Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Association of the Tight Junction Protein Claudin-4 with Lung Function and Exacerbations in Chronic Obstructive Pulmonary Disease
Authors Park S, Lee PH, Baek AR ![]() , Park JS, Lee J
, Park JS, Lee J ![]() , Park SW, Kim DJ
, Park SW, Kim DJ ![]() , Jang AS
, Jang AS ![]()
Received 26 July 2021
Accepted for publication 13 September 2021
Published 5 October 2021 Volume 2021:16 Pages 2735—2740
DOI https://doi.org/10.2147/COPD.S330674
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Shinhee Park,1 Pureun-Haneul Lee,2 Ae Rin Baek,1 Jong Sook Park,1 Junehyuk Lee,1 Sung-Woo Park,1 Do Jin Kim,1 An-Soo Jang1
1Division of Allergy and Respiratory Medicine, Department of Internal Medicine, Soonchunhyang University Bucheon Hospital, Bucheon, Republic of Korea; 2Department of Interdisciplinary Program in Biomedical Science Major, Soonchunhyang Graduate School, Soonchunhyang University Bucheon Hospital, Bucheon, Republic of Korea
Correspondence: An-Soo Jang
Division of Allergy and Respiratory Medicine, Department of Internal Medicine, Soonchunhyang University Bucheon Hospital, 170 Jomaru-ro, Bucheon, 14584, Republic of Korea
Tel +82 32 621 5143
Fax +82 32 621 6950
Email [email protected]
Purpose: Chronic obstructive pulmonary disease (COPD) imposes a major healthcare burden. A tight junction protein, claudin-4 (CLDN4), may play a protective role in acute lung injury, but its role in COPD is unclear. To investigate the relationship between CLDN4 and COPD, we evaluated the association of CLDN4 with the clinical parameters of COPD, including exacerbations.
Patients and Methods: We analyzed a cohort of 30 patients with COPD and 25 healthy controls and evaluated their clinical parameters, including lung function. The plasma CLDN4 level in stable and exacerbated COPD was measured.
Results: The COPD patients were all males and predominantly smokers; their initial lung function was poorer than the healthy controls. The mean CLDN4 plasma level was 0.0219 ± 0.0205 ng/mg in the control group, 0.0086 ± 0.0158 ng/mg in the stable COPD group (COPD-ST) and 0.0917 ± 0.0871 ng/mg in the exacerbated COPD (COPD-EXA) group. The plasma CLDN4 level was significantly lower in the COPD-ST than the control group, but was significantly elevated in the COPD-EXA group. The plasma CLDN4 level was inversely correlated with forced vital capacity and forced expiratory volume in 1 second in the COPD-EXA group (r=0.506, P=0.001 and r=0.527, P< 0.001, respectively).
Conclusion: The plasma CLDN4 level is closely correlated with COPD exacerbations and decreased lung function. This suggests that CLDN4 has potential as a severity marker for COPD.
Keywords: claudin-4, COPD, lung function
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic respiratory disease characterized by airway inflammation and incompletely reversible airflow limitation. It is a major global health burden and the third leading cause of death among non-communicable diseases.1 COPD is a multifactorial disease affected by host susceptibility and various noxious stimuli; smoking is a well-established risk factor.2
The human respiratory system has several defenses against noxious particles and gases on the mucosal surface.3 It acts as a structural barrier and removes inhaled foreign substances by mucociliary clearance, and also serves as a functional barrier that modulates lung inflammation, immunity to infections, and tissue remodeling.4 The epithelial barrier function is maintained by the formation of tight junctions, which consist of several anchoring proteins such as occludin and claudins.5
The formation of airway epithelial junction in patients with COPD is disrupted.6 Cigarette smoking downregulates apical junctional complex-related genes, thus decreasing transepithelial resistance.7 Cigarette smoke extract suppressed the expression of genes encoding various junctional proteins, including claudin, occludin, E-cadherin, and zonula occludens-1.8 Given that smoking is a major risk factor for COPD, disruption of epithelial barrier function may play an important role in the pathogenesis of COPD.9
Claudins are tight junction proteins that regulate paracellular permeability. Among the 27 human claudins, claudin-4 (CLDN4) is present in human alveolar cells types I and II.10 CLDN4 is associated with barrier dysfunction and may protect against acute lung injury.11,12 The plasma CLDN4 level was significantly increased in patients with asthma compared to healthy subjects and was associated with the blood eosinophil count, total immunoglobulin E (IgE), and lung function decline.13 To date, the relationship between CLDN4 and COPD is unclear. We investigated the association of CLDN4 with clinical parameters of COPD, including exacerbations and lung function.
Patients and Methods
Subjects
We enrolled 30 patients with COPD from the cohort of the Genome Research Center for Allergy and Respiratory Diseases in South Korea. The biospecimens and clinical data were provided by the Biobank of Soonchunhyang University Bucheon Hospital, a member of the Korea Biobank Network. This study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Institutional Review Board of the Soonchunhyang University Bucheon Hospital (SCHBC_2015-03-005).
The enrolled patients were adults aged ≥ 40 years with a clinical diagnosis of stable COPD or moderate-to-severe COPD according to the GOLD 2015 criteria.14 Smokers and ex-smokers were included if they had a smoking history of at least 10 pack-years. Diagnosis of COPD was confirmed based on post-bronchodilator spirometry criteria: a forced expiratory volume in 1 second (FEV1) ≥ 30% and < 70% of the predicted normal value, and a FEV1/forced vital capacity (FVC) ratio < 0.7. Subjects who had malignancy within 5 years were excluded. Other exclusion criteria were asthma, respiratory infection at screening, vocal cord dysfunction, obstructive sleep apnea, cardiac dysfunction, allergic bronchopulmonary aspergillosis, Churg-Strauss syndrome, and poor compliance.
Demographic data including age at enrollment, sex, body mass index (BMI), and smoking status were collected. All subjects underwent standardized assessments comprising a complete blood cell count with differentials, chest radiography, and spirometry. In the COPD group, all data except for demographic data were collected twice: at the time of enrollment (ie, during stable disease) and during exacerbations for comparison. An exacerbation was defined as exacerbation of any respiratory symptoms requiring treatment with systemic corticosteroids, antibiotics, or both.
The healthy control subjects were recruited from the general population or the family members of the patients, based on the following criteria: negative responses on a screening questionnaire for respiratory symptoms and other respiratory diseases, FEV1 values > 80% of predicted values, and no abnormal findings on simple chest radiographs. Some of the disease and control group patients were included in prior studies.13,15
Spirometry
Spirometry was performed in accordance with American Thoracic Society/European Respiratory Society guidelines.16 Basal and post-bronchodilator FEV1 and FVC values were measured. Spirometric measurements were obtained in the absence of bronchodilator use within 8 h. We used a Vmax Series 2130 Autobox Spirometer (Sensor Medics, Yorba Linda, CA, USA) and performed a calibration check every morning at 8 AM.
Enzyme-Linked Immunosorbent Assay
Plasma CLDN4 level was measured by enzyme-linked immunosorbent assay (USCN, Wuhan, China). To compare plates, the optical densities (ODs) of samples were adjusted relative to the positive and negative controls in the kits. The mean OD of duplicate wells was calculated. The index value of each serum sample was calculated as follows: Index = (OD of tested serum − OD of negative control) ÷ (OD of positive control − OD of negative control) × 100. The lower limit of detection was set at 0.061 ng/mL for CLDN4, as per the manufacturer’s recommendations. The CLDN4 level was adjusted to the serum protein level.
Statistical Analysis
Data are shown as means ± standard deviation (SD) for continuous variables and as percentages for categorical variables. Statistical analysis was performed using SPSS version 26.0 (IBM Corp., Armonk, NY, USA). The Mann–Whitney U-test was used for analyzing continuous data and Fisher’s exact test was used for categorical data. Correlations were assessed by calculating the Pearson or Spearman correlation coefficient. All P-values are two-sided with statistical significance set at P less than 0.05.
Results
Clinical Characteristics
From March 2015 to May 2016, 30 COPD patients and 25 healthy adults were enrolled. The demographic and clinical data of the subjects are shown in Table 1. The patients with COPD were male and older than the controls. The BMI of the patients with COPD was significantly lower than the healthy controls. Baseline lung function was significantly different between the groups; the mean FVC, presented as a percentage of the predicted value, was 96.5 in healthy controls and 66.9 in patients with COPD (P<0.001), and the mean FEV1 values were 115.4 and 44.4 (P<0.001), respectively. The total IgE was 30.4 kU in the control group and 333.2 kU in patients with COPD (P<0.001). The white blood cell (WBC) count was significantly increased in the patients with COPD compared to the healthy control subjects.
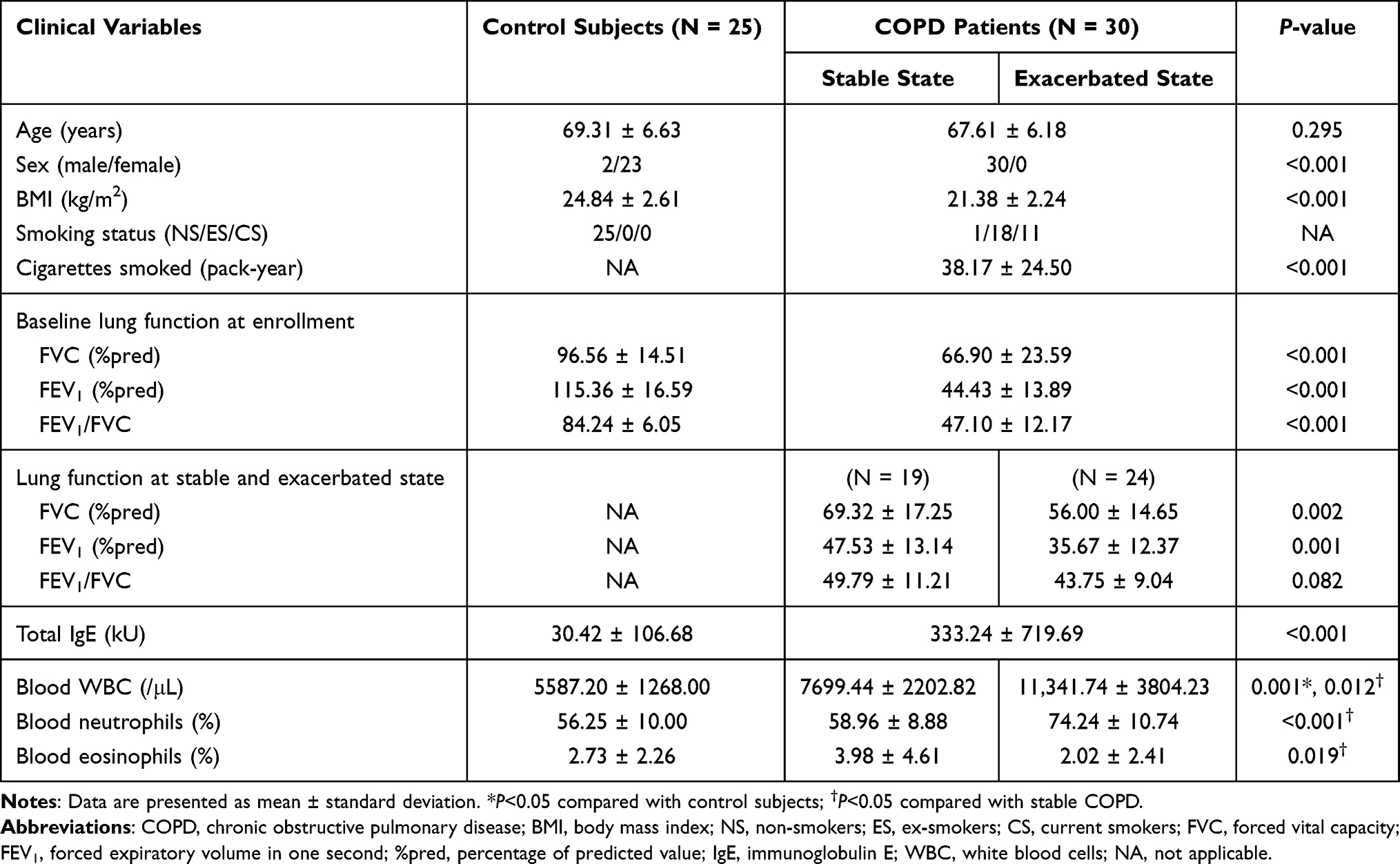 |
Table 1 Clinical Characteristics of the Healthy Control Subjects and the COPD Patients |
Lung function data at exacerbation were available in 24 COPD patients; function was significantly reduced during exacerbations. The WBC and neutrophil counts were significantly elevated in patients with COPD compared to those without exacerbations.
Plasma CLDN4 Level
The mean plasma CLDN4 level was 0.0219 ± 0.0205 ng/mg in the healthy controls, 0.0086 ± 0.0158 ng/mg in patients with stable COPD (COPD-ST) and 0.0917 ± 0.0871 ng/mg in those with exacerbated COPD (COPD-EXA). The plasma CLDN4 level was significantly lower (P=0.011) in the COPD-ST than the control group, but was significantly elevated (P<0.001 for controls and COPD-ST) in the COPD-EXA group (Figure 1).
 |
Figure 1 The plasma claudin-4 (CLDN4) levels of the subjects. The plasma CLDN4 levels of healthy control subjects were compared to those of patients with stable (ST) or exacerbated (EXA) chronic obstructive pulmonary disease (COPD). *P<0.05, control vs COPD-ST; †P<0.05, COPD-ST vs COPD-EXA. |
Associations of the Plasma CLDN4 Level with Clinical Parameters
The plasma CLDN4 level showed negative correlations with FVC and FEV1 in the COPD-EXA group (r=0.506, P=0.001 and r=0.527, P<0.001, respectively; Figure 2). BMI, smoking amount, lung function at baseline, serum IgE level, and the blood eosinophil count were not significantly associated with the plasma CLDN4 level.
 |
Figure 2 Association of plasma claudin-4 (CLDN4) levels and lung function of chronic obstructive pulmonary disease (COPD) patients. (A) Forced vital capacity (FVC), and (B) forced expiratory volume in one second (FEV1) of COPD-stable (ST) was correlated with plasma CLDN5 levels. Abbreviation: EXA, exacerbated. |
Discussion
We evaluated the clinical parameters and plasma CLDN4 levels of patients with COPD and healthy controls. The plasma CLDN4 level in the patients with stable COPD was lower than in the healthy controls, whereas that in patients with exacerbated COPD was higher than in the healthy controls and patients with stable COPD. The plasma CLDN4 level was inversely correlated with lung function (FEV1 and FVC) in patients with COPD. These findings implicate CLDN4 in the pathogenesis of COPD and exacerbations thereof.
The airway epithelium must maintain a proper barrier against outside insults. Airway epithelial cells regulate the airway surface liquid beneath the mucus layer, which is crucial for mucociliary clearance.17 This surface is maintained by plasma-membrane channels and tight junctions. Claudins are tight junction proteins that regulate the paracellular diffusion of solutes and water.18 The disruption of airway microvascular permeability, which is observed in patients with COPD exacerbation and lung injury, leads to disintegration of tight junction molecules and this may promote paracellular leakage of solutes and large molecules that causes airway inflammation.19 CLDN4 is expressed in the normal alveolar epithelium and may protect against lung injury.11,12 Also, CLDN4 expression is decreased in interstitial lung diseases, including idiopathic pulmonary fibrosis, compared to the healthy lung.20
Although the plasma CLDN5 level is decreased in stable COPD and controlled asthma,15,21 the plasma CLDN4 level was decreased in stable COPD compared to healthy controls but increased in controlled and exacerbated asthma,13 suggesting that CLDN4 has different functions in asthma and COPD. Also, plasma CLDN4 elevation during COPD exacerbations could be a response to inflammation, to protect further injury. Further studies of lung tissue, sputum, and bronchoalveolar lavage specimens are needed to uncover the underlying mechanism.
This study had several limitations. First, the cohort was small and not standardized for age, sex, or smoking amount. Second, some FEV1 and FVC data were missing for stable and exacerbated COPD, limiting the statistical power. Third, we lacked longitudinal data after improvement of exacerbations, hampering the determination of causality with respect to the relationship between the plasma CLDN4 level and COPD exacerbations. Larger, well-structured prospective studies are needed to overcome these limitations.
The plasma CLDN4 level is closely correlated with COPD exacerbations and decreased lung function. This suggests that CLDN4 has potential as a severity marker for COPD.
Funding
This study was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2020R1A2C1006506), and the Soonchunhyang University Research Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
1. GBD 2017 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1859–1922. doi:10.1016/s0140-6736(18)32335-3
2. Postma DS, Bush A, van den Berge M. Risk factors and early origins of chronic obstructive pulmonary disease. Lancet. 2015;385(9971):899–909. doi:10.1016/s0140-6736(14)60446-3
3. Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8(4):432–446. doi:10.1046/j.1440-1843.2003.00493.x
4. Hiemstra PS, McCray PB
5. Rezaee F, Georas SN. Breaking barriers. New insights into airway epithelial barrier function in health and disease. Am J Respir Cell Mol Biol. 2014;50(5):857–869. doi:10.1165/rcmb.2013-0541RT
6. Heijink IH, Noordhoek JA, Timens W, van Oosterhout AJ, Postma DS. Abnormalities in airway epithelial junction formation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;189(11):1439–1442. doi:10.1164/rccm.201311-1982LE
7. Shaykhiev R, Otaki F, Bonsu P, et al. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci. 2011;68(5):877–892. doi:10.1007/s00018-010-0500-x
8. Tatsuta M, Kan OK, Ishii Y, et al. Effects of cigarette smoke on barrier function and tight junction proteins in the bronchial epithelium: protective role of cathelicidin LL-37. Respir Res. 2019;20(1):251. doi:10.1186/s12931-019-1226-4
9. Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: role of Cigarette Smoke Exposure. Am J Respir Cell Mol Biol. 2018;58(2):157–169. doi:10.1165/rcmb.2017-0200TR
10. Soini Y. Claudins in lung diseases. Respir Res. 2011;12(1):70. doi:10.1186/1465-9921-12-70
11. Jin W, Rong L, Liu Y, Song Y, Li Y, Pan J. Increased claudin-3, -4 and -18 levels in bronchoalveolar lavage fluid reflect severity of acute lung injury. Respirology. 2013;18(4):643–651. doi:10.1111/resp.12034
12. Kage H, Flodby P, Gao D, et al. Claudin 4 knockout mice: normal physiological phenotype with increased susceptibility to lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307(7):L524–536. doi:10.1152/ajplung.00077.2014
13. Lee PH, Kim BG, Lee SH, et al. Alteration in Claudin-4 Contributes to Airway Inflammation and Responsiveness in Asthma. Allergy Asthma Immunol Res. 2018;10(1):25–33. doi:10.4168/aair.2018.10.1.25
14. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for diagnosis, management, and prevention of COPD. [Internet]; 2015. Available from: https://goldcopd.org.
15. Kim BG, Lee PH, Lee SH, et al. Impact of the endothelial tight junction protein claudin-5 on clinical profiles of patients with COPD. Allergy Asthma Immunol Res. 2018;10(5):533–542. doi:10.4168/aair.2018.10.5.533
16. Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. doi:10.1183/09031936.05.00034805
17. Tarran R, Button B, Boucher RC. Regulation of normal and cystic fibrosis airway surface liquid volume by phasic shear stress. Annu Rev Physiol. 2006;68:543–561. doi:10.1146/annurev.physiol.68.072304.112754
18. Schlingmann B, Molina SA, Koval M. Claudins: gatekeepers of lung epithelial function. Semin Cell Dev Biol. 2015;42:47–57. doi:10.1016/j.semcdb.2015.04.009
19. Suzuki R, Nakamura Y, Chiba S, et al. Mitigation of tight junction protein dysfunction in lung microvascular endothelial cells with pitavastatin. Pulmonary Pharmacol Therapeutics. 2016;38:27–35.
20. Lappi-Blanco E, Lehtonen ST, Sormunen R, Merikallio HM, Soini Y, Kaarteenaho RL. Divergence of tight and adherens junction factors in alveolar epithelium in pulmonary fibrosis. Hum Pathol. 2013;44(5):895–907. doi:10.1016/j.humpath.2012.08.016
21. Moon KY, Lee PH, Kim BG, Park CS, Leikauf GD, Jang AS. Claudin 5 in a murine model of allergic asthma: its implication and response to steroid treatment. J Allergy Clin Immunol. 2015;136(6):1694–1696.e1695. doi:10.1016/j.jaci.2015.08.004
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.