Back to Journals » Infection and Drug Resistance » Volume 15
Association of Mannose-Binding Lectin 2 Gene Polymorphism with Tuberculosis Based on Mycobacterium tuberculosis Lineages
Authors Liu M, Wang Q, Liu H ![]() , Yin C, Mijiti X, Anwaierjiang A, Wan K, Xu M, Li M, Nong S, Li G, Xiao H
, Yin C, Mijiti X, Anwaierjiang A, Wan K, Xu M, Li M, Nong S, Li G, Xiao H
Received 18 October 2021
Accepted for publication 25 January 2022
Published 24 March 2022 Volume 2022:15 Pages 1225—1234
DOI https://doi.org/10.2147/IDR.S344935
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Mengwen Liu,1 Quan Wang,2 Haican Liu,3 Chunjie Yin,1 Xiaokaiti Mijiti,2 Aiketaguli Anwaierjiang,1 Kanglin Wan,3 Miao Xu,2 Machao Li,3 Siqin Nong,4 Guilian Li,3 Hui Xiao1
1School of Public Health, Xinjiang Medical University, Urumqi, 830011, People’s Republic of China; 2The Eighth Affiliated Hospital of Xinjiang Medical University, Urumqi, 830001, People’s Republic of China; 3State Key Laboratory for Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, 102206, People’s Republic of China; 4College of Life Science and Technology, Beijing University of Chemical Technology, Beijing, 100029, People’s Republic of China
Correspondence: Guilian Li; Hui Xiao, Email [email protected]; [email protected]
Purpose: Polymorphisms in MBL2 may contribute to the susceptibility to tuberculosis. The aim of the present study was to determine the associations of the polymorphisms of five loci (rs1800450, rs1800451, rs7096206, rs7095891, and rs11003125) in the MBL2 gene with susceptibility to tuberculosis and specific lineages of Mycobacterium tuberculosis causing tuberculosis in the Uyghur population of Xinjiang, China.
Methods: From January 2019 to January 2020, we enrolled 170 Uyghur tuberculosis patients as the case group and 147 Uyghur staff with no clinical symptoms as the control group from four designated tuberculosis hospitals in southern Xinjiang, China. The polymorphisms of five loci in MBL2 of human were detected by sequencing. Whole-genome sequencing was applied in 68 M. tuberculosis isolates from the case group and the data were used to perform genealogy analysis.
Results: The distributions of allele and genotype frequencies of five loci in MBL2 varied little between the case and control groups and varied little among the groups, including those infected with different lineages of M. tuberculosis and the control (except those of rs11003125), the P values were all > 0.05. The distribution of alleles of rs11003125 was statistically different between patients infected with lineages 3 and 4 M. tuberculosis (χ2=7.037, P=0.008). The C allele and CC genotype of rs11003125 were found to be protective factors against lineage 4 infection when compared to lineage 3 (ORs were 0.190 and 0.158, respectively; 95% confidence intervals were 0.053∼ 0.690 and 0.025∼ 0.999, respectively).
Conclusion: Our results suggested that human’s susceptibility to tuberculosis is affected both by the host genetic polymorphisms and the lineage of the M. tuberculosis that people were exposed to. However, due to the limitation of the sample size in the present study, larger sample size and more rigorous design should be guaranteed in future studies.
Keywords: mannose-binding lectin 2 gene, polymorphism, tuberculosis, Mycobacterium tuberculosis, lineage
Introduction
Tuberculosis is a serious public health problem, and the global COVID-19 pandemic since 2020 may reverse progress in reducing the global tuberculosis burden, allowing for more than 1 million additional tuberculosis cases per year during 2020–2025.1 The World Health Organization (WHO) estimates that the number of new tuberculosis patients in China was 833,000 in 2019, ranking third among the top thirty countries with high tuberculosis burden globally.1 About a quarter of the world’s population are latently infected with Mycobacterium tuberculosis. However, their lifetime risk of reactivation is about 5–10%,1 suggesting that the individual differences in susceptibility to tuberculosis are universal. Another fact far back in 1930 in Germany, noted as the Ramp van Lübeck disaster, also provided clues: among 250 newborns vaccinated with the vaccine contaminated with the virulent M. tuberculosis, 77 died, 127 cases showed tuberculosis symptoms, while the other 47 had no manifestations of M. tuberculosis infection.2–4
In recent years, genes related to tuberculosis susceptibility have been reported continuously through genome-wide association studies (GWAS) and hot single nucleotide polymorphism (SNP) correlation studies.5–7 The human mannose-binding lectin 2 gene (MBL2) is located on chromosome 10q21.1. The MBL2 gene encodes a liver-derived glycoprotein named mannose-binding lectin (MBL), which exists in the serum as a multimeric protein and acts as an important anti-infective factor in the innate immune system. MBL is a member of the Ca2+ dependent C-type lectin superfamily,8 which is involved in the detection of pathogens by recognizing the conserved structures known as pathogen associated molecular patterns (PAMPs) and subsequently promotes the activation of innate immunity.9 For the characteristics that MBL can recognize PAMPs on the surface of pathogens, it is also considered to be a pattern recognition receptor (PRRs).8 MBL attached to the surface of the pathogen can activate the MBL-related serine proteases MASP-1 and MASP-2, which will trigger the proteolytic cascade reaction and induce the activation of the complement system.8,10 Several single nucleotide polymorphisms (SNPs) on the MBL2 gene associated with serum MBL levels and function have been reported, including rs7096206, rs7095891 and rs11003125 in the upstream region, and sites rs5030737, rs1800450 and rs1800451 in the exon 1 region.11,12 As knowledge on the MBL2 immune defense activity and the immunological mechanism against M. tuberculosis is growing, a number of studies were conducted to find hints on the MBL2 gene polymorphisms to clarify the mechanisms of people’s susceptibility to tuberculosis. However, consistency in conclusions was lacking across different groups in terms of race, age and gender.6,13,14
Liu et al performed whole-genome sequencing (WGS) on a large number of individual colonies from 18 patients to characterize the genetic diversity of the M. tuberculosis population at the onset of the disease and reconstruct the evolution of M. tuberculosis within the host, and they found that mutagenesis of M. tuberculosis in vivo is modulated by the host environment.15 Omae et al performed a M. tuberculosis lineage-based GWAS with CD53 gene and found that two SNPs (rs1418425 and rs1494320) are risk factors for the old age tuberculosis onset infected with strains of non-Beijing genotype, and they speculated that the genetic risk of host susceptibility is related to the genotypes of infected strains.16 Therefore, the interactions between the genetic systems of humans and M. tuberculosis should be taken into account in discovering the mechanisms of host susceptibility to tuberculosis. The various lineages of M. tuberculosis defined by molecular epidemiology tools like WGS showed genetic distance may pose different stress on the human immune system. In the present study, we will explore the polymorphisms of rs1800450, rs1800451, rs7096206, rs7095891 and rs11003125 in the MBL2 gene among the Uyghur population with tuberculosis or not, as well as among people infected with different lineages of M. tuberculosis defined by WGS from Xinjiang, China, in order to determine their impact on the susceptibility to tuberculosis.
Methods
The Sources of Subjects
The Case Group
A total of 170 Uyghur tuberculosis patients were enrolled from the Eighth Affiliated Hospital of Xinjiang Medical University, Kashgar Pulmonary Hospital, Kuqa County Infectious Disease Hospital and Wushi County People’s Hospital from January 2019 to January 2020. The inclusion criteria for the case group are as follows: 1) comply with the diagnostic criteria for tuberculosis in the industry standard “WS 288–2017 Diagnosis for pulmonary tuberculosis” issued by the National Health and Family Planning Commission of the People’s Republic of China;17 and 2) both parents of the patients are Uyghur. We excluded the patients with severe damage to organs such as the heart, liver and kidney (primary disease), endocrine system diseases (such as hyperthyroidism, Cushing syndrome, etc.), malignant tumors, hematological diseases, mental diseases and pregnancy.
The Control Group
The control group consisted of 147 Uyghur staff in the Eighth Affiliated Hospital of Xinjiang Medical University with no clinical symptoms. The inclusion criteria for the control group are as follows: 1) they were confirmed to have a negative TSPOT.TB through participating in annual physical examination; and 2) both parents of the controls are Uyghur. The exclusion criteria are the same as those of the case group.
Collection of Specimens and DNA Preparation
After all participants signed the informed consent form and joined the group, 5 mL of venous blood from each participant were collected in EDTA anticoagulant tubes. The nucleic acid extraction kit (magnetic bead method) and AU1001-96 automatic nucleic acid extractor provided by China Wuxi Biotech Biotechnology Co., Ltd were then used to extract DNA. A total of 68 clinical isolates of M. tuberculosis in the case group were collected, and their genomic DNA was extracted through the cetyltrimethylammonium bromide (CTAB) method.18 The quantity and quality of the obtained DNA samples were analyzed spectrophotometrically (NanoDrop 2000, Thermo Scientific, MA, USA).
Polymorphisms of Five SNPs in MBL2
A total of five SNPs in MBL2 were analyzed in the present study (Table 1). According to the NCBI dbSNP database, rs1800450 and rs1800451, rs7096206 and rs7095891 in MBL2 are adjacent to each other, respectively. So, we totally designed three pairs of primers to sequence all of the five SNPs. The primers were designed by Oligo 6.0 software and synthesized by Beijing Tianyi Huiyuan Biotechnology Co., Ltd, Beijing, China. The total volume of the PCR reaction was 25 µL, including 12.5 µL PCR mixture, 1 µL DNA, 10 ng upstream primer, 10 ng downstream primer and 9.5 µL ddH2O. PCR conditions were 95 °C for 3 min, followed by 35 cycles of 95 °C for 30 s, 62.0 °C (rs1800450 and rs1800451) or 61.4 °C (rs7096206 and rs7095891) or 53.9 °C (rs11003125) for 30 s, and 72 °C for 30 s, and finally extension at 72 °C for 7 min (Table 1). All samples were amplified and the PCR products were sent to Beijing Tianyi Huiyuan Biotechnology Co., Ltd, Beijing, China for sequencing on an ABI 3730xl DNA Sequencer (Applied Biosystems, Foster City, CA, USA).
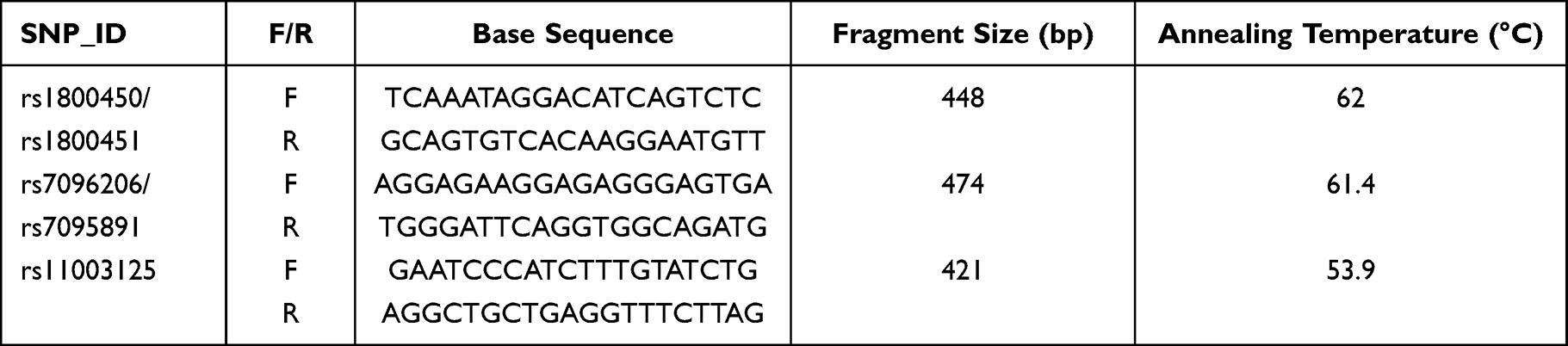 |
Table 1 Primer Information for the Five Loci of the MBL2 Gene |
Whole-Genome Sequencing and Genetic Lineage Analysis of M. tuberculosis Isolates
DNA libraries were prepared with genomic DNA using kits as instructed by the manufacturer. DNA libraries were then selected to perform cluster growth and 150 bp paired-end sequencing on DNB SEQ-2000 instrument (Beijing Genomics Institute, China). Following the quality control process, Snippy software (https://github.com/tseemann/snippy, version: 4.6.0) was used to analyze all of the genome sequence data that met the quantity control criteria. The M. tuberculosis H37Rv genome sequence (NC_000962.3) was used as the reference to identify the core SNPs and the SNPs located in the PE/PPE/PGRS gene regions were filtered out. The lineage information was determined by TB-Profiler (https://github.com/jodyphelan/TBProfiler, version 4.0.3). Then the core SNPs were used for building the phylogeny tree using the FastTree software (http://www.microbesonline.org/fasttree/) and then the lineage information was used for annotating the phylogeny tree by iTOL.19
Statistical Analysis
A nearest neighbor 1:1 matching ratio with propensity scores that fell within a caliper of 0.10 was used to generate the matched group to balance potentially confounding baseline characteristics. Univariate regression analysis and multivariable logistic regression model were then used to study the relationships between alleles or genotypes of MBL2 gene and tuberculosis status or lineages of M. tuberculosis infection as applicable. All analyses were performed by SPSS 25.0 (SPSS Inc., Chicago, IL, USA). The statistical significance was established at P≤0.05.
Results
Hardy–Weinberg Equilibrium Test
According to previous literature reports, if gene polymorphisms are linked to disease susceptibility, the case group may not be in accordance with the Hardy–Weinberg equilibrium.20 It is recommended that the Hardy–Weinberg equilibrium test be performed only in the control group.
Goodness-of-fit test using a chi-square discrepancy measure was employed to compare the distribution of the actual frequency and the theoretical frequency of objective loci to observe the fitting degree. The results demonstrated that the control group’s genetic inheritance followed the Hardy–Weinberg equilibrium (P>0.05) (Table 2), implying that the control group is representative of the research population.
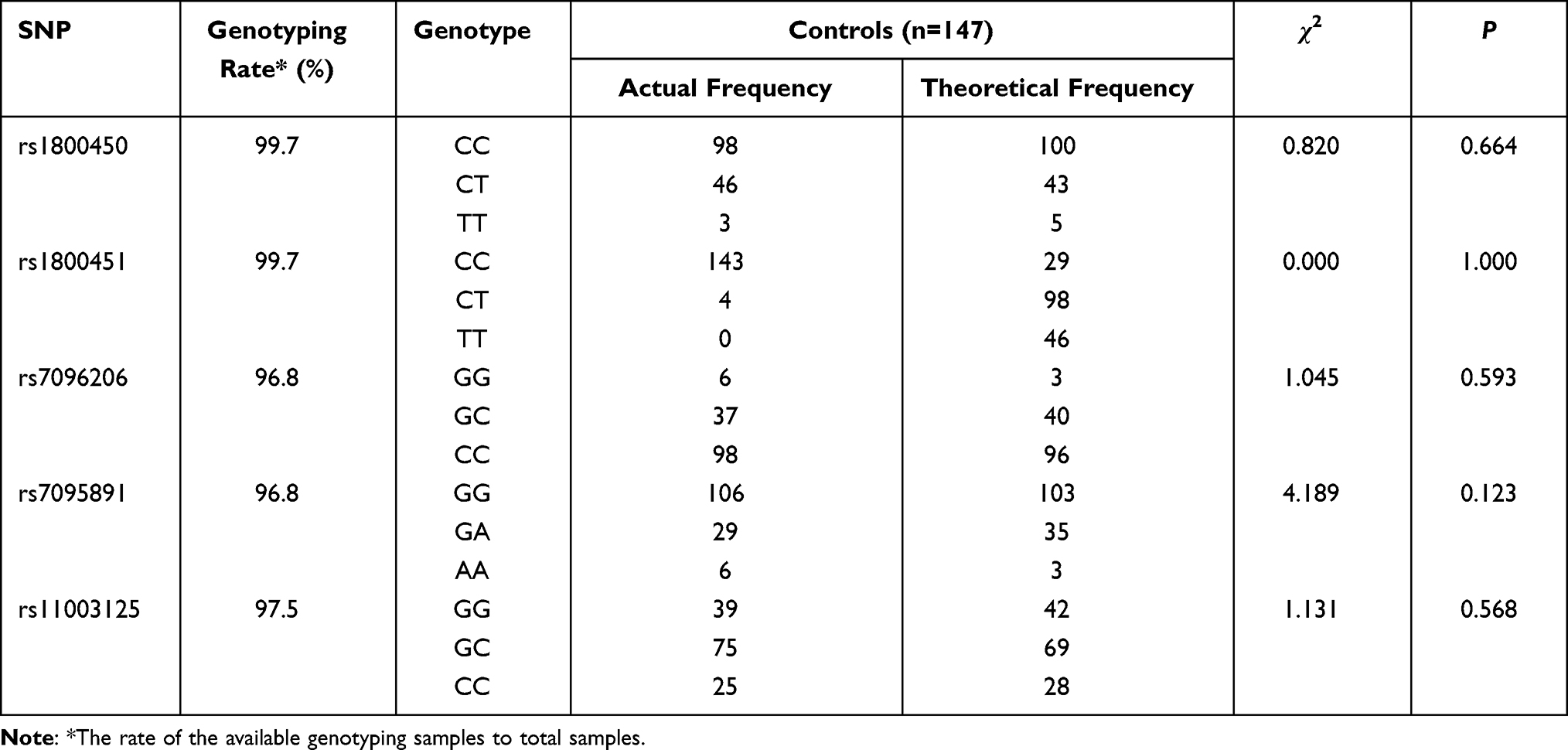 |
Table 2 Distributions of the MBL2 Genotypes and HWE Testing in the Control Group |
Association Analysis of MBL2 Gene Polymorphisms and Susceptibility to tuberculosis
Univariate Regression Analysis on the Associations Between the Alleles or Genotypes of Five Loci and Susceptibility to Tuberculosis
For the whole subjects, it was found that the differences in gender and age between the two groups were statistically significant (both P values <0.05). In addition, none of the alleles or genotypes of the five loci were statistically associated with susceptibility to tuberculosis (Supplementary Table 1).
We used PS with nearest neighbor 1:1 matching to control the effect attributed to age and gender between the two groups, and 103 subjects were included in each group after matching. Among these subjects, the SMD values of each covariate (age and gender) were less than 0.10, indicating a good balance between the groups and good propensity score matching. None of the alleles or genotypes of the five loci were found to statistically associated with susceptibility to tuberculosis, too (Supplementary Table 1).
Multivariable Logistic Regression Analysis
For the whole subjects, age, gender, rs1800451 and rs11003125 genotypes were found with P<0.20 in the univariate regression analysis and were included in the multivariable logistic regression model for further analysis. The results showed that no association was found between rs1800451 and rs11003125 polymorphisms and susceptibility to tuberculosis (Supplementary Table 2).
Lineage Distributions of the M. tuberculosis Isolates
Susceptibility to tuberculosis may depend on the potential interaction of host genetic factors with genetic nature of M. tuberculosis. In the present study, 68 isolates of M. tuberculosis were collected from the case group. The lineage analysis results showed that the lineage 2 occurred most frequently with 34 (50%) cases, followed by lineage 4 with 22 (32.4%) cases, lineage 3 with 8 (11.8%) cases, and other lineages with 4 (5.9%) cases, as shown in Figure 1.
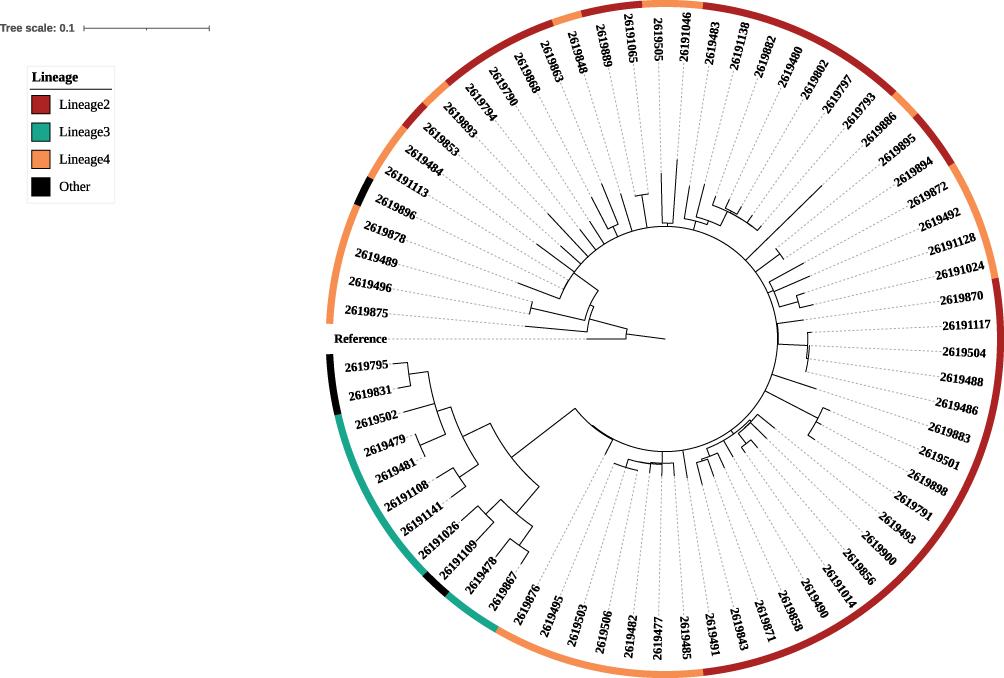 |
Figure 1 The phylogeny tree. |
Association of MBL2 Gene Polymorphisms with Different Lineages of M. tuberculosis
None of the SNPs in the MBL2 gene showed a statistically significant difference of distributions among groups infected with different lineages of M. tuberculosis, while the distribution of rs11003125 alleles showed a possible association with distinct lineages of M. tuberculosis infection (Table 3).
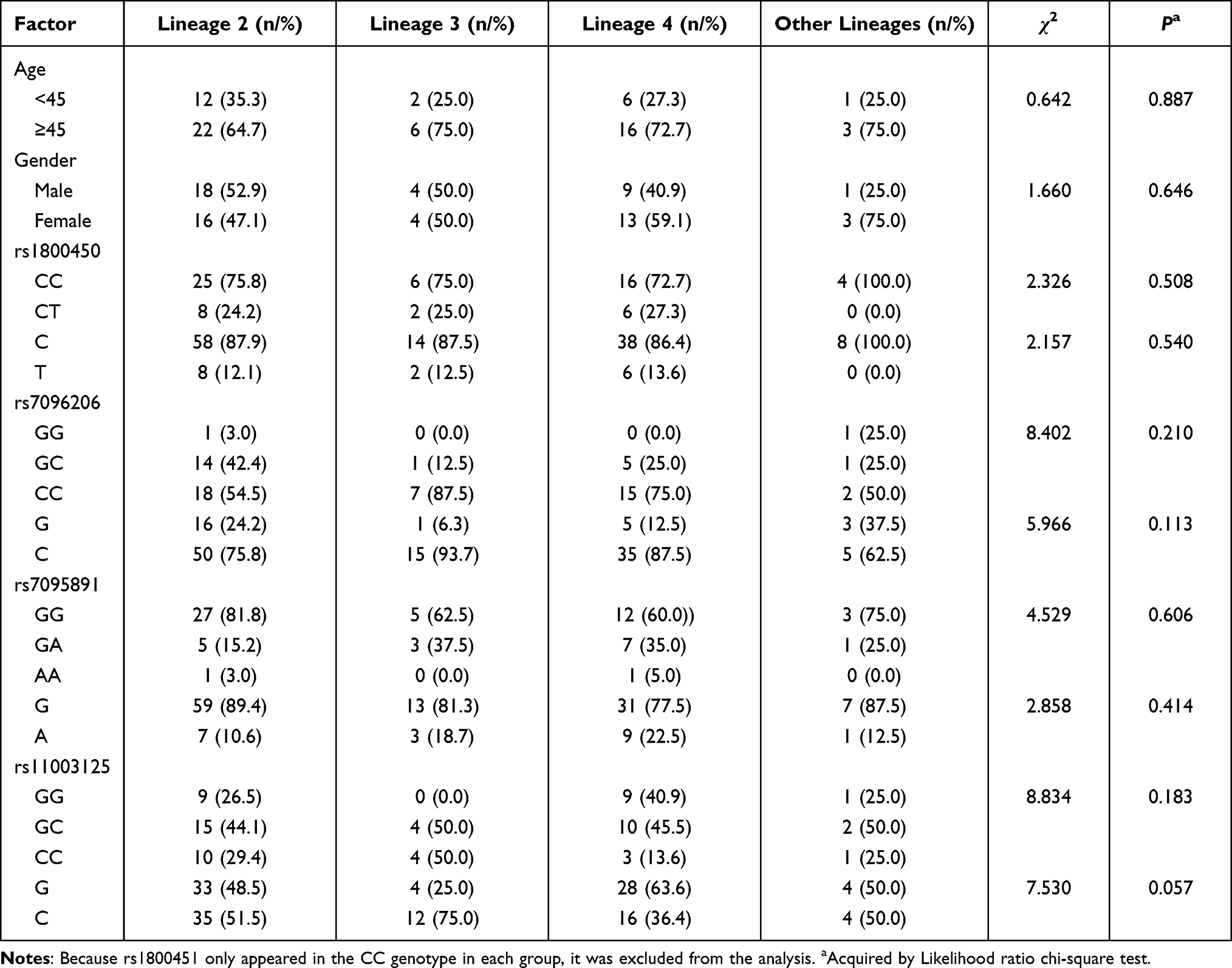 |
Table 3 Association of MBL2 Gene Polymorphisms with Different Lineages of M. tuberculosis Infection |
We next performed a two-by-two comparison and found that with an adjusting α’=0.008, the distribution of the allele of rs11003125 was statistically different between patients infected with lineages 3 and 4 of M. tuberculosis (χ2=7.037, P=0.008). By comparing to lineage 3, the C allele of rs11003125 showed as a protective factor of lineage 4 infection (OR=0.190, 95% confidence interval [CI]: 0.053~0.690), G allele was used as reference) (Table 4). In order to determine which genotype plays the key role in protecting against lineage 4 infection, we then performed a univariate regression analysis (we did not include gender and age, for that their differences between lineage 3 and lineage 4 were not found statistically significant) and found that the CC genotype showed as a protective factor compared to GC&GG genotypes (OR=0.158, 95% CI: 0.025–0.999) (Table 5).
 |
Table 4 Distributions of the Alleles of rs11003125 in MBL2 Gene Between Patients Infected with Lineages 3 and 4 of M. tuberculosis |
 |
Table 5 Univariate Regression Analysis of rs11003125 Polymorphisms and Lineages 4 of M. tuberculosis Infection |
Considering the number of four groups of M. tuberculosis was insufficient, we classified M. tuberculosis into lineage 2- and non-lineage 2-infected groups. And then compared the differences in the distribution of MBL2 gene polymorphisms among these two groups and the control group. The results showed that none of the alleles or genotypes of the four loci were statistically associated with specific lineages of M. tuberculosis infection (Supplementary Table 3).
Discussion
Previous researches have indicated that comprehending the co-evolution of pathogen and host faces many different constraints, and has advised that reciprocal polymorphisms in genes involved in the host–pathogen interaction be identified at the molecular level.21–23 By understanding the genetic architecture of coevolution, we can use population genetic models to predict such events as the expected temporal dynamics of allele frequencies, the likelihood of new mutations arising, and the spread of new variants.24 In the present study, we explore the association of the polymorphisms in rs1800450, rs1800451, rs7096206, rs7095891 and rs11003125 of the MBL2 gene with susceptibility to tuberculosis and their potential interactions with infected M. tuberculosis lineages in the Uyghur population of Xinjiang, China. When merely population genetic factors were considered, however, no significant association was discovered. This is the first report on the association between genetic polymorphisms and the lineages of M. tuberculosis infection in the population of China.
The MBL pathway identifies the peptidoglycan of Gram-positive bacteria through the C-type lectin domain and regulates the release of cytokines and chemokines, making it a key component of the innate immune system in pathogen identification.25 In one study, serum MBL deficiency rates were consistent regardless of the severities of tuberculosis patients, implying that MBL deficiency may predispose patients to M. tuberculosis infection.26 Nevertheless, other researchers have suggested that MBL deficiency may protect patients from tuberculosis infection by restricting pathogen entry into host cells.27 Although the mechanism of MBL2 gene mutation regulating the progression of tuberculosis is not clear, there is no doubt that MBL2 gene plays a significant part in tuberculosis pathogenesis.
In the present study, 170 Uyghur tuberculosis patients and 147 healthy controls were selected to explore the polymorphisms of MBL2 associated with susceptibility to tuberculosis. The results showed that the frequency distributions of alleles and genotypes of rs1800450, rs1800451, rs7096206, rs7095891 and rs11003125 in patients were not statistically different from those of the control group, which was inconsistent with the previously reported results.13,14,28,29 Several studies have been conducted among the Chinese Han population.13,28,29 The rs7096206-GC genotype and the rs7095891-GA genotype were revealed to be risk factors for the progression of spinal tuberculosis by Zheng et al.13 Chen et al discovered that the risk of tuberculosis is higher with the GC genotype than with the CC genotype of rs7096206 after eliminating various confounding factors.29 Guo et al discovered that the YA/YA haplotype was associated with a high serum MBL level, suggesting that it was a key predictor of serum MBL level and could be a protective factor for tuberculosis susceptibility.28 The CC genotype and C allele of rs11003125 were shown to be strongly related with tuberculosis susceptibility (risk factors) in an Iranian population, but the G allele may be a protective factor.14 The variations between the studies could be due to the diversity of geographic origins, although further research is needed to establish this.
According to the National Survey of Drug-Resistant Tuberculosis database from 2007, lineages 1, 2, 3, and 4 of M. tuberculosis isolates were the most prevalent in China.30 Lineage 2 accounted for 50% of all lineages identified using whole-genome sequencing (WGS) in our study, which is lower than the national average of 75% mentioned above.30 Lineage 2 (or East Asian lineage), which is prevalent in East Asian countries, has attracted interest due to its hypervirulence in laboratory models.31,32 In the present study, we divided M. tuberculosis into lineage 2- and non-lineage 2-infected groups for analysis. However, none of the alleles or genotypes of the four loci were observed to statistically associate with lineage 2. Analysis among the four major lineages of isolates, the C allele and the CC genotype of rs11003125 were found to be protective factors in lineage 4 infection by using lineage 3 infection as control, which provides clues for tuberculosis control where lineage 3 and 4 of M. tuberculosis were prevalent and suggested that the host’s susceptibility to tuberculosis differs depending on the lineages of M. tuberculosis challenges they face. Wang et al infected human macrophages and dendritic cells with M. tuberculosis clinical isolates from various mycobacterial families and found that Beijing strains elicited fewer cytokines than other M. tuberculosis strains, indicating that Beijing strains have a different ability to elicit innate and adaptive immune responses than other M. tuberculosis strains.33 Various M. tuberculosis strains have varied pro-inflammatory cytokine profiles, which could explain how biological factors play a role in disease transmission.34 Müller et al compared two geographically distinct cohorts (the South African population of 947 participants and the Ghanaian population of 3311 participants) and discovered that 32 SNPs were statistically significantly associated with risk of infection with various M. tuberculosis strains in the Ghanaian cohort, whist no correlation was found in the South African cohort.35
In the present study, we observed that rs11003125 exhibited a lineage-dependent association in the Uyghur population. These findings imply that combining the pathogen and host genetic nature would assist in the identification of consistent genetic risk factors of tuberculosis in different populations and then contribute to long-term tuberculosis control. MBL is a PRR and has ability of inducing different inflammatory responses to different M. tuberculosis lineages, and potential interaction between MBL2 function and lineages appears to be a viable prospect. One limitation of the present study is that the sample sizes of the host subjects and the patients with M. tuberculosis were small, so larger sample size and more rigorous design should be guaranteed in future studies.
Conclusion
We found that the C allele and CC genotype of rs11003125 were protective factors in the Uyghur population against lineage 4 of M. tuberculosis infection by using lineage 3 infection as control, which provides clues for tuberculosis control where lineage 3 and 4 of M. tuberculosis were prevalent and suggests that human’s susceptibility to tuberculosis is affected both by the host genetic polymorphisms and the lineages of the M. tuberculosis that people are exposed to.
Abbreviations
CI, confidence interval; CTAB, cetyltrimethylammonium bromide; EDTA, ethylene diamine tetraacetic acid; GWAS, genome-wide association studies; HWE, Hardy–Weinberg equilibrium; MBL2, mannose-binding lectin 2; M. tuberculosis, Mycobacterium tuberculosis; PAMPs, pathogen associated molecular patterns; PRRs, pattern recognition receptors; PSM, propensity score matching; SNPs, single nucleotide polymorphisms; WGS, whole-genome sequencing; WHO, World Health Organization.
Data Sharing Statement
Data can be made available through contact with the corresponding author.
Ethics Approval and Informed Consent
The experimental protocol was established, according to the ethical guidelines of the Helsinki Declaration and was approved by the Human Ethics Committee of the Eighth Affiliated Hospital of Xinjiang Medical University (XJMU8HEC-20161215). Written informed consent was obtained from individuals.
Acknowledgments
The authors thank all staffs working in the tuberculosis hospitals or institutes for tuberculosis control of Xinjiang Uygur Autonomous Region Chest Hospital, Kashgar, Kuqa and Wushi for supplying the clinical M. tuberculosis isolates and collecting data.
Funding
This study was supported by grants from the Major Science and Technology Project of Xinjiang Uygur Autonomous Region (The research and application of key technologies for tuberculosis prevention and treatment in southern Xinjiang, 2017A03006-3) and the Ministry of Science and Technology, China (Mega Projects of Research on the Prevention and Control of HIV/AIDS, Viral Hepatitis Infectious Diseases, 2018ZX10103001-003-012).
Disclosure
The authors report no conflicts of interest in this work.
References
1. WorId Health Organization. Global Tuberculosis Report 2020. Geneva: World Health Organization; 2020.
2. Yong Tan S, Kwok E. Albert Calmette (1863–1933): originator of the BCG vaccine. Singapore Med J. 2012;53:433–434.
3. Towey F. Historical Profile Leon Charles Albert Calmette and Jean-Marie Camille Guerin. Lancet Respir Med. 2015;3:186–187. doi:10.1016/S2213-2600(15)00065-X
4. Sakula A. BCG: who were Calmette and Guérin?. Thorax. 1983;38:806–812.
5. Zhang JX, Gong WP, Zhu DL, et al. Mannose-binding lectin 2 gene polymorphisms and their association with tuberculosis in a Chinese population. Infect Dis Poverty. 2020;9:46. doi:10.1186/s40249-020-00664-9
6. Ceylan E, Karkucak M, Coban H, et al. Evaluation of TNF-alpha gene (G308A) and MBL2 gene codon 54 polymorphisms in Turkish patients with tuberculosis. J Infect Public Health. 2017;10:774–777. doi:10.1016/j.jiph.2016.11.003
7. Hu CY. X-Linked Susceptible Genes and Genome-Wide Association Study of Pulmonary Tuberculosis. Beijing: Chinese People’s Liberation Army Academy of Military Sciences; 2015.
8. Dos Santos Silva PM, de Oliveira WF, Albuquerque PBS, et al. Insights into anti-pathogenic activities of mannose lectins. Int J Biol Macromol. 2019;140:234–244.
9. Kilpatrick DC. Animal lectins: a historical introduction and overview. Biochim Biophys Acta. 2002;1572:187–197. doi:10.1016/S0304-4165(02)00308-2
10. Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol. 2003;21:547–578. doi:10.1146/annurev.immunol.21.120601.140954
11. Garred P, Larsen F, Seyfarth J, et al. Mannose-binding lectin and its genetic variants. Genes Immun. 2006;7(2):85–94.
12. Heitzeneder S, Seidel M, Förster-Waldl E, et al. Mannan-binding lectin deficiency - good news, bad news, doesn’t matter?. Clin Immunol. 2012;143(1):22–38. doi:10.1016/j.clim.2011.11.002
13. Zheng M, Shi S, Wei W, et al. Correlation between MBL2/CD14/TNF-α gene poly-morphisms and susceptibility to spinal tuberculosis in Chinese population. Biosci Rep. 2018;38:BSR20171140. doi:10.1042/BSR20171140
14. Amiri A, Sabooteh T, Shahsavar F, et al. Mannose-Binding Lectin (MBL) gene polymorphisms in susceptibility to pulmonary tuberculosis among the lur population of Lorestan Province of Iran. Genom Data. 2017;12:146–150.
15. Liu Q, Wei J, Li Y, et al. Mycobacterium tuberculosis clinical isolates carry mutational signatures of host immune environments. Sci Adv. 2020;6:eaba4901. doi:10.1126/sciadv.aba4901
16. Omae Y, Toyo-Oka L, Yanai H, et al. Pathogen lineage-based genome-wide association study identified CD53 as susceptible locus in tuberculosis. J Hum Genet. 2017;62:1015–1022. doi:10.1038/jhg.2017.82
17. National Health and Family Planning Commission of the People’s Republic of China, WS 288–2017. Diagnosis for Pulmonary Tuberculosis. Beijing: People’s Medical Publishing House; 2017.
18. Honore S, Vincensini JP, Hocqueloux L, et al. Diagnostic value of a nested polymerase chain reaction assay on peripheral blood mononuclear cells from patients with pulmonary and extrapulmonary tuberculosis. Int J Tuberc Lung Dis. 2001;5:754–762.
19. Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–W296. doi:10.1093/nar/gkab301
20. Thakkinstian A, McElduff P, D’Este C, et al. A method for meta-analysis of molecular association studies. Stat Med. 2005;24:1291–1306. doi:10.1002/sim.2010
21. Woolhouse ME, Webster JP, Domingo E, et al. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat Genet. 2002;32:569–577. doi:10.1038/ng1202-569
22. Ebert D, Fields PD. Host-parasite co-evolution and its genomic signature. Nat Rev Genet. 2020;21:754–768.
23. Ahlawat N, Geeta Arun M, Maggu K, et al. Enemies make you stronger: co-evolution between fruit fly host and bacterial pathogen increases postinfection survivorship in the host. Ecol Evol. 2021;11:9563–9574. doi:10.1002/ece3.7774
24. Ebert D. Open questions: what are the genes underlying antagonistic coevolution?. BMC Biol. 2018;16:114. doi:10.1186/s12915-018-0583-7
25. Nadesalingam J, Dodds AW, Reid KB, et al. Mannose-binding lectin recognizes peptidoglycan via the N-acetyl glucosamine moiety, and inhibits ligand-induced proinflammatory effect and promotes chemokine production by macrophages. J Immunol. 2005;175:1785–1794. doi:10.4049/jimmunol.175.3.1785
26. Michelle A. Mannose-binding lectin levels and susceptibility to tuberculosis. Clin Immunol. 2010;135:S79–S79. doi:10.1016/j.clim.2010.03.238
27. Søborg C, Madsen HO, Andersen AB, et al. Mannose-binding lectin polymorphisms in clinical tuberculosis. J Infect Dis. 2003;188:777–782. doi:10.1086/377183
28. Guo YL, Liu Y, Ban WJ, et al. Association of mannose-binding lectin gene polymorphisms with the development of pulmonary tuberculosis in China. BMC Infect Dis. 2017;17:210. doi:10.1186/s12879-017-2310-3
29. Chen M, Liang Y, Li W, et al. Impact of MBL and MASP-2 gene polymorphism and its interaction on susceptibility to tuberculosis. BMC Infect Dis. 2015;15:151. doi:10.1186/s12879-015-0879-y
30. Chen H, He L, Cai C, et al. Characteristics of distribution of Mycobacterium tuberculosis lineages in China. Sci China Life Sci. 2018;61:651–659. doi:10.1007/s11427-017-9243-0
31. Glynn JR, Whiteley J, Bifani PJ, et al. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg Infect Dis. 2002;8(8):843–849. doi:10.3201/eid0808.020002
32. Karmakar M, Trauer JM, Ascher DB, Denholm JT. Hyper transmission of Beijing lineage Mycobacterium tuberculosis: systematic review and meta-analysis. J Infect. 2019;79(6):572–581.
33. Wang C, Peyron P, Mestre O, et al. Innate immune response to Mycobacterium tuberculosis Beijing and other genotypes. PLoS One. 2010;5:e13594. doi:10.1371/journal.pone.0013594
34. Chen YY, Chang JR, Huang WF, et al. The pattern of cytokine production in vitro induced by ancient and modern Beijing Mycobacterium tuberculosis strains. PLoS One. 2014;9:e94296. doi:10.1371/journal.pone.0094296
35. Müller SJ, Schurz H, Tromp G, et al. A multi-phenotype genome-wide association study of clades causing tuberculosis in a Ghanaian- and South African cohort. Genomics. 2021;113:1802–1815. doi:10.1016/j.ygeno.2021.04.024
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.