Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 15
Association Between Selected Single Nucleotide Polymorphisms in Globin and Related Genes and Response to Hydroxyurea Therapy in Ghanaian Children with Sickle Cell Disease
Authors Manu GP, Segbefia C, N'guessan BB, Coffie SA, Adjei GO
Received 3 December 2021
Accepted for publication 17 February 2022
Published 10 March 2022 Volume 2022:15 Pages 205—214
DOI https://doi.org/10.2147/PGPM.S351599
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Gloria Pokuaa Manu,1 Catherine Segbefia,2 Benoit Banga N’guessan,1 Shadrack Asiedu Coffie,3 George Obeng Adjei4
1Department of Pharmacology and Toxicology, School of Pharmacy, College of Health Sciences, University of Ghana, Accra, Ghana; 2Department of Child Health, University of Ghana Medical School, Accra, Ghana; 3Biotechnology Centre, University of Ghana, Accra, Ghana; 4Centre for Tropical, Clinical Pharmacology and Therapeutics, University of Ghana Medical School, College of Health Sciences, University of Ghana, Accra, Ghana
Correspondence: George Obeng Adjei, Email [email protected]
Background: Sickle cell disease (SCD) is a group of genetic disorders affecting the structure and function of haemoglobin. Hydroxyurea (HU) stimulates fetal haemoglobin (HbF) and reduces sickle erythrocyte-endothelial cell interaction. However, the degree of HbF response to HU varies, with HbF expression-associated single nucleotide polymorphisms (SNPs) in quantitative trait loci (QTL) been implicated. We investigated the relationship between four SNPs (rs11886868, rs6706648, rs7606173 and 158C/T Xmn1) in two QTL (B-cell lymphoma 11A (BCL11A) and Xmn1) and HbF levels in children with SCD in Accra, Ghana.
Methods: A total of 110 children with SCD in steady-state, comprising 64 and 46 SCD children treated with HU (HU+) or with no history of HU therapy (HU-), respectively, were recruited. HbF levels were measured in peripheral blood by alkali denaturation and SNPs were genotyped using polymerase chain reaction and restriction fragment length polymorphism.
Results: The presence of SNPs (rs11886868, rs6706648, rs7606173 and − 158C/T Xmn1) was identified. Observed heterozygosity and homozygosity for the derived alleles were 45.7%, 82.6%, 21.7% and 39.1% in rs11886868, rs6706648, rs7606173 and − 158C/T Xmn1 polymorphisms, respectively, for the HU+ population. Observed frequencies of the minor alleles were 0.204, 0.477, 0.171 and 0.190 for rs11886868, rs6706648, rs7606173 and − 158C/T Xmn1 polymorphisms, respectively. The three BCL11A SNPs in the HU+ population showed homozygous individuals for rs11886868 (CC), rs6706648 (CC) and heterozygous or homozygous mutant individuals for rs7606173 (CG/GG) having higher HbF values. The combined effect of the SNPs was associated with variance in HbF levels in the HU+ population. The BCL11A SNP, rs6706648 was strongly associated with HbF levels and the C allele frequency, with significantly elevated HbF levels.
Conclusion: An association between the various variants and combined effect of SNPs and HbF among children with SCD was found and confirms the known association between HU intake and increased HbF in SCD.
Keywords: hydroxyurea, single nucleotide polymorphism, sickle cell disease, haemoglobin F
Introduction
Sickle cell disease (SCD) is a group of autosomal recessive blood disorders affecting the structure of haemoglobin and resulting in acute and chronic complications including recurrent, painful events or vaso-occlusive crises (VOCs), acute chest syndrome, stroke, and organ dysfunction. These complications are major causes of hospitalizations, morbidity, and premature mortality. Repeated cycles of red blood cells (RBCs) sickling leading to haemolysis and the multicellular adhesion process are interlinked mechanisms that contribute to the clinical picture of the disease.1
Hydroxyurea (HU), a ribonucleotide reductase inhibitor, was the first disease-modifying therapy approved for use in adults and children with SCD. HU depletes intracellular deoxynucleotide triphosphate pools required for DNA synthesis and repair.2 One of its main effects is HbF induction leading to significant reductions in VOCs, acute chest syndrome, hospitalizations and need for blood transfusions.2,3
The benefits of treating SCD patients with HU are well described;2,4–6 however, the increase in HbF in response to HU therapy varies considerably among patients. The regulation of HbF is a complex genetic trait that is heritable and genetic/transposable elements linked to the β-globin gene and quantitative trait loci (QTL) on chromosomes govern this regulation.7–9 Additionally, certain polymorphisms of the −158 C/T Xmn1 and the B-cell lymphoma 11A (BCL11A) are capable of modulating HbF.10–13 It is possible that these polymorphisms in QTL also modulate or affect the HbF induction response to HU and may influence response to HU therapy.
Given that HU treatment is lifelong, determining the presence of genetic variations or polymorphisms that may lead to identification of responder and non-responder phenotypes in SCD patients is important. We hypothesized that SNPs in BCL11A and Xmn1 gene affect HbF levels induced by HU therapy in SCD patients and investigated the presence or otherwise of SNPs in BCL11A (rs11886868, rs6706648, rs7606173) and −158C/T Xmn1 among children with SCD and determined associations between HbF and selected SNPs in the −158C/T Xmn1 and BCL11A loci.
Materials and Methods
Study Design, Site and Population
The study was a hospital-based cross-sectional comparative study, conducted at the Paediatric Sickle Cell Clinic (SCC), of the Department of Child Health (DCH), Korle Bu Teaching Hospital (KBTH) in Accra, Ghana. KBTH is the third-largest hospital in Africa and the largest referral centre in Ghana with 2000-bed capacity and seventeen clinical and diagnostic departments, including the DCH. The SCC is a subspecialty outpatient clinic at the DCH with about 5000 SCD children registered and an average weekly attendance of about 60–70 patients.14
The study population consisted of children diagnosed with SCD and registered at the SCC. Children on HU therapy (HU+) for at least 6 months (aged 1 year or older, in steady state, visiting the SCC for their routine scheduled evaluations). Steady state was defined as the absence of an acute crisis, no use of blood therapy and no hospital admission in the preceding three-month period. A comparison group of children with SCD in steady state who had no history of HU therapy (HU-) and visiting the SCC for their routine follow-up visits were also recruited for the purpose of comparing selected haematologic indices and genetic data. Clinical severity criteria were the indication for HU therapy in our study setting at the time the study was conducted. Siblings were excluded to ensure genetic independence. The study was conducted between March and August 2019 and there were about 500 SCD children on HU at the time of conducting the study.
Haematology and Fetal Haemoglobin Level Measurement
Haematology analysis was carried out using the Mindray BC-5300®/TM Auto Haematology Analyzer (Mindray, China). HbF was determined using the alkaline denaturation method as described by Jonxis and Husman15 with slight modification. In brief, haemoglobin was converted to cyanomethaemoglobin, and the cyanomethaemoglobin exposed to alkali. Absorbance of the filtrate containing alkali resistant HbF was taken at 540nm and compared with absorbance of the total haemoglobin at 540 nm (using the 34 Genesys 10S UV-VIS Spectrophotometer (Thermo Scientific). Haematological parameters determined were red blood cell (RBC), white blood cells (WBC), mean corpuscular haemoglobin (MCH), mean corpuscular volume (MCV), mean corpuscular haemoglobin concentration (MCHC), haemoglobin (Hb), fetal haemoglobin (HbF), platelets (PLT) and hematocrit (HCT).
DNA Isolation and Genotyping
Two genetic loci, Xmn1 and B-cell Lymphoma 11A (BCL11A) determined to be associated with variations in HbF levels13,16,17 were investigated. Four SNPs in these loci previously reported were genotyped; three in BCL11A locus (rs11886868, rs7606173, rs6706648) and one in −158 (C>T) Xmn1 (Xmn1).
Molecular analyses were carried out on genomic DNA extracted from whole blood sample using Quick DNA miniprep kit from Zymo Research (USA). SNPs were genotyped using Polymerase Chain Reaction and Restriction Fragment Length Polymorphism (PCR-RFLP) as previously described.11,13 The amplification process was performed using the BIO-RAD icycler thermal cycler (Bio-Rad, USA). RFLP analysis was carried out using AvaII, MboII, AatII and Xmn1 restriction enzymes (New England Biolabs, USA) for rs11886868, rs7606173 and rs6706648 and Xmn1, respectively. To detect rs11886868 (C→T), a 441 bp fragment was amplified and subjected to MboII digestion, which yielded 370bp and 71bp fragments and the presence of three genotypes: CC, CT, and TT were identified. For rs6706648 (C→T), a 333bp fragment was amplified and subjected to AatII enzyme digestion which yielded 193 and 140bp fragments showing the presence of three genotypes: CC, CT, and TT. In the rs7606173 (C→G), a 201bp fragment was amplified and subjected to AvaII digestion which yielded 110bp and 91bp fragments. The presence of three genotypes: CC, CG, and GG, were identified. For the −158Gγ (C→T) Xmn1 polymorphism, it was expected that the C alleles produce 122 bp DNA bands and the T alleles produce 195 bp DNA band and the control, a DNA band of 317 bp. The presence of two genotypes: CC and CT were identified in this study. The presence of the homozygous mutant TT was not detected in our study population.
Statistical Analysis
Statistical analyses were carried out using Statistical Package for the Social Sciences (SPSS) version 20 (USA), Haploview 4.2 (http://www.broad.mit.edu/haploview) and Graph Pad Prism 8.3.0(538) (San Diego, California). Data are presented as mean ±SD and summarized as frequencies and proportions. An independent t-test was used to determine significant differences between the haematology means in HU+ and HU- groups. Mann–Whitney U-test was used to determine the differences between the HbF level distribution according to the presence or absence of the minor allele frequency in SNP. Analysis of variance (ANOVA) with post-hoc testing was done to determine differences between mean HbF of the genotypes. Chi-square was performed to assess if there were differences in the frequency of SNPs among groups. Multiple linear regression was used to investigate associations between SNPs. A p-value < 0.05 was considered statistically significant. Genotype data were inspected using Haploview 4.2. Haploview utilizes EM algorithm to calculate linkage disequilibrium (LD) coefficients (D’).
Results
Demographic and Clinical Data
A total of 110 SCD children in steady state (mean age [±SD] 7.9 [3.1] years) were recruited. A total of 64 (58.2%) were on HU therapy (HU+). The indications for HU therapy were primary stroke prevention in 29 (45%), recurrent painful crises in 16 (25%), acute chest syndrome in 11 (17%). Other reasons included secondary stroke prevention and frequent infections. All participants were receiving routine folic acid supplementation and non-iron containing multivitamins. The median HU dose and therapy duration of the HU+ group was 500mg/day and 12 months, respectively. Selected demographic and haematologic parameters of recruited participants in HU+ and HU- groups are shown (Table 1). The proportion of participants with HbSS genotype was 95.3% and 76.1% in the HU+ and HU- groups, respectively. Among the HU+ population, HbF levels were significantly increased in HbSS patients compared with the HbSC population (P = 0.015; Supplementary Table 1).
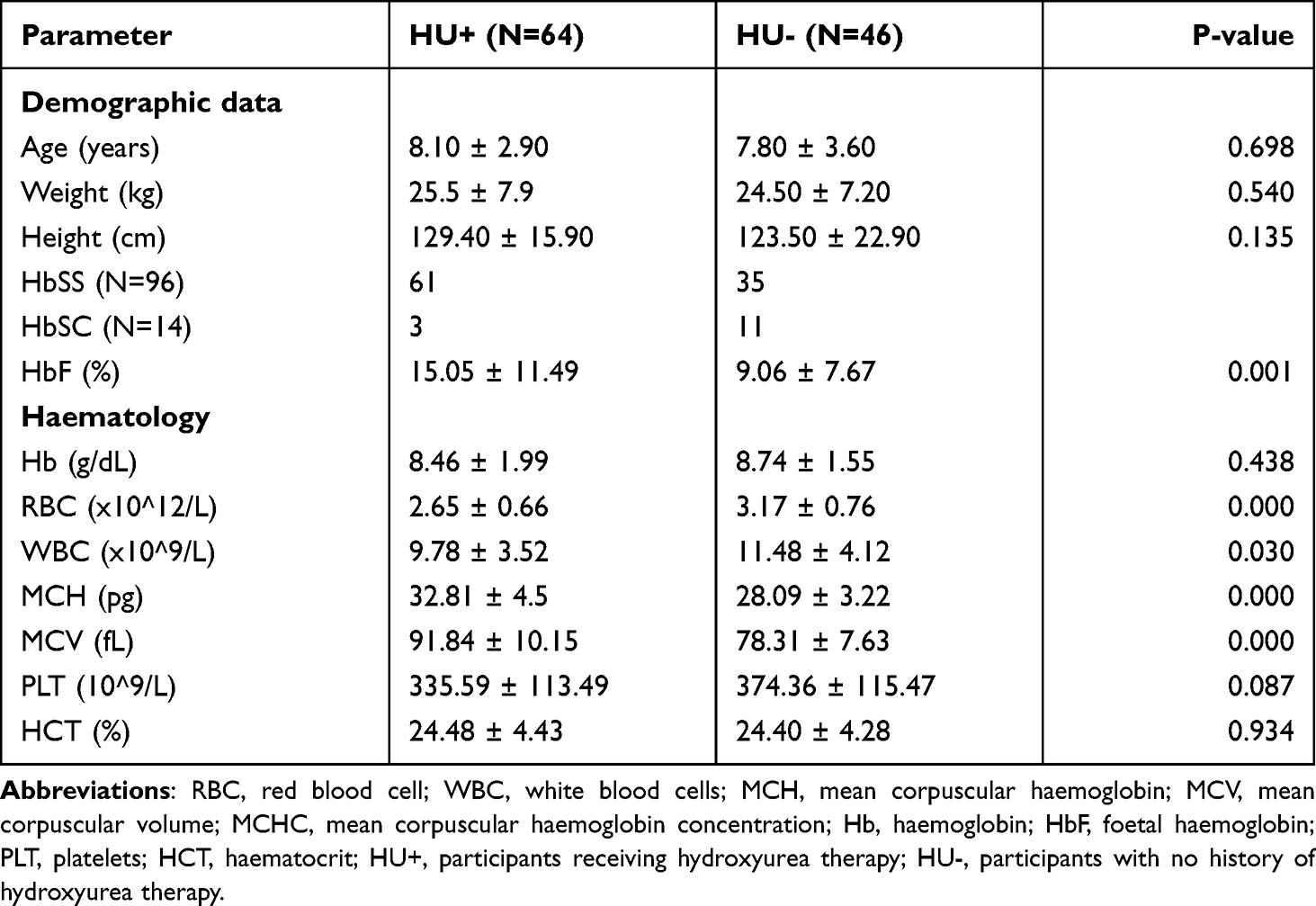 |
Table 1 Demographic and Haematological Parameters of HU+ and HU- Patients at Recruitment |
HbF Distribution Among HU+ and HU- Participants
There were significant differences in HbF between the HU+ and HU- groups (Figure 1, p = 0.004).
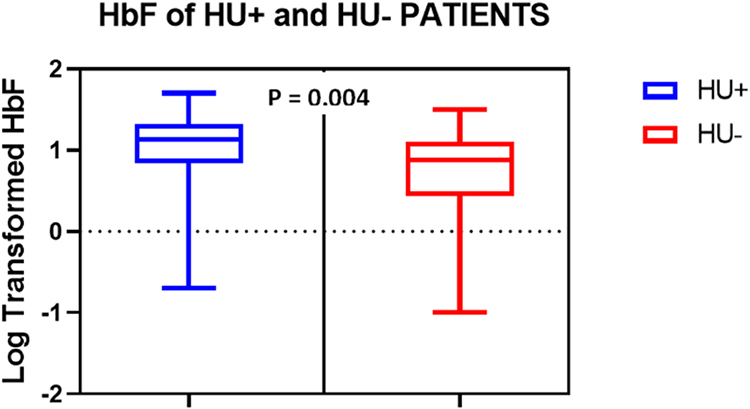 |
Figure 1 Boxplots showing the distribution of HbF levels (log-transformed) among HU+ (n = 64) and HU- (n = 46) groups. Data were compared using Mann–Whitney U-test. |
Allele and Genotype Frequencies and HbF Concentrations
Genotype frequencies of the SNPs investigated were in Hardy-Weinberg equilibrium (χ2 test, p > 0.05, HWpval > 0.001; Table 2). Genotype frequency analysis performed in HU+ and HU- groups showed comparatively higher frequencies in the HU+ group (Table 3).
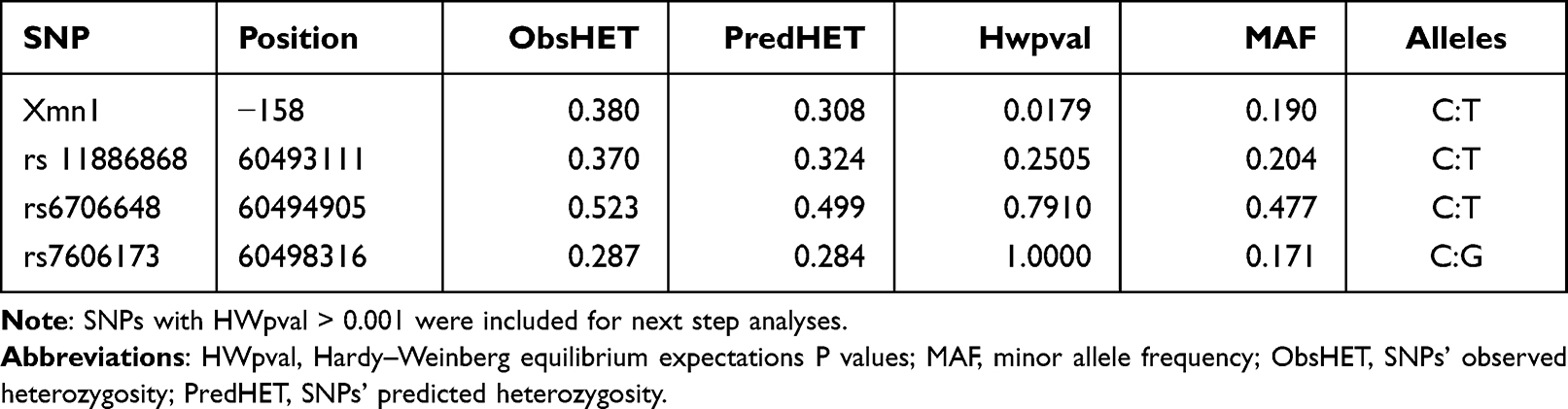 |
Table 2 Description of the SNPs Studied |
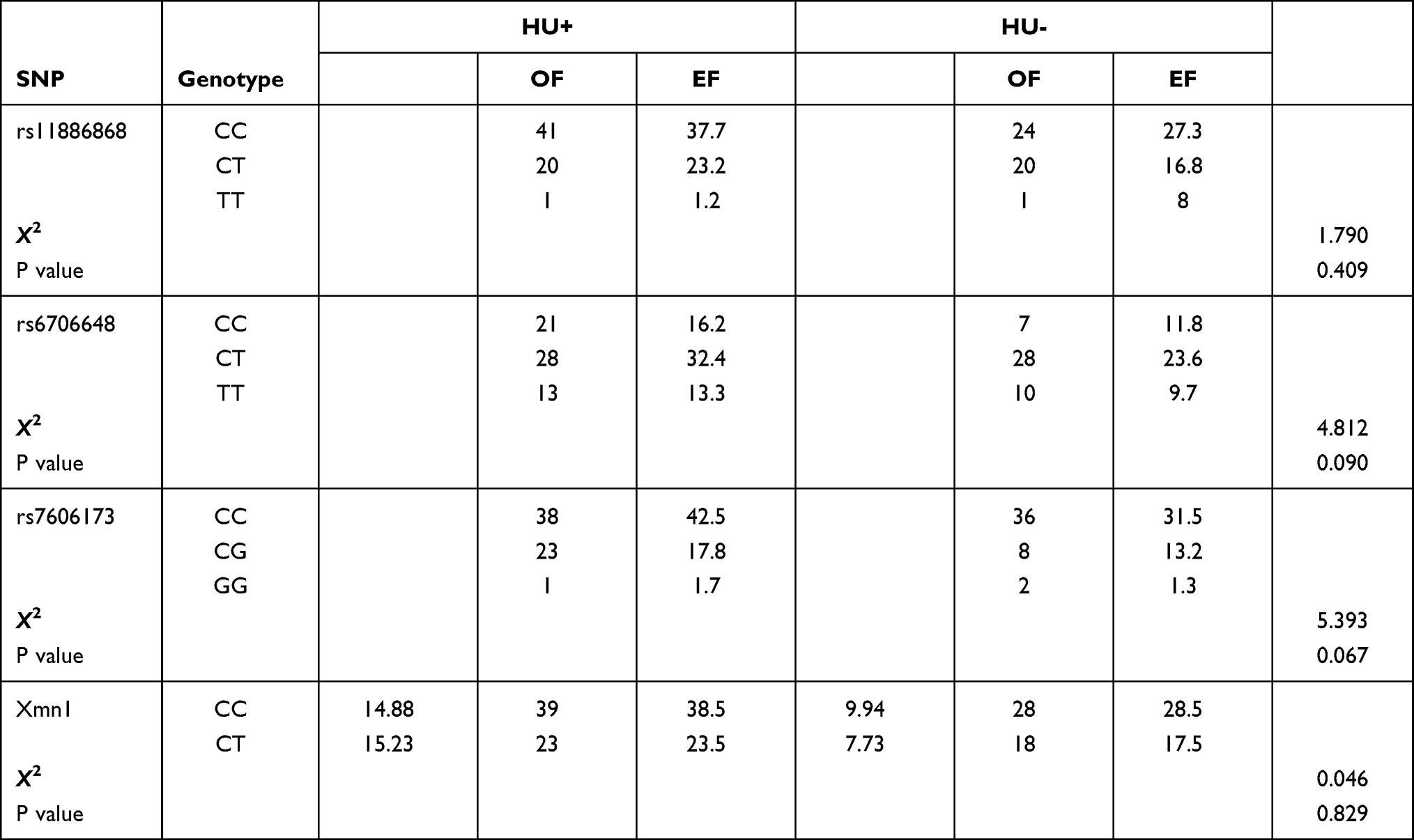 |
Table 3 Genotype Frequencies for SNPs in HU+ and HU- Populations |
There were no statistical differences observed in the frequency of the SNP genotypes among participants based on HU intake, or whether they were homozygous, heterozygous, or homozygous mutant (P > 0.05).
HbF concentrations were similar among the genotypes of rs11886868, rs6706648, rs7606173 and Xmn1 (Supplementary Table 2).
However, analyzing the distribution of HbF values (log-transformed) according to SNP genotypes by using a dominant model-based analysis (ie, genotypes homozygous for the wild type allele versus homozygous and heterozygous for the mutant allele), Rs 6706648 showed a significant difference in observed HbF levels for the derived and the ancestral allele (p<0.05) in the HU+ population (Table 4).
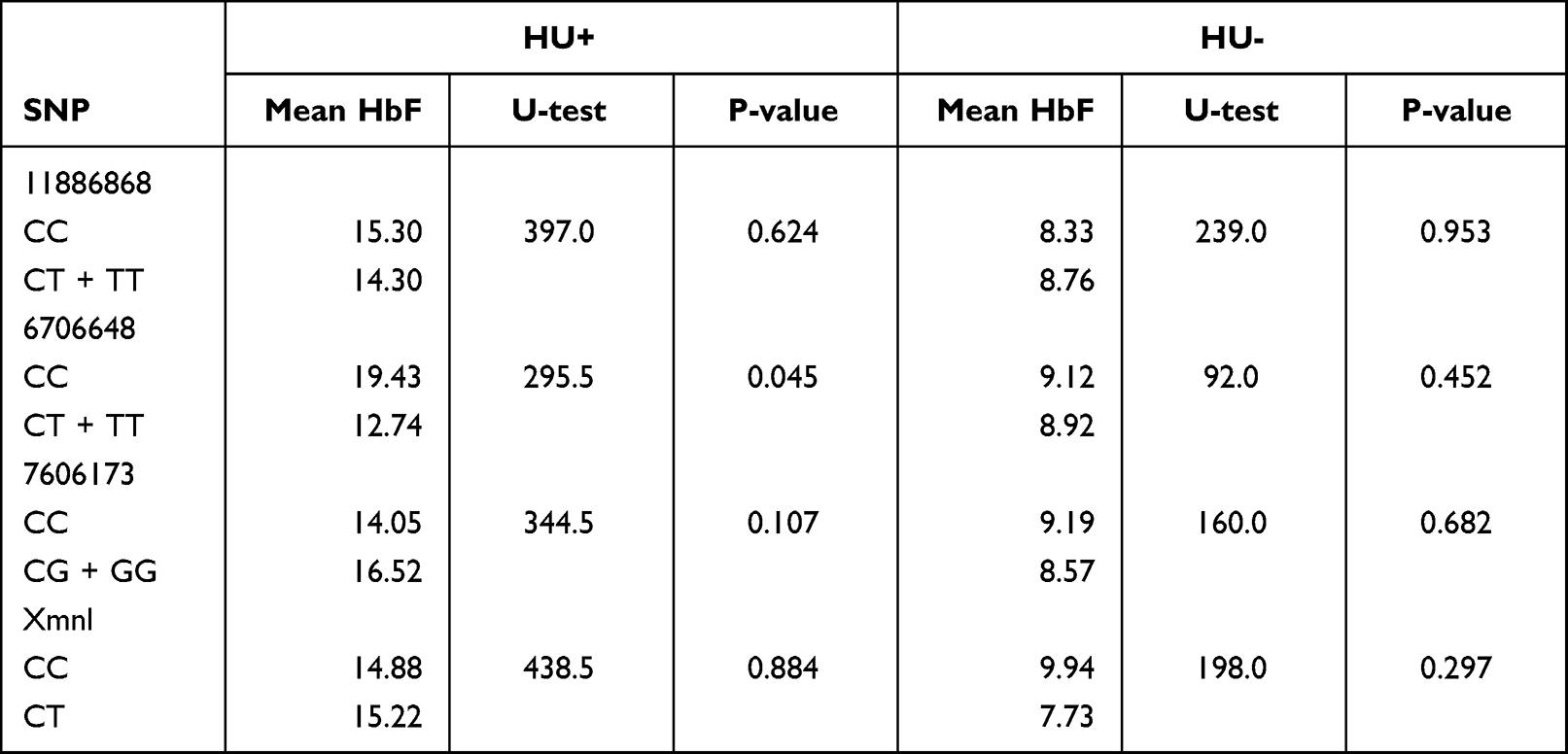 |
Table 4 Distribution of HbF According to SNP Genotypes in HU+ and HU– |
The three BCL11A SNPs in HU+ participant group showed heterozygous individuals for rs11886868 (CT), 6706148 (CT) and homozygous individuals for rs7606173 (CC) with the lowest HbF values. In HU- on the other hand, rs11886868 (CC), rs6706148 (CT), rs7606173 (GG) were observed to have lower HbF levels (Figure 2). For the statistical significance SNP (rs6706648) observed in HU+, individuals homozygous for the wild type allele showed higher HbF levels compared to those with the derived allele (Figure 3).
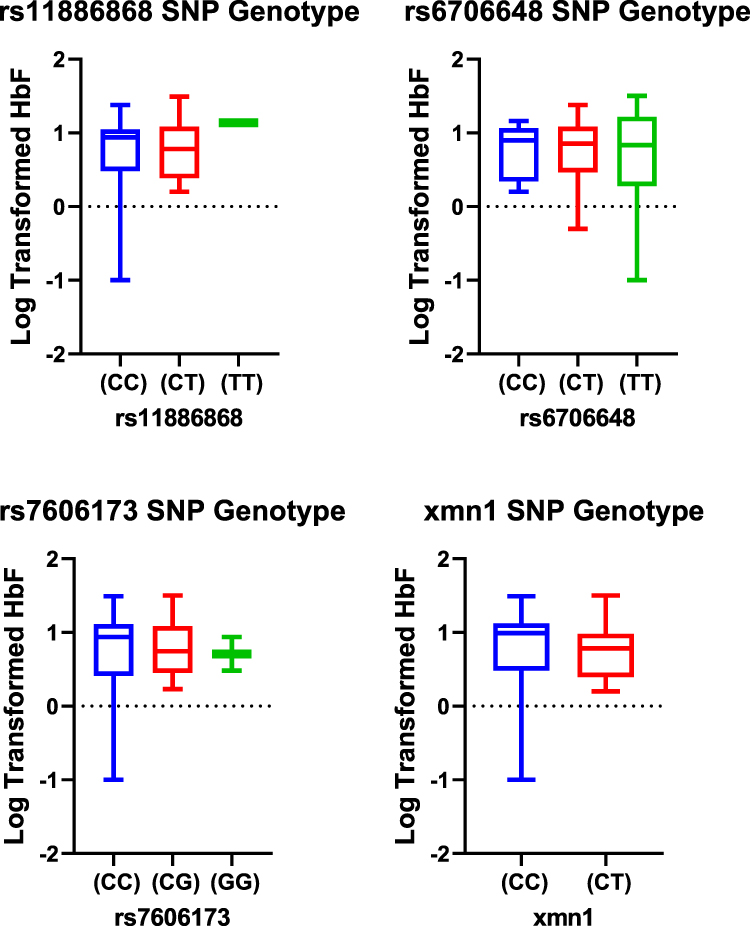 |
Figure 2 Box plots distribution of HbF levels (log-transformed) within the SNP genotypes for HU- group. Data were compared with Mann–Whitney U-test using a dominant model (ie, homozygous genotypes for the wild type allele versus homozygous and heterozygous for the mutant allele). |
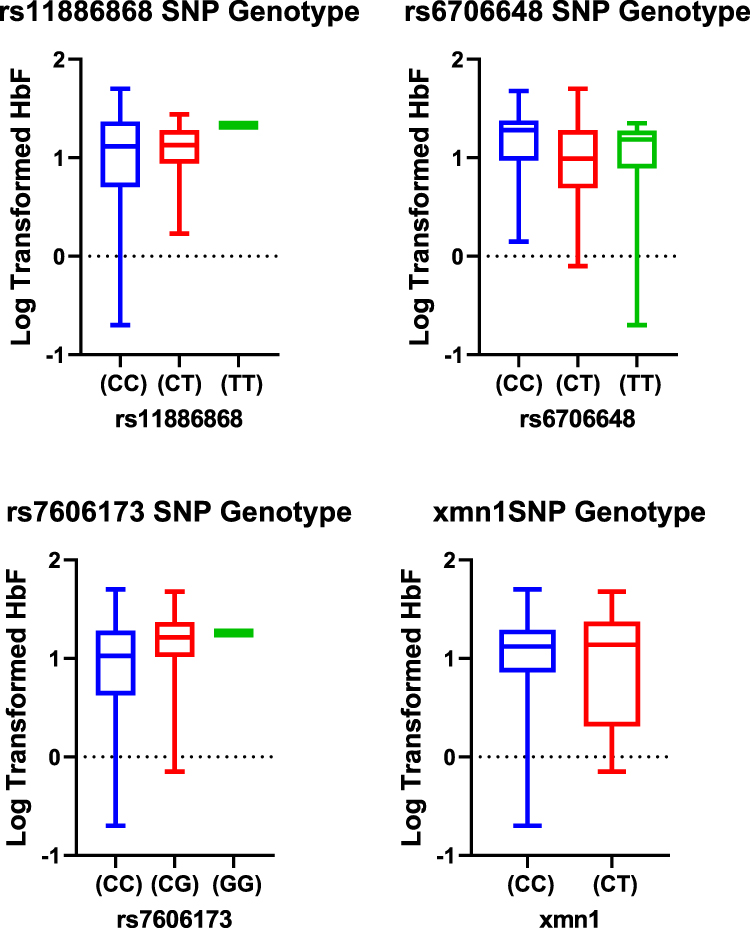 |
Figure 3 Box plots distribution of HbF levels (log-transformed) within the SNP genotypes for HU+ group. Data were compared with Mann–Whitney U-test using a dominant model (ie, homozygous genotypes for the wild type allele versus homozygous and heterozygous for the mutant allele). |
The Xmn1 polymorphism also did not show any significance in the distribution of HbF among the genotypes in both the HU+ and HU- populations, however, heterozygous individuals presented with a higher mean HbF the HU+ group (Table 3; supplementary Table 2).
Association Analysis Between HbF and SNPs
The SNP variables statistically predicted HbF levels in the HU+ group for the three BCL11A SNPs in an additive genetic model (F = 2.945, p = 0.040, R2 = 0.132). The average change in % HbF per allele copy of each individual SNP were also significant in SNP rs11886868 (p = 0.048), and rs6706648 (p = 0.022). Combining all three BCL11A and Xmn1 SNPs in an additive genetic model, a significant association was observed only in HU+ participants [(F = 2.620, p = 0.044, R2 = 0.155; Table 5), where the F-statistic determines the overall significance of the linear regression model.] As standalone SNPs, no significant association were observed with HbF in either HU+ or HU- groups (Supplementary Table 3).
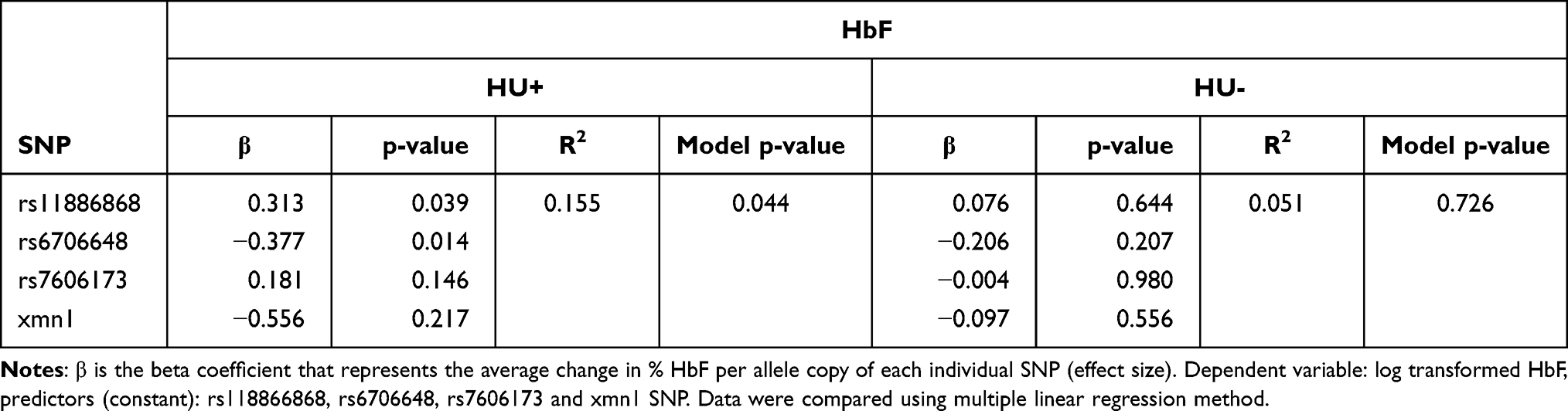 |
Table 5 Multiple Linear Regression Models of Genetic Polymorphisms for Analysis of Association Between SNPs and HbF Levels for HU+ and HU- Participants |
HU intake thus, significantly predicted HbF levels in participants (F = 5.632, p = 0.019, R2 = 0.050) confirming the significantly higher HbF values observed in HU+ participants.
Discussion
Foetal haemoglobin (HbF) increase is of clinical relevance given its role in reducing SCD complications. Consistent with the findings from other studies, the observed reduced WBCs, and platelets in the HU+ participants in our study participants confirms the cytoreductive effect of HU.18,19 The findings of higher percentage HbF levels in the HU-treated participants are also consistent with what is generally known about the genetic modifiers of HbF in SCD.
However, the response to HU therapy is variable and HbF, is known to be influenced by many genetic loci inside or outside the β-globin gene cluster,8,20 with SNPs in QTLs such as Xmn1, and BCL11A playing major roles in HbF level variations in SCD patients.8,16,21–23
We identified the presence of rs11886868, rs6706648, rs7606173 and Xmn1 SNPs among our study population. The frequency of the SNP genotypes was comparable in both HU+ and HU- populations for all the SNPs studied. Consistent with other studies,24 we observed that carriers with double copy of the T (mutant) allele of rs11886868 had higher HbF levels compared to those with at least one copy of the C (wild type or ancestral) allele in both the HU+ and HU- populations. It is known that rs11886868 might affect HbF through linkage disequilibrium (LD) with other nearby SNPs in the BCL11A loci25 and in addition, the region between MYB and HBSIL region.26 Our study was focused on determining SNPs in two major loci (Xmn1 and BCL11A, rs11886868, rs6706648 and rs7606173). For the rs6706648 SNP, carriers of the T (mutant) allele in the HU+ population had lower mean HbF compared to those with the C allele (p = 0.045), while the reverse trend was observed with the HU- population. This is consistent with findings from a recent study in which the CC genotype presented increased HbF (> 15%) whereas CT and T showed decreased HbF,25 and which may suggest the presence of the T (mutant) allele is likely associated with a lower HbF response irrespective of HU intake status, while CC genotype is likely to be associated with a higher HU response. The minor allele (G) of the rs7606173 SNP was found to be associated with higher HbF in the HU+ group, with the homozygous form having the highest level, suggesting the possibility of potentially higher HbF levels in SCD patients with the derived allele. While it remains unclear whether HU directly affects BCL11A expression, it can be speculated that decreased BCL11A expression following HU therapy may be expected to increase HbF levels in developing erythroid cells, as BCK11A is a negative regulator of HbF expression,27 with some types of BCL11A SNP associated with decreased expression and increased HbF production.20,28
The most frequent genotype with the Xmn1 polymorphism observed was homozygosity (CC) for the absence of the Xmnl site in both HU+ and HU- populations. The homozygous mutant (TT), similar to the findings from a recent study29 was also not observed in our study, thus limiting our ability to draw any conclusions on the influence of this genotype on HbF.
In our association studies, the four SNPs (rs11886868, rs6706648, rs7606173 and the Xmn1) when combined in an additive genetic model, predicted HbF levels in the HU+ population, albeit by 15.5%, with the most significant being rs6706648 and rs11886868. The association between HbF levels and polymorphisms presented by SNPs in BCL11A gene and the Xmn1 gene has been demonstrated in different populations.11,16,25–27,29–35 While HbF regulation is highly diversified and genetically controlled, reproducing association studies across different populations, particularly of diverse genetic backgrounds and environmental setting is likely to produce different observations in different studies. Interestingly, we observed a slightly reduced Hb level in the HU+ group and speculate that, this could be due to crenation and subsequent lysing of RBC’s since HU plays a role in RBC differentiation.19
Conclusion
Our data support and confirm the known association between HU intake and HbF increase in SCD patients. The presence of rs 11886868, rs6706648 rs7606173 and −158C/T Xmn1 polymorphism were identified in SCD patients in Ghana, and we have shown that the combined effect of rs 11886868, rs6706648 rs7606173 and −158C/T Xmn1 polymorphism is associated with HbF increase in HU+ patients. As HU use is being expanded for patients with SCD in Ghana, understanding inter-individual variability for HU response is important and therefore, developing a robust predictive model for these genetic modifiers may prove useful in clinical decision-making in SCD.
Data Sharing Statement
The de-identified data of this study are available from the corresponding author ([email protected]) upon reasonable request.
Ethics Approval and Informed Consent
Ethical approval was obtained from the Ethical and Protocol Review Committee of the College of Health Sciences, University of Ghana (CHS/EPRC/JAN/2019, FWA: 000185779, IRB: 00006220, IORG: 0005170) and the Institutional Review Board (IRB), Korle Bu Teaching Hospital (KBTH-IRB/00038/2019). The study was conducted in accordance with the principles of the Declaration of Helsinki. Parents/guardians of all participants provided written informed consent prior to study commencement and assent was obtained for all children age ≥8 years.
Acknowledgment
Our special thanks go to the staff of the SCC and laboratory, DCH, KBTH. We also want to thank the staff of the Biotechnology Centre, Research Laboratory, University of Ghana, for their support during the molecular analysis.
Author Contributions
All authors contributed to the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; have agreed on the journal to which the article was submitted; gave final approval of the version to be published, and agree to be accountable for all aspects of the work described in this article.
Funding
This work was supported by funding from the Building Stronger Universities (BSU) phase III Project, DANIDA Fellowship Centre, to University of Ghana.
Disclosure
Dr Catherine Segbefia reports personal fees from Novartis and Global Blood Therapeutics, outside the submitted work. The authors declare no other potential conflicts of interest in this work.
References
1. Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390(10091):311–323. doi:10.1016/S0140-6736(17)30193-9
2. Agrawal RK, Patel RK, Shah V, Nainiwal L, Trivedi B. Hydroxyurea in sickle cell disease: drug review. Indian J Hematol Blood Transfus. 2014;30(2):91–96. doi:10.1007/s12288-013-0261-4
3. Lebensburger JD, Pestina TI, Ware RE, Boyd KL, Persons DA. Hydroxyurea therapy requires HbF induction for clinical benefit in a sickle cell mouse model. Haematologica. 2010;95(9):1599–1603. doi:10.3324/haematol.2010.023325
4. Pallis FR, Conran N, Fertrin KY, Olalla Saad ST, Costa FF, Franco-Penteado CF. Hydroxycarbamide reduces eosinophil adhesion and degranulation in sickle cell anaemia patients. Br J Haematol. 2014;164(2):286–295. doi:10.1111/bjh.12628
5. Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377:1663–1672. doi:10.1016/S0140-6736(11)60355-3
6. Ware RE. Hydroxycarbamide: clinical aspects. Comptes Rendus - Biol. 2013;336(3):177–182. doi:10.1016/j.crvi.2012.09.006
7. Green NS, Barral S. Genetic modifiers of HbF and response to hydroxyurea in sickle cell disease. Pediatr Blood Cancer. 2011;56(2):177–181. doi:10.1002/pbc.22754
8. Green NS, Ender KL, Pashankar F, et al. Candidate sequence variants and fetal hemoglobin in children with sickle cell disease treated with hydroxyurea. PLoS One. 2013;8(2):1–6. doi:10.1371/journal.pone.0055709
9. Steinberg MH, Lu ZH, Barton FB, Terrin ML, Charache S, Dover GJ; Multicenter Study of Hydroxyurea. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Blood. 1997;89(3):1078–1088. doi:10.1182/blood.V89.3.1078
10. Friedrisch JR, Sheehan V, Flanagan JM, et al. The role of BCL11A and HMIP-2 polymorphisms on endogenous and hydroxyurea induced levels of fetal hemoglobin in sickle cell anemia patients from southern Brazil. Blood Cells Mol Dis. 2016;62:32–37. doi:10.1016/j.bcmd.2016.11.002
11. El Gawhary S, Farid M, Ezzat M, ElAwady H, Mostafa H. XmnI polymorphism in Egyptian patients with β-thalassemia major and its correlation with the HbF level. Gene Reports. 2018;11:69–73. doi:10.1016/J.GENREP.2018.02.005
12. Mikobi TM, Tshilobo lukusa P, Aloni MN, et al. Protective BCL11A and HBS1L-MYB polymorphisms in a cohort of 102 Congolese patients suffering from sickle cell anemia. J Clin Lab Anal. 2018;32:e22207. doi:10.1002/jcla.22207
13. Fanis P, Kousiappa I, Phylactides M, Kleanthous M. Genotyping of BCL11A and HBS1L-MYB SNPs associated with fetal haemoglobin levels: a SNaPshot minisequencing approach. BMC Genom. 2014;15:108. doi:10.1186/1471-2164-15-108
14. Adjei GO, Goka BQ, Enweronu-Laryea CC, et al. A randomized trial of artesunate-amodiaquine versus artemether-lumefantrine in Ghanaian paediatric sickle cell and non-sickle cell disease patients with acute uncomplicated malaria. Malar J. 2014;13(1):369. doi:10.1186/1475-2875-13-369
15. Jonxis JH, Husman TH. The detection and estimation of fetal hemoglobin by means of the alkali denaturation test. Blood. 2002;1956(11):1009–1018.
16. Rujito L, Basalamah M, Siswandari W, et al. Modifying effect of XmnI, BCL11A, and HBS1L-MYB on clinical appearances: a study on β-thalassemia and hemoglobin E/β-thalassemia patients in Indonesia. Hematol Oncol Stem Cell Ther. 2016;9(2):55–63. doi:10.1016/j.hemonc.2016.02.003
17. Ware RE, Despotovic JM, Mortier NA, et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood. 2017;118(18):4985–4992. DOI:10.1182/blood-2011-07-364190.The
18. Cocou S, Alexandre M, Dé Hou Y, et al. Hydroxyurea alters hematological, biochemical and inflammatory biomarkers in Brazilian children with SCA: investigating associations with βS haplotype and α-thalassemia. PLoS One. 2019. doi:10.1371/journal.pone.0218040
19. Karimi M, Jooya P, Haghpanah S, et al. Evaluation of the relationship between Hb F levels and nucleated red blood cells with morbidity in non transfusion-dependent thalassemia patients. Hemoglobin. 2016;40(4):250–256. doi:10.1080/03630269.2016.1183212
20. Lettre G, Sankaran VG, André M, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and NL -globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. PNAS. 2008;105(33):11869–11874. doi:10.1073/pnas.0804799105
21. Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science (80-). 2013;342(6155):253–257. doi:10.1126/science.1242088
22. Cardoso GL, Diniz IG, Martins da Silva ANL, et al. DNA polymorphisms at BCL11A, HBS1L-MYB and Xmn1-HBG2 site loci associated with fetal hemoglobin levels in sickle cell anemia patients from Northern Brazil. Blood Cells Mol Dis. 2014;53(4):176–179. doi:10.1016/j.bcmd.2014.07.006
23. Sebastiani P, Farrell JJ, Alsultan A, et al. BCL11A enhancer haplotypes and fetal hemoglobin in sickle cell anemia. Blood Cells Mol Dis. 2015;54(3):224–230. doi:10.1016/j.bcmd.2015.01.001
24. Adekile A, Menzel S, Gupta R, et al. Response to hydroxyurea among Kuwaiti patients with sickle cell disease and elevated baseline HbF levels. Am J Hematol. 2015;90(7):E138–E139. doi:10.1002/AJH.24027
25. Chaouch L, Moumni I, Ouragini H, et al. rs11886868 and rs4671393 of BCL11A associated with HbF level variation and modulate clinical events among sickle cell anemia patients. Hematology. 2016;21(7):425–429. doi:10.1080/10245332.2015.1107275
26. Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc Natl Acad Sci U S A. 2008;105(5):1620–1625. doi:10.1073/pnas.0711566105
27. Flanagan JM, Steward S, Howard TA, et al. Hydroxycarbamide alters erythroid gene expression in children with sickle cell anaemia. Br J Haematol. 2012;157(2):240–248. doi:10.1111/j.1365-2141.2012.09061.x
28. Stadhouders R, Aktuna S, Thongjuea S, et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J Clin Invest. 2014;124(4):1699–1710. doi:10.1172/JCI71520
29. Moez P, Moftah R, Mahmoud HA. A study on the genotype frequency of −158 Gγ (C→T) Xmn1 polymorphism in a sickle cell trait cohort from Siwa Oasis, Egypt. J Genet. 2018;97(2):505–511. doi:10.1007/s12041-018-0942-8
30. Said F, Abdel-Salam A. XmnI polymorphism: relation to β-thalassemia phenotype and genotype in Egyptian children. Egypt J Med Hum Genet. 2015;16(2):123–127. doi:10.1016/J.EJMHG.2014.12.005
31. Zuhdi Nimer S, Ali W, Khalil H, Nimer SZ. Association of BCL11A genetic polymorphisms with fetal haemoglobin level in Sudanese patients with sickle cell anaemia. J Genom Gene Study. 2019;2:2.
32. Pule GD, Ngo Bitoungui VJ, Chetcha Chemegni B, Kengne AP, Antonarakis S, Wonkam A. Association between variants at BCL11A erythroid-specific enhancer and fetal hemoglobin levels among sickle cell disease patients in Cameroon: implications for future therapeutic interventions. Omi A J Integr Biol. 2015;19(10):627–631. doi:10.1089/omi.2015.0124
33. Hashemi-Soteh MB, Mousavi SS, Tafazoli A. Haplotypes inside the beta-globin gene: use as new biomarkers for beta-thalassemia prenatal diagnosis in north of Iran. J Biomed Sci. 2017;24(1):92. doi:10.1186/s12929-017-0396-y
34. Pandey S, Pandey S, Mishra RM, Saxena R. Modulating effect of the −158 Gγ (C-→T) Xmn1 polymorphism in Indian sickle cell patients. Mediterr J Hematol Infect Dis. 2012;4(1):e2012001. doi:10.4084/MJHID.2012.001
35. Bhagat S, Patra PK, Thakur AS. Association between XmnI polymorphism and HbF level in sickle cell disease patients from Chhattisgarh. Int J Biomed Sci. 2012;8(1):36–39.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.