Back to Journals » Neuropsychiatric Disease and Treatment » Volume 22
Association Between Clinical Symptoms and Inflammatory Markers in First-Episode Unmedicated Patients with Major Depressive Disorder
Authors Guan H, Sun J, Cheng J, Chen L ![]() , Zhao Y, Yang X, Wang D, Zhang X
, Zhao Y, Yang X, Wang D, Zhang X
Received 5 December 2025
Accepted for publication 12 February 2026
Published 23 February 2026 Volume 2026:22 582480
DOI https://doi.org/10.2147/NDT.S582480
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Taro Kishi
Huarui Guan,1– 3 Jingyao Sun,1– 3 Jun Cheng,1 – 3 Long Chen,2,3 Yueming Zhao,1– 3 Xiao Yang,1– 3 DongMei Wang,4 Xulai Zhang1– 3
1School of Mental Health and Psychological Sciences, Anhui Medical University, Hefei, Anhui, People’s Republic of China; 2Department of Psychiatry, Anhui Mental Health Center, Hefei Fourth People’s Hospital, Hefei, Anhui, People’s Republic of China; 3Department of Psychiatry, Anhui Clinical Center for Mental and Psychological Diseases, Hefei, Anhui, People’s Republic of China; 4Mental Health Key Laboratory, Institute of Psychology, Chinese Academy of Sciences, Beijing, People’s Republic of China
Correspondence: Xulai Zhang, Hefei Fourth People’s Hospital, Hefei, Anhui, People’s Republic of China, Email [email protected]
Background: Depression represents a serious psychiatric disorder globally, imposing significant burdens on patients’daily lives. Numerous hypotheses including the inflammatory hypothesis. Changes in inflammatory factors within the bodies of individuals with depression may represent a key biological mechanism. Multiple clinical investigations have demonstrated that pro-inflammatory cytokine levels in the peripheral blood of depressed patients are markedly higher than in healthy controls, with the degree of elevation in these inflammatory markers positively correlating with the severity of depressive symptoms. We shall examine changes in specific pro-inflammatory and anti-inflammatory factors.
Method: We employed the enzyme-linked immunosorbent assay (ELISA) to detect inflammatory cytokine levels in subject’s serum samples. Clinical symptoms were assessed using the Childhood Trauma Questionnaire (CTQ), the 17-item Hamilton Depression Rating Scale (HAMD-17), and the Quick Inventory of Depressive Symptomatology (Self-Rating) (QIDS-SR16).
Results: Compared to the healthy group, the depression group exhibited a significantly higher IFN-γ and IL-17 (p< 0.05) and lower IL-4, MCP-1, MIP-1β, and IL-8 (p< 0.05). Serum IL-4 levels showed significant positive correlations with HAMD-17 score (p=0.03), QIDS-SR 16 scores (p=0.009). IL-17 level was positively correlated with the QIDS-SR16 score (p=0.04). Several markers showed significant discriminatory power. IL-4 demonstrated the highest AUC of 0.72 (P < 0.01), with a sensitivity of 87.10% and specificity of 51.61%, indicating a moderate predictive value for depression.
Conclusion: The distinct inflammatory profile—characterized by elevated IFN-γ/IL-17 and reduced IL-4/MCP-1/IL-8/MIP-1β—in first-episode MDD patients underscores the role of immune imbalance in early depression pathophysiology. The strong association of IL-4 with symptom severity and its high sensitivity for disease identification highlight its potential as a valuable clinical biomarker. These findings support further investigation into immune-based stratification of MDD patients, which could pave the way for more personalized diagnostic and therapeutic strategies targeting specific inflammatory pathways”.
Keywords: depression, inflammatory cytokine, IL-4, IL-17, MDD
Introduction
Depression is a common mental disorder characterized by a range of symptoms, including low mood, anhedonia, fatigue, anxiety, irritability, insomnia, appetite changes, and suicidal ideation. These symptoms severely impact patients’ health and quality of life. According to the Global Burden of Disease study, depression ranks among the top ten most disabling conditions worldwide in terms of years lived with disability (YLDs).1 With a global lifetime prevalence of approximately 10%–15%, it stands as the leading cause of YLDs globally.
The inflammatory hypothesis of depression has progressively emerged as a significant theoretical framework for elucidating its pathological mechanisms. This hypothesis posits that chronic low-grade inflammatory responses, by activating both central and peripheral immune systems, induce neurotransmitter metabolic disturbances, impaired neural plasticity, and dysfunction of the hypothalamic-pituitary-adrenal (HPA) axis, thereby promoting the onset and maintenance of depressive disorders.2,3 Specifically, pro-inflammatory cytokines such as interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α) and C-reactive protein (CRP) are significantly elevated in the peripheral blood of patients with depression, with their levels positively correlated with disease severity.4,5 Inflammatory cytokines may promote a metabolic shift from tryptophan towards kynurenine by activating the indoleamine 2,3-dioxygenase (IDO) pathway, thereby reducing serotonin (5-HT) synthesis whilst generating neurotoxic kynurenine metabolites, leading to neuronal dysfunction.6 Persistently elevated peripheral pro-inflammatory factors (IL-6, TNF-α, CRP) may cross the compromised blood-brain barrier into the central nervous system, activating microglia, inducing neurotransmitter depletion and HPA axis hyperactivity, ultimately leading to depressed mood and cognitive impairment.7,8 Depression arises from the combined effects of genetic susceptibility and environmental stressors, leading to multisystem dysregulation. At the molecular level, reduced levels of monoamine neurotransmitters, such as serotonin and dopamine, are associated with sustained activation of the hypothalamic-pituitary-adrenal (HPA) axis. This activation triggers persistent cortisol secretion, which can damage the hippocampus and prefrontal cortex, thereby impairing memory, attention, and emotional regulation.9 Furthermore, recent studies have revealed that chronic low-grade inflammation and reduced levels of the neurotrophic factor BDNF,10 further inhibit neuroplasticity, potentially trapping emotional circuits in a negative feedback loop. Additionally, psychosocial factors—such as negative cognition and lack of social support—act as exacerbating factors, interacting bidirctionally with these biological alterations to ultimately trigger clinically observable depressive episodes. The monoamine neurotransmitter imbalance hypothesis remainsthe predominant framework in current clinical antidepressant drug development. However, approximately half of patients fail to achieve sustained remission after standardized treatment, suggesting that the underlying pathological mechanisms extend beyond monoaminergic system dysfunction.11 This has directed attention to the role of Inflammation in the pathophysiology of major depressive disorder (MDD).12 Over the past two decades, the “depression-inflammation hypothesis” has gained support, demonstrating that peripheral and central immune activation can induce depressive-like behaviors viamechanismsincluding the tryptophan-kynurenine pathway, HPA axis hyperactivation, and impaired neuroplasticity.10 Notably, the hypothalamic-pituitary-adrenal (HPA) axis is activated during inflammatory responses, with cytokines such as IL-1, IL-6, and tumor necrosis factor (TNF) playing key roles in this circuit.13 In a subset of depressed patients, this immune dysregulation is evidence, characterized by elevated levels of inflammatory mediators includingIL-6, CRP, IL-1β, and TNF-α.14 Furthermore, an earlier study demonstrated that depression severity correlates with inflammatory markers levels and may even exhibits a dose-response relationship.15 This inflammatory process is dynamically regulated by a network of pro- and anti-inflammatory factors; however, the specific roles of different factors across disease stages, symptom dimensions, and treatment responses remain unclear, presenting a critical bottleneck for current research.16 Although previous studies have separately reported alterations in MCP-1 (CCL2) or IL-8 (CXCL8) in depression, there remains a lack of research combining the detection of CC-type chemokines (CCL2, CCL4, CCL5) with CXC-type chemokines (CXCL8), particularly studies synchronising this analysis with Th1/Th2/Th17 cytokines and growth factors within the same cohort. Therefore, this study will systematically examine the co-expression profiles of the aforementioned chemokines and immunomodulatory factors to investigate their potential synergistic effects in depression. Based on the inflammation hypothesis of depression and the contradictory phenomena observed in previous research analyses, the study aimed to investigate differences in inflammatory cytokine levels between patients with first-episode, untreated depression and healthy controls, and to assess their correlation with clinical symptoms. Specifically, we verified the following hypothesis: Certain cellular inflammatory mediators exhibit elevated or reduced levels in individuals with depression. These mediators correlate with depressive symptoms and may predict the onset of depression. By quantifying the dose-response relationship between inflammatory markers and clinical symptoms, this study elucidates the pathways through which biological factors contribute to the pathogenesis of psychosocial stress, offering new insights into the mechanisms of mind-body interaction.
Materials and Methods
Study Participate
The study enrolled patients who met the following criteria: (1) a diagnosis of major depressive disorder according to the DSM-5 criteria;17 Through comprehensive clinical interviews, combined with DSM-5 diagnostic criteria, we identified patients meeting the criteria for major depressive disorder; (2) no history of any antidepressant or anti-inflammatory therapy; (3) availability of complete clinical data. Key exclusion criteria included the presence of concurrent malignant tumors, severe infectious diseases, significant dysfunction of vital organs (eg, cardiopulmonary), cerebral hemorrhage, or other organic brain lesions. All participants were fully informed about the study procedure and provided written informed consent prior to enrollment.
General Information
This study employed a case-control design. We enrolled patients meeting the criteria for first-episode, treatment-naive depression from Hefei Fourth People’s Hospital between 2023 and 2025. A total of 62 participants were included, Aged 18 to 65 years old, comprising 31 patients with major depressive disorder (12 males, 19 females) and 31 healthy controls (13 males, 18 females). All participants provided written informed consent. The study protocol was approved by the Ethics Committee of Hefei Fourth People’s Hospital, The trial registration number was HFSY-IRB-YJ-KYXM-CL (2024–064-001).
Methods
Data Collection
Sociodemographic data, including height, weight, age, and gender, were collected from participants (both patients and healthy controls).
Measurement of Serum Inflammatory Markers Using the ELISA
Venous blood samples (5mL each) were collected using all participants using vacuum blood collection tubes. After collection, the samples were allowed to clot at room temperature for 30 minutes and then centrifuged at 2000 rpm for 10 minutes. The serum was aliquoted and stored at −80°C until subsequent analysis. The serum levels of the following inflammatory markers were quantified using enzyme-linked immunoscorbent assay (ELISA) according to the manufacturers’ protocols: IFN-γ, IL-17, IL-4 (Th2 cytokines), MCP-1 (CC chemokine: CCL2), MIP-1β (CC chemokine: CCL4), IL-8 (CXC chemokine: CXCL8), TNF-α (tumor necrosis factor), IL-2 (T-cell growth factor), IL-1β (pro-inflammatory factor), IL-13 (Th2 cytokine), RANTES (CC chemokine: CCL5), VEGF (vascular endothelial growth factor), and PDGF-BB (platelet-derived growth factor).
Assessment
Clinical symptoms were assessed using the following scales: the 17-item Hamilton Depression Rating Scale (HAMD-17), the Childhood Trauma Questionnaire (CTQ), the Hamilton Anxiety Rating Scale (HAMA), and the 16-item Quick Inventory of Depressive Symptomatolgy (Self-Report) (QIDS-SR16). The HAMD-17 assesses severity across several domains:core depression factors, anxiety/agitation factors, sleep disturbance factors, and somatic symptoms. Depressive symptoms in patients who have experienced childhood trauma represent a common trauma response. We employed the Childhood Trauma Scale to assess the relationship between their trauma and depressive symptoms.17 The CTQ evaluates five types of childhood trauma: Emotional Abuse (EA), Physical Abuse (PA), Sexual Abuse (SA), Emotional Neglect (EN), and Physical Neglect (PN). Individuals with depression frequently experience comorbid anxiety symptoms, and sleep disturbances are common throughout the course of the illness.18 The HAMA is used to measure anxiety severity, includes core depression factors, anxiety factors, sleep disturbance factors, and somatic nutrition factors; The QIDS-SR16 covers multiple symptom dimensions: sleep disturbance dimension, vegetative/physicalsymptoms, affective-cognitive symptoms, and psychomotor changes.
Statistical Analysis
Data analysis was performed using SPSS 27.0 (IBM, USA). Continuous data are presented as mean ± standard deviation for normally distributed variables or as median (interquartile range) for nonnormally variables (See Tables 1 and 2 for details). Intergroup comparisons of inflammatory factors between the experimental and control groups were conducted using the non-parametric U-test (See Figure 1 for details). Employing Spearman correlation analysis to examine the relationship between specific inflammatory markers and clinical scores (See Figure 2 for details). The diagnostic utility of serum inflammatory factors for depression severity was evaluated by operating characteristic (ROC) curves (See Table 3 and Figure 3 for details). Figures were generated using GraphPad Prism (GraphPad Software, USA). A two-sided p-value of less than 0.05 was considered statistically significant. The results of sensitivity and specificity adjusted for the sociodemographics or other factors. The ROC curves to evaluate the diagnostic utility of serum inflammatory factors for depression severity were generated using the standard non-parametric method (also known as the empirical method). The key steps were as follows: 1. Outcome: Depression severity was binarized using a clinical cutoff on the [eg Hamilton Depression Rating Scale]. 2. Analysis: For each inflammatory factor (treated as a continuous predictor), we plotted the true positive rate (sensitivity) against the false positive rate (1 − specificity) across all its observed concentration values. The Area Under the Curve (AUC) and its 95% confidence interval were calculated directly from this empirical curve. 3. Optimal Cut-off, Sensitivity, and Specificity: The optimal diagnostic cut-off concentration for each biomarker was selected as the value that maximized Youden’s Index (J = Sensitivity + Specificity − 1). The sensitivity and specificity reported correspond to this optimal threshold. 4. Software: This analysis was performed using GraphPad Prism 9.
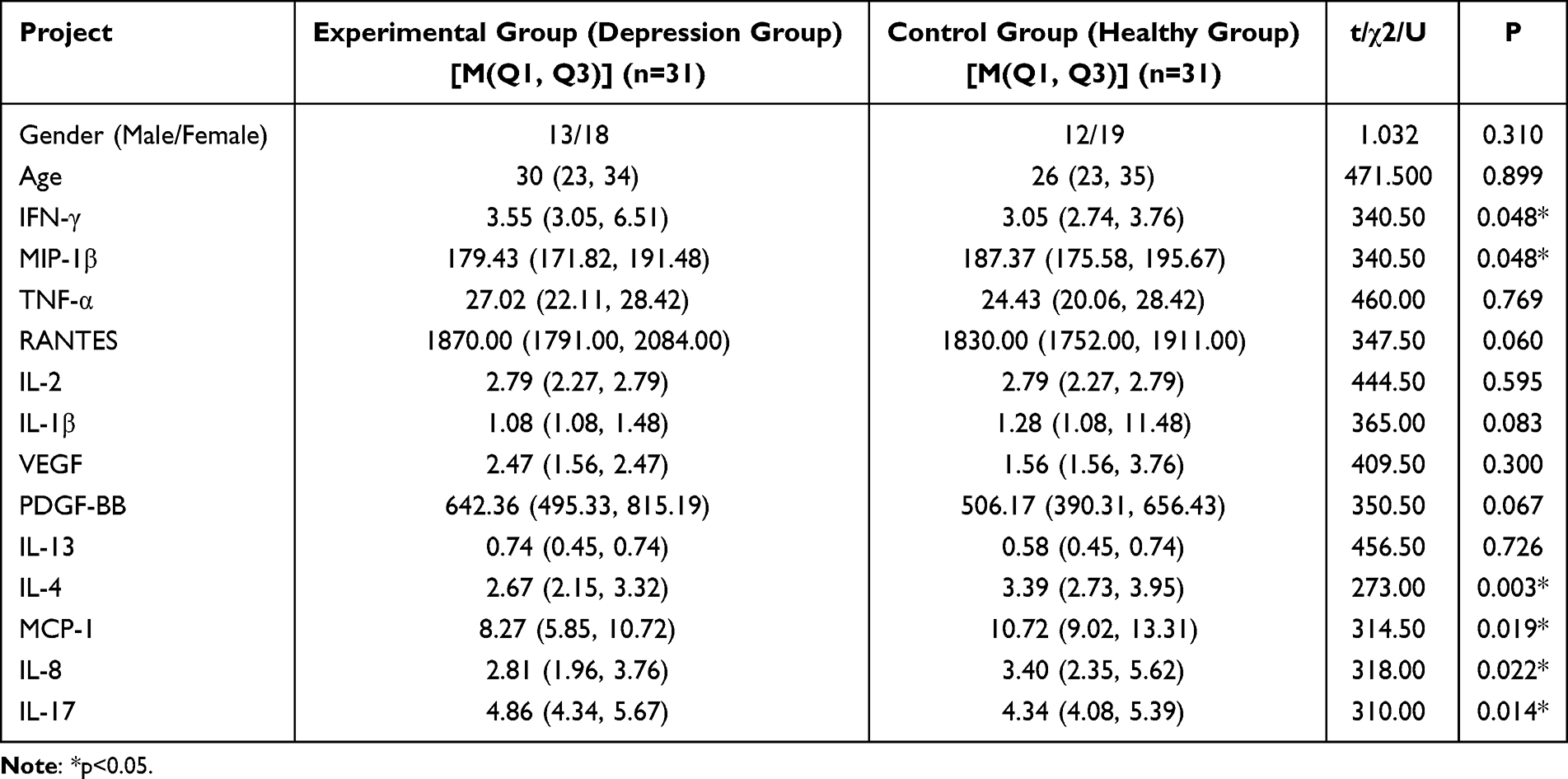 |
Table 1 Comparison of Demographic Characteristics, Inflammatory Cytokine Levels Among All Participants |
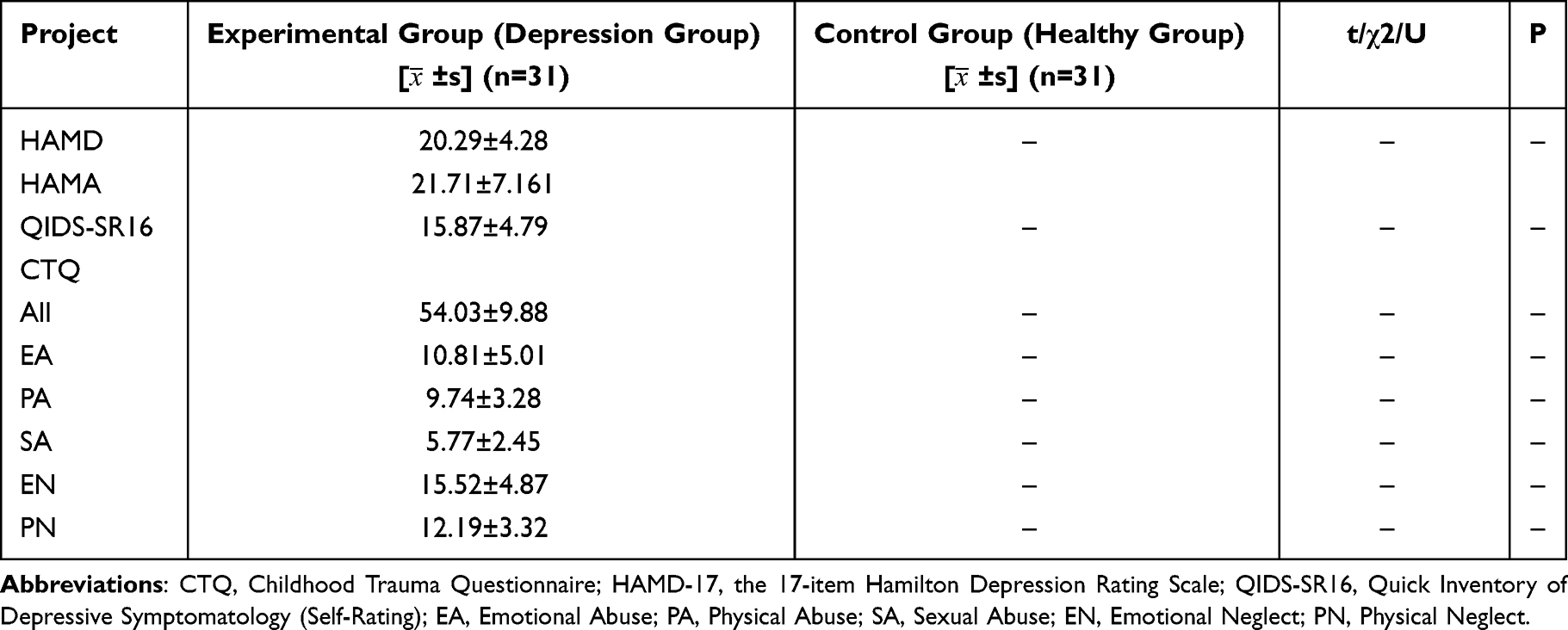 |
Table 2 Comparison of Clinical Data Among All Participants |
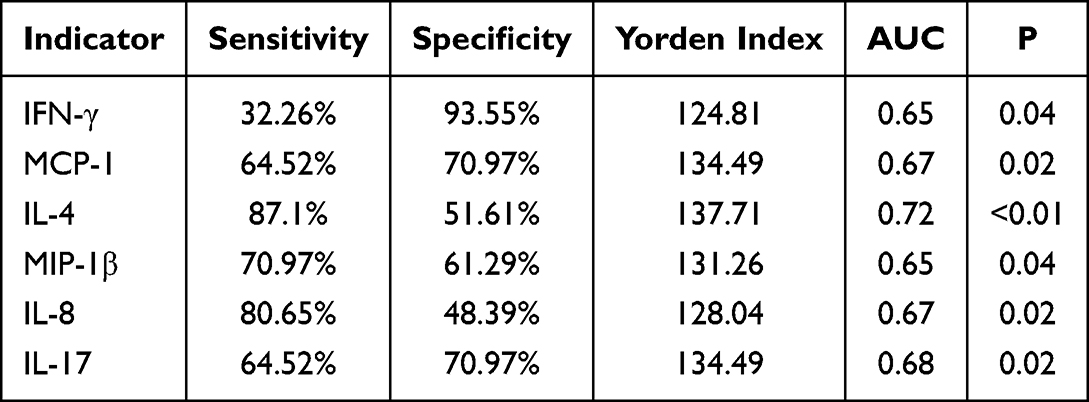 |
Table 3 ROC Curve Assessment of the Diagnostic Potential of Serum Inflammatory Cytokines for Depression |
 |
Figure 1 Comparative analysis of inflammatory markers between the depression group and the healthy control group. Abbreviations: HC, healthy; MDD, major depression disorder. Notes: *P<0.05, **P<0.01. |
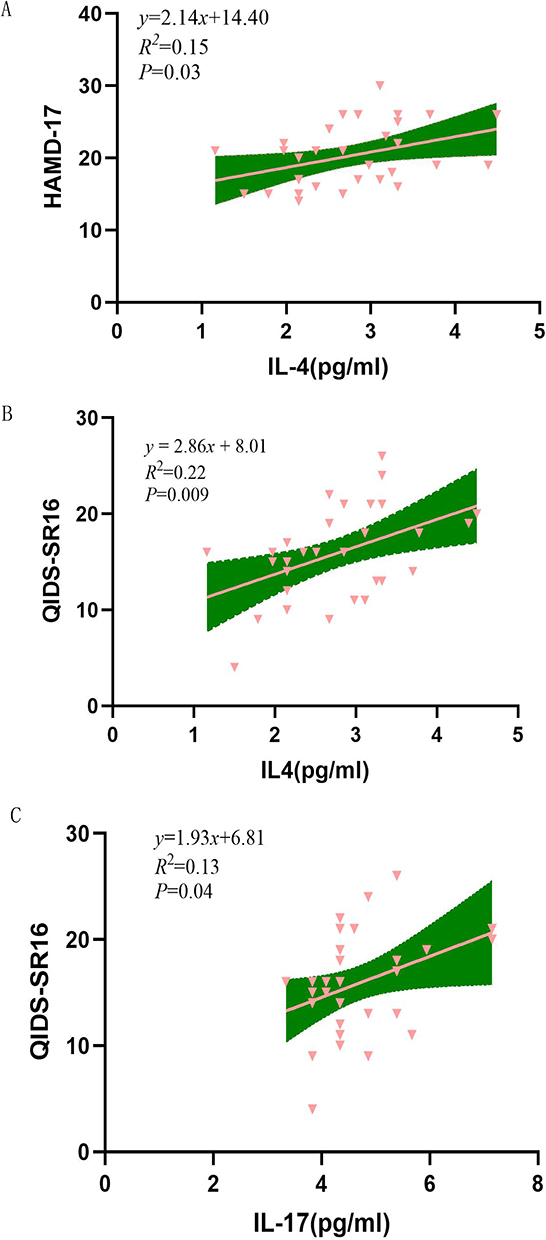 |
Figure 2 Correlation between specific inflammatory markers and clinical scores. (A) Correlation analysis between IL-4 levels and HAMD-17 scores. Pink triangles denote individual sample data points, the pink line represents the linear regression fit, and the green area indicates the 95% confidence interval for the fit. The regression equation is y = 2.14x + 14.40, with R2 = 0.15 and P = 0.03. (B) Correlation analysis between IL-4 levels and QIDS-SR16. Pink triangles denote individual sample data points, the pink line represents the linear regression fit line, and the green area indicates the 95% confidence interval for the fit line. The regression equation is y = 2.86x + 8.01, R2 = 0.22, P = 0.009. (C) Correlation analysis between IL-17 levels and QIDS-SR16. Pink triangles denote individual sample data points, the pink line represents the linear regression fit line, and the green area indicates the 95% confidence interval for the fit line. The regression equation is y = 1.93x + 6.81, R2 = 0.13, P = 0.04. Abbreviations: HAMD-17, the 17-item Hamilton Depression Rating Scale; QIDS-SR16, Quick Inventory of Depressive Symptomatology (Self-Rating). |
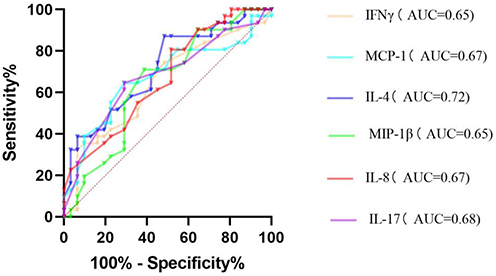 |
Figure 3 ROC curve for predicting onset in the depression group using inflammatory markers. |
Results
Demographic and Clinical Characteristics of All Subjects
The demographic and clinical characteristics of all participants are summarized in Tables 1 and 2. A total of 31 patients with depression and 31 healthy controls were included in this study. No significant differences were observed in age or gender between the two groups (P > 0.05).
Inflammatory Cytokine Concentrations
Compared to the healthy control group, the depression group exhibited a significantly higher IFN-γ and IL-17 (p<0.05) and lower IL-4, MCP-1, MIP-1β, and IL-8 (p<0.05). In contrast, no significant differences were found in the levels of TNF-α, IL-2, IL-1β, IL-13, RANTES, VEGF, and PDGF-BB between the two groups (all P > 0.05; Figure 1).
Correlation Between Inflammatory Cytokine Levels and Depression Symptoms
Spearman correlation analysis revealed significant positive correlation between specific inflammatory markers and clinical scores. Serum IL-4 levels showed significant positive correlations with HAMD-17 score (p=0.03), QIDS-SR 16 scores (p=0.009). Similarly, IL-17 level was positively correlated with the QIDS-SR16 score (p=0.04). Several markers showed significant discriminatory power.
To further investigate these relationships, linear regression analysis were performed. IL-4 was significant positive predictor of the HAMD-17 score (β = 0.389, p = 0.03, standardized coefficient = 2.14), accounting for 15.1% of the variance (adjusted R2 = 0.151), IL-4 was also a significant positive predictor of the QIDS-SR16 score (β = 0.464, p = 0.009, standardized coefficient = 2.86), explaining 22% of variance (adjusted R2 = 0.22). Furthermore, IL-17 was a significant positive predictor of the QIDS-SR16 (β = 0.452, p = 0.04, standardized coefficient = 1.93), explaining 13% of the variance (adjusted R2 = 0.13). These results support the hypothesis that IL-4 and IL-17 influence depressive symptomatology. See Figure 2 for details.
Diagnostic Value of Inflammatory Cytokines for Depression
The diagnostic potential of serum inflammatory cytokines for depression was evaluated using receiver operating characteristic (ROC) curve analysis, with disease status as the state variable. Key performance metrics, including the area under the curve (AUC), sensitivity, specificity, and Youden’s index, are summarized in Table 3. Several markers showed significant discriminatory power. IL-4 demonstrated the highest AUC of 0.72 (P < 0.01), with a sensitivity of 87.10% and specificity of 51.61%, indicating a moderate predictive value for depression. Other significant markers included MCP-1 (AUC = 0.67, P = 0.02), IL-8 (AUC = 0.67, P = 0.02), IL-17 (AUC = 0.68, P = 0.02), IFN-γ (AUC = 0.65, P = 0.04), and MIP-1β (AUC = 0.65, P = 0.04). See Figure 3 for details.
The novelty of our work lies in three key aspects: First, by focusing on first-episode, medication-naïve patients, we captured the intrinsic immune profile at depression onset, free from chronicity or treatment confounders. Second, we identified a distinct and somewhat counterintuitive signature—elevated IFN-γ/IL-17 co-occurring with reduced IL-4 and key chemokines—supporting the concept of an immune-dysregulated subtype beyond simple “inflammation”. Third, we highlight IL-4’s dual role as both a sensitive diagnostic marker (87.1% sensitivity) and a severity correlate, underscoring its unique translational potential for patient stratification.
Discussion
Our findings add to the growing yet complex literature on immune dysregulation in MDD. The observed elevation of the pro-inflammatory cytokines IFN-γ and IL-17 in our cohort of first-episode, unmedicated patients aligns with a substantial body of evidence supporting the inflammatory hypothesis of depression. Peripheral inflammatory mediators can influence the central nervous system by crossing the blood-brain barrier or the vagus nerve, thereby participating in the pathology of depression.19 For instance, the elevated Th1/Th17 profile we observed is consistent with recent studies and meta-analyses reporting increased IFN-γ and IL-17 in MDD patients, linking them to neuroinflammation and HPA axis dysregulation.20–22 A 2023 review article also indicated that individuals with first-episode depression exhibit autoimmune hyperactivation, characterised by elevated serum IL-17 levels, which positively correlate with Hamilton Depression Rating Scale (HAMD) score.21 This consistency strengthens the notion that a pro-inflammatory shift is a relevant biological feature in at least a subset of patients during the acute, untreated phase of illness.
However, our results regarding IL-4, MCP-1, MIP-1β, and IL-8 appear to contradict several previous reports. While many studies describe elevated levels of these mediators in depression, we found them to be significantly lower in patients compared to healthy controls. This discrepancy is not without precedent. Our findings are congruent with studies by Myung et al23 and Proma et al,24 who also reported reduced MCP-1 in depression, suggesting a potential association with the neuroprotective functions of neurotrophic factor.25,26 This contradiction likely stems from critical methodological and phenotypic heterogeneity across studies. Key factors include: (1) clinical heterogeneity: Depression is not a unitary disorder. The “low-inflammation” subtype hypothesis, as suggested by Iob et al27 may explain our findings. In this subtype, chronic HPA axis hyperactivity and elevated cortisol could suppress the production of certain chemokines and anti-inflammatory cytokines via glucocorticoid-mediated pathways,28 Researchers have observed that serum MCP-1 levels in patients with major depressive disorder (MDD) are significantly lower than in healthy controls and negatively correlated with HAMD scores,24 potentially reflecting chronic stress-induced hyperactivation of the HPA axis. Our sample of first-episode, treatment-naïve patients might be enriched for such a subtype; (2) Methodological Variations: Differences in assay sensitivity, sample processing, and the timing of blood collection (given the circadian rhythm of cytokine29) can significantly influence measured levels; (3) confounding comorbidities: As highlighted by Dias et al30 conditions like metabolic syndrome or diabetes can be primary drivers of inflammation. The absence of such comorbidities in our relatively young cohort could result in a different inflammatory profile compared to studies of older or comorbid populations.
The significant positive correlation between IL-4 and depression severity scores, despite its lower absolute level, presents a particularly intriguing finding. It suggests that within the depressed state, even a relative deficiency of this anti-inflammatory, neuroprotective cytokine is functionally linked to symptom burden. This aligns with mechanistic studies showing IL-4 is critical for promoting resilience through microglia-mediated BDNF release and neurogenesi.31 Zhang J’s study indicate that IL4-driven, Arg1-highly-expressed microglia regulate adult neurogenesis in the hippocampal dentate gyrus. Reducing IL4 receptor on microglia decreases Arg1⁺ microglia, thereby inhibiting neurogenesis and increasing stress susceptibility; conversely, enhancing IL4 signaling amplifies this cell population, restoring neurogenesis and antidepressant capacity. Mechanistically, IL-4-Arg1⁺ microglia promote neurostem cell proliferation and differentiation by secreting BDNF, thereby alleviating chronic stress-induced depressive-like behavior,31 In a rat model of depressive-like behavior, IL-4 was found to suppress IL-1β-induced central glial activation and neurotransmitter alterations, thereby modulating, depressive behavior. IL-4 reduced central and systemic inflammatory activation while reversing IL-1β-induced neurotransmitter changes, suggesting this pathway may be therapeutically effective against IL-1β-induced depressive behavior and neuroinflammation.32 Additionally, the expression of IFN-γ in depression appears complex and context-dependent. For instance, in a chronic restraint stress-induced depressive rat model, IFN-γ expression was decreased in thyroid tissue—a trend distinct from other inflammatory factors—indicating potentially different regulatory mechanisms across tissues.33 This biphasic shift may be associated with disease stage and treatment response. During the acute phase, untreated MDD patients predominantly exhibit a Th1-dominant inflammatory profile, where IFN-γ exacerbates serotonin depletion and neurotoxicity by activating indoleamine 2,3-dioxygenase (IDO) and the glutamate-kynurenine pathway, thereby worsening symptoms.34 In contrast, during chronic stress or following pharmacological intervention, the immune may shifts toward Th2 dominance, and a decrease in IFN-γ levels can be regarded as an indicator of successful antidepressant treatment.35 Nonetheless, other studies have observed a potential decrease in IFN-γ levels among depressed patients,36 highlighting the heterogeneous dynamics of t IFN-γ in depression.
Potential Solutions and Future Directions: To reconcile conflicting findings and advance the field, future research must move beyond cross-sectional designs. Large-scale, longitudinal studies tracking patients from the first episode through treatment are essential. These studies should rigorously stratify patients based on potential subtypes (eg using clinical biomarkers like CRP or childhood trauma history) and strictly control for confounding factors (comorbidities, medication, circadian rhythm). Integrating multi-omics approaches with neuroimaging and detailed clinical phenotyping will help disentangle distinct biological pathways underlying depressive syndromes.
Policy and Clinical Implications: Our study underscores the critical need for a precision medicine approach in psychiatry. The heterogeneous nature of immune findings argues against a one-size-fits-all “anti-inflammatory” treatment for depression. Instead, healthcare policy should support the development and implementation of affordable biomarker panels to identify patients with specific immune profiles (eg “high IL-17” or “low IL-4”). This stratification can guide targeted interventions, such as selecting patients most likely to benefit from immunomodulatory therapies in clinical trials. Furthermore, our results reinforce the importance of managing systemic inflammatory conditions (eg obesity, diabetes) as part of comprehensive psychiatric care, as these comorbidities may fundamentally alter the pathophysology of depressive symptoms. Ultimately, integrating immune biomarkers into diagnostic and treatment frameworks holds promise for improving patient outcomes through more personalized and effective strategies.
Conclusion
In summary, individuals with depression exhibit reduced levels of the anti-inflammatory factor IL-4 compared to healthy individuals, while pro-inflammatory factor levels are relatively elevated and closely correlated with disease severity. This suggests that pro-inflammatory factors may constitute a key component in the pathogenesis of depression, suggesting they may represent a pivotal component in the pathogenesis of depression. Inflammatory responses not only influence central nervous system function through multiple pathways but may also contribute to the development of specific subtypes (such as inflammatory depression) and affect treatment response. Consequently, inflammatory cytokines hold promise as important biomarkers for the diagnosis, classification, and efficacy assessment of depression. They also provide a theoretical basis for developing anti-inflammatory treatment strategies, thereby advancing the development of personalised precision medicine.
Abbreviations
CTQ, Childhood Trauma Questionnaire; HAMD-17, the 17-item Hamilton Depression Rating Scale; QIDS-SR16, Quick Inventory of Depressive Symptomatology (Self-Rating); EA, Emotional Abuse; PA, Physical Abuse; SA, Sexual Abuse; EN, Emotional Neglect; PN, Physical Neglect; MDD, Major Depressive Disorder.
Data Sharing Statement
The data that support the findings of this study are available from Hefei Fourth People’ Hospital but restrictions apply to the availability of those data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of Hefei Fourth People’ Hospital. To obtain the data in this study, the researchers may be contacted at [email protected].
Ethics Approval and Consent to Participate
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of Hefei Fourth People’s Hospital. Informed consent was obtained from all the subjects. The trial registration number was HFSY-IRB-YJ-KYXM-CL (2024-064-001). All procedures carried out in studies conformed to the 1964 Helsinki Declaration and its subsequent amendments or similar ethical standards.
Acknowledgments
We would like to thank the support of Hefei Fourth People’s Hospital.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the STI2030-Major Project (2021ZD0200600) and the National Clinical Key Specialty Construction Project of China, the Anhui Clinical Medical Research Center for Mental and Psychological Diseases, and the Healthcare Application Medical Research Project of Hefei City (Hwk2024yb003).
Disclosure
All authors declare no conflict of interest.
References
1. Suda K, Matsuda K. How microbes affect depression: underlying mechanisms via the gut-brain axis and the modulating role of probiotics. Int J Mol Sci. 2022;23(3):1172. doi:10.3390/ijms23031172
2. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732–12. doi:10.1016/j.biopsych.2008.11.029
3. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. doi:10.1038/nrn2297
4. Khandaker GM, Pearson RM, Zammit S, Lewis G, Jones PB. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiatry. 2014;71(10):1121–1128. doi:10.1001/jamapsychiatry.2014.1332
5. Young JJ, Bruno D, Pomara N. A review of the relationship between proinflammatory cytokines and major depressive disorder. J Affect Disord. 2014;169:15–20. doi:10.1016/j.jad.2014.07.032
6. Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25(2):181–213. doi:10.1016/j.bbi.2010.10.015
7. Beurel E, Toups M, Nemeroff CB. The bidirectional relationship of depression and inflammation: double trouble. Neuron. 2020;107(2):234–256. doi:10.1016/j.neuron.2020.06.002
8. Lucido MJ, Bekhbat M, Goldsmith DR, et al. Aiding and abetting anhedonia: impact of inflammation on the brain and pharmacological implications. Pharmacol Rev. 2021;73(3):1084–1117. doi:10.1124/pharmrev.120.000043
9. Clinical Assessment and Diagnosis-Treatment. Guidance for Melancholic/Anhedonic Depression.
10. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16(1):22–34. doi:10.1038/nri.2015.5
11. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. doi:10.1176/ajp.2006.163.11.1905
12. Liu Y, Ho RCM, Mak A. Interleukin (IL)-6, tumour necrosis factor alpha (TNF-α) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: a meta-analysis and meta-regression. J Affect Disord. 2012;139(3):230–239. doi:10.1016/j.jad.2011.08.003
13. Besedovsky HO, Del Rey A, Klusman I, Furukawa H, Monge Arditi G, Kabiersch A. Cytokines as modulators of the hypothalamus-pituitary-adrenal axis. J Steroid Biochem Mol Biol. 1991;40(4–6):613–618. doi:10.1016/0960-0760(91)90284-C
14. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27(1):24–31. doi:10.1016/j.it.2005.11.006
15. Miller GE, Stetler CA, Carney RM, Freedland KE, Banks WA. Clinical depression and inflammatory risk markers for coronary heart disease. Am J Cardiol. 2002;90(12):1279–1283. doi:10.1016/S0002-9149(02)02863-1
16. Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB. Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry. 2015;2(3):258–270. doi:10.1016/S2215-0366(14)00122-9
17. Vibhakar V, Allen LR, Gee B, Meiser-Stedman R. A systematic review and meta-analysis on the prevalence of depression in children and adolescents after exposure to trauma. J Affect Disord. 2019;255:77–89. doi:10.1016/j.jad.2019.05.005
18. Chen Z, Tang Y, Liu X, et al. Edge-centric connectome-genetic markers of bridging factor to comorbidity between depression and anxiety. Nat Commun. 2024;15(1):10560. doi:10.1038/s41467-024-55008-0
19. Köhler CA, Freitas TH, Maes M, et al. Peripheral cytokine and chemokine alterations in depression: a meta-analysis of 82 studies. Acta Psychiatr Scand. 2017;135(5):373–387. doi:10.1111/acps.12698
20. Nava RG, Adri AS, Filgueiras IS, et al. Modulation of neuroimmune cytokine networks by antidepressants: implications in mood regulation. Transl Psychiatry. 2025;15(1):314. doi:10.1038/s41398-025-03532-y
21. Lu Y, Zhang P, Xu F, Zheng Y, Zhao H. Advances in the study of IL-17 in neurological diseases and mental disorders. Front Neurol. 2023;14:1284304. doi:10.3389/fneur.2023.1284304
22. Lu L, Hu X, Jin X. IL-4 as a potential biomarker for differentiating major depressive disorder from bipolar depression. Medicine. 2023;102(15):e33439. doi:10.1097/MD.0000000000033439
23. Myung W, Lim SW, Woo HI, et al. Serum cytokine levels in major depressive disorder and its role in antidepressant response. Psychiatry Invest. 2016;13(6):644–651. doi:10.4306/pi.2016.13.6.644
24. Proma MA, Daria S, Nahar Z, Ashraful Islam SM, Bhuiyan MA, Islam MR. Monocyte chemoattractant protein-1 levels are associated with major depressive disorder. J Basic Clin Physiol Pharmacol. 2022;33(6):735–741. doi:10.1515/jbcpp-2021-0132
25. Madrigal JLM, Leza JC, Polak P, Kalinin S, Feinstein DL. Astrocyte-derived MCP-1 mediates neuroprotective effects of noradrenaline. J Neurosci. 2009;29(1):263–267. doi:10.1523/JNEUROSCI.4926-08.2009
26. de Haas AH, van Weering HRJ, de Jong EK, Boddeke HWGM, Biber KPH. Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol Neurobiol. 2007;36(2):137–151. doi:10.1007/s12035-007-0036-8
27. Iob E, Kirschbaum C, Steptoe A. Persistent depressive symptoms, HPA-axis hyperactivity, and inflammation: the role of cognitive-affective and somatic symptoms. Mol Psychiatry. 2020;25(5):1130–1140. doi:10.1038/s41380-019-0501-6
28. Ehrchen JM, Roth J, Barczyk-Kahlert K. More than suppression: glucocorticoid action on monocytes and macrophages. Front Immunol. 2019;10:2028. doi:10.3389/fimmu.2019.02028
29. Poletti S, Mazza MG, Benedetti F. Inflammatory mediators in major depression and bipolar disorder. Transl Psychiatry. 2024;14(1):247. doi:10.1038/s41398-024-02921-z
30. Dias NS, Teixeira AL, Diniz BS, Vieira EL, de M Viana B, Barbosa IG. Higher IL-6 and IL-4 plasma levels in depressed elderly women are influenced by diabetes mellitus. Trends Psychiatry Psychother. 2024;46:e20220466. doi:10.47626/2237-6089-2022-0466
31. Zhang J, Rong P, Zhang L, et al. IL4-driven microglia modulate stress resilience through BDNF-dependent neurogenesis. Sci Adv. 2021;7(12):eabb9888. doi:10.1126/sciadv.abb9888
32. Park HJ, Shim HS, An K, Starkweather A, Kim KS, Shim I. IL-4 inhibits IL-1β -induced depressive-like behavior and central neurotransmitter alterations. Mediators Inflamm. 2015;2015(1):941413. doi:10.1155/2015/941413
33. Wu X, Gao Y, Liu S, Meng L, Wang S, Song C. Effects of depression on thyroid function, pathology and ultrasonography in rats. J Mol Histol. 2025;56(5):293. doi:10.1007/s10735-025-10584-3
34. Liu M, Tang J, Xu G, et al. Investigating the relationship between inflammatory cytokines and adolescent depression: a comparative analysis. Front Psychiatry. 2025;16:1524015. doi:10.3389/fpsyt.2025.1524015
35. Catalogna M, Saporta N, Nathansohn-Levi B, et al. Mobile application leads to psychological improvement and correlated neuroimmune function change in subjective cognitive decline. NPJ Digit Med. 2025;8(1):359. doi:10.1038/s41746-025-01765-1
36. Serafini G, Costanza A, Aguglia A, et al. The role of inflammation in the pathophysiology of depression and suicidal behavior: implications for treatment. Med Clin North Am. 2023;107(1):1–29. doi:10.1016/j.mcna.2022.09.001
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.