Back to Journals » Drug Design, Development and Therapy » Volume 11
Astragaloside IV protects rat retinal capillary endothelial cells against high glucose-induced oxidative injury
Received 26 September 2017
Accepted for publication 22 November 2017
Published 13 December 2017 Volume 2017:11 Pages 3567—3577
DOI https://doi.org/10.2147/DDDT.S152489
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Yuan Qiao, Chun-Lan Fan, Min-Ke Tang
School of Chinese Materia Medica, Beijing University of Chinese Medicine, Chaoyang District, Beijing, People’s Republic of China
Aim: Diabetic retinopathy is a microvascular complication of diabetes that leads to blindness. Hyperglycemia causes oxidative stress, which is an important cause in the pathogenesis of microangiopathy. The aim of this study was to investigate the potential protective effects of astragaloside IV (AS-IV) in retinal capillary endothelial cells (RCECs) incubated with high glucose conditions.
Methods and results: Based on rat RCECs cultured with high glucose (30 mM) in vitro, a significant increase in cell viability in rat RCECs incubated with both AS-IV and high glucose for 48 or 72 h by MTT assay. The increased viability was accompanied by decreased glucose transporter-1 expression using immunofluorescent assay. Meanwhile, AS-IV reduced intracellular hydrogen peroxide and superoxide, decreased mitochondrial reactive oxygen species in rat RCECs with high glucose by the fluorescent probes, and lowered malondialdehyde levels. In addition, AS-IV increased the activities of total superoxide dismutase, MnSOD, catalase, and glutathione peroxidase. The glutathione content also increased after AS-IV treatment. Furthermore, AS-IV reduced NADPH oxidase 4 expression by western blot method.
Conclusion: These results suggest that the main mechanism underlying the protective effects of AS-IV in high glucose-injured RCECs may be related to its antioxidative function.
Keywords: astragaloside IV, retinal capillary endothelial cells, oxidative stress, Glut1, Nox4
Introduction
Diabetic retinopathy, one of the major microvascular complications of diabetes, is considered the most feared complication among young adults, and it is the leading cause of acquired blindness in working-age adults. Hyperglycemia is known to be the primary triggering factor for the progression of diabetic retinopathy.1 In the pathogenesis of diabetic retinopathy, retinal microvascular cells (endothelial cells and pericytes) are lost before other histopathology is detectable or before the loss of vision is evident.2–4 The capillaries of the retina are lined with endothelial cells that are responsible for maintaining the blood–retinal barrier (BRB) and are supported with an equal number of pericytes that help provide tone to the vessels.
As previously shown, hyperglycemia is associated with endothelial cell dysfunction in diabetic retinopathy. In the retina, glucose transport across the retinal capillary endothelial cells (RCECs) of the inner BRB and the retinal pigment epithelium of the outer BRB is mediated exclusively by sodium-independent glucose transporter-1 (GLUT1), which is the first isoform cloned of the GLUT family,5 and it is expressed in high density in the membranes of human erythrocytes and is characteristically expressed in the blood tissues.6 Since GLUT1 represents a unique portal for entry of glucose into the endothelial cells of the inner BRB, changes in retinal endothelial cell GLUT1 expression and glucose transport may have a major impact on providing substrate for the various pathogenic processes thought to underlie the development of diabetic retinopathy.
Additionally, the retina is highly susceptible to oxidative stress because of its high consumption of oxygen, its proportion of polyunsaturated fatty acids, and its exposure to visible light. It has been suggested that oxidative stress and the production of reactive oxygen species (ROS), including superoxide (O2−) and hydrogen peroxide (H2O2), induced by hyperglycemia play a key role in diabetic retinopathy.7,8 The cellular response to oxidative stress involves the elimination of, protection against, and repair of damage caused by ROS. Scavenger antioxidant enzymes, including superoxide dismutase (SOD) and catalase (CAT), are responsible for the direct elimination of ROS, whereas systems that reconstitute antioxidants (eg, glutathione [GSH] and GSH peroxidase [GSH-PX]) can indirectly reduce ROS.
Astragaloside IV (AS-IV) is a small molecular saponin found in Astragalus membranaceus (Fisch) Bge, a herb widely used in traditional medicine in China. Recent studies have shown that the molecule has diverse pharmacological activities, including anti-inflammatory, antihypertensive, antidiabetic, and myocardial protective properties, both in vitro and in vivo.9–12 Meanwhile, AS-IV has been identified as one of the main active compounds of saponins, and saponins can modulate purinergic receptors such as the P2X7 receptor,13 which represents an attractive pharmacological target to manage the early phase of diabetic retinopathy.14 It has been reported that AS-IV has protective effects on db/db mice with diabetic retinopathy15 or diabetic nephropathy.16 In addition, AS-IV has been shown to have an antioxidant effect, can ameliorate ischemia reperfusion-induced injury in the brain17 and heart,18 and can protect retinal ganglion cells from high glucose-induced damage.19 However, the protective effects of AS-IV on oxidative stress in RCECs have not been investigated. In this study, the rat RCECs elicited by high glucose (30 mM) exposure were chosen as an in vitro model, which is also a good in vitro model to study the inflammatory pathway,20 and we examined the effect of AS-IV on the ROS and malonaldehyde (MDA) contents by evaluating the activities of the total SOD, MnSOD, CAT, GSH-PX, and GSH contents as well as the expressions of NADPH oxidase 4 (Nox4) and GLUT1. The present findings provide evidence for a functional role of AS-IV in RCECs for the prevention of diabetic retinopathy.
Materials and methods
Materials
AS-IV (Figure 1), as a reference compound (purity >98%), was obtained from the Chengdu Must Bio-Technology Co., Ltd. (Chengdu, China). The molecular formula for AS-IV is C41H68O14, and its molecular weight is 784.97 g/mol. AS-IV is a white to pale yellow powder with a melting point of 295.0°C–296.0°C, and it is soluble in methanol, ethanol, and acetone. Endothelial cell medium (ECM) was purchased from ScienCell (San Diego, CA, USA). 2,7′-Dichlorofluorescindiacetate (DCFH-DA), dihydroethidium (DHE), dimethylsulfoxide (DMSO), MTT, and type II collagenase were products of Sigma–Aldrich Co. (St Louis, MO, USA). Mito Tracker Red CM-H2XRos, DAPI, and gelatin were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Rabbit anti-vWf, rabbit anti-CD31, and rabbit anti-Nox4 were products of Santa Cruz Biotechnology Inc. (Dallas, TX, USA); rabbit anti-GLUT1 was purchased from Abcam (Cambridge, MA, USA); fluorescein isothiocyanate (FITC)-labeled secondary antibody (goat anti-rabbit IgG/FITC) was purchased from Zhongshanjinqiao Co. (Beijing, China). MDA, SOD, MnSOD, CAT, and GSH-PX kits were the products of Nanjing Jiancheng Bioengineering Institute (Nanjing, China). GSH and glutathione disulfide (GSSG) measurement kits were purchased from Beyotime Biotech Inc. (Shanghai, China). All other chemicals and regents were of analytical grade. In this study, all experimental protocols were approved by the Review Committee for the Use of Human or Animal Subjects of Beijing University of Chinese Medicine.
 | Figure 1 Chemical structure of AS-IV. |
Animals
Male Sprague Dawley rats weighing 180–200 g were purchased from the Vital River Laboratory Animal Technology Co., Ltd, Beijing, China. The certificate number was SCXK (Jing) 2012-0001. The Principles of Laboratory Animal Care Guidelines, approved by the Animal Ethics Committee at the Beijing University of Chinese Medicine, were strictly followed. All efforts were made to minimize animal suffering and to reduce the number of animals used for the experiments.
Cell culture
RCECs were isolated from Sprague Dawley rats. Briefly, rats were sacrificed, and whole retinas were removed from the eyes. The retinas were minced into small pieces and washed with cold PBS. The retinal fragments were retained on a 100 μm cell strainer. Following harvesting, the fragments were transferred from the sieve to a 15 mL tube containing 0.1% type II collagenase. After incubation at 37°C for 30 min, the retinal fragment suspensions were filtered through a 70 μm cell strainer. After centrifugation at 174× g for 5 min, the pellets containing microvessel fragments were suspended in ECM. The above suspension was transferred to a T-25 cm2 flask precoated with 1% gelatin, maintained in the ECM, and cultured at 37°C in a 5% CO2 humidified incubator. The formation of cells was checked daily, and 50% of the medium was changed every 2–3 days. The cells used in this study were passaged 3–5 times and stained with primary antibodies recognizing rabbit anti-vWf (1:100) and rabbit anti-CD31 (1:100). The images were captured using a fluorescence microscope.
Cell viability assay
The cell viability was assessed using an MTT colorimetric assay. The rat RCECs were seeded at a density of 1×105 cells/mL in gelatin-coated 96-well plates, allowed to attach overnight, and then incubated with either 5.5 mM of glucose or 30 mM of glucose with or without varying concentrations of AS-IV (2, 5, 10, and 20 μM) for 24, 48, and 72 h. After incubation, MTT (0.5 mg/mL) was added into each well. The plates were incubated for 4 h at 37°C, the supernatants were removed, and 150 μL of DMSO was added into each well for 10 min to dissolve the formazan crystals. The absorbance value (A) was determined at 540 nm with an enzyme-linked immunosorbent assay reader (Thermo Labsystems, Helsinki, Finland).
The cell viability (percentage) from the MTT assay was calculated using the following equation:
|
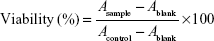
where A is the absorbance value.
Measurement of cellular GLUT1 expression
Cells were seeded at a density of 5×105 cells/cm2 in gelatin-coated 24-well plates, allowed to attach overnight, and then exposed to the experimental conditions for 72 h. After treatments, cells were fixed in 4% paraformaldehyde for 30 min at room temperature. After fixation, the cells were washed with PBS and treated with 3% H2O2-methanol solution for 30 min. Nonspecific binding sites were blocked with normal goat serum for 30 min. Cells were incubated with primary antibody (rabbit polyclonal anti-GLUT1, 1:50) at 4°C overnight. Cells were then washed with PBS and incubated with FITC-labeled secondary antibody (goat anti-rabbit IgG/FITC, 1:100) for 1 h at 37°C. DAPI was used to stain the nuclei in the final step, and then the cells were observed under an ECLIPSE Ti laser confocal microscope (Nikon, Tokyo, Japan). The images were analyzed using ImageJ software.
Measurement of intracellular ROS production
The production of intracellular H2O2 and O2− as well as mitochondrial ROS was determined using the fluorescent probes DCFH-DA, DHE, and Mito Tracker Red CM-H2XRos according to the manufacturer’s instructions. Briefly, rat RCECs were plated in confocal laser special disks at a density of 5×105 cells/cm2 and exposed to varying experimental conditions. After 72 h of incubation, cells were loaded with DCFH-DA (final concentration of 10 μM for 30 min), DHE (final concentration of 5 μM for 30 min), and MitoTracker Red CM-H2XRos (final concentration of 2.5 μM for 20 min) at 37°C, washed with ice cold PBS, and then observed under an ECLIPSE Ti laser confocal microscope (Nikon) equipped with ImageJ software to process the image.
Measurement of cellular MDA content
Rat RCECs were incubated with varying experimental conditions for 72 h at a density of 1×106 cells/cm2. The cellular MDA content was measured using a kit. Briefly, MDA reacts with thiobarbituric acid (TBA) at 90°C–100°C under acidic conditions. The reaction yielded a pink MDA-TBA conjugate, which was measured at a wavelength of 532 nm. The cellular MDA was expressed as nanomoles per milligram of cellular protein.
Measurement of the activities of antioxidant enzymes
Rat RCECs were incubated with varying experimental conditions for 72 h at a density of 1×106 cells/cm2. Cellular extracts were prepared for the detection of SOD, MnSOD, CAT, and GSH-PX activities by the corresponding kits. Briefly, the SOD activity was measured according to its ability to inhibit the production of a water-soluble formazan dye. The CAT activity was determined by the conversion of H2O2 into H2O and O2. The GSH-PX activity was determined by measuring the rate of oxidation of GSH to GSSG by the dismutation of cumene hydroperoxide, which is catalyzed by GSH-PX. The total SOD, MnSOD, CAT, and GAH-PX activities were expressed as units per milligram of protein.
Measurement of cellular GSH and GSSG contents
Rat RCECs were seeded in gelatin-coated T-25 cm2 flasks and incubated with varying experimental conditions for 72 h at a density of 1×106 cells/cm2. The cells were harvested and prepared for GSH and GSSG measurements using a kit. The GSH assay is based on the chemical conjugation of GSH with 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB). The total GSH was measured by first reducing oxidized GSH using GSH reductase and b-NADPH followed by conjugation with DTNB. To measure the GSSG, GSH was first covalently reacted with 2-vinylpyridine, and then GSSG was measured as described earlier. GSH and GSSG levels were expressed in nanomoles per milligram of protein.
Measurement of Nox4 expression
The expression of cellular Nox4 was determined by Western blot assay. Rat RCECs were seeded at a density of 1×106 cells/cm2 in gelatin-coated T-25 cm2 flasks and then exposed to various experimental conditions. For the Western blot assay, cells were harvested in lysis buffer (50 mM Tris [pH=7.5], 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 1 mM EDTA, and 0.1% SDS), and the protein concentrations were determined by a bicinchoninic acid protein assay. The protein (8 μg) underwent electrophoresis on a 10% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and incubated with primary antibody (rabbit polyclonal anti-Nox4, 1:500) at 4°C overnight. After washing, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 40 min. The immune reactive bands were visualized using an enhanced chemiluminescence kit. The relative densities of the bands were determined by image analysis with Image-Pro Plus software.
Data analysis and statistics
All data are expressed as mean ± standard deviation (SD). One-way ANOVA, followed by a Student–Newman–Keuls test was used to analyze all the data. Values of P<0.05 and P<0.01 were considered significant. The statistical analyses were performed using SPSS 17.0 software (SPSS Inc., Chicago, IL, USA).
Results
Validation of rat RCECs
In this study, the migration of cells from fragments of retinal microvessels began 10 days after plating. The cells showed a cobble-stone morphology and formed a contact-inhibited monolayer (Figure 2A). To confirm that these cells were endothelial cells, the expressions of the endothelial cell-specific markers vWF and CD31 were detected by immunofluorescence analysis. Figure 2B shows that the cells express high levels of vWF, and Figure 2C shows that the cells express high levels of CD31.
 | Figure 2 Verification of rat RCECs. (A) Cells were cultured in endothelial cell medium for 10 days, and the characteristic cobble-stone morphology of RCECs was observed, scale bar: 100 μm, magnification: ×100. Immunocytochemical staining showed that the rat RCECs expressed vWf (B, green fluorescent dye) and CD31 (C, green fluorescent dye), scale bar: 40 μm, magnification: ×200. All nuclei were stained with DAPI (blue fluorescent dye). |
AS-IV increases cell viability in rat RCECs treated with high glucose
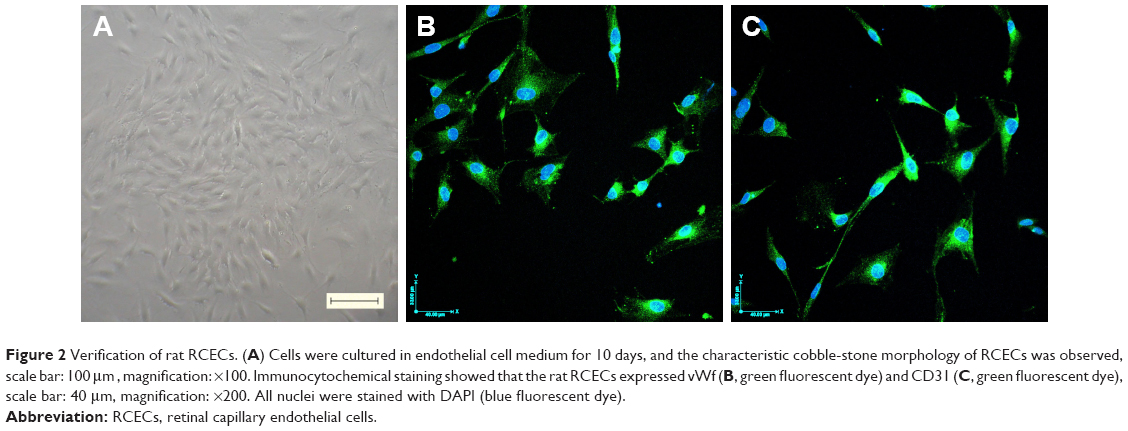
As shown in Figure 3, we investigated the cell viability in different experimental conditions. Compared with 5.5 mM glucose-treated cells, the cell viability was significantly decreased in the cells treated with 30 mM glucose for 24, 48, and 72 h (P<0.01). After 48 h or 72 h of treatment, AS-IV at 5, 10, and 20 μM concentrations significantly increased the cell viability (P<0.01). However, AS-IV at 20 μM compromised the protection effect of AS-IV against “hyperglycemia”. Based on these findings, 10 μM AS-IV was selected as the final treatment concentration. These data suggest that AS-IV may reverse the inhibition of cell viability by high glucose.
 | Figure 3 AS-IV increased cell viability in rat RCECs exposed to high glucose. Seeded in 96-well plates, the rat RCECs were incubated with varying concentrations of AS-IV (2, 5, 10, and 20 μM) in high glucose (30 mM glucose). The MTT assay was used to examine cell viability after culturing for 24 h (A), 48 h (B), and 72 h (C). The results showed that 5, 10, and 20 μM of AS-IV increased cell viability after 48 and 72 h, respectively. The experiment was repeated three times. Data represent mean ± SD (n=15). ##P<0.01 and #P<0.05 versus 5.5 mM glucose; **P<0.01 versus 30 mM glucose. |
AS-IV inhibits high glucose-induced GLUT1 expression in rat RCECs
Immunofluorescence was used to assay the change in the GLUT1 levels released by cultured rat RCECs in vitro in high glucose and AS-IV (10 μM) for 72 h. As shown in Figure 4, the expression of GLUT1 in rat RCECs in vitro was enhanced significantly under high glucose (P<0.01), and it was mainly expressed in the membranes of cells. Treatment with AS-IV (10 μM) can decrease the expression of GLUT1 in the presence of high glucose (P<0.01) and inhibit the translocation of GLUT1 from the cytoplasm to the plasma membrane, where it may participate in mediating glucose entry into the endothelial cell and protect rat RCECs from injury by high glucose. In addition, there was a significant difference in GLUT1 expression between the groups treated with 5.5 mM glucose and AS-IV (P<0.05).
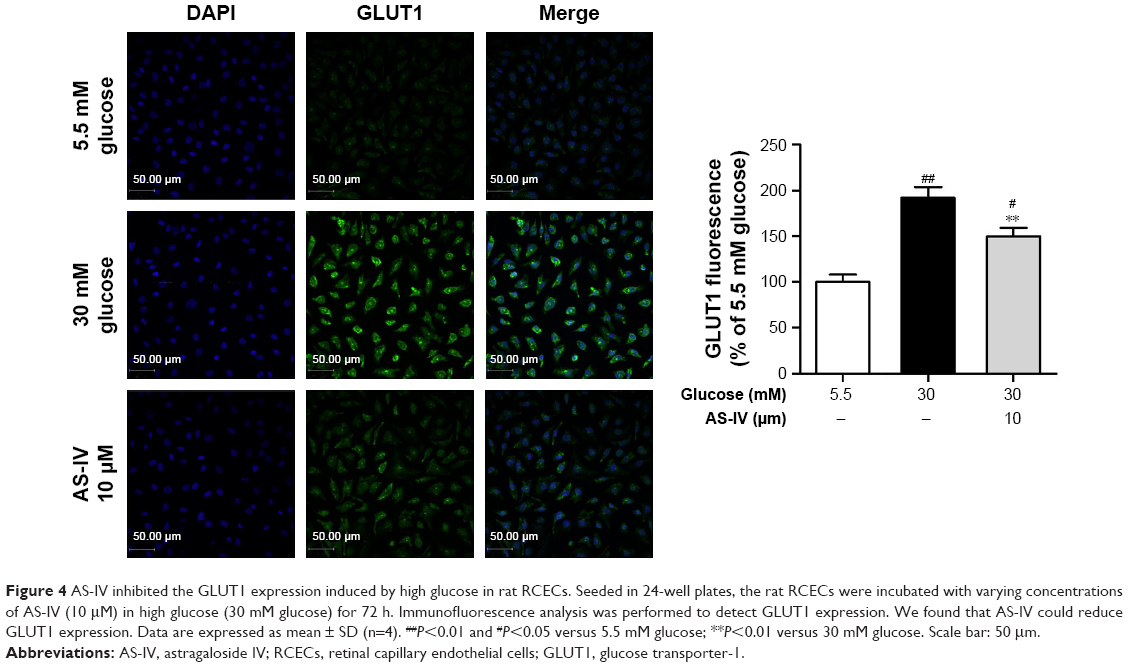 | Figure 4 AS-IV inhibited the GLUT1 expression induced by high glucose in rat RCECs. Seeded in 24-well plates, the rat RCECs were incubated with varying concentrations of AS-IV (10 μM) in high glucose (30 mM glucose) for 72 h. Immunofluorescence analysis was performed to detect GLUT1 expression. We found that AS-IV could reduce GLUT1 expression. Data are expressed as mean ± SD (n=4). ##P<0.01 and #P<0.05 versus 5.5 mM glucose; **P<0.01 versus 30 mM glucose. Scale bar: 50 μm. |
AS-IV inhibits the ROS and MDA generation induced by high glucose in rat RCECs
In our study, compared with 5.5 mM glucose-treated cells, the production of intracellular H2O2 and O2− as well as mitochondrial ROS were significantly enhanced (P<0.01) in rat RCECs treated with 30 mM glucose. These results were observed by staining with three different fluorogenic probes: DCFH-DA, DHE, and Mito Tracker Red CM-H2XRos. AS-IV at 10 μM significantly reduced the 30 mM glucose-induced increase in intracellular H2O2 and O2− as well as mitochondrial ROS (P<0.01) (Figure 5A and B). We then measured MDA, a product generated from lipid peroxidation. After 72 h of 30 mM glucose treatment, the MDA content was significantly increased (P<0.01) compared to cells treated with 5.5 mM glucose. The MDA content in rat RCECs treated with 10 μM AS-IV was marginally decreased (P<0.01) in comparison to 30 μM glucose treatment (Figure 5C). In addition, there were significant differences in the production of intracellular H2O2 and O2− as well as mitochondrial ROS between the groups treated with 5.5 mM glucose and AS-IV (P<0.05). The results suggest that AS-IV may reduce the levels of ROS and MDA, showing an effective attenuation of high glucose-induced oxidative injury.
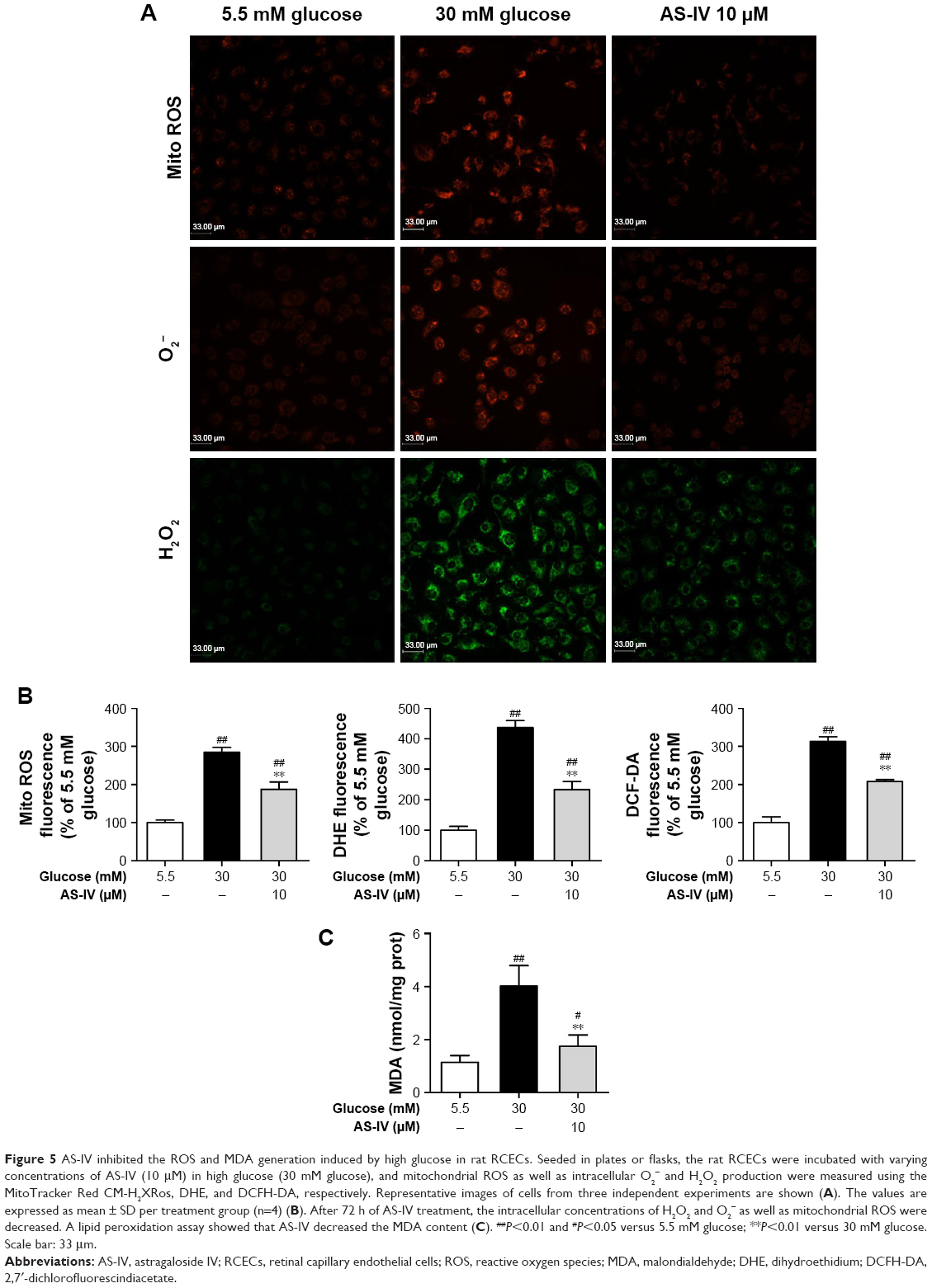 | Figure 5 AS-IV inhibited the ROS and MDA generation induced by high glucose in rat RCECs. Seeded in plates or flasks, the rat RCECs were incubated with varying concentrations of AS-IV (10 μM) in high glucose (30 mM glucose), and mitochondrial ROS as well as intracellular O2− and H2O2 production were measured using the MitoTracker Red CM-H2XRos, DHE, and DCFH-DA, respectively. Representative images of cells from three independent experiments are shown (A). The values are expressed as mean ± SD per treatment group (n=4) (B). After 72 h of AS-IV treatment, the intracellular concentrations of H2O2 and O2− as well as mitochondrial ROS were decreased. A lipid peroxidation assay showed that AS-IV decreased the MDA content (C). ##P<0.01 and #P<0.05 versus 5.5 mM glucose; **P<0.01 versus 30 mM glucose. Scale bar: 33 μm. |
AS-IV increases the activity of antioxidant enzymes and regulates the GSH redox system in rat RCECs exposed to high glucose
When cells were stimulated with 30 μM glucose, significant decreases in the activities of SOD, MnSOD, CAT, and GSH-PX were observed (P<0.01). However, treatment with 10 μM AS-IV significantly elevated the activities of SOD, MnSOD, CAT, and GSH-PX (P<0.01), as shown in Figure 6A. When exposed to 30 mM glucose, the GSH levels in cells markedly decreased while the GSSG levels increased, subsequently reducing the GSH/GSSG ratio (P<0.01). Treatment with AS-IV increased the GSH levels, decreased the GSSG levels, and enhanced the GSH/GSSG ratio (P<0.01) (Figure 6B). In addition, there were significant differences in the activities of SOD, MnSOD, CAT, and GSH-PX (all P<0.05) as well as the levels of GSH and GSSG (P<0.05) between the groups treated with 5.5 mM glucose and AS-IV (P<0.05). These data suggest that AS-IV shows significant antioxidative effects against high glucose-induced oxidative stress in RCECs.
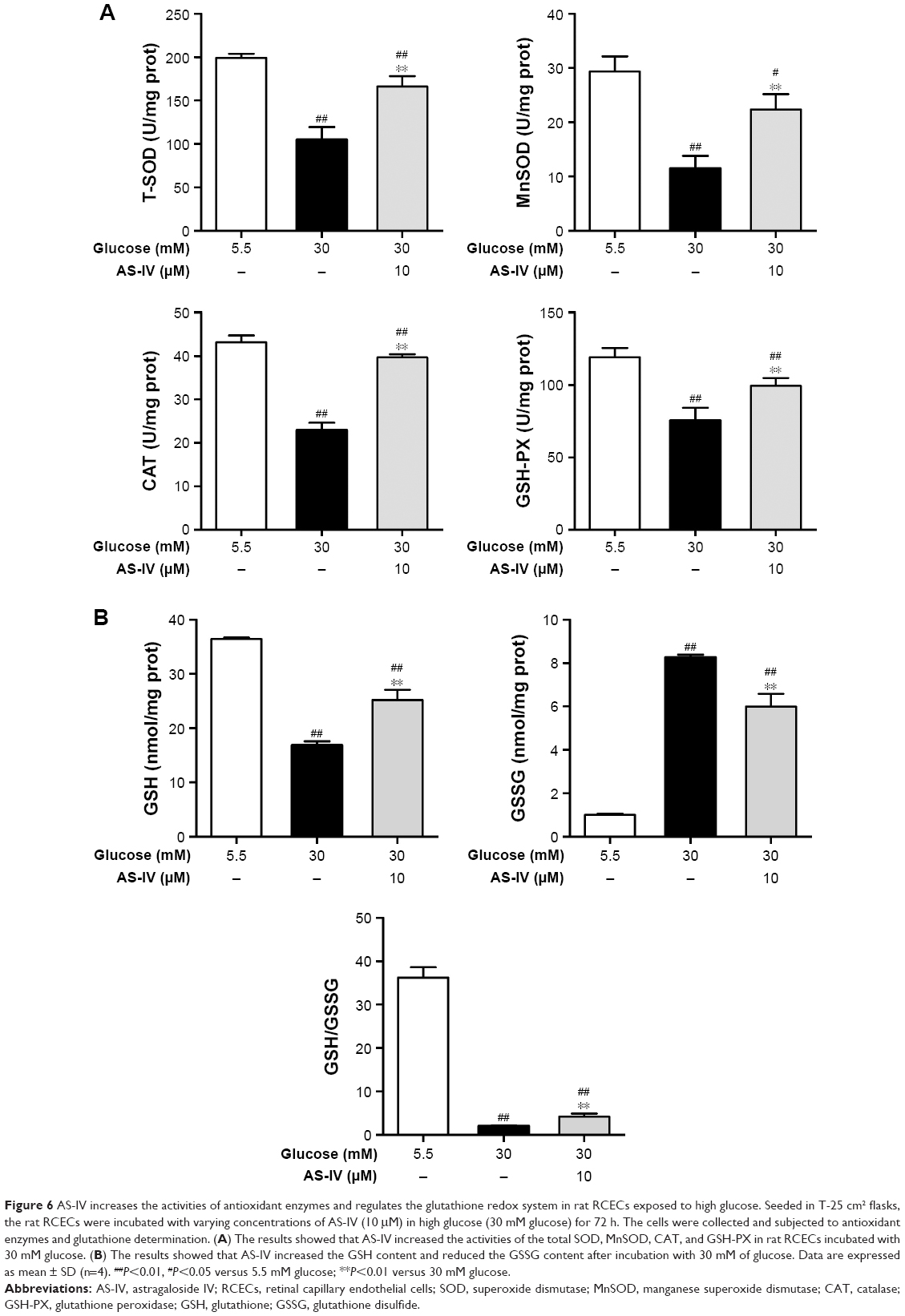 | Figure 6 AS-IV increases the activities of antioxidant enzymes and regulates the glutathione redox system in rat RCECs exposed to high glucose. Seeded in T-25 cm2 flasks, the rat RCECs were incubated with varying concentrations of AS-IV (10 μM) in high glucose (30 mM glucose) for 72 h. The cells were collected and subjected to antioxidant enzymes and glutathione determination. (A) The results showed that AS-IV increased the activities of the total SOD, MnSOD, CAT, and GSH-PX in rat RCECs incubated with 30 mM glucose. (B) The results showed that AS-IV increased the GSH content and reduced the GSSG content after incubation with 30 mM of glucose. Data are expressed as mean ± SD (n=4). ##P<0.01, #P<0.05 versus 5.5 mM glucose; **P<0.01 versus 30 mM glucose. |
AS-IV inhibits the Nox4 expression induced by high glucose in rat RCECs
As shown in Figure 7, 30 mM glucose significantly enhanced the expression of Nox4 protein in rat RCECs (P<0.01), determined by Western blot assay analysis. AS-IV at 10 μM markedly inhibited the high glucose-induced increase of Nox4 expression (P<0.05). In addition, there was a significant difference in Nox4 expression between the groups treated with 5.5 mM glucose and AS-IV (P<0.05). These data suggest that AS-IV regulates Nox4 expression to maintain a balance between the production of ROS and the antioxidant defense system.
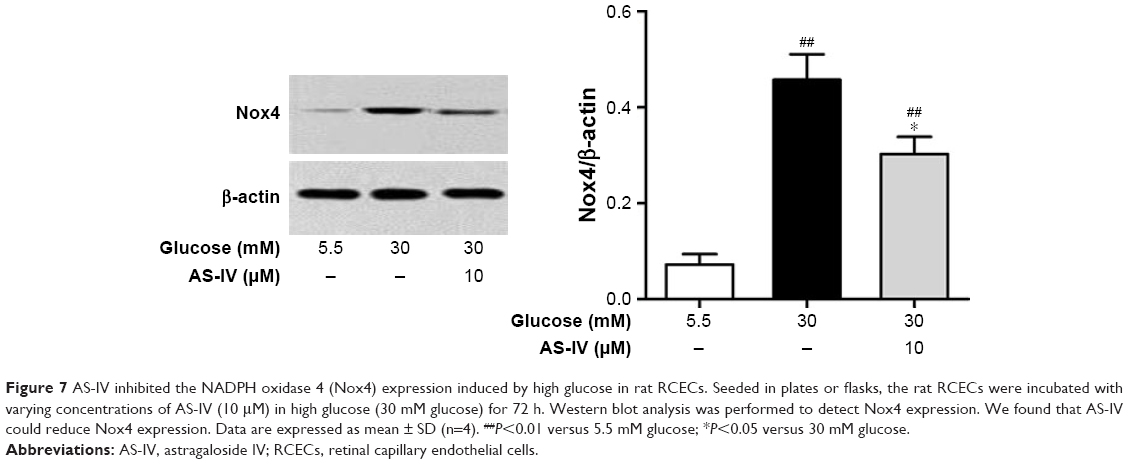 | Figure 7 AS-IV inhibited the NADPH oxidase 4 (Nox4) expression induced by high glucose in rat RCECs. Seeded in plates or flasks, the rat RCECs were incubated with varying concentrations of AS-IV (10 μM) in high glucose (30 mM glucose) for 72 h. Western blot analysis was performed to detect Nox4 expression. We found that AS-IV could reduce Nox4 expression. Data are expressed as mean ± SD (n=4). ##P<0.01 versus 5.5 mM glucose; *P<0.05 versus 30 mM glucose. |
Discussion
In the present study, we demonstrated for the first time that AS-IV is capable of protecting RCECs from the oxidative stress caused by high glucose concentrations. Treatment with AS-IV promoted the antioxidant capacity of RCECs, inhibited lipid peroxidation, and reduced ROS generation.
Endothelial cells are key participants in retinal diseases,21 and the loss of endothelial cells in the retinal capillaries occurs early in diabetic retinopathy. Indeed, several reports have shown that retinal endothelial cells undergo apoptosis in the pathogenesis of diabetes.22–24 When assayed in culture, the exposure of human,25 rat,26 and bovine27 retinal endothelial cells to high glucose causes dysfunction, which is reflected by altered cellular activities. To determine the extent of injury elicited in RCECs by high glucose and to investigate whether AS-IV would effectively ameliorate damage, cell viability was examined in rat RCECs cultured in high glucose concentrations (30 mM), simulating a diabetic microenvironment in vivo. Our results revealed that the incubation of rat RCECs with high glucose (30 mM) for 48 and 72 h produced a consistent reduction in cell viability. These results confirmed the detrimental role of hyperglycemia in the functionality of RCECs. However, at the same time, coincubation with AS-IV significantly improved the viability of RCECs.
As is known, glucose transport across the RCECs of the inner BRB is mediated exclusively by the sodium-independent glucose transporter GLUT1,28 and changes in the endothelial glucose transport and GLUT1 abundance in the barrier of the retina may have profound effects on diabetic retinopathy. In this study, we demonstrate the upregulation of GLUT1 protein expression in rat RCECs exposed to high glucose, and that it is mainly expressed in the membranes of cells, as previously reported by others.29,30 AS-IV treatment can downregulate the expression of GLUT1 with high glucose treatment and inhibit the translocation of GLUT1 from the cytoplasm to the plasma membrane, where it may participate in mediating glucose entry into the endothelial cell. Taken together, this study suggested that AS-IV can cure the diabetic retinopathy by decreasing the expression of GLUT1 in a high glucose solution.
Oxidative stress, a hallmark of high glucose-induced endothelial dysfunction, was significantly attenuated by AS-IV treatment in the current study. There is growing evidence that oxidative stress is involved in the pathogenesis of diabetic retinopathy, and oxidative stress has been associated with the increase of apoptosis in retinal endothelial cells exposed to hyperglycemic conditions.31 Several studies have reported that the overproduction of ROS is responsible for hyperglycemia-induced oxidative damage.32,33 Our results revealed that the mitochondrial ROS as well as intracellular H2O2 and O2− were significantly increased when RCECs were cultured in a high glucose (30 mM) medium. To confirm whether increases in the ROS due to high glucose cause oxidative stress in RCECs, we measured MDA, a product generated from lipid peroxidation. Indeed, high glucose caused an increase in MDA generation. Thus, hyperglycemia appears to increase oxidative damage in RCECs, an observation that is in agreement with previous studies.34,35 Interestingly, we found that AS-IV treatment inhibited the high glucose-induced MDA and ROS formation, suggesting a protective role of AS-IV in high glucose-induced RCECs. In addition, we found that the increase in ROS production observed in the RCECs treated with high glucose was associated with an increase in the generation of Nox4. NADPH oxidase of the Nox family is a major source of ROS, and the overexpression of Nox4 can cause an imbalance between the production of ROS and the antioxidant defense system.36 AS-IV significantly reduced the Nox4 expression levels. These data suggest that AS-IV has significant protective antioxidative effects against high glucose-induced injury in RCECs.
As is known, in diabetes, the antioxidant defense enzymes responsible for scavenging free radicals and maintaining redox homeostasis such as SOD, GSH-PX, and CAT are diminished in the retina.37,38 SOD is considered as first-line defense against ROS, and SOD is present in nearly all cells and converts O2− into H2O2. However, intramitochondrial O2− does not readily cross mitochondrial membranes. MnSOD, a superoxide scavenging enzyme in mitochondria, converts intramitochondrial O2− to H2O2, which can diffuse out of the mitochondria. As H2O2 can react with ROS, it is further degraded by one of two antioxidant enzymes, GSH-PX or CAT.39,40 In our study, AS-IV-treated RCECs inhibited the SOD, MnSOD, GSH-PX, and CAT activities induced by 30 mM glucose. Furthermore, the cell is equipped with GSH, an intracellular antioxidant that is one of the most important defense enzymes in the cell. Our results indicated that AS-IV treatment caused a significant enhancement of the GSH content. Therefore, it is reasonable to conclude that reducing the ROS overload, coupled with the activities of the antioxidant enzymes and GSH redox system, may be the main protective mechanisms of AS-IV in RCECs.
In conclusion, this study describes the cellular protective effects of AS-IV against high glucose-induced oxidative injury in RCECs. This protection was associated with the preservation of cell function by the stimulation of cellular antioxidant defenses. These findings provide an important basis for further studies to address the protective effect of AS-IV against diabetic retinopathy.
Acknowledgment
This research is supported by Grant “Major Innovative drug development Project (2011ZX11201) from the Ministry of Science and Technology of China”.
Disclosure
The authors report no conflicts of interest in this work.
References
Chew EY, Davis MD, Danis RP, et al. The effects of medical management on the progression of diabetic retinopathy in persons with type 2 diabetes: the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Eye Study. Ophthalmology. 2014;121(12):2443–2451. | ||
Kowluru RA. Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxi Redox Signal. 2005;7(11–12):1581–1587. | ||
Kowluru RA, Odenbach S. Effect of long-term administration of alpha-lipoic acid on retinal capillary cell death and the development of retinopathy in diabetic rats. Diabetes. 2005;53(12):3233–3238. | ||
Hammes HP. Pericytes and the pathogenesis of diabetic retinopathy. Horm Metab Res. 2005;37(1):39–43. | ||
Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects of Med. 2013;34(2–3):121. | ||
Silva Filho TJD, Moura IDS, Gonzaga AKG, et al. Importance of GLUT1 in differential diagnosis of vascular anomalies. J Vascular Brasileiro. 2015;14(2):168–176. | ||
Evans JL, Goldfine ID, Maddux BA, et al. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23(5):599–622. | ||
Kassab A, Piwowar A. Cell oxidant stress delivery and cell dysfunction onset in type 2 diabetes. Biochimie. 2012;94(9):1837–1848. | ||
Ren S, Zhang H, Mu Y, et al. Pharmacological effects of Astragaloside IV: a literature review. J Tradit Chin Med. 2013;33(3):413. | ||
He Y, Du M, Gao Y, et al. Astragaloside IV attenuates experimental autoimmune encephalomyelitis of mice by counteracting oxidative stress at multiple levels. PLoS One. 2013;8(10):e76495. | ||
Zhang N, Wang XH, Mao SL, et al. Astragaloside IV improves metabolic syndrome and endothelium dysfunction in fructose-fed rats. Molecules. 2011;16(5):3896–3907. | ||
Gui D, Huang J, Guo Y, et al. Astragaloside IV ameliorates renal injury in streptozotocin-induced diabetic rats through inhibiting NF-κB-mediated inflammatory genes expression. Cytokine. 2013;61(3):970–977. | ||
Helliwell RM, Shioukhuey CO, Dhuna K, et al. Selected ginsenosides of the protopanaxdiol series are novel positive allosteric modulators of P2X7 receptors. Br J Pharmacol. 2015;172(13):3326–3340. | ||
Cbm P, Giurdanella G, Di PL, et al. P2X7 receptor antagonism: implications in diabetic retinopathy. Biochem Pharmacol. 2017;15(138):130–139. | ||
Ding Y, Yuan S, Liu X, et al. Protective effects of astragaloside IV on db/db mice with diabetic retinopathy. PLoS One. 2014;9(11):112207. | ||
Sun H, Wang W, Han P, et al. Astragaloside IV ameliorates renal injury in db/db mice. Sci Rep. 2016;6:32545. | ||
Zhang WD, Chen H, Zhang C, et al. Astragaloside IV from Astragalus membranaceus shows cardioprotection during myocardial ischemia in vivo and in vitro. Planta Med. 2006;72(1):4–8. | ||
Chen J, Gui D, Chen Y, et al. Astragaloside IV improves high glucose-induced podocyte adhesion dysfunction via alpha3beta1 integrin upregulation and integrin-linked kinase inhibition. Biochem Pharmacol. 2008;76(6):796–804. | ||
Hao M, Kuang HY, Fu Z, et al. [Protective effects of astragaloside IV on high-glucose-induced damage of retinal ganglion cells]. Zhonghua Yi Xue Za Zhi. 2012;92(30):2104. Chinese. | ||
Giurdanella G, Lazzara F, Caporarello N, et al. Sulodexide prevents activation of the PLA2/COX-2/VEGF inflammatory pathway in human retinal endothelial cells by blocking the effect of AGE/RAGE. Bio Pharmacol. 2017;14(142):145–154. | ||
Bharadwaj AS, Appukuttan B, Wilmarth PA, et al. Role of the retinal vascular endothelial cell in ocular disease. Prog Retin Eye Res. 2013;32(1):102–180. | ||
Dagher Z, Park YS, Asnaghi V, et al. Studies of rat and human retinas predict a role for the polyol pathway in human diabetic retinopathy. Diabetes. 2004;53(9):2404–2411. | ||
Du Y, Miller CM, Kern TS. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic Biol Med. 2003;35(11):1491–1499. | ||
Du Y, Smith MA, Miller CM, et al. Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J Neurochem. 2002;80(5):771. | ||
Williams KP, Steinle JJ. Maintenance of beta-adrenergic receptor signaling can reduce Fas signaling in human retinal endothelial cells. Exp Eye Res. 2009;89(4):448. | ||
Panjala SR, Steinle JJ. Insulin and β-adrenergic receptors inhibit retinal endothelial cell apoptosis through independent pathways. Neurochem Res. 2011;36(4):604–612. | ||
Kowluru RA, Kowluru A, Kanwar M. Small molecular weight G-protein, H-Ras, and retinal endothelial cell apoptosis in diabetes. Mol Cell Biochem. 2007;296(1–2):69–96. | ||
Takata K, Hirano H. Mechanism of glucose transport across the human and rat placental barrier: a review. Micro Res Tech. 2010;38(1–2):145–152. | ||
Fernandes R, Suzuki K, Kumagai AK. Inner blood-retinal barrier GLUT1 in long-term diabetic rats: an immunogold electron microscopic study. Invest Ophthalmol Vis Sci. 2003;44(7):3150. | ||
Fernandes R, Carvalho AL, Kumagai A, et al. Downregulation of retinal GLUT1 in diabetes by ubiquitinylation. Mol Vis. 2004;10(74–75):618. | ||
Grammas P, Riden M. Retinal endothelial cells are more susceptible to oxidative stress and increased permeability than brain-derived endothelial cells. Microvasc Res. 2003;65(1):18–23. | ||
Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. | ||
Kiritoshi S, Nishikawa T, Sonoda K, et al. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: potential role in diabetic nephropathy. Diabetes. 2003;52(10):2570. | ||
Kowluru RA. Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxid Redox Signal. 2005;7(11–12):1581. | ||
Nishikawa T, Araki E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal. 2007;9(3):343–353. | ||
Griendling KK. Novel NAD(P)H oxidase in the cardiovascular system. Heart. 2007;90(5):491–493. | ||
Raffaele M, Di PD, Chiara V, et al. Lipid peroxidation and total antioxidant capacity in vitreous, aqueous humor, and blood samples from patients with diabetic retinopathy. Mol Vis. 2011;17(145):1298–1304. | ||
Matough FA, Budin SB, Hamid ZA, et al. The role of oxidative stress and antioxidants in diabetic complications. Sultan Qaboos Univ Med J. 2012;12(1):5–18. | ||
Haskins K, Bradley B, Powers K, et al. Oxidative stress in type 1 diabetes. Ann N Y Acad Sci. 2003;1005(1):43–54. | ||
Kowluru RA, Tang J, Kern TS. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. Diabetes. 2001;50(8):1938–1942. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.