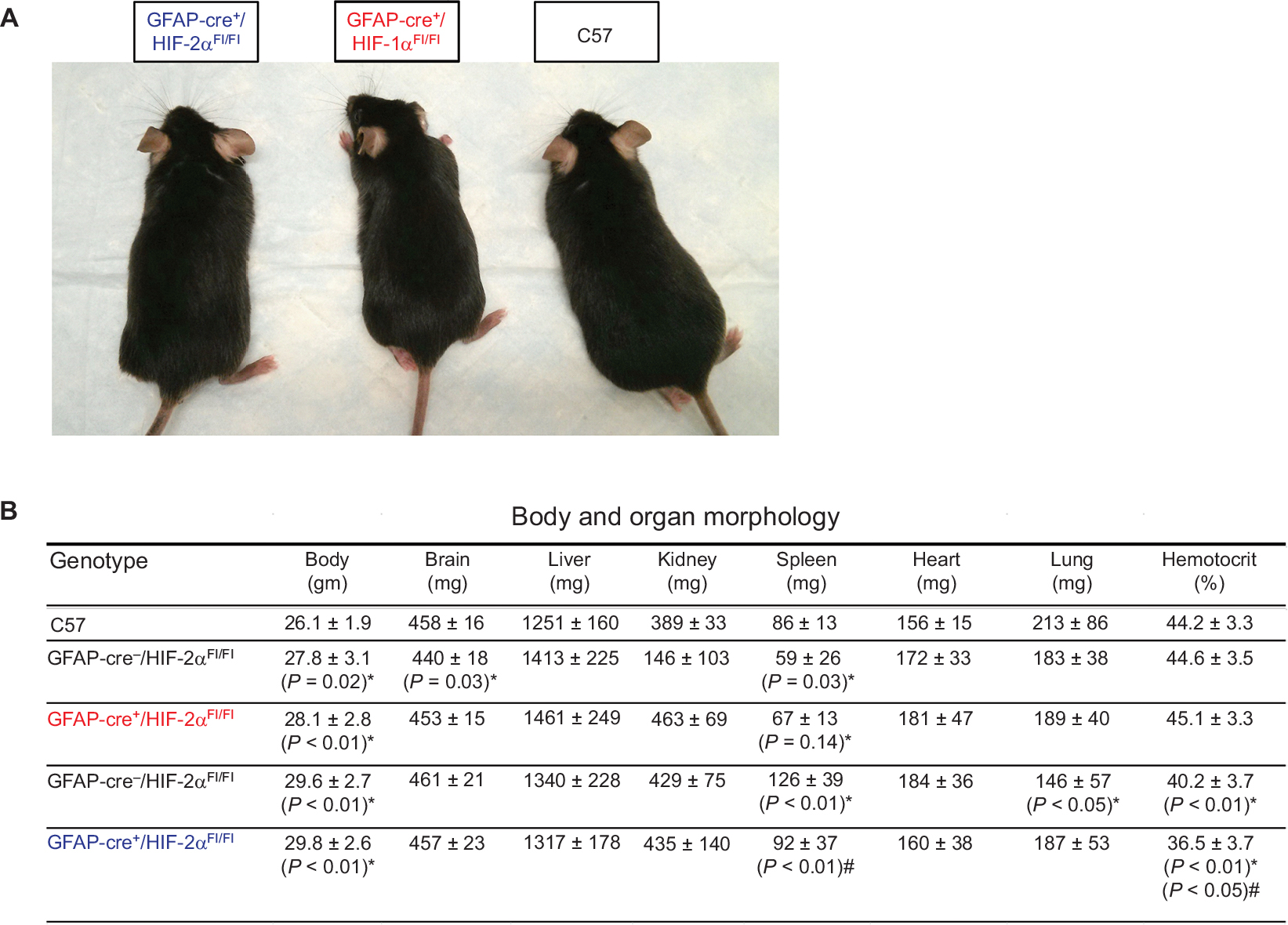 | Figure 2 HIF-1α and HIF-2α knock-out animals show similar somatic physiological characteristics to C57 animals. Notes: (A) Gross appearance of C57, GFAP-Cre+/HIF-1αFl/Fl, and GFAP-Cre+/HIF-2αFl/Fl males at the age of 3–6 months show no overt differences. (B) Body and organ morphology measurements for all genotypes: C57, GFAP-Cre-/HIF-1αFl/Fl, GFAP-Cre+/HIF-1αFl/Fl, GFAP-Cre-/HIF-2αFl/Fl, and GFAP-Cre+/HIF-2αFl/Fl. Body, brain, liver, kidney, spleen, heart, and lung weights are depicted in addition to hematocrit. *Compared to C57; #significance compared to GFAP-Cre-/HIF-αFl/Fl counterpart (n=8–24/group). Data are presented as mean ± SD. Abbreviation: GFAP, glial fibrillary acidic protein. |
Behavioral phenotyping
In addition to functioning during pathological hypoxia, HIF1/2 has been described to regulate embryonic development,27 as well as neural stem cell survival and differentiation93,94 during development. To determine if HIF-1α or HIF-2α KOs resulted in any neurodevelopmental delay that may affect performance on behavioral tests, we performed behavioral phenotyping at baseline (no prior shock or hypoxic stimuli) by measuring performance in the following three tests: OF, RR, and PA.
The OF apparatus is best known as a test of general activity measured by total path, locomotor activity measured by speed, and anxiety measured by time in center/time near wall (outer zone). OF total path length and speed (Figure 3A and B) differed significantly among GFAP-Cre-/HIF-1αFl/Fl, GFAP-Cre+/HIF-1αFl/Fl, GFAP-Cre-/HIF-2αFl/Fl, and GFAP-Cre+/HIF-2αFl/Fl when compared with C57. We compared the performance of GFAP-Cre-negative vs -positive HIF-1αFl/Fl, as well as GFAP-Cre-negative vs -positive HIF-2αFl/Fl (termed “within-group” comparisons), and found no significant differences in total path length, speed, percentage of time in center middle and outer zones when tested within groups (Figure 3A–D). Nor were there any significant differences in any of these measurements between HIF-1αFl/Fl or HIF-2αFl/Fl KOs. OF analyses further revealed no significant differences between any group in the percentage of time spent in the center middle or outer zone (Figure 3C and D). Together, these results show that GFAP-Cre+/HIF-1αFl/Fl and GFAP-Cre+/HIF-2αFl/Fl perform at lower speeds than C57 (Figure 3B), resulting in a reduced path length (Figure 3A), but demonstrate no difference in anxiety levels, as assessed by the percentage of time spent in the center and outer zones (Figure 3C and D).
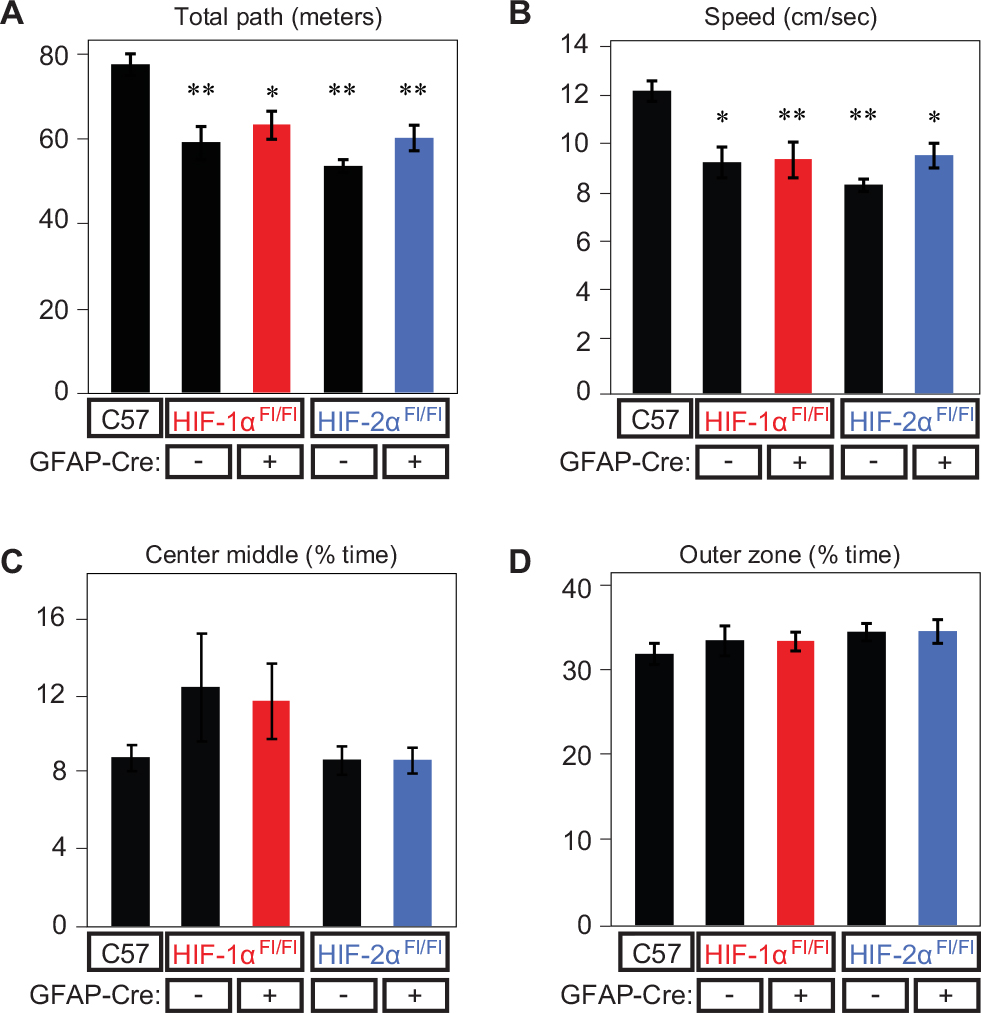 | Figure 3 Open-field performance at baseline. Notes: Open-field performance at baseline (21% O2 – room air) for all genotypes as measured by (A) total path length (m), (B) speed (cm/s), (C) center middle (% time), and (D) outer zone (% time). *P<0.01 and **P<0.001 (compared to WT). Abbreviations: GFAP, glial fibrillary acidic protein; WT, wild type. |
To extend these findings to a second assessment, we used the RR apparatus, which tests for challenged motor activity, learning, and fitness (see “Materials and methods” section). While latency differed significantly between all HIF-Cre-negative controls and HIF KOs when compared with C57 (Figure 4A), there were no significant differences when tested within groups. Furthermore, there were no significant differences in latency between GFAP-Cre+/HIF-1αFl/Fl and GFAP-Cre+/HIF-2αFl/Fl mice. RR measurements also revealed no within- or between-group differences in maximal rotational speed on days 1 and 3 of testing (Figure 4B). Only GFAP-Cre+/HIF-1αFl/Fl demonstrated a difference in speed from C57 and to its Cre-negative control, which occurred on day 2 only.
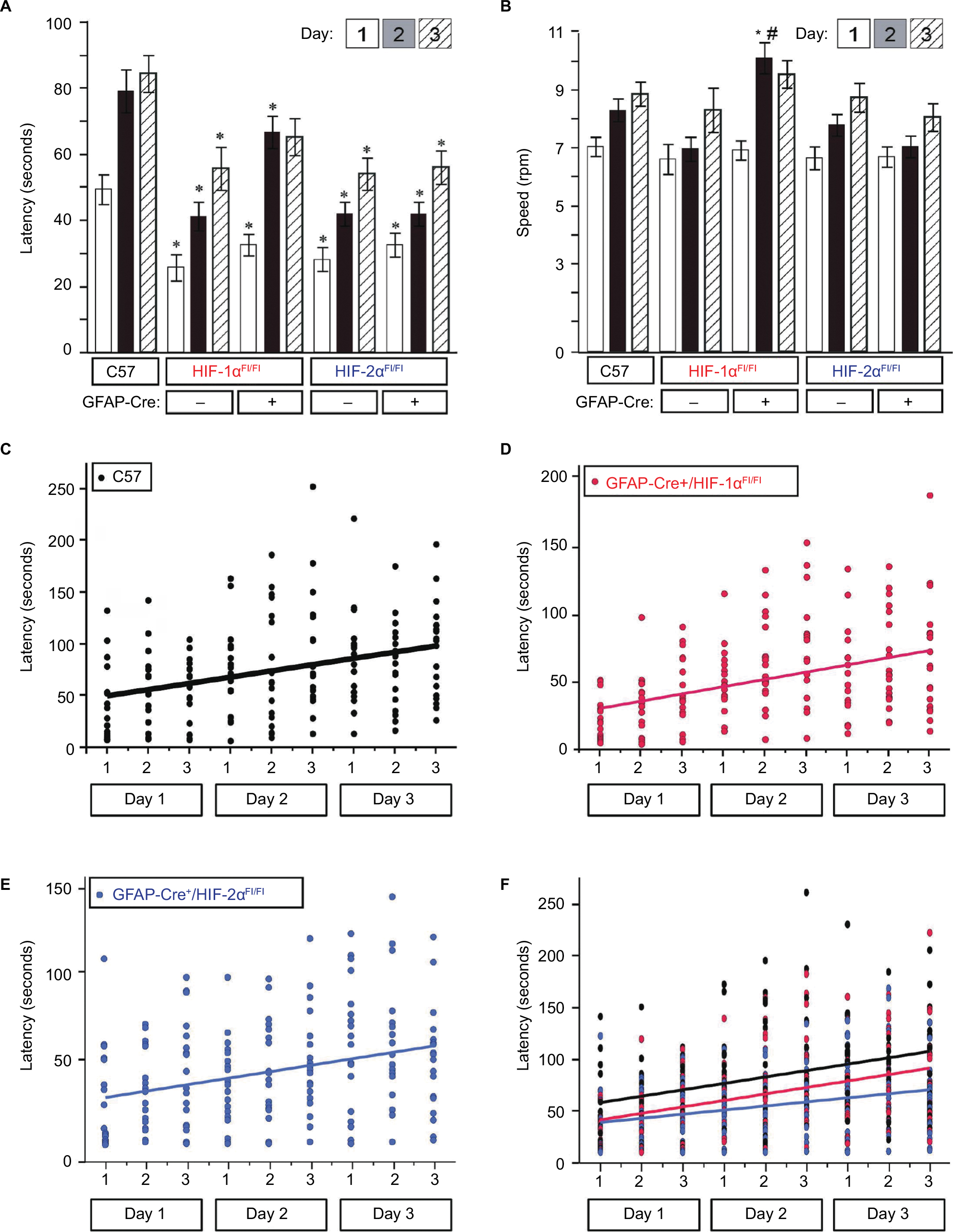 | Figure 4 Rotarod performance at baseline. Notes: Rotarod performance was measured daily, for 3 consecutive days, with three trials per day. (A) Mean latency by group for all genotypes. (B) Mean speed by group (*P<0.05 and #P<0.01 in comparison to C57) for all genotypes. Slopes depicting change in latency by day and trial for (C) C57, (D) GFAP-Cre+/HIF-1αFl/Fl, and (E) GFAP-Cre+/HIF-2αFl/Fl. (F) Superimposed slopes of change in latency for all groups. No differences between genotype groups were identified (GFAP-Cre+/HIF-1αFl/Fl vs C57, P=0.80; GFAP-Cre+/HIF-2αFl/Fl vs C57, P=0.22; GFAP-Cre+/HIF-1αFl/Fl vs GFAP-Cre+/HIF-2αFl/Fl, P=0.31). Abbreviation: GFAP, glial fibrillary acidic protein. |
Analysis of improvement in peak latency from days 1 to 3 on the RR apparatus revealed no differences in the rate of learning between groups, as depicted by the nearly parallel slopes of all groups (Figure 4C–F) and statistically tested via comparison of the group–day interaction effect between groups (GFAP-Cre+/HIF-1αFl/Fl vs C57, P=0.80; GFAP-Cre+/HIF-2αFl/Fl vs C57, P=0.22; GFAP-Cre+/HIF-1αFl/Fl vs GFAP-Cre+/HIF-2αFl/Fl, P=0.31). However, there was a significant diminished peak latency (time to fall from rotating drum on day 3) in GFAP-Cre+/HIF-2αFl/Fl (P=0.002, Figure 4A, indicated by *) but not in GFAP-Cre+/HIF-1αFl/Fl (P=0.19) when compared with C57. There was no significant difference between GFAP-Cre+/HIF-2αFl/Fl and GFAP-Cre+/HIF-1αFl/Fl in peak latency on day 3 (P=0.16) nor peak speed (P=0.051, Figure 4A and B). In general, GFAP-Cre+/HIF-2αFl/Fl performed at significantly reduced latencies and speeds relative to C57 throughout days 1–3 of testing and relative to GFAP-Cre+/HIF-1αFl/Fl on day 2. Taken together, these results suggest that HIF-1α and HIF-2α KOs are similar in mobility and have similar motor ability as indicated by RR performance.
We next assessed learning ability using the PA apparatus, a chamber divided into light (illuminated) and dark (nonilluminated) compartments. A single animal placed into the light compartment will normally follow its instinct to enter the dark compartment and, where upon an aversive stimulus (foot shock), will learn to avoid the dark chamber despite the habitual tendency (Figure 5A). We tested all animal groups at baseline and found no significant difference in latency between GFAP-Cre-/HIF-1αFl/Fl and GFAP-Cre+/HIF-1αFl/Fl when compared with C57 (Figure 5B). Latency was diminished in GFAP-Cre-/HIF-2αFl/Fl and GFAP-Cre+/HIF-2αFl/Fl compared to C57 (Figure 5B); however, performance within the two strains was not significant. Thus, there was no evidence that HIF-1α or HIF-2α KO in astrocytes resulted in a significant neurodevelopmental delay in any of the KOs when compared with respective GFAP-Cre-negative littermates in the tests we performed. With this information in hand, we consolidated our studies to C57, GFAP-Cre+/HIF-1αFl/Fl, and GFAP-Cre+/HIF-2αFl/Fl and conducted between-group analyses, as we observed that the HIF-α KOs performed equally well as control littermates at baseline, indicating that the absence of each HIF-α isoform does not explain the control and KO differences observed relative to C57. With the knowledge of these between-group differences in performance at baseline, we then also performed within-group analysis of the impact of hypoxia on the consolidated groups.
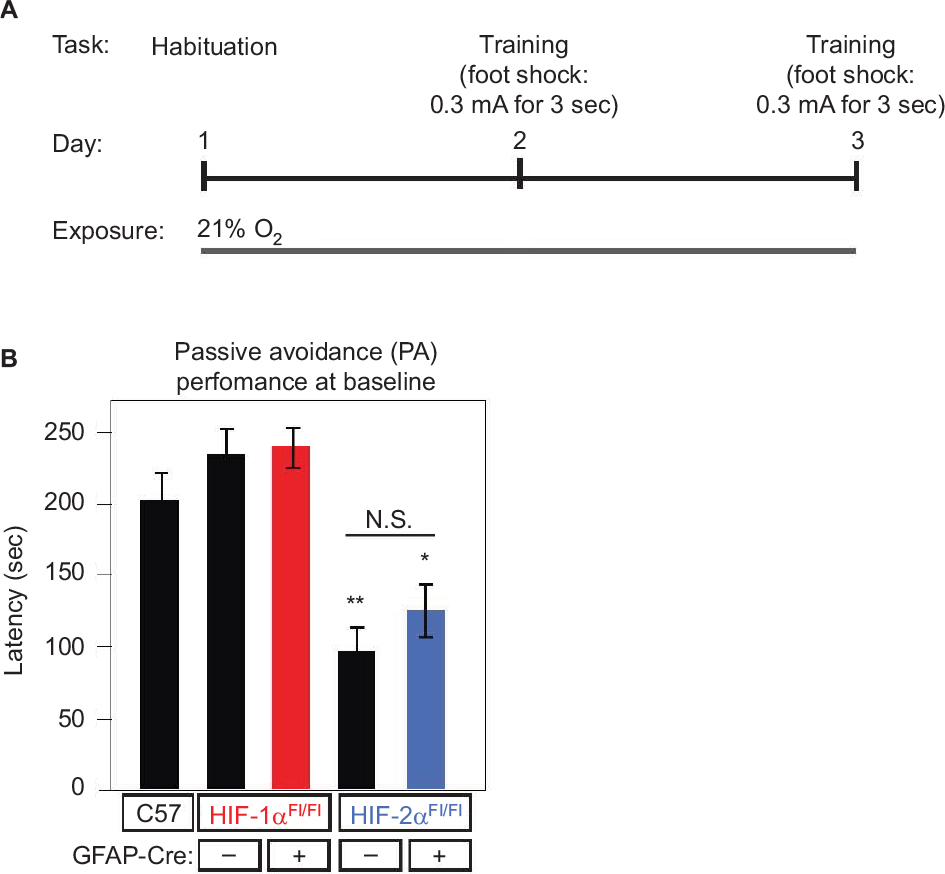 | Figure 5 Baseline learning performance on the PA learning paradigm. Notes: The PA test consists of a light and dark chamber. Animals are placed into the chamber in the light side but quickly move into the dark side due to habitual preference for the dark. (A) Schematic representation depicting the protocol, where on day 1, animals are allowed to roam freely in the dark and light sides to habituate. On training day (day 2), they were placed in the light side, and when they enter the dark side, they are given a foot shock. On testing day (day 3), testing is done where the time to enter the dark side is calculated as latency. Animals were exposed to normoxic conditions (21% O2) at all times. (B) Latency during the PA paradigm is plotted for the five groups. Differences in performance between GFAP-Cre-/HIF-1αFl/Fl, GFAP-Cre+/HIF-1αFl/Fl, GFAP-Cre-/HIF-2αFl/Fl, GFAP-Cre+/HIF-2αFl/Fl, and C57, all under air, are indicated as follows: *P<0.05 and **P<0.01. There was NS within-group differences in performance in GFAP-Cre-/HIF-1αFl/Fl vs GFAP-Cre+/HIF-1αFl/Fl, or GFAP-Cre-/HIF-2αFl/Fl vs GFAP-Cre+/HIF-2αFl/Fl. Abbreviations: GFAP, glial fibrillary acidic protein; NS, no significant; PA, passive avoidance. |
Astrocyte HIF-2α, but not HIF-1α, supports learning under mild–moderate hypoxia
To determine the contribution of astrocyte HIF proteins on learning upon mild–moderate hypoxia, the PA performance of each genotype upon exposure to either 21% O2 (room air) or 12% O2 (Figure 6A) was modeled in Cox proportional hazard curves of latency (seconds) by task success rate in each genotype (Figure 6B). This analysis demonstrates that exposure to 12% O2 impaired learning in all groups, even in C57. However, the magnitude of the impact of 12% O2 upon learning was greatest and only significant in the GFAP-Cre+/HIF-2αFl/Fl group, P=0.015 (Figure 6C, blue lines). Importantly, no animals died following the hypoxic exposures.
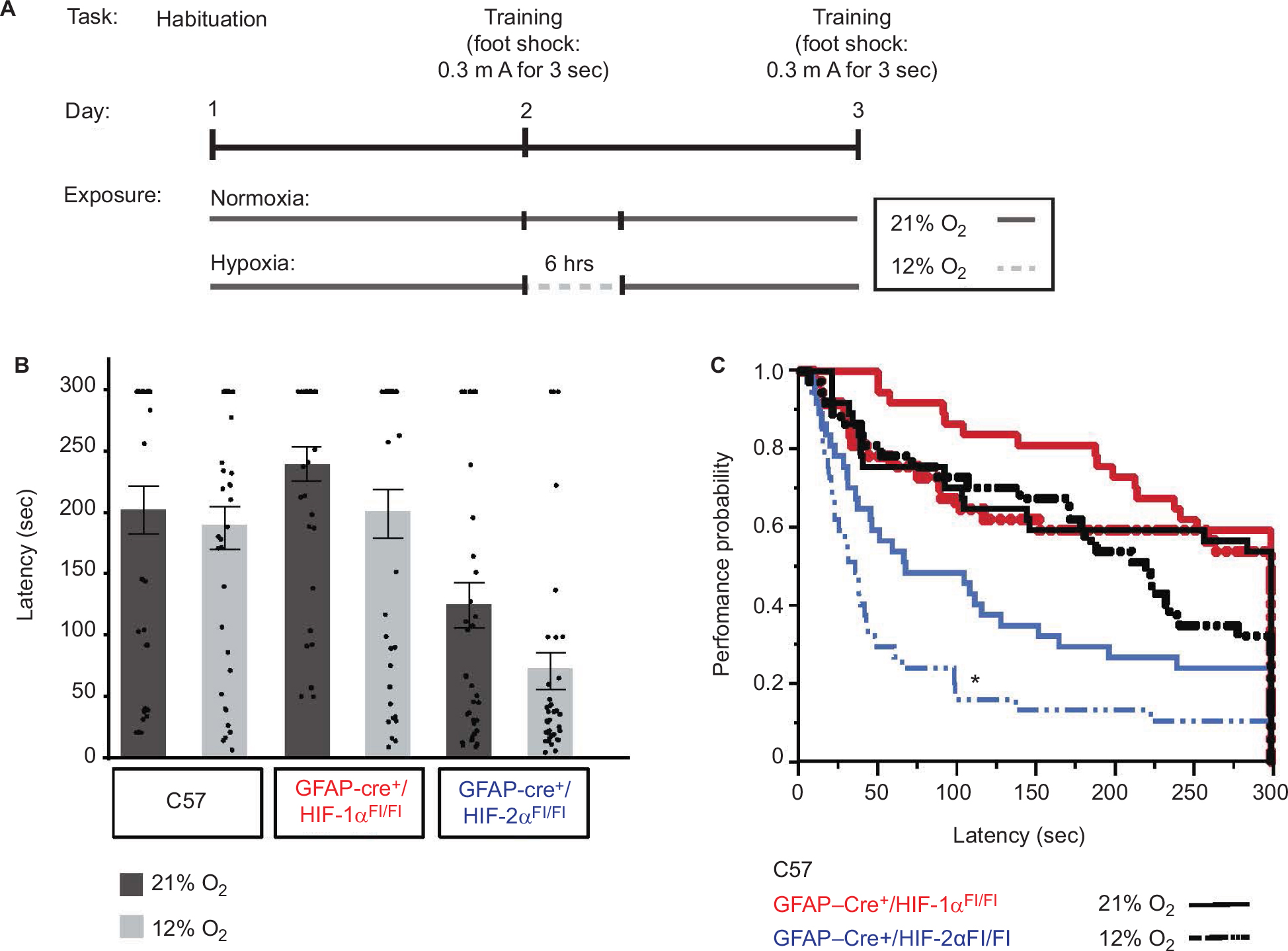 | Figure 6 HIF-2α knock-out in GFAP-expressing cells results in impaired learning. Notes: (A) Similar to the testing protocol described in Figure 5A, animals are habituated on day 1. On testing day (day 2), animals are given a foot shock when they enter the dark compartment and are immediately exposed to either air (21% O2) or hypoxia (12% O2) for 6 hours. A total of 24 hours after training (day 3), they are tested for the time to enter the dark side (latency). (B) Bar graphs and scatter plots depicting mean latency per group at 21% O2 (dark gray bars) vs 12% O2 (light gray bars). A ceiling latency of 300 seconds was enforced. The mean latency for C57 was at 21% O2 =202±119 and at 12% O2 =188±106, that for GFAP-Cre+/HIF-1αFl/Fl was at 21% O2 =240±85 and at 12% O2 =199±120, and that for GFAP-Cre+/HIF-2αFl/Fl at 21% O2 =124±113 and at 12% O2=71±90. (C) Cox proportional hazard analysis of mean latency for C57 (black), GFAP-Cre+/HIF-1αFl/Fl (red), and GFAP-Cre+/HIF-2αFl/Fl (blue) was 21% O2 (solid lines) vs 12% O2 (dashed lines) (N=37/group). Only the risk ratio comparison between 21% O2 and 12% O2 within the GFAP-Cre+/HIF-2αFl/Fl group (blue) showed statistical significance (*P=0.015). Abbreviation: GFAP, glial fibrillary acidic protein. |
Astrocyte HIF-2α, but not HIF-1α, supports LTP under mild–moderate hypoxia
To determine if the performance differences in the PA test were due to altered synaptic function in the GFAP-Cre+/HIF-2αFl/Fl mice upon hypoxic challenge, we tested LTP. C57, GFAP-Cre+/HIF-1αFl/Fl, and GFAP-Cre+/HIF-2αFl/Fl mice underwent 6-hour exposure to 21% O2 or 12% O2 to activate HIF and the expression of HIF-controlled genes and proteins, followed immediately by euthanasia and hippocampal slice recovery for LTP studies. No animals died due to the hypoxic exposures. We found that LTP measured 1 hour after tetanus-induced stimulation was not significantly reduced by the 6-hour hypoxic exposure compared with 21% O2 in the C57 group (average slope of fEPSPs to baseline 50–60 minutes after tetanus [21% O2, 151±6%; n=11 slices vs 6 hours 12% O2, 147±4%; n=10 slices]; P=0.06) (Figure 7A and D). In GFAP-Cre+/HIF-1αFl/Fl, hypoxia did not significantly alter LTP either (21% O2, 133±6%, n=11 slices vs 6 hours 12% O2, 137 ± 7%, n=10; P=0.15) (Figure 7B and D). However, LTP was significantly reduced in the GFAP-Cre+/HIF-2αFl/Fl hypoxia-treated animals (6 hours 12% O2, 146±8%, n=8 slices vs 21% O2, 193±10%, n=9 slices; P<0.0001) (Figure 7C and D). Interestingly, LTP in GFAP-Cre+/HIF-2αFl/Fl (193±10%) was significantly higher in 21% O2-treated animals than in C57 (151±6%; P≤0.0002) or GFAP-Cre+/HIF-1αFl/Fl (137±7%; P≤0.0002). Taken together, the PA and LTP results demonstrate an effect of astrocyte HIF-2α but not HIF-1α in supporting learning and synaptic plasticity under mild–moderate hypoxia.
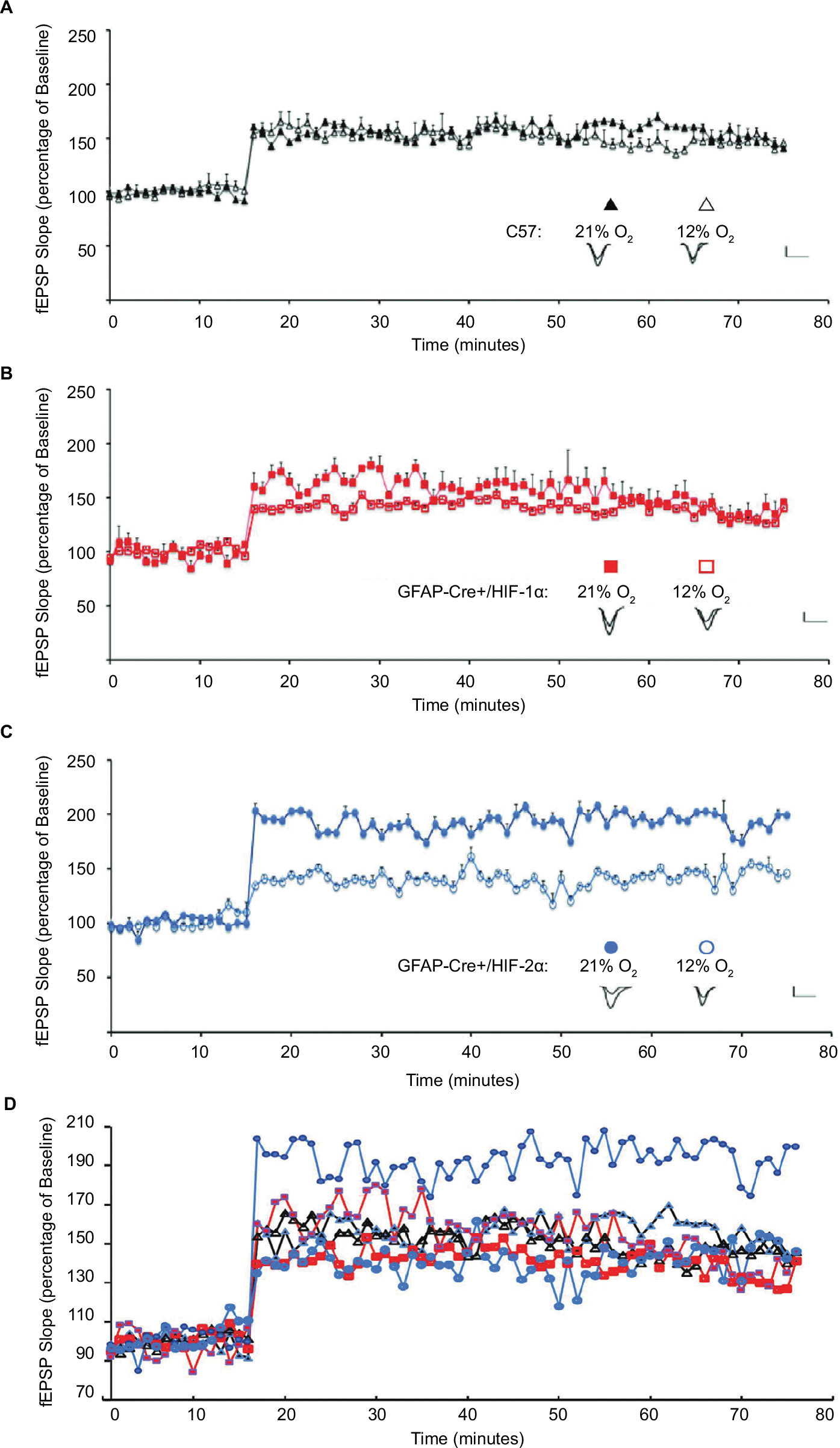 | Figure 7 LTP is altered upon hypoxic exposure in GFAP-Cre+/HIF-2αFl/Fl mice. Notes: Hippocampal slices recovered from animals after exposure to 6 hours of either 21% (colored shape) or 12% O2 (empty shape). fEPSP slope is plotted as the percentage of mean fEPSP baseline (mean ± SD) for (A) C57 (triangle), (B) GFAP-Cre+/HIF-1αFl/Fl (square), (C) GFAP-Cre+/HIF-2αFl/Fl (circle), and (D) all. The mean LTP during the 50–60 minutes period after tetanus was compared within groups. Inset: representative traces from animals exposed to 21% O2 (left) or 6 hours of 12% O2 (right), recorded immediately before and after LTP induction. Scale bars: 2 mV, 10 ms. Hypoxia did not significantly change LTP in C57 mice (147±4% in 12% O2, n=10 slices, 10 mice, vs 151±6% in 21% O2, n=10 slices, 11 mice; P>0.06) or GFAP-Cre+/HIF-1αFl/Fl mice (137±7% in 12% O2, n=10 slices, 10 mice, exposure to 21% O2, vs 133±6% at 21% O2, n=11 slices, 11 mice, P=0.15). In GFAP-Cre+/HIF-2αFl/Fl mice, LTP is significantly reduced in slices from animals exposed to 6 hours of 12% O2 compared with slices from animals maintained in 21% O2 (146±8% in 12% O2, n=8 slices, eight mice, vs 193±10% at 21% O2, n=9 slices, nine mice, P<0.0001). Abbreviations: fEPSP, field excitatory postsynaptic potential; GFAP, glial fibrillary acidic protein; LTP, long-term potentiation. |
To determine whether this effect upon learning or LTP was due to cell death elicited by hypoxia, C57, GFAP-Cre+/HIF-1αFl/Fl, and GFAP-Cre+/HIF-2αFl/Fl mice were exposed to 3 seconds, 0.3 mA foot shock, and immediately placed in either room air or 12% O2 for 6 hours. After the 6-hour exposure, the animals were either euthanized at 6 hours, or kept at 21% O2 for 18 hours for recovery, and then euthanized at the end of the 18 hours (24 hours total postfoot shock) (Figure 8A). Brains were immunohistochemically processed for the detection of cleaved caspase-3 (CC3) to determine if apoptosis had occurred in the CA1 region of the hippocampus. Distinct cellular puncta discriminate the CC3-positive cells in the CA1 region of the hippocampus in positive control brain tissue (Figure 8B, red arrows). In addition, widespread cell death in the surrounding cortical and dentate tissue is positive for CC3, a definitive sign of cell death. The same tissue treated with no primary anti-CC3 antibody and only biotinylated antirabbit secondary served as the negative control. In this condition, no noticeable cell death is identifiable in any areas of the hippocampus or surrounding cortex. Similarly, CC3-positive cells and puncta were not identified or detected in any of the C57, GFAP-Cre+/HIF-1αFl/Fl, and GFAP-Cre+/HIF-2αFl/Fl tissues, in any of the hypoxic or room air conditions tested at the 6- and 24-hour time points in the CA1 region or any of the areas observed on the tissue processed (Figure 8C). We qualitatively assessed cell death in the same manner in other more rostral and caudal regions of the brain and also did not identify cell death (data not shown). It is unlikely that necrosis occurred as there were no signs of tissue degeneration, debris, and leukocyte infiltration and as necrosis would only be expected as a result of ischemia due to chronic and/or prolonged hypoxic exposure that was not the case in this experimental exposure. This indicates that 12% O2 exposure is not sufficient to induce cell death and that this mechanism does not play a role in the cognitive deficiency exhibited by GFAP-Cre+/HIF-2αFl/Fl.
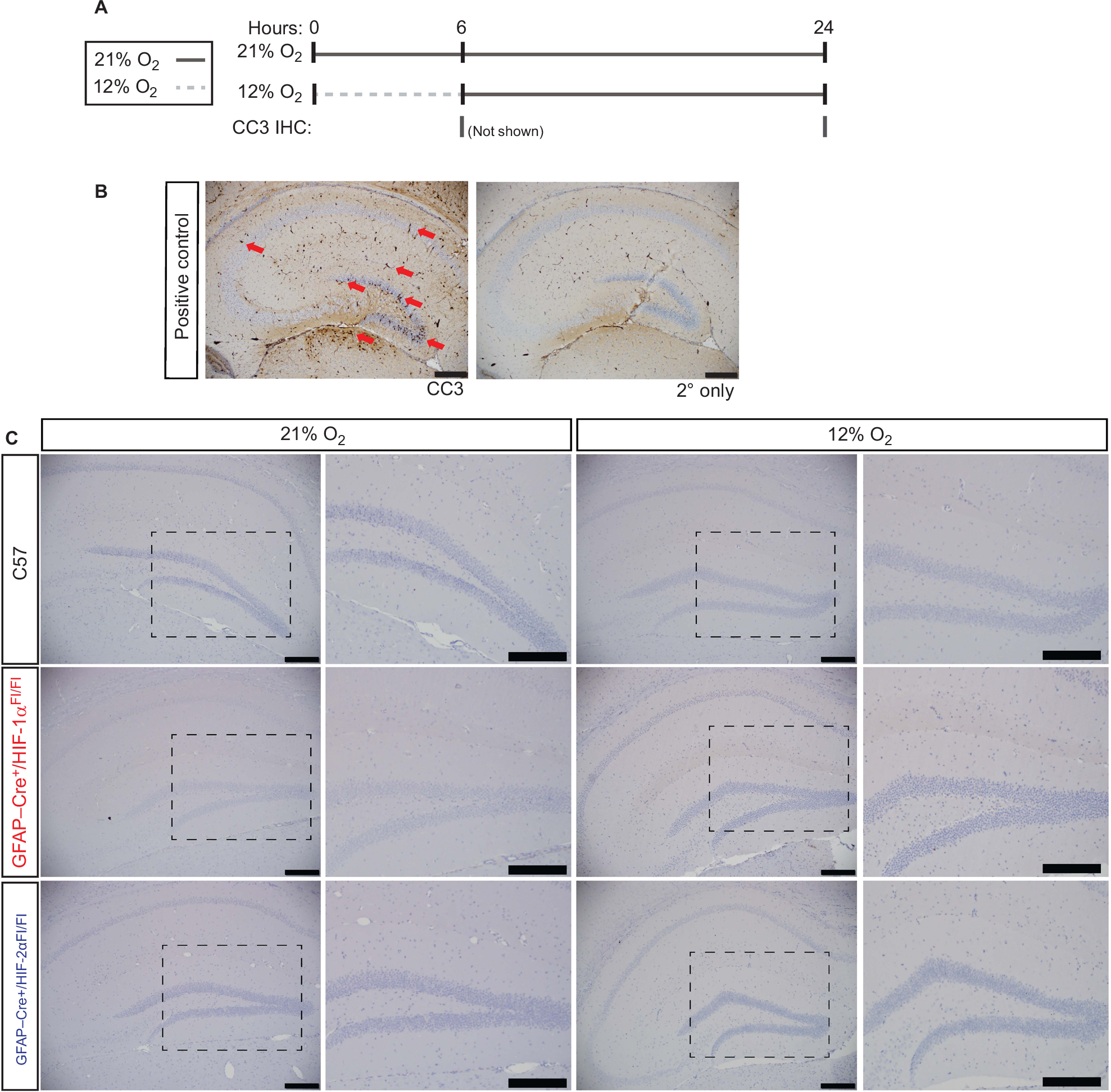 | Figure 8 Cell death in the hippocampus does not occur upon 12% O2 exposure. Notes: (A) Schematic representation depicting exposure to 21% O2 and 12% O2 for 6 hours, both followed by 18-hour recovery, and tissue collection time points at 6 and 24 hours. (B) Immunohistochemical detection of CC3 in postnatal day 8 pups injected with ethanol to induce widespread cell death in the hippocampus is marked by red arrows and serves as positive control tissues. A secondary antibody only control shows no background or nonspecific staining. (C) Animals exposed for 6 hours of 21% O2 or 12% O2 show no CC3 detection 18 hours after exposure, demonstrating that apoptosis was not induced. Tissues were also analyzed at 6 hours after the exposure and also showed no detectable CC3 staining (not shown). N=3 per condition. Images at 5× magnification, followed by insets indicated by dashed areas at 20× magnification. Scale bars =200 µm. Abbreviations: CC3, cleaved caspase-3; GFAP, glial fibrillary acidic protein; IHC, immunohistochemistry. |
HIF-1α, but not HIF-2α, mediates the expression of lactate metabolism regulating gene transcripts in astrocytes upon hypoxia
To determine the underlying basis of the observed impaired learning and LTP in GFAP-Cre+/HIF-2αFl/Fl mice, we looked for mechanisms through which HIF-2α could potentially disrupt synaptic function. The astrocyte–neuron lactate shuttle (ANLS) is a key metabolic pathway that serves to provide neurons with lactate, a necessary energy source to maintain normal LTP.95 This mechanism links the breakdown of glucose absorbed through the vasculature by receiving astrocytes through the Glut-1. Glucose then enters glycolysis, is intermediately processed into pyruvate, and is then metabolized into lactate by LDHA. Lactate is then shuttled into the extracellular space via MCT-4 and quickly taken up by neurons for use in many key energy-requiring functions involved in synaptic function. Interestingly, the expression of the transporters and enzymes responsible for the conversion of absorbed glucose into lactate is directly regulated by the HIF transcription factors, raising the question of whether HIF-2α null animals may have altered LTP at least in part due to insufficient metabolic support at the synapse upon hypoxia.
We preliminarily tested this hypothesis by assessing the transcript expression of ANLS signatures upon acute chronic hypoxic exposure in primary astrocytes via the following two approaches: 1) astrocytes isolated from GFAP-Cre-negative or -positive HIF-1αFl/Fl or HIF-2αFl/Fl (as described in Figure 1C and D) and 2) knock-down of HIF-1α, HIF-2α, or both in primary WT astrocytes using siRNAs. The first approach would allow us to test cells derived from the animal model tested and these results would be complemented by information about the contribution of both isoforms upon simultaneous knock-down in the second approach (Figure 9A).
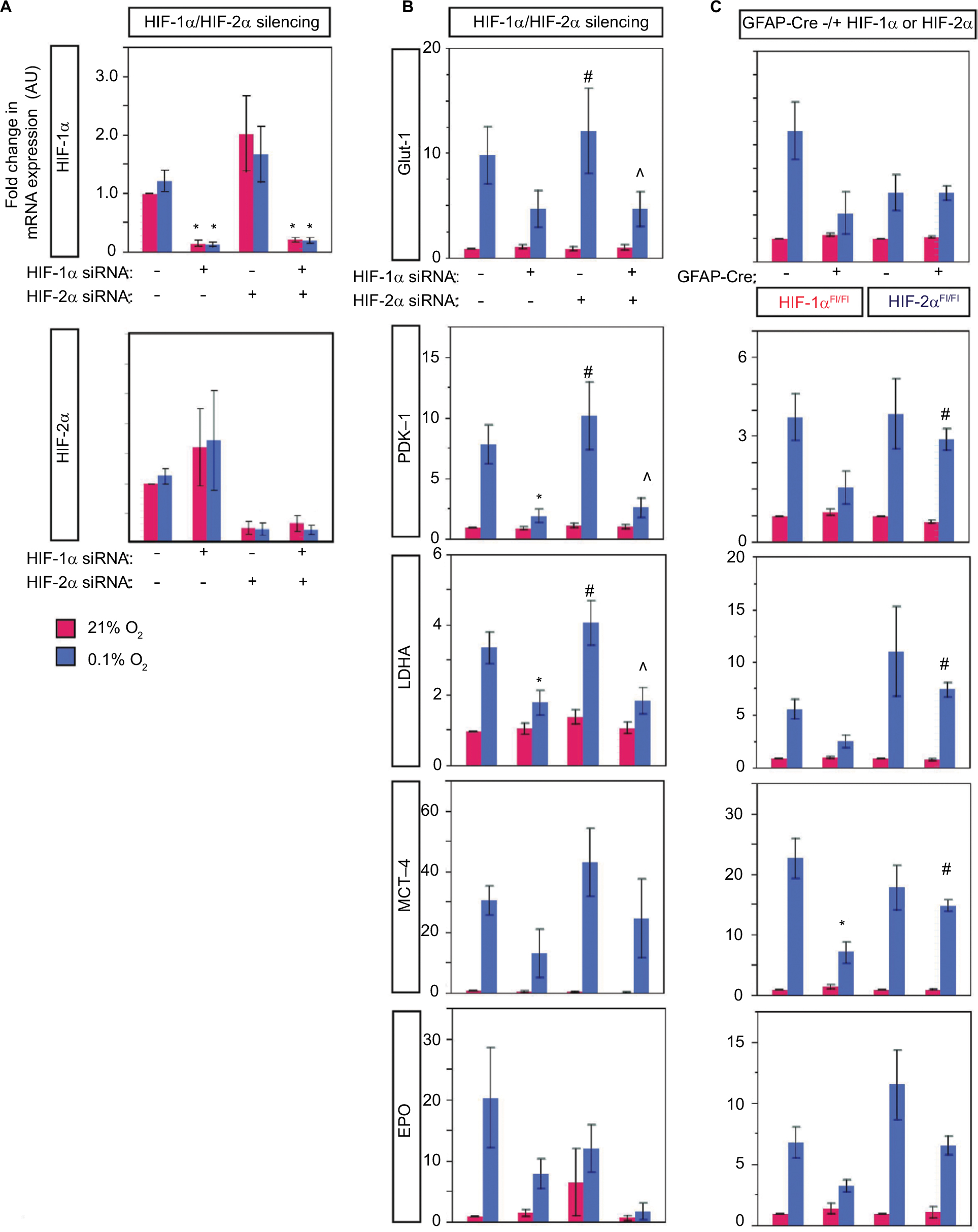 | Figure 9 Astrocyte HIF-2α does not target genes involved in metabolism and EPO induction in vitro. Notes: Gene expression changes in HIF-1α- and/or HIF-2α-deficient astrocytes after chronic hypoxic challenge in vitro. (A and B) Astrocytes were transfected with control, HIF-1α, HIF-2α, or both HIF-1α and HIF-2α targeting siRNAs for knock-down. In (A), verification of knock-down by the assessment of HIF transcripts shows significantly decreased HIF proteins upon respective siRNA treatment. (C) Astrocytes isolated from GFAP-Cre-negative or -positive HIF-1αFl/Fl or HIF-2αFl/Fl neonatal brains, n=5–8/group. In (A–C), astrocytes were exposed to either 21% O2 or 0.1% O2 for 6 hours. Mean ± SE are plotted for transcript fold changes in Glut-1, PDK-1, LDHA, MCT-4, and EPO relative to 18S. In (A and B), *P<0.05 relative to the control without silencing, #P<0.05 relative to HIF-1α silencing, and ^P<0.05 relative to HIF-2α silencing. In (C), *P<0.05 relative to Cre-/HIF-αFl/Fl of same genotype under the same exposure and #P<0.05 relative to GFAP-Cre+/HIF-1αFl/Fl under the same exposure. Abbreviations: AU, arbitrary units; EPO, erythropoietin; GFAP, glial fibrillary acidic protein; Glut-1, glucose transporter 1; LDHA, lactate dehydrogenase A; MCT-4, monocarboxylate transporter 4; PDK-1, pyruvate dehydrogenase kinase-1. |
Assessment of glucose and lactate transporters (Glut-1 and MCT-4), PDK-1 (inhibitor of pyruvate dehydrogenase), and LDHA showed a trend in which they appeared to be regulated by HIF-1α and not HIF-2α (Figure 9B and C), suggesting that insufficient metabolic adaptation in the HIF-2α null animals may not be the cause of the observed learning and LTP impairment.
The potent trophic and angiogenic stimulator, EPO, has been linked to memory function through its role in LTP enhancement.96 Furthermore, astrocytes have been shown to be the main producers of EPO in the brain and EPO is a hypoxia response gene, the expression of which is regulated by the HIF transcription factors. Astrocytes have been shown to elevate EPO levels in a HIF-2α-dependent mechanism.53 For this reason, we tested whether EPO expression is affected in knock-down or KO of HIF transcription factors in astrocytes upon hypoxic challenge in culture. We found that both HIF-1 and HIF-2 knock-down or KO resulted in a decreased EPO transcription, although the changes did not reach significance (Figure 9B and C). This suggests that EPO may also not be the main cause of the observed learning deficit in the GFAP-Cre+/HIF-2αFl/Fl mice.
Discussion
In this study, we have uncovered a novel role for the astrocyte hypoxic response in supporting synaptic plasticity and learning under mild hypoxic stress. This, in combination with prior documentation that HIF activity declines with aging, and the fact that aging is accompanied by disease processes that accelerate hypoxic stress, point to the potentially critical role that astrocyte HIF may play in promoting impaired cognitive performance with aging, highlighting the importance of additional work to understand this phenomenon.
The goal of this study’s experimental design was to mimic the loss of astrocyte HIF activity to model loss of HIF activity, as has been observed during aging, and to determine how this affects cognitive function. We first investigated how animals performed using baseline behavioral testing and found significant decreases in speed and path length in both HIF-1α and HIF-2α controls and KOs relative to C57 in the OF test, but no significant differences in time spent in the center or outer zone, suggesting no differences in anxiety-like behavior. The RR test results also showed that the HIF-1α and HIF-2α controls and KO animals displayed decreased performance relative to C57 but did not show differences in the rate of learning over the 3-day testing period. It is possible that the deficit in performance observed by the HIF-α control and KO animals on the RR test compared to C57 was due to the decreased speed observed. However, there was no change between the control and KO animals within groups. In the baseline measurements of the PA test, the GFAP-HIF-2α control and KO animals displayed significant decreased latencies relative to C57, but the KO was not different than the floxed control, indicating that HIF-2α depletion is not the cause for this.
With this information, we proceeded to determine how a modest hypoxic challenge, such as is presented to humans during acute exacerbations in heart or pulmonary failure, would affect learning. We found that only the HIF-2α KO group displayed deficient performance in the PA test, suggesting that the response to hypoxia in this animal group is altered. PA is a classic approach for the study of episodic memory,97 yet is far from complete in its ability to assess the multiple aspects of memory. However, the PA test is considered a valid testing paradigm with which to start one’s investigation98 and provides valuable information about learning capacity. As we have observed locomotor effects displayed by the HIF-2α control and KO animals, future studies could take advantage of the novel object recognition test, which is more likely to be free of biases based on mobility.99 This test is ideal to assess whether the behavior observed in PA is a function of avoidance vs escape.100
We hypothesized that the deficient PA performance by the GFAP-HIF-2α KOs could be at least in part explained by defective synaptic plasticity. We performed LTP induction studies and observed that both C57 and GFAP-HIF-1α showed nearly identical responses (fEPSP values) to LTP induction independent of O2 exposure, suggesting their ability to sustain homeostatic synaptic plasticity during the hypoxic challenge. However, the GFAP-HIF-2α KO group presented the following two notable differences: 1) the fEPSP induction values were higher than those of C57 and GFAP-HIF-1α KO at 21% O2 and 2) the induction upon 12% O2 exposure was significantly decreased relative to 21% O2 and was similar to that of C57 and GFAP-HIF-1α.
As HIF-2α is found maximally expressed at baseline in aging brains under normoxic conditions46 but appears to be nonfunctional, GFAP-HIF-2α KO may be recapitulating an important LTP characteristic previously shown in aging, where enhanced LTP may be partly compensatory.101 Interestingly, enhanced LTP is displayed in healthy aging and is associated with defective performance on several cognitive tests,102–105 suggesting that enhanced LTP does not just represent synaptic strengthening but potentially overexcitation or inadequate inhibitory signaling that could involve N-methyl-D-aspartate receptors.101 These studies and our LTP results raise the consideration that perhaps defective cognitive functions in the aged are a result of disrupted synaptic functions that manifest as enhanced LTP for compensation. Further studies would require a more thorough characterization of both the excitatory and inhibitory circuits in addition to expanding to other regions of the brain to garner a more global assessment of astrocyte HIF function at baseline and upon hypoxia as a function of age.
If at 21% O2, loss of HIF-2α results in higher LTP induction, it points to a role for HIF-2α during normoxia. While HIF-2α functions during normoxia have not yet been reported, they have been for HIF-1α.106 We did not detect HIF-2α in astrocytes grown in culture during normoxia. However, whether HIF-2α functions during normoxia in vivo is unknown and could occur due to local changes in oxygen consumption and availability or any of the mechanisms previously described for HIF-1α that include dysregulation of HIF degradation enzymes (prolyl hydroxylases and von Hippel–Lindau factor) or conferred post-translational modifications.107,108 This also suggests that an experimental model that mimics HIF-2α stabilization may complement and inform the current and future studies.
The LTP studies were conducted on hippocampal slices and provided insight about the function of astrocyte-HIF-2α in this brain region. While we know that the PA paradigm integrates circuit activation of the hippocampus, amygdala, perirhinal, and prefrontal cortices,88 additional studies could probe astrocyte HIF functions in these other regions to gain a more global understanding of the pathways involved.
The expression pattern of HIF-1α, HIF-2α, and related gene targets within neurons and astrocytes may be co-dependent, with HIF and HIF-controlled target proteins synthesized in cells exerting a paracrine function109 upon the other. Indeed, compensatory roles for both HIF-1α and HIF-2α have been described,110,111 raising the question of how neurons are compensating by activating their own HIF-response system and if this is also affected by aging. Additionally, the balance of HIF-1α vs HIF-2α expression may shift when one or the other is diminished; however, we did not identify this in primary astrocytes in culture. Nevertheless, HIF-2α is known to be stabilized at higher oxygen levels than HIF-1α in vitro,112 has been shown to be active under mild hypoxia (~5% O2),113 and appears to drive responses to more chronic hypoxia.38 Conversely, HIF-1α is most active during short periods (2–24 hours) of intense hypoxia (<0.1% O2). This supports the notion that during moderate hypoxia, astrocyte HIF-2α may be the primary HIF isoform responsible for the adaptation and maintenance of cognitive performance.
Astrocyte HIF could modulate learning through its control over several mechanisms that would involve direct effects on synaptic plasticity. Neurovascular coupling114–116 and the tripartite synapse117 are instances in which the astrocyte integrates physical and chemical processes such as blood flow,118 nutrient uptake and catabolism (ie, glucose),119 and neurotransmitter recycling and synthesis65 to directly support neuronal function. These, along with many other processes, have a molecular root in the programs mediated by HIF transcriptional effects. Cerebrovascular dynamics are an important aspect of neuronal function that is in part modulated by astrocytes through neurovascular coupling.118 VEGF and EPO have been shown to regulate vascular dynamics in organ systems that include the CNS,120 and astrocytes potentially use similar mechanisms in addition to others to perform this function. HIF can regulate NO-producing enzymes121 and the expression of VEGF83 and EPO,122 all with the purpose to increase blood flow. It is therefore possible that some of these mechanisms are altered upon loss of HIF-2α in astrocytes.
Astrocyte HIF may support learning under hypoxia in several ways, including trophic and metabolic mechanisms. HIF-2α is synthesized in all brain cells but appears to be predominantly synthesized within astrocytes, while HIF-1α is predominantly synthesized within neurons.53 HIF-2α appears to exert greater control over EPO synthesis,24 while neuronal HIF-1α may control its receptor, EPO-R.24 EPO is one of the perhaps many HIF targets that may support synaptic plasticity and is one of the best studied. Supplemental EPO has been found to enhance memory in humans123 and mice96 under normoxia. Adamcio et al,96 demonstrated a 25% increase in LTP in response to long-term systemic administration of EPO, under normoxia. In young rats exposed to chronic intermittent hypoxia, EPO reduced spatial learning deficits.124,125 EPO may support synaptic plasticity and memory through several mechanisms, which include enhanced BDNF synthesis and promoting CREB phosphorylation, both events which are crucial for synaptic plasticity, LTP, and long-term memory.126–130 EPO has also been reported to be primarily controlled by HIF-2α in the brain,53 kidney, and liver.131,132 We found, however, that both HIF-1α and HIF-2α had the ability to regulate the transcription of EPO, as EPO transcription was not eliminated by HIF-1α or HIF-2α KO, and EPO transcription was most severely suppressed in the presence of both HIF-1α and HIF-2α knock-down. Others have found that HIF-1α and HIF-2α bind to the hypoxia response elements on the EPO gene,110,133 supporting compensatory roles for both HIF-2α and HIF-1α in controlling EPO transcription. This suggests that the mechanism through which the astrocyte HIF-2α KO results in impaired LTP may be due to the lack of genes induced by HIF that HIF-1α cannot compensate for in the absence of HIF-1α.
One proposed mechanism could involve the contribution of HIF-2a to the support of protein synthesis under hypoxia.134 Interestingly, astrocyte processes, the primary sites of contact between astrocytes and neurons/synapses, have been shown to regulate local translation resulting in the expression of necessary synaptogenic proteins (eg, Hevin and SPARC).135 It is therefore possible that loss of astrocyte HIF-2α upon hypoxia results in depressed local protein synthesis and consequently attenuated expression of these necessary components, altering normal LTP and resulting in poor learning ability.
Finally, while the GFAP promoter is expressed most strongly in astrocytes, low levels of GFAP or GFAP-like immunoreactivity have been described in several other cell types, to include reactive retinal Muller cells, nonmyelinating Schwann cells,136–138 and glial progenitor cells (radial glia) that give rise to astrocytes, neurons, and oligodendrocytes during development.139–141 Embryonic expression of GFAP is first detected at E13.5–14.5 and is intensely expressed by E18.5.142,143 The GFAP-Cre-driven conditional model of HIF deletion that we employed was chosen based on findings that it produces animals at normal birth rates, with normal fertility, and with grossly normal behavior and normal life spans.71 The strain of origin for the HIF-1αFl/Fl animals ([129×1/SvJ ×129S1/Sv] F1- Kitl<+>) and HIF-2αFl/Fl (129×1/SvJ) are similar, and both are maintained commercially on C57B6/J backgrounds. The lack of gross developmental abnormalities in this inducible model may be driven by low recombinase activity in cells that only weakly express GFAP.144 We did not identify overt phenotypes such as those previously identified in the nestin-Cre HIF-1α KO, suggesting that the GFAP-Cre model employed does not KO HIF-1α in neural stem cells.93 An alternate model employing an inducible GFAP-Cre-driven deletion was also considered. However, such inducible systems can display recombination rates as low as 30%.140,145 We characterized HIF expression in primary astrocytes derived from control and KO mice and found that, upon hypoxic challenge in vitro, only the untargeted HIF isoform was stabilized, confirming KO of the correct HIF target.
Conclusion
The key finding of this study is that learning impairment can occur under mild–moderate hypoxia when astrocyte HIF-2α activity is blunted, as occurs with aging. Of note, this occurs in the absence of hippocampal neuronal cell death. Thus, decreasing HIF activity with aging may be mechanistically linked to age-related cognitive impairment in a host of disease states accompanied by mild–moderate hypoxia.
Acknowledgments
The authors thank Ying Wang, PhD, for help in the characterization of the physiology and recombination of the animal models described herein, thank Ted Abel, PhD, at the University of Pennsylvania, Smilow Center for Translational Research (Philadelphia, PA, USA) for consultation on the passive avoidance paradigm and for helpful conversations on the other behavioral and LTP tests, and specially thank Laurie Levine at the Division for Laboratory Animal Resources at SBU for help in animal maintenance, breeding, and caretaking. The authors would like to thank Lucia Roa-Peña, MD, and Yan Ji, BS, MBA, from the Histopathology Core at the Department of Pathology, Stony Brook University, for help in processing tissue and optimizing the cleaved caspase-3 antibody and immunohistochemistry conditions. The authors would like to thank the laboratory of Dr. Holly Colognato for the development of protocols to establish mixed glial cultures used for generation of primary cortical astrocytes. This research was sponsored by the Department of Anesthesiology, Stony Brook University, and the Research Foundation of the State University of New York at Stony Brook, Stony Brook, NY, USA.
Author contributions
TFF, CVL, and EC designed the research strategy. EC, KC, JS, JP, and MC performed gross physiological assessments, behavioral tests (open field, rotarod, and passive avoidance), and characterized recombination in the animal models described herein. EC and JS performed the electrophysiological measurements of LTP. All data were analyzed by TFF and CVL who discussed the interpretation with all investigators. CVL in cooperation with KM, DMM, AD, and BC cultured astrocyte primary cell cultures for the analysis of HIF expression from the animal models described. CVL and AC cultured and analyzed astrocyte primary cell cultures for HIF target gene expression. CVL and MK optimized astrocyte in vitro cultures and transfection protocols. CVL and RL tested cell death via cleaved caspase-3 staining. CVL managed mouse colonies. TFF, CVL, and MK wrote, edited, and constructed the manuscript with input and discussion from all other authors. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
CVL was partly supported by a postdoctoral fellowship awarded by the National Science Foundation’s workforce development program, the Alliance for Graduate Education in the Professoriate-Transformation grant (HRD-1311318), through the Stony Brook University Center for Inclusive Education, Stony Brook, NY, USA. The authors report no other conflicts of interest in this work.
References
Pittman RN. Regulation of Tissue Oxygenation. San Rafael, CA: Morgan & Claypool Life Sciences; 2011. | ||
Bruce AS, Aloia MS, Ancoli-Israel S. Neuropsychological effects of hypoxia in medical disorders. In: Grant I, Adams KM, editors. Neuropsychological Assessment of Neuropsychiatric and Neuromedical Disorders. 3rd ed. Oxford, New York: Oxford University Press; 2009:336–349. | ||
Cannon JA, Moffitt P, Perez-Moreno AC, et al. Cognitive Impairment and Heart Failure: Systematic Review and Meta-Analysis. J Card Fail. 2017;23:464–475. | ||
Yohannes AM, Chen W, Moga AM, Leroi I, Connolly MJ. Cognitive Impairment in Chronic Obstructive Pulmonary Disease and Chronic Heart Failure: A Systematic Review and Meta-analysis of Observational Studies. J Am Med Dir Assoc. 2017;18(5):451.e1–451.e11. | ||
Schega L, Peter B, Törpel A, Mutschler H, Isermann B, Hamacher D. Effects of intermittent hypoxia on cognitive performance and quality of life in elderly adults: a pilot study. Gerontology. 2013;59(4):316–323. | ||
Qiu C, Fratiglioni L. A major role for cardiovascular burden in age-related cognitive decline. Nat Rev Cardiol. 2015;12(5):267–277. | ||
Popa-Wagner A, Buga A-M, Popescu B, Muresanu D. Vascular cognitive impairment, dementia, aging and energy demand. A vicious cycle. J Neural Transm. 2015;122(S1):47–54. | ||
Lahousse L, Tiemeier H, Ikram MA, Brusselle GG. Chronic obstructive pulmonary disease and cerebrovascular disease: A comprehensive review. Respir Med. 2015;109(11):1371–1380. | ||
Daulatzai MA. Evidence of neurodegeneration in obstructive sleep apnea: Relationship between obstructive sleep apnea and cognitive dysfunction in the elderly. J Neurosci Res. 2015;93(12):1778–1794. | ||
Bressler J, Knopman DS, Sharrett AR, et al. Incident Heart Failure and Cognitive Decline: The Atherosclerosis Risk in Communities Study. J Card Fail. 2017;23(1):47–55. | ||
Suzuki H, Matsumoto Y, Ota H, et al. Hippocampal Blood Flow Abnormality Associated With Depressive Symptoms and Cognitive Impairment in Patients With Chronic Heart Failure. Circ J. 2016;80(8):1773–1780. | ||
Kurella Tamura M, Vittinghoff E, Yang J, et al. Anemia and risk for cognitive decline in chronic kidney disease. BMC Nephrol. 2016;17(1):13. | ||
Dlugaj M, Winkler A, Weimar C, et al. Anemia and Mild Cognitive Impairment in the German General Population. J Alzheimers Dis. 2016;49:1031–1042. | ||
Schneider ALC, Jonassaint C, Sharrett AR, et al. Hemoglobin, Anemia, and Cognitive Function: The Atherosclerosis Risk in Communities Study. J Gerontol A Biol Sci Med Sci. 2016;71(6):772–779. | ||
Weiskopf RB, Kramer JH, Viele M, et al. Acute Severe Isovolemic Anemia Impairs Cognitive Function and Memory in Humans. Anesthesiology. 2000;92(6):1646–1652. | ||
Weiskopf RB, Feiner J, Hopf HW, et al. Oxygen Reverses Deficits of Cognitive Function and Memory and Increased Heart Rate Induced by Acute Severe Isovolemic Anemia. Anesthesiology. 2002;96(4):871–877. | ||
Hornbein TF. The high-altitude brain. J Exp Biol. 2001;204(Pt 18):3129–3132. | ||
Wilson MH, Newman S, Imray CH. The cerebral effects of ascent to high altitudes. Lancet Neurol. 2009;8(2):175–191. | ||
Neuhaus C, Hinkelbein J. Cognitive responses to hypobaric hypoxia: implications for aviation training. Psychol Res Behav Manag. 2014;7:297–302. | ||
Malle C, Quinette P, Laisney M, et al. Working Memory Impairment in Pilots Exposed to Acute Hypobaric Hypoxia. Aviat Space Environ Med. 2013;84(8):773–779. | ||
Tran PV, Carlson ES, Fretham SJB, Georgieff MK. Early-Life Iron Deficiency Anemia Alters Neurotrophic Factor Expression and Hippocampal Neuron Differentiation in Male Rats. J Nutr. 2008;138(12):2495–2501. | ||
Hennigan A, Callaghan CK, Kealy J, Rouine J, Kelly Áine M.. Deficits in LTP and recognition memory in the genetically hypertensive rat are associated with decreased expression of neurotrophic factors and their receptors in the dentate gyrus. Behav Brain Res. 2009;197(2):371–377. | ||
Jänicke B, Schulze G. Influence of normobaric hypoxia on learning capacity of different aged rats. Neurobiol Aging. 1987;8(6):495–500. | ||
Li M, Bertout JA, Ratcliffe SJ, Eckenhoff MF, Simon MC, Floyd TF. Acute Anemia Elicits Cognitive Dysfunction and Evidence of Cerebral Cellular Hypoxia in Older Rats with Systemic Hypertension. Anesthesiology. 2010;113(4):845–858. | ||
Gozal D, Row BW, Kheirandish L, et al. Increased susceptibility to intermittent hypoxia in aging rats: changes in proteasomal activity, neuronal apoptosis and spatial function. J Neurochem. 2003;86(6):1545–1552. | ||
Ingraham JP, Forbes ME, Riddle DR, Sonntag WE. Aging Reduces Hypoxia-Induced Microvascular Growth in the Rodent Hippocampus. J Gerontol A Biol Sci Med Sci. 2008;63(1):12–20. | ||
Dunwiddie TV. Age-related differences in the in vitro rat hippocampus. Development of inhibition and the effects of hypoxia. Dev Neurosci. 1981;4:165–175. | ||
Larson J, Drew KL, Folkow LP, Milton SL, Park TJ. No oxygen? No problem! Intrinsic brain tolerance to hypoxia in vertebrates. J Exp Biol. 2014;217(7):1024–1039. | ||
Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–5514. | ||
Sharp FR, Bergeron M, Bernaudin M. Hypoxia-inducible factor in brain. Adv Exp Med Biol. 2001;502:273–291. | ||
Lahiri S, Roy A, Baby SM, Hoshi T, Semenza GL, Prabhakar NR. Oxygen sensing in the body. Prog Biophys Mol Biol. 2006;91(3):249–286. | ||
Kaelin WG, Ratcliffe PJ. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol Cell. 2008;30(4):393–402. | ||
Prabhakar NR, Semenza GL. Oxygen Sensing and Homeostasis. Physiology (Bethesda). 2015;30:340–348. | ||
Mclaren AT, Mazer CD, Zhang H, Liu E, Mok L, Hare GM. A potential role for inducible nitric oxide synthase in the cerebral response to acute hemodilution. Can J Anaesth. 2009;56(7):502–509. | ||
Powell FL, Kim BC, Johnson SR, Fu Z. Oxygen sensing in the brain-invited article. Adv Exp Med Biol. 2009;648:369–376. | ||
Lamanna JC. Hypoxia in the central nervous system. Essays Biochem. 2007;43:138–152. | ||
Knabe W, Knerlich F, Washausen S, et al. Expression patterns of erythropoietin and its receptor in the developing midbrain. Anat Embryol. 2004;207(6):503–512. | ||
Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117(23):e207–e217. | ||
Wenger RH. Mammalian oxygen sensing, signalling and gene regulation. J Exp Biol. 2000;203(Pt 8):1253–1263. | ||
Semenza GL. Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Annu Rev Pathol. 2014;9(1):47–71. | ||
Rivard A, Berthou-Soulie L, Principe N, et al. Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J Biol Chem. 2000;275(38):29643–29647. | ||
Chavez JC, Lamanna JC. Hypoxia-inducible factor-1alpha accumulation in the rat brain in response to hypoxia and ischemia is attenuated during aging. Adv Exp Med Biol. 2003;510:337–341. | ||
Giulio CD, Bianchi G, Cacchio M, et al. Oxygen and life span: chronic hypoxia as a model for studying HIF-1α, VEGF and NOS during aging. Respir Physiol Neurobiol. 2005;147(1):31–38. | ||
Bosch-Marce M, Okuyama H, Wesley JB, et al. Effects of Aging and Hypoxia-Inducible Factor-1 Activity on Angiogenic Cell Mobilization and Recovery of Perfusion After Limb Ischemia. Circ Res. 2007;101(12):1310–1318. | ||
Katschinski D. Is there a molecular connection between hypoxia and aging? Exp Gerontol. 2006;41(5):482–484. | ||
Ndubuizu OI, Chavez JC, Lamanna JC. Increased prolyl 4-hydroxylase expression and differential regulation of hypoxia-inducible factors in the aged rat brain. Am J Physiol Regul Integr Comp Physiol. 2009;297(1):R158–R165. | ||
Rapino C, Bianchi G, di Giulio C, et al. HIF-1alpha cytoplasmic accumulation is associated with cell death in old rat cerebral cortex exposed to intermittent hypoxia. Aging Cell. 2005;4(4):177–185. | ||
Frenkel-Denkberg G, Gershon D, Levy AP. The function of hypoxia-inducible factor 1 (HIF-1) is impaired in senescent mice. FEBS Lett. 1999;462(3):341–344. | ||
Kim H, Lee D-K, Choi J-W, Kim J-S, Park SC, Youn H-D. Analysis of the effect of aging on the response to hypoxia by cDNA microarray. Mech Ageing Dev. 2003;124(8-9):941–949. | ||
Benderro GF, Lamanna JC. Hypoxia-induced angiogenesis is delayed in aging mouse brain. Brain Res. 2011;1389:50–60. | ||
Rabie T, Kunze R, Marti HH. Impaired hypoxic response in senescent mouse brain. Int J Dev Neurosci. 2011;29(6):655–661. | ||
Anderson J, Sandhir R, Hamilton ES, Berman NEJ. Impaired Expression of Neuroprotective Molecules in the HIF-1α Pathway following Traumatic Brain Injury in Aged Mice. J Neurotrauma. 2009;26(9):1557–1566. | ||
Chavez JC, Baranova O, Lin J, Pichiule P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J Neurosci. 2006;26(37):9471–9481. | ||
Schousboe A, Sickmann HM, Bak LK, et al. Neuron-glia interactions in glutamatergic neurotransmission: Roles of oxidative and glycolytic adenosine triphosphate as energy source. J Neurosci Res. 2011;89(12):1926–1934. | ||
Dienel GA. Astrocytic energetics during excitatory neurotransmission: What are contributions of glutamate oxidation and glycolysis? Neurochem Int. 2013;63(4):244–258. | ||
Harris JJ, Jolivet R, Attwell D. Synaptic Energy Use and Supply. Neuron. 2012;75(5):762–777. | ||
Lu B, Nagappan G, Lu Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. In: Lewin GR, Carter BD, editors. Neurotrophic Factors. Berlin: Springer. 2014:223–250. | ||
Lum JJ, Bui T, Gruber M, et al. The transcription factor HIF-1 plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007;21(9):1037–1049. | ||
Jelkmann W. Regulation of erythropoietin production. J Physiol. 2011;589(6):1251–1258. | ||
Nakazawa MS, Keith B, Simon MC. Oxygen availability and metabolic adaptations. Nat Rev Cancer. 2016;16(10):663–673. | ||
Sardinha VM, Guerra-Gomes S, Caetano I, et al. Astrocytic signaling supports hippocampal-prefrontal theta synchronization and cognitive function. Glia. 2017;65(12):1944–1960. | ||
Holthoff K, Witte OW. Directed spatial potassium redistribution in rat neocortex. Glia. 2000;29(3):288–292. | ||
Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129(4):1043–1054. | ||
Ota Y, Zanetti AT, Hallock RM. The Role of Astrocytes in the Regulation of Synaptic Plasticity and Memory Formation. Neural Plast. 2013;2013(1):11–11. | ||
Allen NJ. Astrocyte Regulation of Synaptic Behavior. Annu Rev Cell Dev Biol. 2014;30(1):439–463. | ||
Christopherson KS, Ullian EM, Stokes CCA, et al. Thrombospondins Are Astrocyte-Secreted Proteins that Promote CNS Synaptogenesis. Cell. 2005;120(3):421–433. | ||
Risher WC, Eroglu C. Thrombospondins as key regulators of synaptogenesis in the central nervous system. Matrix Biology. 2012;31(3):170–177. | ||
Pannasch U, Vargová L, Reingruber J, et al. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci U S A. 2011;108(20):8467–8472. | ||
Stogsdill JA, Eroglu C. The interplay between neurons and glia in synapse development and plasticity. Curr Opin Neurobiol. 2017;42:1–8. | ||
Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci U S A. 2007;104(7):2301–2306. | ||
Weidemann A, Kerdiles YM, Knaup KX, et al. The glial cell response is an essential component of hypoxia-induced erythropoiesis in mice. J Clin Invest. 2009;119:3373–3383. | ||
Liu R, Suzuki A, Guo Z, Mizuno Y, Urabe T. Intrinsic and extrinsic erythropoietin enhances neuroprotection against ischemia and reperfusion injury in vitro. J Neurochem. 2006;96(4):1101–1110. | ||
Petzold GC, Murthy VN. Role of Astrocytes in Neurovascular Coupling. Neuron. 2011;71(5):782–797. | ||
Tarantini S, Tran CHT, Gordon GR, Ungvari Z, Csiszar A. Impaired neurovascular coupling in aging and Alzheimer’s disease: Contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp Gerontol. 2017;94:52–58. | ||
Wang M, He Y, Sejnowski TJ, Yu X. Brain-state dependent astrocytic Ca2+ signals are coupled to both positive and negative BOLD-fMRI signals. Proc Natl Acad Sci U S A. 2018;115(7):E1647–E1656. | ||
Bolaños JP, Cidad P, Garcı́a-Nogales P, Delgado-Esteban M, Fernández E, Almeida A. Regulation of glucose metabolism by nitrosative stress in neural cells. Mol Aspects Med. 2004;25(1-2):61–73. | ||
Ryan HE, Poloni M, Mcnulty W, et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–4015. | ||
Sakr HF. Abbas AM and El Samanoudy AZ. Effect of vitamin E on cerebral cortical oxidative stress and brain-derived neurotrophic factor gene expression induced by hypoxia and exercise in rats. J Physiol Pharmacol. 2015;66:191–202. | ||
Bell M, Thake CD, Wolff CB. Effect of inspiration of 12% O2 (Balance N2) on cardiac output, respiration, oxygen saturation, and oxygen delivery. In: LaManna JC, editor. Oxygen Transport to Tissue New York. Vol. (XXXII). Berlin, Germany: Springer; 2011:327–332. | ||
Group TL-TOTTR. A Randomized Trial of Long-Term Oxygen for COPD with Moderate Desaturation. New England Journal of Medicine. 2016;375:1617–1627. | ||
Kim C-H. The effects of hypoxia and exercise on physiological and cognitive performance. 2011. Available from: https://etd.ohiolink.edu/pg_10?0::NO:10:P10_ACCESSION_NUM:kent1311015088. Accessed September 05, 2018. | ||
Gray A, Goodacre S, Newby DE, Masson M, Sampson F, Nicholl J. Noninvasive Ventilation in Acute Cardiogenic Pulmonary Edema. N Engl J Med Overseas Ed. 2008;359(2):142–151. | ||
Forsythe JA, Jiang BH, Iyer NV, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16(9):4604–4613. | ||
Cable GG. In-flight hypoxia incidents in military aircraft: causes and implications for training. Aviat Space Environ Med. 2003;74:169–172. | ||
Stroka DM, Burkhardt T, Desbaillets I, et al. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. Faseb J. 2001;15(13):2445–2453. | ||
Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report. GOLD Executive Summary. Am J Respir Crit Care Med. 2017;195(5):557–582. | ||
Cranston JM, Crockett A, Moss J, Alpers JH, Cochrane Airways Group. Domiciliary oxygen for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2005;14(5). | ||
Burman MA, Simmons CA, Hughes M, Lei L. Developing and validating trace fear conditioning protocols in C57BL/6 mice. J Neurosci Methods. 2014;222:111–117. | ||
Mccarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85(3):890–902. | ||
Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res. 2002;133(2):115–126. | ||
Lamberty Y, Gower AJ. Age-related changes in spontaneous behavior and learning in NMRI mice from maturity to middle age. Physiol Behav. 1990;47(6):1137–1144. | ||
Jahn-Eimermacher A, Lasarzik I, Raber J. Statistical analysis of latency outcomes in behavioral experiments. Behav Brain Res. 2011;221(1):271–275. | ||
Tomita S, Ueno M, Sakamoto M, et al. Defective Brain Development in Mice Lacking the Hif-1 Gene in Neural Cells. Mol Cell Biol. 2003;23(19):6739–6749. | ||
Cy K, Tsai MY, Tseng WF, et al. Integration of CNS survival and differentiation by HIF2alpha. Cell Death Differ. 2011;18:1757–1770. | ||
Suzuki A, Stern SA, Bozdagi O, et al. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell. 2011;144(5):810–823. | ||
Adamcio B, Sargin D, Stradomska A, et al. Erythropoietin enhances hippocampal long-term potentiation and memory. BMC Biol. 2008;6(1):37. | ||
Ögren SO, Kuteeva E, Elvander-Tottie E, Hökfelt T. Neuropeptides in learning and memory processes with focus on galanin. Eur J Pharmacol. 2010;626(1):9–17. | ||
Detrait ER, Hanon E, Dardenne B, Lamberty Y. The inhibitory avoidance test optimized for discovery of cognitive enhancers. Behav Res Methods. 2009;41(3):805–811. | ||
Antunes M, Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process. 2012;13(2):93–110. | ||
Krypotos A-M, Effting M, Kindt M, Beckers T. Avoidance learning: a review of theoretical models and recent developments. Front Behav Neurosci. 2015;9:189. | ||
Pinho J, Vale R, Batalha VL, et al. Enhanced LTP in aged rats: Detrimental or compensatory? Neuropharmacology. 2017;114:12–19. | ||
Costenla AR, de Mendonça A, Ribeiro JA. Adenosine modulates synaptic plasticity in hippocampal slices from aged rats. Brain Res. 1999; 851(1-2):228–234. | ||
Kumar A, Foster TC. Enhanced Long-Term Potentiation During Aging Is Masked by Processes Involving Intracellular Calcium Stores. J Neurophysiol. 2004;91(6):2437–2444. | ||
Huang Y-Y, Kandel ER. Age-related enhancement of a protein synthesis-dependent late phase of LTP induced by low frequency paired-pulse stimulation in hippocampus. Learn Mem. 2006;13(3):298–306. | ||
Diógenes MJ, Fernandes CC, Sebastião AM, Ribeiro JA. Activation of adenosine A2A receptor facilitates brain-derived neurotrophic factor modulation of synaptic transmission in hippocampal slices. J Neurosci. 2004;24(12):2905–2913. | ||
Kuschel A, Simon P, Tug S. Functional regulation of HIF-1α under normoxia-is there more than post-translational regulation? J Cell Physiol. 2012;227(2):514–524. | ||
Pagé EL, Chan DA, Giaccia AJ, Levine M, Richard DE. Hypoxia-inducible factor-1alpha stabilization in nonhypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol Biol Cell. 2008;19(1):86–94. | ||
Baek SH, Kim KI. Regulation of HIF-1α stability by lysine methylation. BMB Rep. 2016;49(5):245–246. | ||
Mamczur P, Borsuk B, Paszko J, et al. Astrocyte-neuron crosstalk regulates the expression and subcellular localization of carbohydrate metabolism enzymes. Glia. 2015;63(2):328–340. | ||
Tsuboi I, Yamashita T, Nagano M, Kimura K, To’a Salazar G, Ohneda O. Impaired expression of HIF-2α induces compensatory expression of HIF-1α for the recovery from anemia. J Cell Physiol. 2015;230(7):1534–1548. | ||
Jha MK, Kim JH, Song GJ, et al. Functional dissection of astrocyte-secreted proteins: Implications in brain health and diseases. Prog Neurobiol. 2018;162:37-69. | ||
Zhao J, du F, Shen G, Zheng F, Xu B. The role of hypoxia-inducible factor-2 in digestive system cancers. Cell Death Dis. 2015;6:e1600. | ||
Holmquist-Mengelbier L, Fredlund E, Löfstedt T, et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell. 2006;10(5):413–423. | ||
Filosa JA, Blanco VM. Neurovascular coupling in the mammalian brain. Exp Physiol. 2007;92(4):641–646. | ||
Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100(1):328–335. | ||
Iadecola C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron. 2017;96(1):17–42. | ||
Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32(8):421–431. | ||
Koehler RC, Roman RJ, Harder DR. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009;32(3):160–169. | ||
Marina N, Turovsky E, Christie IN, et al. Brain metabolic sensing and metabolic signaling at the level of an astrocyte. Glia. 2018;66(6):1185-1199. | ||
Vallon M, Chang J, Zhang H, Kuo CJ. Developmental and pathological angiogenesis in the central nervous system. Cell Mol Life Sci. 2014;71(18):3489–3506. | ||
Jung F, Palmer LA, Zhou N, Johns RA. Hypoxic regulation of inducible nitric oxide synthase via hypoxia inducible factor-1 in cardiac myocytes. Circ Res. 2000;86(3):319–325. | ||
Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proceedings of the National Academy of Sciences. 1991;88(13):5680–5684. | ||
Esmaeili Tazangi P, Moosavi SM, Shabani M, Haghani M. Erythropoietin improves synaptic plasticity and memory deficits by decrease of the neurotransmitter release probability in the rat model of Alzheimer’s disease. Pharmacol Biochem Behav. 2015;130:15–21. | ||
Dayyat EA, Zhang SX, Wang Y, Cheng ZJ, Gozal D. Exogenous erythropoietin administration attenuates intermittent hypoxia-induced cognitive deficits in a murine model of sleep apnea. BMC Neurosci. 2012;13:77. | ||
Al-Qahtani JM, Abdel-Wahab BA, Abd El-Aziz SM. Long-term moderate dose exogenous erythropoietin treatment protects from intermittent hypoxia-induced spatial learning deficits and hippocampal oxidative stress in young rats. Neurochem Res. 2014;39(1):161–171. | ||
Viviani B, Bartesaghi S, Corsini E, et al. Erythropoietin protects primary hippocampal neurons increasing the expression of brain-derived neurotrophic factor. J Neurochem. 2005;93(2):412–421. | ||
Dzietko M, Felderhoff-Mueser U, Sifringer M, et al. Erythropoietin protects the developing brain against N-methyl-D-aspartate receptor antagonist neurotoxicity. Neurobiol Dis. 2004;15(2):177–187. | ||
Mahmood A, Lu D, Qu C, et al. Treatment of traumatic brain injury in rats with erythropoietin and carbamylated erythropoietin. J Neurosurg. 2007;107(2):392–397. | ||
Sönmez A, Kabakçi B, Vardar E, et al. Erythropoietin attenuates neuronal injury and potentiates the expression of pCREB in anterior horn after transient spinal cord ischemia in rats. Surg Neurol. 2007;68(3):297–303. | ||
Zhong J, Zhang T, Bloch LM. Dendritic mRNAs encode diversified functionalities in hippocampal pyramidal neurons. BMC Neurosci. 2006;7:17. | ||
Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013;27(1):41–53. | ||
Kapitsinou PP, Liu Q, Unger TL, et al. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood. 2010;116(16):3039–3048. | ||
Yeo EJ, Cho YS, Kim MS, Park JW. Contribution of HIF-1alpha or HIF-2alpha to erythropoietin expression: in vivo evidence based on chromatin immunoprecipitation. Ann Hematol. 2008;87(1):11–17. | ||
Uniacke J, Holterman CE, Lachance G, et al. An oxygen-regulated switch in the protein synthesis machinery. Nature. 2012;486(7401):126–129. | ||
Sakers K, Lake AM, Khazanchi R, et al. Astrocytes locally translate transcripts in their peripheral processes. Proc Natl Acad Sci U S A. 2017;114(19):E3830–E3838. | ||
Jessen KR, Mirsky R. Nonmyelin-forming Schwann cells coexpress surface proteins and intermediate filaments not found in myelin-forming cells: a study of Ran-2, A5E3 antigen and glial fibrillary acidic protein. J Neurocytol. 1984;13(6):923–934. | ||
Sarthy PV, Fu M. Transcriptional activation of an intermediate filament protein gene in mice with retinal dystrophy. DNA. 1989;8(6):437–446. | ||
Feinstein DL, Weinmaster GA, Milner RJ. Isolation of cDNA clones encoding rat glial fibrillary acidic protein: expression in astrocytes and in Schwann cells. J Neurosci Res. 1992;32(1):1–14. | ||
Doetsch F. The glial identity of neural stem cells. Nat Neurosci. 2003;6(11):1127–1134. | ||
Casper KB, Jones K, Mccarthy KD. Characterization of astrocyte-specific conditional knockouts. Genesis. 2007;45(5):292–299. | ||
Chow LM, Zhang J, Baker SJ. Inducible Cre recombinase activity in mouse mature astrocytes and adult neural precursor cells. Transgenic Res. 2008;17(5):919–928. | ||
Bovolenta P, Liem RK, Mason CA. Glial filament protein expression in astroglia in the mouse visual pathway. Brain Res. 1987;430(1):113–126. | ||
Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22(14):5100–5113. | ||
Lee Y, Messing A, Su M, Brenner M. GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia. 2008;56(5):481–493. | ||
Hirrlinger PG, Scheller A, Braun C, Hirrlinger J, Kirchhoff F. Temporal control of gene recombination in astrocytes by transgenic expression of the tamoxifen-inducible DNA recombinase variant CreERT2. Glia. 2006;54(1):11–20. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.