Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Associations of autophagy with lung diffusion capacity and oxygen saturation in severe COPD: effects of particulate air pollution
Authors Lee K, Chiang L, Ho S, Liu W ![]() , Chen T, Feng P, Su C, Chuang K, Chang C, Chuang H
, Chen T, Feng P, Su C, Chuang K, Chang C, Chuang H
Received 22 March 2016
Accepted for publication 21 May 2016
Published 11 July 2016 Volume 2016:11(1) Pages 1569—1578
DOI https://doi.org/10.2147/COPD.S108993
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Kang-Yun Lee,1,2,* Ling-Ling Chiang,1,3,* Shu-Chuan Ho,1,3 Wen-Te Liu,1–3 Tzu-Tao Chen,1 Po-Hao Feng,1,2 Chien-Ling Su,1,3 Kai-Jen Chuang,4,5 Chih-Cheng Chang,1,2 Hsiao-Chi Chuang1–3
1Division of Pulmonary Medicine, Department of Internal Medicine, Shuang Ho Hospital, 2Department of Internal Medicine, School of Medicine, 3School of Respiratory Therapy, 4Department of Public Health, School of Medicine, College of Medicine, 5School of Public Health, College of Public Health and Nutrition, Taipei Medical University, Taipei City, Taiwan
*These authors contributed equally to this work
Abstract: Although traffic exposure has been associated with the development of COPD, the role of particulate matter <10 µm in aerodynamic diameter (PM10) in the pathogenesis of COPD is not yet fully understood. We assessed the 1-year effect of exposure to PM10 on the pathogenesis of COPD in a retrospective cohort study. We recruited 53 subjects with COPD stages III and IV and 15 healthy controls in a hospital in Taiwan. We estimated the 1-year annual mean levels of PM10 at all residential addresses of the cohort participants. Changes in PM10 for the 1-year averages in quintiles were related to diffusion capacity of the lung for carbon monoxide levels (r=−0.914, P=0.029), changes in the pulse oxygen saturation (ΔSaO2; r=−0.973, P=0.005), receptor for advanced glycation end-products (r=−0.881, P=0.048), interleukin-6 (r=0.986, P=0.002), ubiquitin (r=0.940, P=0.017), and beclin 1 (r=0.923, P=0.025) in COPD. Next, we observed that ubiquitin was correlated with ΔSaO2 (r=−0.374, P=0.019). Beclin 1 was associated with diffusion capacity of the lung for carbon monoxide (r=−0.362, P=0.028), ΔSaO2 (r=−0.354, P=0.032), and receptor for advanced glycation end-products (r=−0.471, P=0.004). Autophagy may be an important regulator of the PM10-related pathogenesis of COPD, which could cause deterioration in the lung diffusion capacity and oxygen saturation.
Keywords: 6-minute walk distance, air pollution, beclin 1, lung function, receptor for advanced glycation end-products
Introduction
COPD is an environmental-related health problem and is the fourth leading cause of death worldwide.1 It is estimated that this condition will become the third leading cause of death worldwide by 2020. COPD currently affects ~10% of the population older than 45 years and 50% of heavy smokers.2 Notably, the Global Initiative for Chronic Obstructive Lung Disease (GOLD) reported that high levels of air pollution indoors/outdoors are deleterious to individuals with existing pulmonary disease.1 However, the mechanisms of the responses to exposure to outdoor air pollution involved in the development of COPD remain unclear.1 Therefore, an investigation of the mechanisms of the effects of air pollution on patients with COPD is crucial for understanding the pathogenesis of COPD.
Air pollutants such as particulate matter <10 μm in aerodynamic diameter (PM10) at levels typically produced from combustion processes have been linked to adverse health effects, including exacerbation of preexisting respiratory diseases such as COPD.3 For example, the physiological effects of PM10 on lung function have been studied for several decades, and a number of studies reported an association of exposure to PM10 with COPD. Schikowski et al4 demonstrated chronic effects of PM10 on the prevalence and incidence of COPD. Kumar et al3 further showed that the risk of acute exacerbation of COPD increased by 2.3% with a unit increase in exposure to PM2.5 (particulate matter <2.5 μm in aerodynamic diameter), and the exposed groups (who experienced exposure to PM2.5 of >15.4 μg/m3) were 54% more likely than the reference group to be admitted for acute exacerbation of COPD. The lungs are the primary organ of entry for airborne agents; therefore, clearance of deposited foreign materials from the lungs is critical for whole-body defense. When particles are inhaled into the lungs, several biological mechanisms may be altered, particularly oxidative stress and inflammation.
COPD is not fully reversible and is associated with pathological changes in the proximal and peripheral airways, lung parenchyma, and pulmonary vasculature.1 Chronic inflammation in the lungs of COPD patients results in structural changes and narrowing of the small airways. The inflammatory responses and narrowing of peripheral airways in COPD cause a reduction in the forced expiratory volume in the first second (FEV1). Emphysema-driven parenchymal destruction contributes to airflow limitations.5 Repair and defense mechanisms become abnormal during the progression of COPD. For example, in patients with COPD, chronic inflammation resulting from repeated injury and repair and an increased number of inflammatory cell types are observed in the airways, lung parenchyma, and pulmonary vasculature. With regard to the pathogenesis of COPD, several mechanisms contribute to the persistence of lung inflammation after smoking cessation.6 However, possible mechanisms underlying PM10-driven COPD development/progression remain unclear.
Autophagy is considered to be an important inflammatory pathway involved in the development of COPD.7 Cigarette smoke was identified as an important determinant of increasing oxidative damage to the epithelium, and this process involves autophagic pathways.8 Gannon et al9 reported that significant mitochondrial damage and an increase in beclin 1 protein expression were induced in mice after exposure to cigarette smoke. Furthermore, an in vitro study showed that PM10-driven oxidative damage was associated with an increase in beclin 1 protein expression,10 suggesting that autophagy plays an important role in regulating particle cytotoxicity; however, the potential mechanisms underlying PM-induced autophagy in COPD remain unclear.
The objective of this study was to investigate PM10-related pathogenesis of COPD. First, clinically relevant indicators/biomarkers for identifying the progression of COPD (such as lung function, oxygen saturation, oxidative stress, and inflammation) were correlated with corresponding PM10 levels in patients with severe COPD. Biomarkers for the ubiquitin-proteasome system and autophagy were related to PM10 in COPD. Finally, associations of ubiquitin and autophagy with lung function, oxygen saturation, and inflammatory biomarkers were determined in order to understand the potential pathogenic effects of COPD caused by PM10.
Methods
Ethical considerations
The ethics committees of Taipei Medical University-Joint Institutional Review Board approved the study protocol (no 201310027). This study was registered in The Chinese Clinical Trial Register (ChiCTR-OCC-13004025). The methods were carried out in accordance with the the approved protocols and relevant regulation (Human Research Act, Human Research Ethics Policy Guidelines, the Medical Care Act, the Physicians Act, the Guidance on the Collection and Use of Tissue Samples for Research Uses and the Taiwan Good Clinical Practice [GCP] Code issued by Taiwan Central Government and the Ministry of Health and Welfare) or guidelines (The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Participants in Research). All subjects received written and oral information prior to inclusion and provided informed consent.
Study design and population
This was a retrospective study evaluating the effects of particulate air pollution on pulmonary-to-systemic responses in patients with COPD. COPD patients at GOLD stages III and IV and healthy controls (nonsmokers and smokers) were recruited in a medical center in New Taipei City, Taiwan, between January 2013 and December 2014. All subjects were between 40 years and 80 years of age at the time of inclusion. All COPD subjects were informed of the diagnosis and exhibited postbronchodilation FEV1/forced vital capacity (FVC) ratios of <70%. Subjects without COPD exhibited an FEV1/FVC ratio of ≥75% and FEV1 of ≥80% of the predicted value. Definitions of COPD GOLD stages were FEV1 of ≥80% predicted for stage I (mild COPD), FEV1 of 50%–79% predicted for stage II (moderate COPD), FEV1 of 30%–49% predicted for stage III (severe COPD), and FEV1 of <30% predicted for stage IV (very severe COPD).1 Patients diagnosed with current cancer or active inflammatory disease or who experienced exacerbation during 4 weeks prior to the study were excluded. Prior to recruitment in the study, a physical examination was performed, and a clinical interview was carried out regarding smoking, comorbidities, and medication use. All COPD subjects continued with a stable regimen of medications throughout the study.
Lung function test and oxygen saturation
Spirometry, body plethysmography, and lung diffusion capacity testing were carried out according to the European Respiratory Society.11 Subjects performed the lung function test at the time of recruitment using a MasterScreen™ body plethysmograph (CareFusion, San Diego, CA, USA). Postbronchodilation measurements for FEV1 and FVC were taken, and the ratio of FEV1/FVC was calculated. The ratio of the residual volume to total lung capacity (RV/TLC) and diffusion capacity of the lung for carbon monoxide (DLCO) were obtained and presented as percentages of predicted values. Exercise capacity was assessed with the 6-minute walking distance (6-MWD) test, according to American Thoracic Society guidelines.12 The scale was validated and shown to be reliable for evaluating the exercise capacity of persons with COPD.13 Patients were instructed to walk as far as possible but were allowed to stop and rest when necessary. Before and after the 6-MWD test, arterial oxygen saturation (SaO2) was analyzed using blood gas analyze. The difference in SaO2 before and after the 6-MWD (ΔSaO2) was obtained. Serum samples were obtained at the time of recruitment and stored at −80°C for later analysis.
PM10 exposure assessment
Corresponding PM10 data for each subject were obtained from 25 monitoring stations (operated by the Taiwan Environmental Protection Administration, Taiwan). Daily concentrations of PM10 were obtained from the Environmental Protection Administration, Taiwan (http://taqm.epa.gov.tw/taqm/tw/default.aspx). If a subject resided within 10 km of multiple monitoring stations, a weighted average was used to calculate PM10 levels. PM10 data were used to estimate 1-year effects of PM10 on COPD.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assays (ELISAs) were used to determine the serum (circulating) levels of 8-isoprostane (Cayman Chemical Company, Ann Arbor, MI, USA), interleukin (IL)-6 (R&D System, Inc., Minneapolis, MN, USA), receptor for advanced glycation end-products (RAGE; R&D System, Inc.), carbonyl oxidation (Cell Biolabs, San Diego, CA, USA), ubiquitin (R&D System, Inc.), proteasome activity (Enzo, Farmingdale, ST, USA), and beclin 1 (MyBioSource, San Diego, CA, USA) according to the manufacturer’s instructions.
Statistical analysis
All enzyme-linked immunosorbent assay experiments were performed at least three times. Data are expressed as the mean ± standard deviation (SD). For multiple-comparison analyses, a one-way analysis of variance with Tukey’s post hoc test was used. Spearman’s rank correlation coefficient was used to examine the correlation of quintiles of PM10 with FVC, FEV1/FVC, RV/TLC, DLCO, ΔSaO2, 8-isoprostane, IL-6, RAGE, carbonyl oxidation, ubiquitin, proteasome, and beclin 1, as well as correlations of ubiquitin and beclin 1 with DLCO, ΔSaO2, IL-6, and RAGE. Statistical analyses were performed using GraphPad Version 5 (GraphPad Software, Inc., La Jolla, CA, USA) for Windows. The level of significance was set to P<0.05.
Results
Characterization of the study population
Forty-three subjects with COPD stage III or IV and 15 healthy controls (including both nonsmokers and smokers) were enrolled in the study. Detailed baseline characteristics of the 58 subjects in the study population are presented in Table 1. Ages of the healthy controls and COPD patients were 63.8±11.6 years and 70.3±9.0 years, respectively. The majority of COPD subjects were men (93.0%), whereas the healthy controls consisted of 53.3% men. The body mass index ranged 23.0–23.6 kg/m2. The current smoking percentage among COPD subjects was 9.3%. The 1-year exposure to PM10 of the healthy controls and COPD subjects was 47.6±8.3 μg/m3 and 44.1±9.1 μg/m3, respectively. The percent predicted FEV1 values for healthy controls and COPD subjects were 95.4%±15.3% and 33.1%±10.0%, respectively. The RV/TLC and DLCO values for COPD subjects were 65.8±14.0 and 63.3±23.5, respectively. ΔSaO2 values were −5.0±0.6 for healthy controls and −12.7±5.6 for COPD subjects. Levels of 8-isoprostane, IL-6, RAGE, carbonyl oxidation, ubiquitin, proteasome, and beclin 1 for COPD subjects were 13.6±3.1 pg/mL, 3.9±3.1 pg/mL, 0.8±0.3 μg/mL, 10.4±5.5 nmol/mg, 0.7±0.5 ng/mL, 1.0±0.9 μg/mL, and 7.4±4.3 ng/mL, respectively.
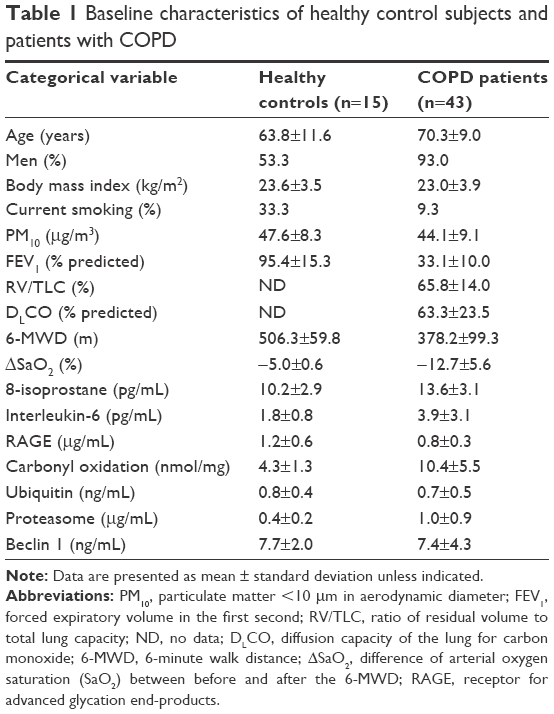 | Table 1 Baseline characteristics of healthy control subjects and patients with COPD |
PM10 and lung function
Changes in PM10 for the 1-year averages in quintiles were related to FVC, FEV1/FVC, RV/TLC, and DLCO in COPD subjects (Figure 1). The 1-year PM10 average was correlated with DLCO levels (r=−0.914, P=0.029); however, there was no correlation of PM10 with FVC (r=−0.554, P=0.333), FEV1/FVC (r=0.892, P=0.062), and RV/TLC (r=−0.599, P=0.286).
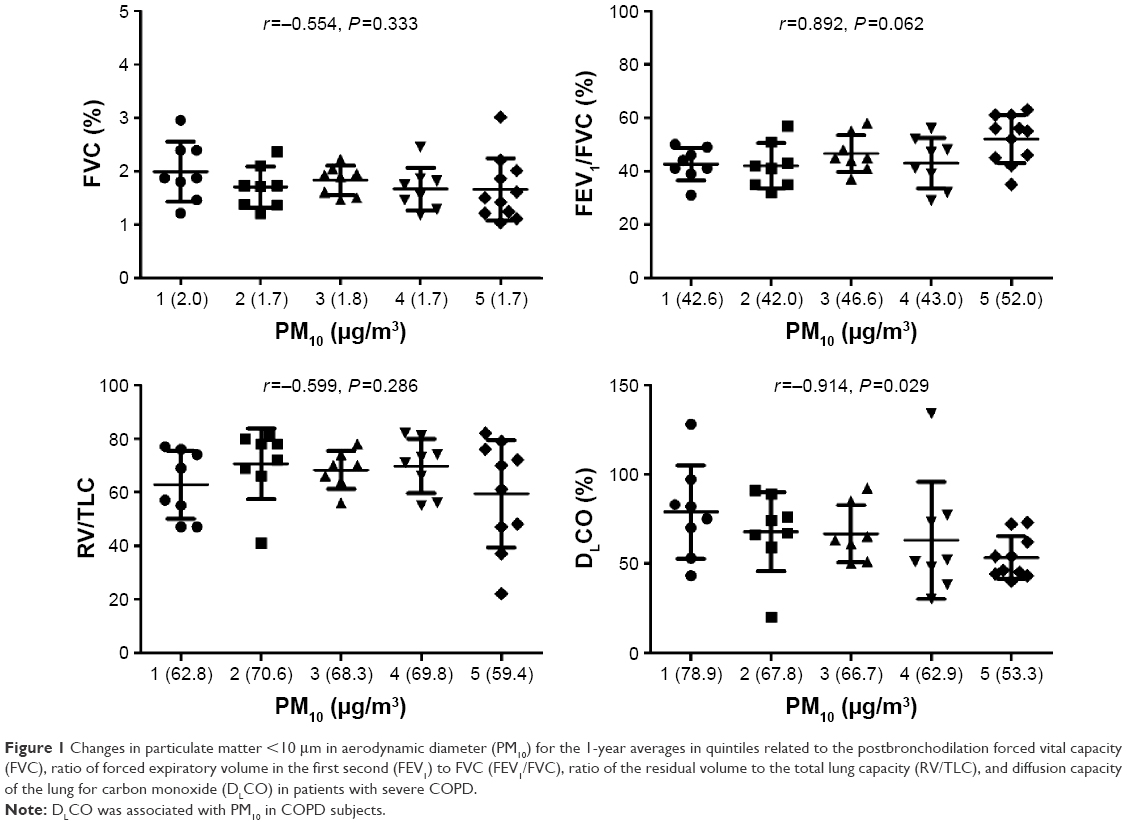 | Figure 1 Changes in particulate matter <10 μm in aerodynamic diameter (PM10) for the 1-year averages in quintiles related to the postbronchodilation forced vital capacity (FVC), ratio of forced expiratory volume in the first second (FEV1) to FVC (FEV1/FVC), ratio of the residual volume to the total lung capacity (RV/TLC), and diffusion capacity of the lung for carbon monoxide (DLCO) in patients with severe COPD. |
PM10 and oxygen saturation, 8-isoprostane, IL-6, and RAGE
The indexes of oxygen saturation (ΔSaO2), 8-isoprostane, IL-6, and RAGE were correlated with changes in PM10 for the 1-year averages in quintiles (Figure 2). Levels of ΔSaO2 (r=−0.973, P=0.005) and RAGE (r=−0.881, P=0.048) were significantly reduced along with an increase in PM10 levels in quintiles. IL-6 was positively correlated with the 1-year PM10 average (r=0.986, P=0.002). There was no correlation between PM10 and 8-isoprostane (r=0.837, P=0.077) in COPD subjects.
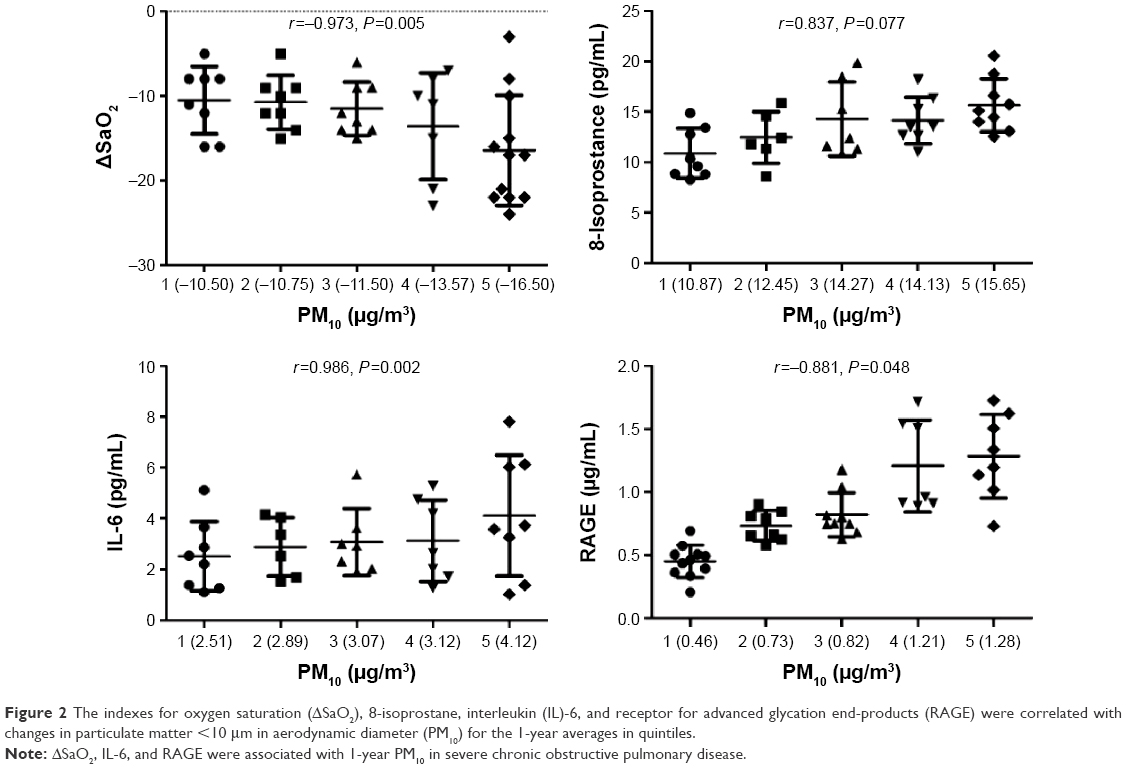 | Figure 2 The indexes for oxygen saturation (ΔSaO2), 8-isoprostane, interleukin (IL)-6, and receptor for advanced glycation end-products (RAGE) were correlated with changes in particulate matter <10 μm in aerodynamic diameter (PM10) for the 1-year averages in quintiles. |
PM10 and carbonyl oxidation, ubiquitin, proteasome, and beclin 1
Figure 3 shows associations of changes in PM10 for the 1-year averages in quintiles with carbonyl oxidation, ubiquitin, proteasome, and beclin 1. The 1-year PM10 averages were positively correlated with levels of ubiquitin (r=0.940, P=0.017) and beclin 1 (r=0.923, P=0.025) in COPD subjects. PM10 levels were not significantly associated with carbonyl oxidation (r=−0.698, P=0.199) or proteasome (r=−0.233, P=0.707) in COPD subjects.
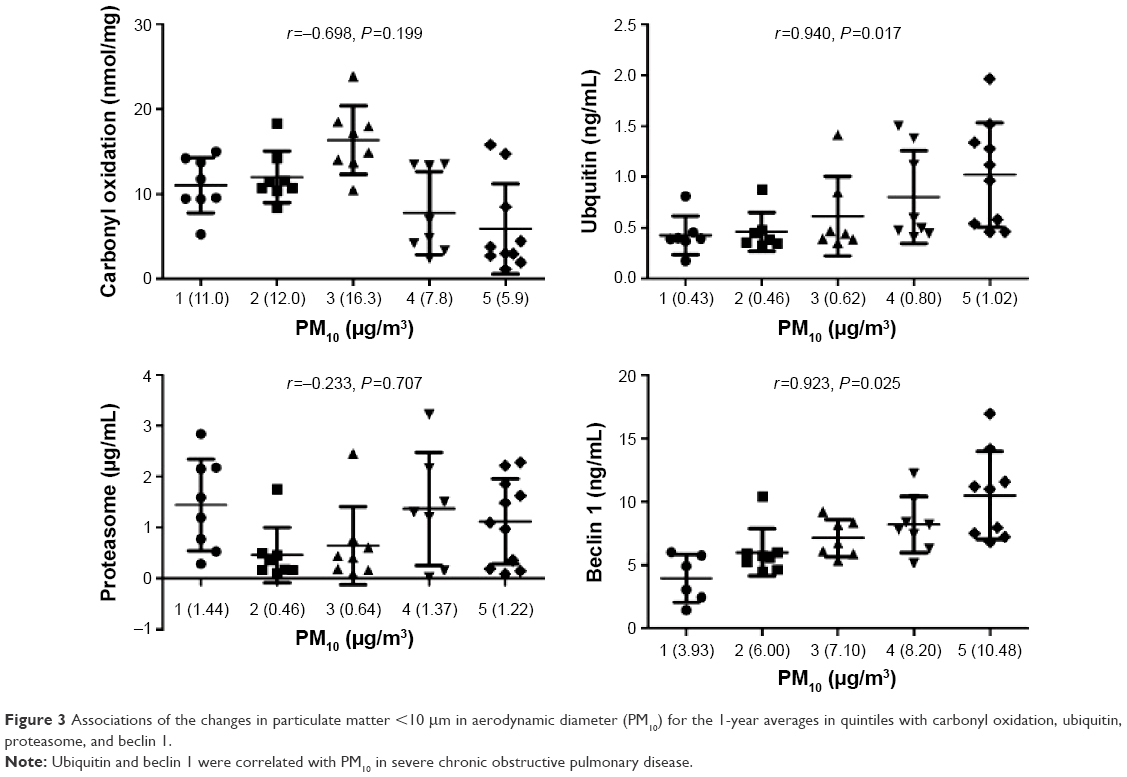 | Figure 3 Associations of the changes in particulate matter <10 μm in aerodynamic diameter (PM10) for the 1-year averages in quintiles with carbonyl oxidation, ubiquitin, proteasome, and beclin 1. |
Associations of ubiquitin and beclin 1 with DLCO, ΔSaO2, IL-6, and RAGE
Spearman’s rank correlation coefficients were used to further determine associations of PM10-related ubiquitin and beclin 1 with the four PM10-related index/markers (ie, DLCO, ΔSaO2, IL-6, and RAGE; Figure 4). We observed that ubiquitin was correlated with ΔSaO2 (r=−0.374, P=0.019), but there were no correlations observed with DLCO (r=−0.010, P=0.950), IL-6 (r=0.006, P=0.973), or RAGE (r=−0.261, P=0.119). Beclin 1 was associated with DLCO (r=−0.362, P=0.028), ΔSaO2 (r=−0.354, P=0.032), and RAGE (r=−0.471, P=0.004). Beclin 1 was not related to IL-6 levels (r=0.011, P=0.953).
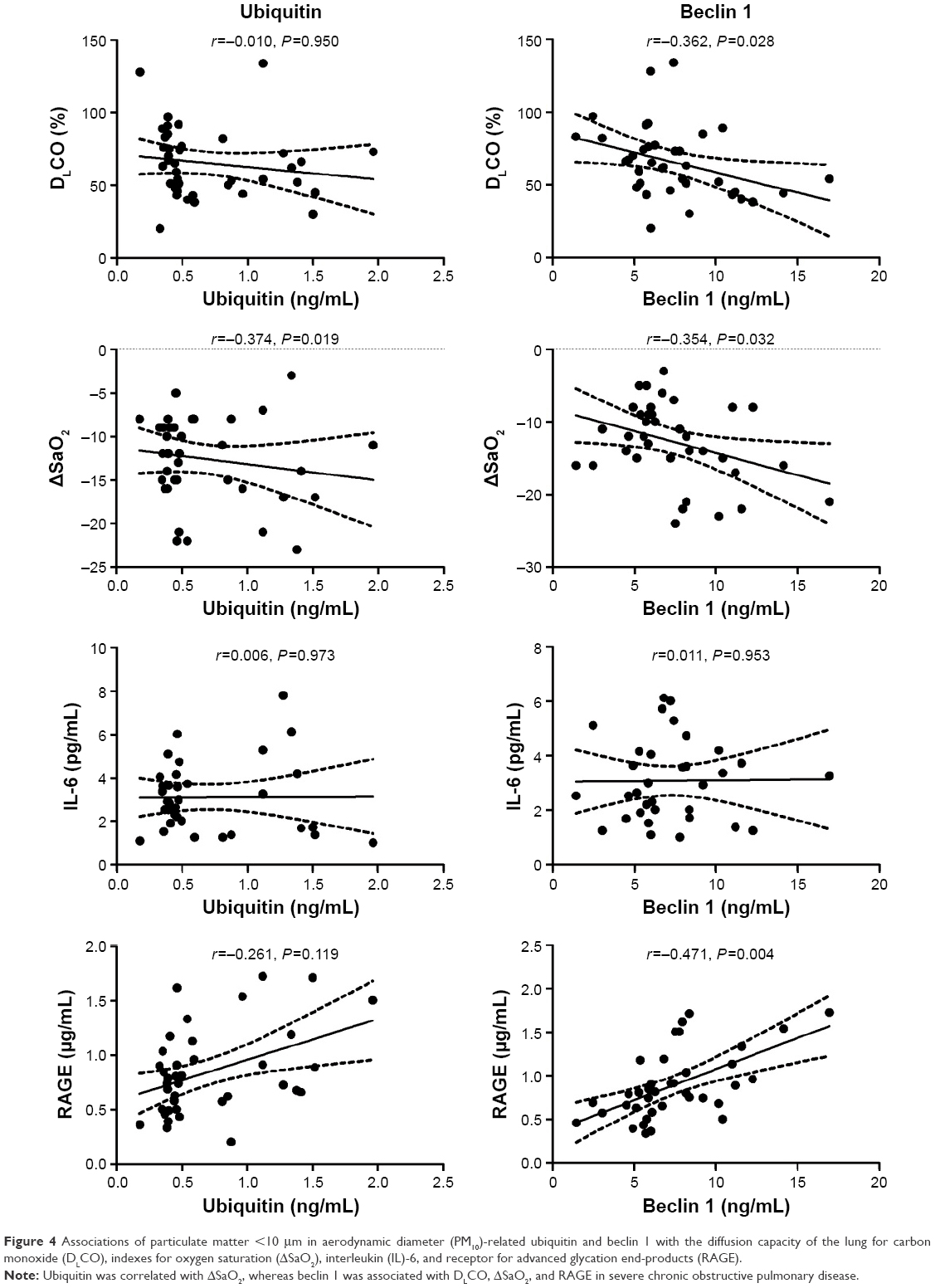 | Figure 4 Associations of particulate matter <10 μm in aerodynamic diameter (PM10)-related ubiquitin and beclin 1 with the diffusion capacity of the lung for carbon monoxide (DLCO), indexes for oxygen saturation (ΔSaO2), interleukin (IL)-6, and receptor for advanced glycation end-products (RAGE). |
Discussion
Chronic pulmonary exposure to traffic-related air pollution has been linked to the development of COPD;14 however, the air pollution-associated pathogenesis of COPD is not yet fully understood. This study showed a pathogenic association of autophagy with the lung diffusion capacity and oxygen saturation regulated by particulate air pollution in patients with severe COPD. Three major findings are reported in the present study: 1) reductions in the lung diffusion capacity and oxygen saturation occurred after exercise in COPD subjects due to PM10, 2) expressions of IL-6, RAGE, ubiquitin, and beclin 1 in COPD subjects were altered by PM10, and 3) PM10-related beclin 1 was associated with the lung diffusion capacity, oxygen saturation, and RAGE levels in COPD subjects.
Increasing numbers of epidemiological and clinical studies have highlighted concerns over the potential adverse health effects of particulate air pollutants. Particulate air pollutants (eg, PM10) are associated with increased hospital admissions and mortality due to cardiopulmonary disorders.15 Air pollution is recognized to be a potential risk factor for COPD; however, its exact role in the development of COPD is difficult to demonstrate. Mehta et al16 evaluated the association between occupational exposure to PM and the incidence of COPD in the Swiss Cohort Study on Air Pollution and Lung and Heart Diseases in Adults. They showed that the incidence of COPD severity (at least moderate severity; GOLD stage II) was significantly associated with exposure to PMs. Therefore, in the present study, subjects with severe COPD (stages III and IV) were recruited to determine the adverse effects due to exposure to PM10. Additionally, healthy controls were enrolled in this study. The 1-year PM10 exposure of the control group was compared to that of the COPD group, suggesting that 1-year PM10 levels were similar between the groups. Additionally, COPD subjects had higher levels of 8-isoprostane, IL-6, and carbonyl oxidation in serum than those in the controls, suggesting that oxidative status and inflammation were persistent in COPD individuals.
COPD features air flow limitations that are not fully reversible and pathological changes in the proximal and peripheral airways, lung parenchyma, and pulmonary vasculature.1 A decline in lung function is recognized to be a critical pathophysiological outcome caused by PM10.17 The present study further identified the effects of PM10 on RV/TLC and DLCO in COPD subjects. We found that DLCO declined along with an increase in PM10 levels, reflecting that PM10 reduced the lung diffusion capacity of COPD patients. This finding is consistent with previous reports that DLCO reduction occurred with acute exposure to particulates in healthy smokers.18 Generally, deposition of PM2.5 in the lungs is higher in COPD patients and smokers compared to healthy nonsmokers, which could increase particle deposition and was proportional to the severity of obstruction.18 The lung diffusion capacity, DLCO, provides a quantitative and qualitative assessment of pulmonary gas exchange across the alveolar–capillary membrane. In COPD patients, the DLCO can reflect alveolar–capillary membrane damage subsequent to emphysema and increased pressure in the pulmonary circulation or secondary to heart failure.19 PM10-reduced DLCO in COPD may be due to more particle deposition in the airways and alveolar region, which causes transient interstitial edema to deteriorate in COPD subjects.18 PM10-induced thickening of the blood–gas barrier can be explained by the reduction in DLCO in COPD; however, further investigation of the mechanisms underlying PM10-reduced DLCO is required in the future.
The DLCO is associated with impairments of exercise capacity and oxygen saturation and elevated inflammation in COPD.20 Previous reports demonstrated that a decrease in the resting oxygen saturation was caused by exposure to concentrated ambient particles in healthy subjects, but the results were inconsistent in COPD patients.21 The inconsistency could have been due to impairment of oxygen delivery in COPD; thus, determining the levels of oxygen desaturation in COPD may be appropriate for examining the effects of PMs. Measurement of exercise-induced oxygen desaturation is associated with the prognosis of COPD patients.22 The 6-MWD test, for example, was suggested as a preferred measure for identifying COPD patients with exercise-induced oxygen desaturation.23 Notably, we observed a trend of ΔSaO2 decreasing with increased PM10 levels during exercise in COPD subjects. Our findings suggest that inhaled particles interacted with small airways and/or the alveoli and subsequently damaged compensation of the oxygen transport. Chronic oxygen desaturation induced by PM10 could cause systemic inflammation, elevating circulating IL-6 levels. We also found that circulating RAGE levels decreased with PM10 exposure in COPD patients. RAGE is highly expressed only in the lungs at readily measurable levels but increases quickly at sites of inflammation, largely on inflammatory and epithelial cells.24 Cheng et al25 showed that lower circulating RAGE levels were associated with emphysema severity in COPD. RAGE expression was also linked to increasing levels of diesel particles and cigarette smoke;26 however, we observed that circulating RAGE concentrations were negatively correlated with PM10. More experimental studies are required to determine the RAGE mechanisms in response to PM10 exposure.
We observed that levels of circulating ubiquitin and beclin 1 were significantly associated with PM10 in patients with severe COPD. COPD is a persistent oxidative inflammatory disease, which causes reactive oxygen species (ROS) imbalances that consequently oxidize/modify proteins.27 Particles have the potential to pass through the lung environments, and en route, cause ROS imbalances due to particulate physicochemistry. These ROS-related parameters cause inflammation via disrupting the function of mitochondria or nicotinamide adenine dinucleotide phosphate-oxidase and activation of inflammatory cells.28 Free radicals and oxidants on particle surfaces can traverse to nuclei or initiate free radical chain reactions that damage surrounding proteins.29 Some forms of oxidized proteins are degraded by the ubiquitin-proteasome pathway.30 An accurate and functional ubiquitin-proteasome system is required for cells to mitigate higher oxidative stress. Moreover, the activity of the ubiquitin-proteasome system is modulated by the cellular redox status.31 A previous study showed that cigarette smoke induced accumulation of polyubiquitinated proteins in soluble and insoluble protein fractions from the lungs, which suggested impairment of proteasome activity in the lungs of mice and in human epithelial cells.32 Also, defective proteasomal protein quality control could exacerbate the detrimental effects of cigarette smoke in the lungs.32 Mild or transient ROS activates the ubiquitination system and proteasome activity, and activation of the ubiquitin-proteasome system transiently enhances intracellular proteolysis.33 Severe or sustained ROS may cause dysfunction of the ubiquitin-proteasome system, resulting in decreased intracellular proteolysis.33 Thus, increased levels of ubiquitin conjugates in cells may be related to the degree of oxidative stress or severity in COPD. We found that only ubiquitin was associated with PM10 in COPD, but this effect was not observed with proteasomes; the results imply that PM10 can cause proteasome activity to deteriorate in severe COPD.
Ubiquitination function is a general tag for selective autophagy in mammalian cells. Autophagy is a regulated cellular mechanism for turning over proteins that is mediated by lysosomal-dependent processing. Beclin 1 (the mammalian homolog of yeast Atg6) is a major autophagic regulator that is associated with phosphatidylinositol 3-kinase (Vps34).34 Alterations in autophagy were linked to COPD progression and daily activities in COPD patients.35 Experimental in vitro studies also showed that PM upregulated autophagy expression such as beclin 1 in A549 cells.36 Our study presented interactions between PM10 and COPD that PM10 triggers beclin 1 levels in severe COPD; however, the effects of PM10 on ubiquitin and beclin 1 in COPD remain unclear.
To understand the effects of PM10 on ubiquitin and autophagy in severe COPD, we then linked the PM10-related ubiquitin and beclin 1 to the DLCO, ΔSaO2, IL-6, and RAGE. The associations suggested that beclin 1 could be more correlated with the pathogenesis of COPD such as are DLCO, ΔSaO2, and RAGE. Poon et al37 observed that autophagy may be a cellular mechanism that promotes airway remodeling and declines in lung function in asthma, consequently leading to inactivity due to oxygen desaturation.35 In environments with limited oxygenation or nutrients, perpetual signaling through RAGE-induced survival pathways results in enhanced autophagy, and diminished apoptosis and necrosis.38 RAGE and its isoforms have important roles in regulating metabolism, inflammation, and epithelial survival in stressful settings. Understanding the molecular structure and function of RAGE and its ligands in a setting of inflammation is critically important for understanding the role of the PM-related pathogenesis of COPD.
Conclusion
Autophagy may be an important regulator of PM10-related pathogenesis of COPD, which could cause deterioration of the lung diffusion capacity and oxygen saturation. Furthermore, RAGE may be another pathway regulating PM10-attributed severity of COPD. The limitations of the present study are the sex distribution (male dominated) of study subjects and the small sample size. Moreover, we used a weighted average to obtain 1-year PM10 exposure based on the distance of the residence to PM monitoring stations, which may potentially underestimate exposure for patients who lived close to busy roads, or have other personal exposure from automobile traffic. Future studies are required to investigate the potential mechanisms underlying PM10 exposure and determine the role of RAGE in PM10-phenotype COPD.
Acknowledgments
The authors wish to thank Miss Yi-Syuan Lin and Mrs Ching-Lin Li for technical assistance with this research. This study was funded by the Ministry of Science and Technology of Taiwan (MOST103-2314-B-038-018, MOST104-2621-M-038-002-MY3, and MOST102-2314-B-038-055-MY2), Taipei Medical University and Shuang Ho Hospital (103TMU-SHH-01-1 and 104TMU-SHH-05), and Shuang Ho Hospital (105-SHH-HCP-11).
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. 2013;144(1):266–273. | ||
Kumar N, Liang D, Comellas A, Chu AD, Abrams T. Satellite-based PM concentrations and their application to COPD in Cleveland, OH. J Expo Sci Environ Epidemiol. 2013;23(6):637–646. | ||
Schikowski T, Mills IC, Anderson HR, et al. Ambient air pollution – a cause for COPD? Eur Respir J. 2014;43(1):250–263. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454. | ||
Ryter SW, Chen ZH, Kim HP, Choi AM. Autophagy in chronic obstructive pulmonary disease: homeostatic or pathogenic mechanism? Autophagy. 2009;5(2):235–237. | ||
Verschuere S, Allais L, Bracke KR, et al. Cigarette smoke and the terminal ileum: increased autophagy in murine follicle-associated epithelium and Peyer’s patches. Histochem Cell Biol. 2012;137(3):293–301. | ||
Gannon AM, Stämpfli MR, Foster WG. Cigarette smoke exposure elicits increased autophagy and dysregulation of mitochondrial dynamics in murine granulosa cells. Biol Reprod. 2013;88(3):63. | ||
Deng X, Zhang F, Rui W, et al. PM2.5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol In Vitro. 2013;27(6):1762–1770. | ||
Miller MR, Crapo R, Hankinson J, et al. General considerations for lung function testing. Eur Respir J. 2005;26(1):153–161. | ||
ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166(1):111–117. | ||
Butland RJ, Pang J, Gross ER, Woodcock AA, Geddes DM. Two-, six-, and 12-minute walking tests in respiratory disease. Br Med J (Clin Res Ed). 1982;284(6329):1607–1608. | ||
Andersen ZJ, Hvidberg M, Jensen SS, et al. Chronic obstructive pulmonary disease and long-term exposure to traffic-related air pollution: a cohort study. Am J Respir Crit Care Med. 2011;183(4):455–461. | ||
Atkinson RW, Carey IM, Kent AJ, van Staa TP, Anderson HR, Cook DG. Long-term exposure to outdoor air pollution and the incidence of chronic obstructive pulmonary disease in a national English cohort. Occup Environ Med. 2015;72(1):42–48. | ||
Mehta AJ, Miedinger D, Keidel D, et al. Occupational exposure to dusts, gases, and fumes and incidence of chronic obstructive pulmonary disease in the Swiss Cohort Study on Air Pollution and Lung and Heart Diseases in Adults. Am J Respir Crit Care Med. 2012;185(12):1292–1300. | ||
Imboden M, Kumar A, Curjuric I, et al. Modification of the association between PM10 and lung function decline by cadherin 13 polymorphisms in the SAPALDIA cohort: a genome-wide interaction analysis. Environ Health Perspect. 2015;123(1):72–79. | ||
Hazucha MJ, Bromberg PA, Lay JC, et al. Pulmonary responses in current smokers and ex-smokers following a two hour exposure at rest to clean air and fine ambient air particles. Part Fibre Toxicol. 2013;10(1):58. | ||
Minai OA, Chaouat A, Adnot S. Pulmonary hypertension in COPD: epidemiology, significance, and management: pulmonary vascular disease: the global perspective. Chest. 2010;137(6 suppl):39S–51S. | ||
Farkhooy A, Janson C, Arnardóttir RH, Malinovschi A, Emtner M, Hedenstrom H. Impaired carbon monoxide diffusing capacity is the strongest predictor of exercise intolerance in COPD. COPD. 2013;10(2):180–185. | ||
Gong H, Linn WS, Terrell SL, et al. Exposures of elderly volunteers with and without chronic obstructive pulmonary disease (COPD) to concentrated ambient fine particulate pollution. Inhal Toxicol. 2004;16(11–12):731–744. | ||
Takigawa N, Tada A, Soda R, et al. Distance and oxygen desaturation in 6-min walk test predict prognosis in COPD patients. Respir Med. 2007;101(3):561–567. | ||
Knower MT, Dunagan DP, Adair NE, Chin R Jr. Baseline oxygen saturation predicts exercise desaturation below prescription threshold in patients with chronic obstructive pulmonary disease. Arch Intern Med. 2001;161(5):732–736. | ||
Sparvero LJ, Asafu-Adjei D, Kang R, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med. 2009;7:17. | ||
Cheng DT, Kim DK, Cockayne DA, et al. Systemic soluble receptor for advanced glycation endproducts is a biomarker of emphysema and associated with AGER genetic variants in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(8):948–957. | ||
Robinson AB, Johnson KD, Bennion BG, Reynolds PR. RAGE signaling by alveolar macrophages influences tobacco smoke-induced inflammation. Am J Physiol Lung Cell Mol Physiol. 2012;302(11):L1192–L1199. | ||
Hackett TL, Scarci M, Zheng L, Tan W, Treasure T, Warner JA. Oxidative modification of albumin in the parenchymal lung tissue of current smokers with chronic obstructive pulmonary disease. Respir Res. 2010;11:180. | ||
Risom L, MØller P, Loft S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat Res. 2005;592(1–2):119–137. | ||
Lee K-Y, Wong CK-C, Chuang K-J, et al. Methionine oxidation in albumin by haze fine particulate matter: an in vitro and in vivo study. J Hazard Mater. 2014;274:384–391. | ||
Poppek D, Grune T. Proteasomal defense of oxidative protein modifications. Antioxid Redox Signal. 2006;8(1–2):173–184. | ||
Kriegenburg F, Poulsen EG, Koch A, Kruger E, Hartmann-Petersen R. Redox control of the ubiquitin-proteasome system: from molecular mechanisms to functional significance. Antioxid Redox Signal. 2011;15(8):2265–2299. | ||
van Rijt SH, Keller IE, John G, et al. Acute cigarette smoke exposure impairs proteasome function in the lung. Am J Physiol Lung Cell Mol Physiol. 2012;303(9):L814–L823. | ||
Shang F, Taylor A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic Biol Med. 2011;51(1):5–16. | ||
Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762): 672–676. | ||
Costes F, Gosker H, Feasson L, et al. Impaired exercise training-induced muscle fiber hypertrophy and Akt/mTOR pathway activation in hypoxemic patients with COPD. J Appl Physiol (1985). 2015;118(8):1040–1049. | ||
Deng X, Zhang F, Wang L, et al. Airborne fine particulate matter induces multiple cell death pathways in human lung epithelial cells. Apoptosis. 2014;19(7):1099–1112. | ||
Poon A, Eidelman D, Laprise C, Hamid Q. ATG5, autophagy and lung function in asthma. Autophagy. 2012;8(4):694–695. | ||
Kang R, Tang D, Lotze MT, Zeh HJ 3rd. RAGE regulates autophagy and apoptosis following oxidative injury. Autophagy. 2011;7(4):442–444. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.