Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Association of peripheral neuropathy with sleep-related breathing disorders in myotonic dystrophies
Authors Banach M, Antczak J ![]() , Rola R
, Rola R
Received 5 October 2016
Accepted for publication 21 November 2016
Published 12 January 2017 Volume 2017:13 Pages 133—140
DOI https://doi.org/10.2147/NDT.S123908
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Marta Banach,1,* Jakub Antczak,1,* Rafał Rola2
1Department of Clinical Neurophysiology, 2First Department of Neurology, Institute of Psychiatry and Neurology, Warsaw, Poland
*These authors contributed equally to this work
Background: Myotonic dystrophy (DM) type 1 and type 2 are inherited diseases characterized by myotonia and myopathy. Additional symptoms include, among others, peripheral neuropathy and sleep-related breathing disorders (SRBDs). There is growing evidence for a complex association between DM1 and DM2, which was described in patients with diabetes mellitus and in the general population. In this study, we investigated whether there is an association between peripheral neuropathy and SRBDs also in the population of patients with DM.
Methods: The study included 16 patients with DM1 (mean age, 37.9±14.1 years; 20–69 years) and eight patients with DM2 (mean age, 47.6±14.1 years; 20–65 years), who underwent a sensory and motor nerve conduction study (NCS) and diagnostic screening for SRBDs. In both groups, the NCS parameters were correlated with respiratory parameters.
Results: In both groups, the amplitude of the ulnar sensory nerve action potential (SNAP) correlated with the mean arterial oxygen saturation (SaO2). In addition, in the DM2 group, the median SNAP correlated with the mean SaO2. In the DM1 group, the median SNAP and the distal motor latency (DML) of the ulnar nerve correlated with the apnea–hypopnea index, while the oxygen desaturation index correlated with the DML of the tibial nerve and with conduction velocity in the sural nerve.
Conclusion: Our results indicate a complex association between neuropathy and SRBDs in DM1 and DM2. Axonal degeneration may contribute to nocturnal hypoxemia and vice versa. Neuropathy may contribute to muscle weakness, which in turn may cause respiratory events.
Keywords: myotonic dystrophy, SRBD and neuropathy with AHI, SNAP, CMAP
Introduction
Myotonic dystrophy (DM) type 1 and type 2 are dominantly inherited, progressive diseases, considered to be the most common muscular dystrophies in adults.1 DM1 is caused by an unstable (CTG)n repeat expansion in the DMPK gene located on chromosome 19q13.3,2 while DM2 is related to the CCTG repeat expansion in the ZNF9 gene located on chromosome 3q21.3.3 The clinical picture of both diseases is characterized by myotonia. Other symptoms include muscle weakness and wasting, cardiac arrhythmias, pulmonary dysfunction, gastrointestinal disorders, endocrine dysfunction, as well as symptoms resulting from involvement of the central nervous system4 and neuropathy.5
The mechanism of the development of neuropathy in DM is unclear. Some authors described it as a primary syndrome, related directly to a genetic mechanism,5,6 while others reported it to be a complication of DM-associated diabetes7 or a side effect of drug treatment.8 Other possible etiological factors are sleep-related breathing disorders (SRBDs), which occur in 16%–75% of patients with DM19,10 and probably in about 60% of those with DM2.11 SRBDs reported in DM include obstructive and central apneas as well as nocturnal hypoventilation and hypoxemia.9,12,13 They are considered to be the consequence of muscle weakness, myotonia,13 and impaired central respiratory drive.14 Importantly, an association of neuropathy with SRBDs has been documented in patients with diabetes mellitus15 and Charcot–Marie–Tooth disease,16 as well as in the general population.17 Some authors reported that polyneuropathy resulted from hypoxic damage of peripheral nerves due to repetitive nocturnal hypoxemia,15,17 while others considered SRBD to be the result of neuropathy due to respiratory muscle weakness16 or pharyngeal reflexes.18 Finally, some authors suggested that both conditions might be affected by one another.19 Considering that there is still insufficient knowledge about these associations, we decided to investigate whether there is in fact a relationship between neuropathy and SRBD in patients with DM, including a possible causal relationship.
Methods
Patients
The study included 16 patients with DM1 (eight females, eight males; mean age, 37.9±14.1 years; 20–69 years) and eight patients with DM2 (four females, four males; mean age, 47.6±14.1 years; 20–65 years), who were recruited from an outpatient clinic at our hospital. Examinations were performed between January 2010 and December 2012. The study was performed according to the 1975 Declaration of Helsinki for Human Research, and the protocol was approved by the Ethics Committee of the Institute of Psychiatry and Neurology in Warsaw, Poland. All subjects gave their written informed consent prior to the inclusion in the study. The exclusion criteria were a pharmacologic treatment that could significantly affect the results of the nerve conduction study (NCS; eg, anticonvulsive cell membrane stabilizers or neuropathy-inducing agents) and active psychiatric disorders. During the study, none of the patients received mexiletine or other pharmacotherapy for myotonic symptoms. Four patients had received mexiletine before it was withdrawn from the Polish market (because of cardiotoxic effects) one year before the beginning of the study.
Clinical evaluation
The height and weight of each patient were recorded. The severity of muscle weakness was assessed with the Medical Research Council (MRC) Scale for Testing Muscle Strength.20 The evaluation with the MRC grading scale included the assessment of the strength of hand extension (which involved mainly the extensor digitorum, carpi ulnaris, and carpi radialis longus and brevis), forearm flexion (biceps brachii, brachialis, brachioradialis), arm abduction (deltoideus, supraspinatus), thigh flexion (iliopsoas, rectus femoris), lower leg extension (quadriceps femoris), and dorsal foot flexion (tibialis anterior, extensor digitorum longus, extensor hallucis). The assessments were made on both sides. According to the MRC scale, the strength of every movement was rated as follows: 0= paralyzed; 1= minimal muscle contraction (visible or on palpation); 2= movement with gravity eliminated; 3= movement against gravity; 4= weakness; and 5= normality.20 After examination, a combined MRC score was calculated by summing up the subscore from every muscle tested. The maximum possible value of the combined score was 60 points. Patients with a combined score of 50 points or higher were considered to have mildly advanced disease; those with a score of 40–49, moderately advanced disease; and the remaining patients, severely advanced disease. The examination with the MRC grading scale was performed by a neurologist with experience in neuromuscular diseases and blinded to the results of the assessment of nocturnal respiration (MB).
For an objective assessment of neuropathic changes, the NCS was performed with a Viking IV electromyograph (Nicolet Biomedical Incorporated, Madison, WI, USA) and included the examination of the sensory fibers of the left median, right ulnar, left sural, and right peroneal nerves. The sensory nerve action potentials (SNAPs) were recorded at standard sites.21 The NCS of the motor fibers was performed for the left median, right ulnar, left tibial, and right peroneal nerves, with the recording of the compound muscle action potential (CMAP) from the left abductor pollicis brevis, right abductor digiti minimi, left abductor hallucis, and right extensor digitorum brevis, respectively. For the recording of the SNAP and CMAP in all nerves, the surface cup electrodes were used, with a distance between the active and reference electrodes of about 3 cm. For stimulation of the sensory fibers, 20 repetitive, supramaximal stimuli were given at a rate of 2 Hz, and for stimulation of the motor fibers, single, supramaximal, and rectangular stimuli were used. The sites of the stimulation were chosen according to the widely accepted recommendations.21 The stimulation of the median and ulnar nerves was done with two ring electrodes, and of other nerves, with a bar electrode (surface electrodes of 5 mm in diameter, placed 2 cm apart). The antidromic technique was used for the NCS of the sensory fibers of the peroneal and sural nerves. The orthodromic stimulation was applied in all the other nerves. In line with previously published criteria, patients were diagnosed with polyneuropathy when the distal motor latency (DML) was prolonged and/or motor conduction velocity (CV) was slowed in at least two separate nerves and/or there was a decrease in the amplitude of the SNAP or a decrease in sensory CV in at least two separate nerves.21 The only exception for applying the mentioned criteria was that the decrease of the amplitude of the CMAP was not taken into account because it might have resulted from myopathy.
Assessment of SRBD
Nocturnal respiration was assessed with an Embletta PDS® (Embla Systems, Inc., Thronton, CO, USA) screening device, which allows the recording of the nasal airflow through the nasal cannula, the respiratory effort through piezoinductive bands placed around the thorax and abdomen, as well as blood oxygen saturation with a finger oximeter. The recorded signals were analyzed according to the guidelines of the American Academy of Sleep Medicine.22 Apnea was scored when there was a drop in the airflow peak signal excursion by 90% or more of the pre-event baseline for 10 seconds or longer. Hypopnea was scored when the airflow peak signal excursions dropped by 30% of the pre-event baseline for 10 seconds or longer and were associated with arterial oxygen desaturation of 3% or higher. The differentiation of central and obstructive apneas was based on a respiratory effort signal provided by both piezoinductive bands. The assessment of SRBDs included a calculation of the apnea–hypopnea index (AHI; defined as the number of apneas and hypopneas per hour of the recording), oxygen desaturation index (ODI; defined as the number of significant [≥3%] desaturations per hour of the recording), and the mean and minimum arterial oxygen saturation during the whole recording (mean SaO2 and minimum SaO2, respectively).
Statistical analysis
The CVs, SNAP amplitudes, and DMLs of all studied nerves were correlated with the AHI, ODI, as well as mean and minimum SaO2 in both groups. The correlations were not assessed for the amplitude of the CMAP because it might have changed due to both neuropathic and myogenic processes. The NCS parameters were also correlated with age. The results were expressed as the mean and standard deviation. The significance level was set at a P-value of less than 0.05. For statistical analysis, the StatSoft, Inc. (2008) STATISTICA software (www.statsoft.com), version 8.0, was used.
Results
The demographic and clinical parameters of individual patients are summarized in Table 1. One patient (male, 58 years of age) was diagnosed with type 2 diabetes and received oral treatment. The remaining patients had no coexistent metabolic or endocrine disorders or significant cardiac disease. Moreover, none of the patients reported symptoms suggesting neuropathy, such as numbness, tingling, or burning sensation. The body mass index (BMI) and MRC score did not differ between patients with DM1 and those with DM2. DM1 was moderately advanced (MRC, 40–49 points) in five patients, and DM2, in one patient. In the remaining patients, the disease was mildly advanced (MRC, ≥50 points). The age showed a trend to be lower in the DM1 group than in the DM2 group (37.9±14.1 vs 47.6±14.1, P=0.07). None of the patients received supplemental O2, continuous positive airway pressure, or noninvasive ventilation.
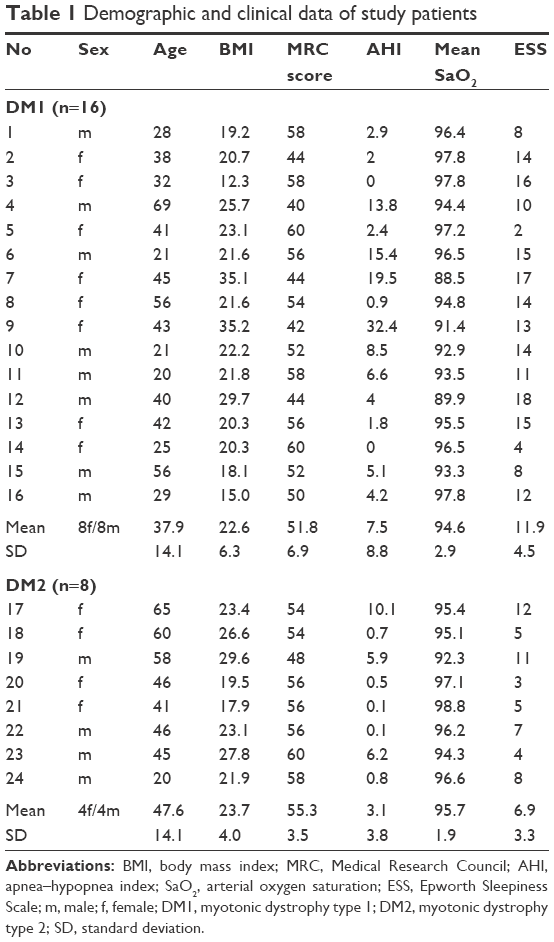 | Table 1 Demographic and clinical data of study patients |
NCS
The results of the NCS are presented in Table 2. The results were normal in six patients. In another six patients (three with DM1 and three with DM2), the changes fulfilled the criteria for polyneuropathy, and in the remaining patients, the abnormalities were shown only for a few nerves, which was not enough to diagnose polyneuropathy.
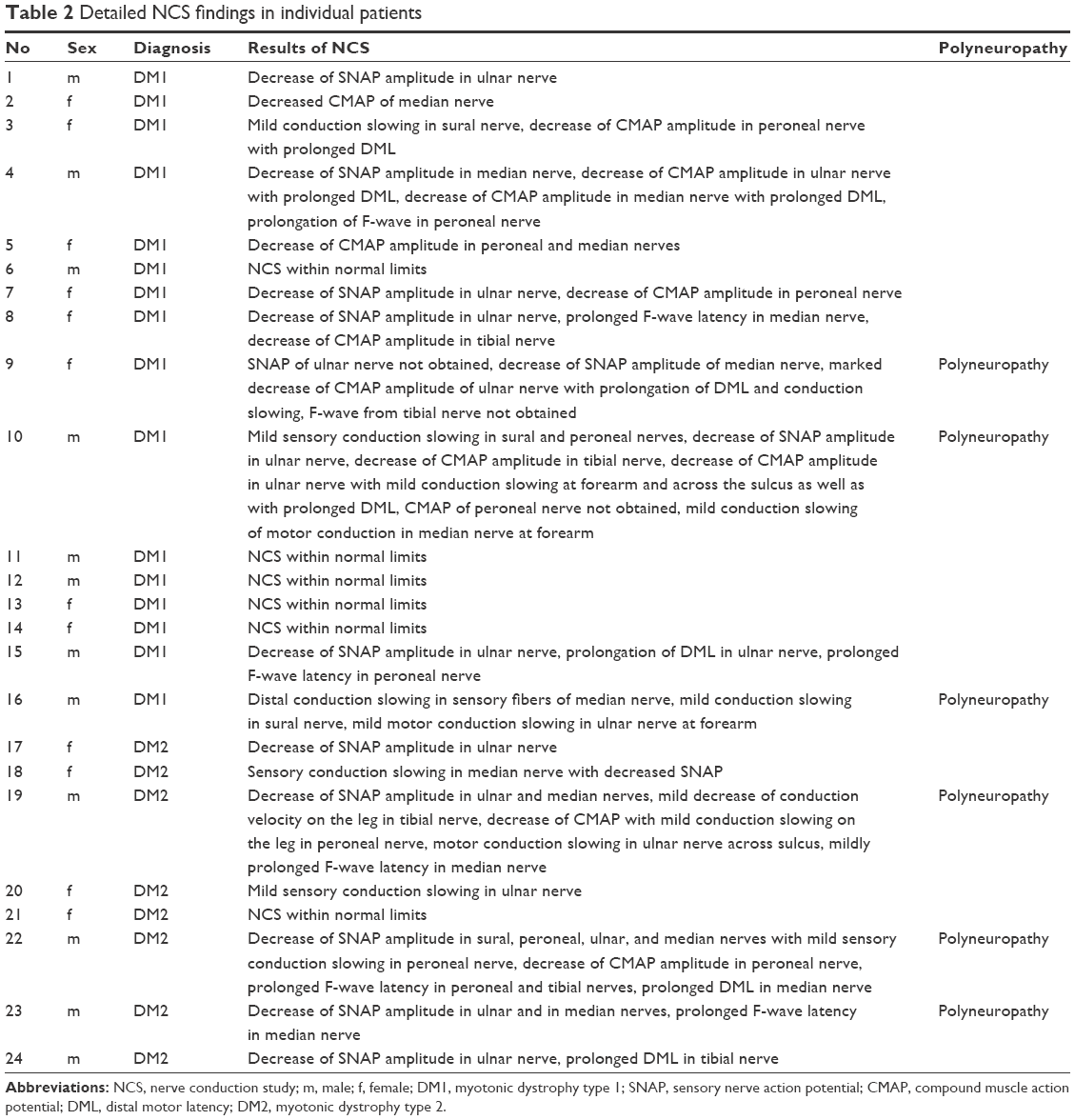 | Table 2 Detailed NCS findings in individual patients |
The amplitude of the CMAP of the ulnar nerve was lower in patients with DM1 than in those with DM2 (9.8±4.4 vs 13.7±2.3 mV, P=0.006), and the motor CV in the forearm ulnar nerve was slower in patients with DM1 than in those with DM2 (54.6±82 vs 64.4±8.4 m/s, P=0.016). The comparison of other NCV parameters showed no significant differences.
SRBD
Mild SRBDs as defined by an AHI of 5–14 were present in seven patients (27%) (four patients [25%] with DM1 and three patients [38%] with DM2). Medium and severe SRBDs (AHI, ≥15) were present in three patients (13%), all of whom had DM1. Two patients (both with DM1) had predominantly central respiratory events, and the remaining eight patients, mainly obstructive respiratory events. There was one patient (male, 40 years of age, DM1) with normal AHI (AHI, 4) but with nocturnal hypoxemia as reflected by a mean SaO2 of 89.9%. Moreover, in the group of patients with an AHI of 5 or higher, there was one patient (female, 45 years of age, DM1) with a mean SaO2 of less than 90% (Table 1). The AHI, ODI, and mean and minimum SaO2 did not differ between the groups. At a cutoff point for significant daytime sleepiness of 11,23 there were 11 patients (69%) with DM1 and two patients (25%) with DM2 who had sleepiness. The Epworth Sleepiness Scale score was also higher in patients with DM1 than in those with DM2 (11.9±4.5 vs 6.9±3.3, P=0.011).
Correlation of nocturnal respiratory parameters and age with NCS
In the DM1 group, the SNAP amplitude of the median nerve correlated with the AHI (R=−0.61, P=0.013, Figure 1); the SNAP amplitude of the ulnar nerve correlated with the mean SaO2 (R=0.53, P=0.036, Figure 2); and the DML of the ulnar nerve correlated with the AHI (R=0.56, P=0.024). The ODI correlated with the DML of the tibial nerve (R=−0.55, P=0.028) and with the CV in the sural nerve (R=0.051, P=0.042). The minimum SaO2 showed no correlations. Age correlated with the amplitude of the peroneal and sural SNAP (R=−0.75, P=0.001 and R=−0.55, P=0.027, respectively).
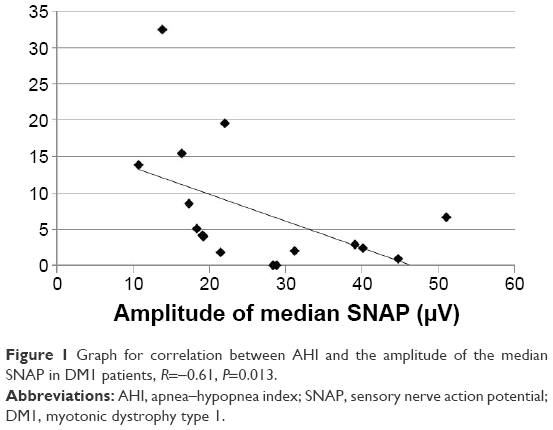 | Figure 1 Graph for correlation between AHI and the amplitude of the median SNAP in DM1 patients, R=−0.61, P=0.013. |
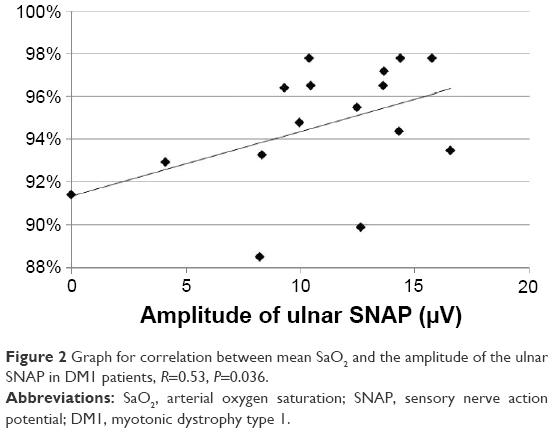 | Figure 2 Graph for correlation between mean SaO2 and the amplitude of the ulnar SNAP in DM1 patients, R=0.53, P=0.036. |
In the DM2 group, the amplitudes of the ulnar and median SNAP correlated with the mean SaO2 (R=0.76, P=0.028, Figure 3, and R=0.73, P=0.037, Figure 4, respectively). The ODI, AHI, minimum SaO2, and age showed no significant correlations with NCS parameters.
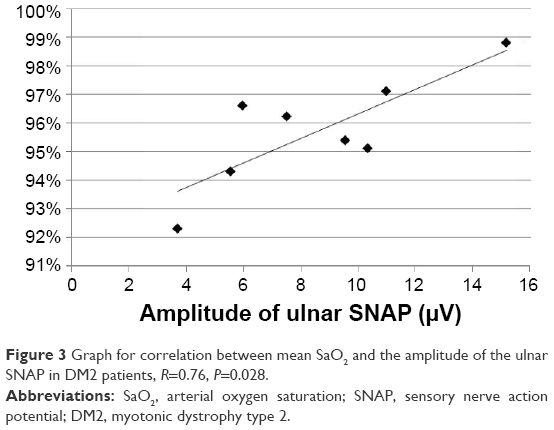 | Figure 3 Graph for correlation between mean SaO2 and the amplitude of the ulnar SNAP in DM2 patients, R=0.76, P=0.028. |
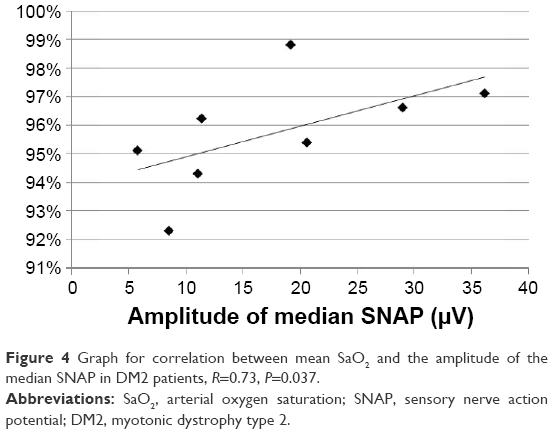 | Figure 4 Graph for correlation between mean SaO2 and the amplitude of the median SNAP in DM2 patients, R=0.73, P=0.037. |
Of the six patients (three with DM1 and three with DM2) that fulfilled the criteria for polyneuropathy, four had SRBDs (two with DM1 and two with DM2): one patient had mild central sleep apnea, two patients had mild obstructive sleep apnea, and one patient had severe obstructive sleep apnea.
Discussion
Our results indicate a possible association between neuropathy and SRBDs in DM. The correlation of several NCS parameters with the mean SaO2 as well as with the AHI and ODI suggests that neuropathy may be related to SRBD in a complex way, whereby the two conditions may affect one another by causing ischemia due to nocturnal desaturation as well as apneas and hypopneas due to the weakness of pharyngeal and respiratory muscles.
Study sample
We included a relatively homogenous group of patients with mild-to-moderate severity of the core clinical symptoms, according to the MRC grading score. The age of both groups, DM1 and DM2, was similar to that in a previous study, with about 10-year difference between the subtypes.24 The sex distribution was slightly different from that in the previous study in patients with DM1, while it was comparable in those with DM2.24 We decided to include patients with diabetes because this condition is significantly more prevalent among patients with DM, and a causal link between DMPK and ZNF9 mutations and insulin resistance has been documented.25
NCS
Similarly to previous studies,24,26 our patients did not report symptoms suggestive of polyneuropathy. Changes in the NCS were also similar to those previously described,7,27 but the prevalence of abnormal results of NCS may seem higher in our study, which might have resulted from a relatively high number of the nerves tested. The comparison of NCS results between the DM1 and DM2 groups showed minor differences, limited to motor conduction of the ulnar nerve. A recent study reported differences in a higher number of nerves, but it also included a significantly higher number of patients.24
SRBD
We documented SRBD in 40% of our patients which is somewhere in the middle of a wide range of the reported prevalence (16%–75%).9–11 In accordance with recent studies, the obstructive events prevailed.10,28 The severity of SRBD was comparable in both types of DM, which is also in line with a previous report.29
Correlation of NCS with respiratory parameters
In both groups, the correlation between NCS parameters and SRBDs was found mainly in the sensory fibers of the ulnar and median nerves. In the DM2 group, the amplitude of the ulnar and median SNAP correlated with the mean SaO2. In the DM1 group, NCS parameters correlated with the mean SaO2, AHI, and ODI. We believe that the correlation with the mean SaO2 may indicate that repetitive nocturnal hypoxemia associated with SRBD plays a causative role in the development of neuropathy in DM. On the other hand, the correlation of the amplitude of the median SNAP and DML of the ulnar CMAP with the AHI allowed us to speculate that neuropathy in DM contributes to the weakness of respiratory and pharyngeal muscles and thus to the development of the obstructive form of SRBDs. Our data also allow us to hypothesize that the causative role of desaturations may be especially important in DM2, while the contribution of neuropathy to respiratory events may be associated more with DM1. However, these tendencies need to be reproduced on a larger sample of patients.
The causative role of nocturnal hypoxemia in inducing neuropathy may rely on the repetitive reoxygenation after the arousal from apnea causes an ischemia–reperfusion injury with the generation of reactive oxygen species and induction of oxidative stress. Oxidative stress activates inflammatory processes with increased expression of the target genes such as tumor necrosis factor alpha, interleukin 6, as well as adhesion molecules such as intracellular adhesion molecule 1 and other pro-inflammatory cytokines. This may in turn lead to increased interaction of blood cells with the endothelium, leading to microangiopathy and nerve degeneration.30,31 Additionally, thickening of the perineurium occurs under hypoxic conditions, which can impair the transport of oxygen and other metabolic substrates and, as a result, enhance axonal degeneration.17 Once neuropathy becomes severe, it can impair the patency of the upper airways or decrease the inspiratory volume due to muscle weakness, which is the common cause of respiratory disturbances in neuromuscular diseases.32 Apart from muscle weakness, the involvement of autonomic fibers in neurodegeneration may disturb the proper pattern of respiratory movements, as it was documented in patients with multisystem atrophy.33 Autonomic neuropathy may also reduce the afferent information from the lungs and decrease the sensitivity of the respiratory center to CO2. This in turn may contribute to the development of central apneas and hypopneas.34
In this study, we documented the association between SRBD and impairment mainly of the sensory fibers. According to a previous study,18 damage to sensory fibers is sufficient to cause repetitive occlusions of the upper airways. This may occur due to the damage of the pharyngeal sensory nerve, which forms an afferent of the protective reflexes, responsible for an increase in the pharyngeal muscle tonus during inspiration.18 On the other hand, another study showed a clear correlation between the decrease of the median CMAP and severity of respiratory events.35 This may also be the case in our patients, and impairment of the motor fibers is likely to be involved in the development of SRBD. However, as mentioned, we did not analyze the CMAP amplitude because it reflects both the neuropathic and myopathic processes in DM.
SRBD in patients with polyneuropathy
Four of six patients (67%) with polyneuropathy in our study showed SRBDs. Therefore, the prevalence of SRBDs in this subgroup may be higher than that in the whole study group (40%) and may constitute further evidence for the detrimental effect of nocturnal hypoxemia on all peripheral nerves. However, this finding has to be confirmed in a larger sample of patients. The direct association between DM and neuropathy has been documented in transgenic DMSXL mice carrying large CTG expansions.36
Effect of age on NCS
Our results confirmed the previous finding that age is associated with changes in NCS.37 The amplitude of the SNAP showed an inverse correlation with age. However, the correlation was found in the nerves of the lower extremities, while the correlation of NCS and SRBDs was found predominantly in the nerves of the upper extremities. Furthermore, the association of age and NCS was found only in the DM1 group. This is probably because the DM2 group was smaller and, except one younger patient, included only middle-aged patients (45–65 years). Thus, the age affected NCS parameters in a different way than SRBDs did.
Daytime sleepiness in DM1 and DM2 groups
Similarly to previous reports, daytime sleepiness was more pronounced in patients with DM1.38 This difference was not associated with respective differences in the severity of SRBDs, BMI, or muscle weakness, the factors that may limit the restorative function of sleep and thus indirectly provoke excessive sleepiness. Therefore, we rather attribute this finding to the primary mechanisms of the disease, which seem to affect the nervous structures responsible for alertness more severely in DM1 than in DM2. This is in agreement with studies documenting the loss of serotoninergic neurons in the dorsal raphe nucleus and decreased hypocretin levels in cerebrospinal fluid in patients with DM1.39,40
Limitations
We studied patients with mild-to-moderate form of the disease, which might obscure the observed association between neuropathy and SRBDs. With the exception of the ulnaris, the studied nerves were normal in a great majority of patients. The inclusion of patients with more advanced neuropathy would probably provide a clearer picture of the relationship between SRBDs and nerve damage.
Another limitation of the study is a relatively small number of patients, especially in the DM2 group. The inclusion of a higher number of patients would probably help us draw more precise conclusions. Finally, our study lacks the assessment of lung function. Changes in lung function as a consequence of the myopathic process may be a significant confounder of the observed relationship between sleep apnea and neuropathy.
Conclusion
Our preliminary results indicate the association between neuropathy and SRBDs, which has been previously observed in other conditions and may also be present in patients with DM1 and DM2. This relation is probably complex and works both ways: axonal degeneration may contribute to nocturnal hypoxemia and vice versa. The neuropathy may in turn contribute to muscle weakness, which may trigger respiratory events.
Disclosure
The authors report no conflicts of interest in this work.
References
Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11(10):891-905. | ||
Harley HG, Brook JD, Rundle SA, et al. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature. 1992;355(6360):545-546. | ||
Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293(5531):864-867. | ||
Banach M, Rakowicz M, Antczak J, Rola R, Witkowski G, Waliniowska E. Zagrożenia kardiologiczne i oddechowe oraz zaburzenia snu u pacjentów z dystrofią miotoniczną. [Cardiac, respiratory and sleep disorders in patients with myotonic dystrophy]. Przegl Lek. 2009;66(12):1065-1068. Polish. | ||
Bae JS, Kim SG, Lim JC, Chung EJ, Kim OK. Peripheral nerve axon involvement in myotonic dystrophy type 1, measured using the automated nerve excitability test. J Clin Neurol. 2011;7(2):90-95. | ||
McComas AJ, Sica RE, Toyonaga K. Incidence, severity, and time-course of motoneurone dysfunction in myotonic dystrophy: their significance for an understanding of anticipation. J Neurol Neurosurg Psychiatry. 1978;41(10):882–893. | ||
Olson ND, Jou MF, Quast JE, Nuttall FQ. Peripheral neuropathy in myotonic dystrophy. Relation to glucose intolerance. Arch Neurol. 1978;35(11):741-745. | ||
Paramesh K, Smith BH, Kalyanaraman K. Early onset myotonic dystrophy in association with polyneuropathy. J Neurol Neurosurg Psychiatry. 1975;38(11):1136–1139. | ||
Cirignotta F, Mondini S, Zucconi M, et al. Sleep related breathing impairment in myotonic dystrophy. J Neurol. 1987;235(2):80-85. | ||
Laberge L, Bégin P, Dauvilliers Y, et al. A polysomnographic study of daytime sleepiness in myotonic dystrophy type 1. J Neurol Neurosurg Psychiatry. 2009;80(6):642-646. | ||
Shepard P, Lam EM, St Louis EK, Dominik J. Sleep disturbances in myotonic dystrophy type 2. Eur Neurol. 2012;68(6):377-380. | ||
Broughton R, Stuss D, Kates M, Roberts J, Dunham W. Neuropsychological deficits and sleep in myotonic dystrophy. Can J Neurol Sci. 1990;17(4):410-415. | ||
Bégin P, Mathieu J, Almirall J, Grassino A. Relationship between chronic hypercapnia and inspiratory-muscle weakness in myotonic dystrophy. Am J Respir Crit Care Med. 1997;156(1):133-139. | ||
Ono S, Kanda F, Takahashi K, et al. Neuronal loss in the medullary reticular formation in myotonic dystrophy: a clinicopathological study. Neurology. 1996;46(1):228-231. | ||
Tahrani AA, Ali A, Raymond NT, et al. Obstructive sleep apnea and diabetic neuropathy: a novel association in patients with type 2 diabetes. Am J Respir Crit Care Med. 2012;186(5):434-441. | ||
de Carvalho Alcântara M, Nogueira-Barbosa MH, Fernandes RM, et al. Respiratory dysfunction in Charcot-Marie-Tooth disease type 1A. J Neurol. 2015;262(5):1164–1171. | ||
Lüdemann P, Dziewas R, Sörös P, Happe S, Frese A. Axonal polyneuropathy in obstructive sleep apnoea. J Neurol Neurosurg Psychiatry. 2001;70(5):685-687. | ||
Lévy P, Pépin JL, Dematteis M. Pharyngeal neuropathy in obstructive sleep apnea: where are we going? Am J Respir Crit Care Med. 2012;185(3):241-243. | ||
Martínez Cerón E, Casitas Mateos R, García-Río F. Sleep apnea-hypopnea syndrome and type 2 diabetes. A reciprocal relationship? Arch Bronconeumol. 2015;51(3):128-139. | ||
Dyck PJ, Boes CJ, Mulder D, et al. History of standard scoring, notation, and summation of neuromuscular signs. A current survey and recommendation. J Peripher Nerv Syst. 2005;10(2):158-173. | ||
Oh SJ. Clinical Electromyography: Nerve Conduction Studies. 3rd ed. Philadelphia: Lippincott William & Wilkins; 2003. | ||
Berry RB, Budhiraja R, Gottlieb DJ, et al; American Academy of Sleep Medicine. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8(5):597-619. | ||
Patel SR, White DP, Malhotra A, Stanchina ML, Ayas NT. Continuous positive airway pressure therapy for treating sleepiness in a diverse population with obstructive sleep apnea: results of a meta-analysis. Arch Intern Med. 2003;163(5):565–571. | ||
Nojszewska M, Łusakowska A, Szmidt-Sałkowska E, et al. Peripheral nerve involvement in myotonic dystrophy type 2 – similar or different than in myotonic dystrophy type 1? Neurol Neurochir Pol. 2015;49(3):164–170. | ||
Savkur RS, Philips AV, Cooper TA, et al. Insulin receptor splicing alteration in myotonic dystrophy type 2. Am J Hum Genet. 2004;74(6):1309-1313. | ||
Hermans MC, Faber CG, Vanhoutte EK, et al. Peripheral neuropathy in myotonic dystrophy type 1. J Peripher Nerv Syst. 2011;16(1):24-29. | ||
Pfeilsticker BH, Bertuzzo CS, Nucci A. Electrophysiological evaluation in myotonic dystrophy: correlation with CTG length expansion. Arq Neuropsiquiatr. 2001;59(2-A):186-191. | ||
Pincherle A, Patruno V, Raimondi P, et al. Sleep breathing disorders in 40 Italian patients with Myotonic dystrophy type 1. Neuromuscul Disord. 2012;22(3):219-224. | ||
Bianchi ML, Losurdo A, Di Blasi C, et al. Prevalence and clinical correlates of sleep disordered breathing in myotonic dystrophy types 1 and 2. Sleep Breath. 2014;18(3):579–589. | ||
Arnardottir ES, Mackiewicz M, Gislason T, Teff KL, Pack AI. Molecular signatures of obstructive sleep apnea in adults: a review and perspective. Sleep. 2009;32(4):447-470. | ||
Lavie L. Oxidative stress – a unifying paradigm in obstructive sleep apnea and comorbidities. Prog Cardiovasc Dis. 2009;51(4):303-312. | ||
Dhand UK, Dhand R. Sleep disorders in neuromuscular diseases. Curr Opin Pulm Med. 2006;12(6):402–408. | ||
Munschauer FE, Loh L, Bannister R, Newsom-Davis J. Abnormal respiration and sudden death during sleep in multiple systematrophy with autonomic failure. Neurology. 1990;40(4):677–679. | ||
Rees PJ, Prior JG, Cochrane GM, Clark TJ. Sleep apnoea in diabetic patients with autonomic neuropathy. J R Soc Med. 1981;74(3):192–195. | ||
Dematteis M, Pépin JL, Jeanmart M, Deschaux C, Labarre-Vila A, Lévy P. Charcot-Marie-Tooth disease and sleep apnoea syndrome: a family study. Lancet. 2001;357(9252):267–272. | ||
Panaite PA, Kielar M, Kraftsik R, Gourdon G, Kuntzer T, Barakat-Walter I. Peripheral neuropathy is linked to a severe form of myotonic dystrophy in transgenic mice. J Neuropathol Exp Neurol. 2011;70(8):678–685. | ||
Tavee JO, Polston D, Zhou L, Shields RW, Butler RS, Levin KH. Sural sensory nerve action potential, epidermal nerve fiber density, and quantitative sudomotor axon reflex in the healthy elderly. Muscle Nerve. 2014;49(4):564–569. | ||
Schneider-Gold C, Bellenberg B, Prehn C, et al. Cortical and subcortical grey and white matter atrophy in myotonic dystrophies type 1 and 2 is associated with cognitive impairment, depression and daytime sleepiness. PLoS One. 2015;10(6):e0130352. | ||
Ono S, Takahashi K, Jinnai K, et al. Loss of serotonin-containing neurons in the raphe of patients with myotonic dystrophy: a quantitative immunohistochemical study and relation to hypersomnia. Neurology. 1998;50(2):535–538. | ||
Martínez-Rodríguez JE, Lin L, Iranzo A, et al. Decreased hypocretin-1 (orexin-A) levels in the cerebrospinal fluid of patients with myotonic dystrophy and excessive daytime sleepiness. Sleep. 2003;26(3):287–290. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.