Back to Journals » Vascular Health and Risk Management » Volume 22
Association Between Apelin Gene −1860T>C (rs56204867) Polymorphism and Coronary Artery Disease in a Syrian Cohort
Authors Abd-Alkareem MH ![]() , Shibli HEM, Alquobaili FA
, Shibli HEM, Alquobaili FA
Received 2 February 2026
Accepted for publication 11 June 2026
Published 26 June 2026 Volume 2026:22 600632
DOI https://doi.org/10.2147/VHRM.S600632
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Konstantinos Tziomalos
Maisaa Hassan Abd-Alkareem,1 Hussam Eddin Mohammed Shibli,2 Faizeh Ali Alquobaili1
1Department of Biochemistry and Microbiology, Faculty of Pharmacy, Damascus University, Damascus, Syrian Arab Republic; 2Department of Internal Medicine, Faculty of Medicine, Damascus University, Damascus, Syrian Arab Republic
Correspondence: Maisaa Hassan Abd-Alkareem, Department of Biochemistry and Microbiology, Faculty of Pharmacy, Damascus University, Damascus, Syrian Arab Republic, Tel +963-33335140, Email [email protected]
Purpose: Apelin is a secreted peptide hormone involved in vasodilation, fluid homeostasis, and angiogenesis, and is therefore considered an important regulator of cardiovascular physiology. The APLN T-1860C (rs56204867) polymorphism is a genetic variation in the promoter region of the apelin gene (APLN) that is associated with increased susceptibility to cardiovascular risk factors. This study aimed to elucidate the relationship between the apelin gene − 1860T>C single-nucleotide polymorphism, plasma apelin levels, and the risk of coronary artery disease (CAD) in a Syrian population.
Patients and Methods: A case-control study was conducted, comprising 108 CAD patients and 114 healthy controls. Plasma apelin levels were quantified using an enzyme-linked immunosorbent assay (ELISA). The apelin − 1860T>C gene polymorphism was analyzed by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP).
Results: Our findings reveal that mean plasma apelin levels were significantly lower in CAD patients (2737.81 ± 1205.01 pg/mL) compared to controls (4258.73 ± 2119.12 pg/mL; p < 0.001). Notably, these levels did not correlate with any of the other anthropometric parameters studied. In females, the frequencies of the TC genotype (30.5% vs 10.5%, p = 0.041, odds ratio [OR] = 3.75) and the mutant C allele (15.2% vs 5.2%, p = 0.043, OR = 3.256) were significantly higher in CAD patients than in controls. Whereas no significant differences were observed in male subjects. Although the plasma apelin level was lower in patients and controls with the TC genotype, this difference was not statistically significant (p > 0.05).
Conclusion: Our findings indicate that the APLN − 1860T>C polymorphism is associated with a higher risk of CAD in the Syrian population, particularly among females, suggesting a sex-specific genetic predisposition. Furthermore, reduced plasma apelin levels in patients suggest that apelin may play an independent role in the development of CAD, warranting further exploration. The infographic illustrates the association between the Apelin gene -1860T>C SNP and coronary artery disease (CAD) risk. It shows a comparison between CAD patients (n = 108) and control group (n = 114), highlighting factors like smoking and hypertension. A bar graph compares Apelin concentration in control and CAD groups, with a significant difference marked by an asterisk (p < 0.05). A table lists genotypes: TT (70 percent CAD, 90 percent control), TC (31 percent CAD, 11 percent control) and CC (0 percent for both). The association section indicates that TC and CC genotypes are linked to increased CAD risk, with the C allele associated with lower Apelin levels. The diagram includes visual representations of DNA and arteries to emphasize genetic and health implications.Infographic on Apelin gene SNP and CAD risk in patients and controls.
Keywords: apelin − 1860T>C gene polymorphism, coronary artery disease, single nucleotide polymorphisms, SNP, Syrian population, PCR-RFLP, plasma apelin levels
Introduction
Coronary artery disease (CAD), a multifaceted condition, is the primary contributor to atherosclerosis, which is the leading cause of global mortality.1,2 The development of CAD is attributed to an intricate interplay of inflammatory responses to various arterial wall injuries, culminating in acute coronary events and cardiovascular complications.1,3 Notably, genetic factors play a substantial role in CAD pathogenesis, with an estimated 40% to 70% of disease risk attributed to heritable components.4 Within the realm of genetic determinants, single-nucleotide polymorphisms (SNPs) have garnered significant attention, with numerous studies demonstrating associations between specific SNPs and CAD susceptibility.5
Apelin, an adipokine produced by adipocytes, endothelial cells, vascular smooth muscle cells, and cardiac cells, serves as an endogenous ligand binding to the G protein-coupled receptor APJ. The apelin/apelin receptor system is ubiquitously expressed in various tissues, including the myocardium and vascular endothelial cells.1,6 The apelin peptide has been implicated in a myriad of physiological processes, including the regulation of blood pressure, maintenance of fluid balance, modulation of endocrine stress responses, heart muscle contraction, angiogenesis, and energy metabolism.7 Notably, its involvement extends to various pathological conditions, such as heart failure, myocardial infarction, atherosclerosis, obesity, and diabetes, underscoring the significance of apelin in both physiological and pathological contexts.8,9 The apelin gene, located on the X chromosome, encodes preproapelin-77, which is subsequently processed into proapelin-55 and further cleaved into circulating apelin peptide isoforms such as apelin-13, apelin-17 and apelin-36, which differ in receptor affinity and physiological effects. Among these isoforms, apelin-13 predominates in human plasma and heart tissue.10,11 These peptides are predominantly found within endocardial and vascular endothelial cells, suggesting a tissue-specific origin of circulating apelin.12
Accumulating evidence suggests that apelin plays a pivotal role in cardiovascular disease physiology, with alterations in its secretion levels implicated in the development of chronic inflammatory diseases. Animal studies have shown that decreased expression of apelin receptor in the heart tissues of ischemic heart disease may contribute to the pathogenesis of the disease.13 In humans, the expression levels of apelin and APJ, in the smooth muscle cells of the aorta decrease during atherosclerosis, which may contribute to increased vulnerability of the plaques.14 Laboratory studies have demonstrated that apelin exerts vasodilatory effects, lowers blood pressure, enhances cardiac contractility, and modulates antioxidant components, thereby influencing atherosclerosis.3 However, the anti-atherosclerotic properties of apelin/APJ in humans remain to be definitively established.15–17 Experimental studies have demonstrated that apelin stimulates the proliferation of vascular smooth muscle cells, while a reduction in APJ levels offers protection against the development of atherosclerotic plaques.12,18 Another study revealed that the absence of apelin promotes atherosclerosis, whereas treatment with it reduces angiotensin II–induced atherosclerosis.19
The −1860T>C variant is located in the promoter region of the APLN gene, an area critically involved in regulating gene transcription and, consequently, apelin expression levels. It was first described in the Han Chinese population,20 where it was associated with susceptibility to cardiovascular risk factors, including essential hypertension.21 Subsequent studies have shown that this polymorphism may influence plasma apelin levels and cardiovascular disease risk in different ethnic groups.22 In Turkish patients with coronary artery disease, the CC genotype and C allele at −1860T>C were significantly more frequent in CAD patients than in controls. They were associated with altered apelin levels and increased CAD risk.23 In addition, this variant has been linked to metabolic and hypertensive-related phenotypes, including obesity related traits,24 further supporting its role in apelin-regulated cardiovascular and metabolic pathways.
A multitude of modifiable and non-modifiable cardiovascular risk factors contribute to the development of atherosclerosis, including unhealthy diet, physical inactivity, dyslipidemia, hyperglycemia, hypertension, obesity, sex, age, and smoking.2 Against this backdrop, the current study is the first to investigate the association between the apelin gene −1860T>C polymorphism and CAD risk in a Syrian cohort. Specifically, we evaluate the genotype and allele distribution of this SNP and measure plasma apelin levels to explore their potential interplay in disease susceptibility.
Materials and Methods
Study Design and Participant Recruitment
This case-control study was conducted between January 2020 and August 2022 at two medical institutions in Damascus: The National University Hospital and the Cardiac Surgery Center at Al-Mouwasat Hospital. The study protocol was approved by the Ethics Committee of Damascus University of Medical Sciences (Approval No. 9/A.Kh, dated 23/7/2019) and registered on ClinicalTrials.gov (Identifier: NCT05562687). Written informed consent was obtained from all participants before their enrollment.
The study population comprised 222 subjects, divided into two groups: a CAD group and a control group. The CAD group consisted of 108 patients (85 males and 23 females) with a diagnosis of CAD, as confirmed by coronary angiography, demonstrating at least 70% stenosis in any coronary artery. The control group consisted of 114 subjects (47 males and 67 females) who presented with chest pain but had normal coronary angiography findings (0% stenosis), and they did not have any chronic inflammatory diseases such as diabetes and nephritis. The participants in the study were classified according to their body mass index into normal weight, overweight, and obese. To minimize potential confounding factors, individuals with valvular heart disease, cardiomyopathy, non-coronary atherosclerotic lesions, chronic kidney disease, diabetes, and inflammatory disease were excluded from the study. Care was taken to match the control group participants to the CAD group in terms of age and gender to ensure a balanced comparison.
Specimen Collection and Laboratory Analysis
This study employed a comprehensive approach to collect and analyze samples from participants, thereby ensuring a robust correlation analysis between the Apelin gene −1860T>C polymorphism and CAD risk in a Syrian cohort.
Demographic and Clinical Data Collection
A standardized questionnaire was administered to gather detailed demographic and clinical information from participants, including medical history, family history of CAD, and lifestyle habits. BMI was calculated using the widely accepted formula: BMI = weight (kg)/height (m2).25 Hypertension was diagnosed based on the average of three consecutive measurements of systolic and diastolic blood pressure (≥140 mmHg and/or ≥90 mmHg, respectively) or the use of antihypertensive medications.26 Hyperlipidemia was defined as total cholesterol (TC) ≥200 mg/dL and triglycerides (TG) >150 mg/dL.27
Blood Sample Collection and Processing
Venous blood samples (5 mL) were drawn from each participant after an overnight fast and prior to angiography. The blood was divided into two aliquots: one in a heparinized tube for apelin quantification and the other in a heparinized tube for biochemical parameter measurement (TG, TC, high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C). The plasma was separated from the blood by centrifugation at 1000g for 10 minutes (apelin analysis) and 3000g for 10 minutes (biochemical parameter analysis). The resulting supernatant plasma was stored at −75°C to −80°C until assay performance. Additionally, 3 mL of whole blood was collected in an EDTA-containing tube and stored at −20°C for genomic DNA extraction.
Biochemical Profiling
Total apelin protein levels in plasma were quantified using a commercially available human ELISA kit (Cusabio, China),23 as per the manufacturer’s instructions, on an ELISA microplate reader (BioTek, USA).
The intra-assay and inter-assay Precision were less than 8% and 10%, respectively, and the lowest detection limit was 31.25 pg/mL. Meanwhile, TC, LDL-C, and HDL-C levels were determined using colorimetric enzymatic assays on a Hitachi 902 Automatic Analyzer (Roche, Swiss).28
Genotyping of Apelin −1860T>C Polymorphism
In this study, we performed a rigorous genotyping approach to investigate the correlation between the apelin −1860T>C polymorphism and CAD risk in a Syrian cohort. We employed a polymerase chain reaction-restriction fragment length polymorphism (RFLP-PCR) approach.
DNA Extraction and Quality Control
Genomic DNA extraction from peripheral blood leukocytes was performed using the QIAGEN MINI AMP Kit, which utilizes spin columns to ensure high-quality DNA isolation. The extracted DNA was quantified and assessed for purity using a Nano Drop 2000 spectrophotometer (Thermo Scientific, USA), with absorbance readings taken at 260 nm and 280 nm wavelengths.
To confirm the quality and integrity of the extracted DNA, we employed electrophoresis on a 1% agarose gel, which allowed for visualization of the DNA samples. Following quality control, the DNA samples were stored in TE buffer, as provided with the kit, at −20°C to ensure optimal preservation until polymerase chain reaction (PCR) was performed.
PCR Amplification
PCR amplification of the apelin gene −1860T>C polymorphism was performed using a T100TM Thermal Cycler (BIO-RAD, Germany). The amplification reaction mixture consisted of specific primers (forward primer 5’-GGG GAA CAG TGA AGG GAG AAT GGT-3’ and reverse primer 5’-AGA AGC GGG TCC TGA AGT TGT TTG −3’).20,23,24 The thermal cycling conditions were as follows: initial denaturation at 95°C for 5 minutes, followed by 30 cycles of denaturation at 95°C for 40 seconds, annealing at 58°C for 40 seconds, and extension at 72°C for 40 seconds. A final extension step was performed at 72°C for 5 minutes. The PCR amplification product (732 bp) was verified by electrophoresis on a 1% agarose gel using 1x TBE running buffer, and the results were visualized under a UV transilluminator.
Restriction Enzyme Digestion
The PCR product (10 μL) was subjected to overnight digestion at 37°C with XhoI restriction enzyme (Thermo Scientific, USA), which recognizes the sequence CTCGAG in the amplification product. The digestion products were analyzed by gel electrophoresis on a 2.5% agarose gel. The RFLP pattern was interpreted as follows: a single DNA band of 732 bp indicated the TT genotype, two fragments (624 bp and 108 bp) suggested the CC genotype, and the presence of three zones indicated heterozygosity (TC).
Statistical Analysis and Data Interpretation
All statistical analyses were performed using SPSS version 25.0 (Statistical Package for Social Sciences, Chicago, IL, USA). To ensure the validity of our results, we employed a range of methods to analyze and interpret our data. Normality tests, specifically the Kolmogorov–Smirnov and Shapiro–Wilk tests, were used to verify the normal distribution of study variables. Descriptive statistics, including frequency tables, percentages, means, standard deviations, medians, and Interquartile Range (IQR), were generated to summarize the characteristics of our sample.
To compare quantitative variables among study groups, we utilized parametric tests (independent sample t-test) when variables were normally distributed and non-parametric tests (Mann–Whitney U-test) when variables were not normally distributed. In addition, the chi-square test was used to compare study groups in categorical variables. Variables were expressed as mean ± standard deviation (SD) for normally distributed data, and as median (interquartile range) for non-normally distributed data. The relationships between plasma apelin levels and other studied variables were evaluated with Spearman’s rank correlation coefficient (ρ).
To evaluate the independent determinants of plasma apelin levels, a multivariable linear regression model was constructed with continuous plasma apelin as the dependent variable. This model was adjusted for established cardiovascular risk factors, including Age (years), BMI (kg/m2), Sex, Smoking status, Hypertension, and Family History of CAD. Furthermore, TG, HDL, and LDL levels (mg/dL) were incorporated as continuous variables to account for the potential influence of metabolic parameters.
Conversely, to determine the independent effect of the APLN −1860T>C polymorphism on CAD risk, two separate binomial logistic regression models were performed (one for the allele-based model and one for the genotype-based model) with CAD status as the binary dependent variable. For the subgroup analyses, the logistic regression models were restricted by sex and adjusted for Age, BMI, Smoking status, Hypertension, Family History of CAD, TG, HDL, and LDL levels, while Sex was omitted as a covariate in the female-only and male-only strata. For the total study population, Sex was reintroduced into the covariates block. All regression models were constructed using the Akaike information criterion method to include clinically relevant covariates. Multicollinearity across all models was evaluated through the Variance Inflation Factor (VIF) values, which were <2.0, suggesting no significant redundancy. The stability and fit of the logistic models were evaluated using deviance residuals, while potential influential data points across all regression analyses were monitored via Cook’s distance (all values <1.0), ensuring that the calculated linear coefficients and logistic odds ratios were not influenced by significant outliers.
Throughout our analysis, two-sided p-values < 0.05 were considered statistically significant. Finally, graphical representations were created using GraphPad Prism 9 software to facilitate the visualization and interpretation of our findings.
The total sample size (n=222) was determined using G*Power 3.1. To achieve a power (1-β) of 0.95 with an α error of 0.05 for a Chi-square test (df=2), the required sample size was calculated as 172. Our total cohort of 222 exceeds this requirement. However, due to the clinical distribution of CAD, an imbalance was observed in the female subgroup (n=90 total; 23 patients and 67 controls). A post-hoc power analysis for the female subgroup revealed a power of 77% for genotype comparisons (df=2). Notably, for allele frequency analysis (df=1), the post-hoc power exceeded 80% (85%), suggesting that the study remained sufficiently powered to detect significant allelic associations even within the female stratum.
Results
Demographic and Clinical Characteristics of the Study Population
The descriptive statistics for the demographic, clinical, and biochemical parameters of the CAD patients and control subjects are presented in Table 1. Notably, the mean age of the CAD patients (58.63 ± 9.05 years) was significantly higher than that of the control group (52.15 ± 8.68 years), with a p-value of < 0.001. Moreover, the median BMI of the patient subjects [28.05 (24.86–31.11) kg/m2] was significantly elevated compared to the control group [25.35 (24.38–27.09) kg/m2] (p < 0.001).
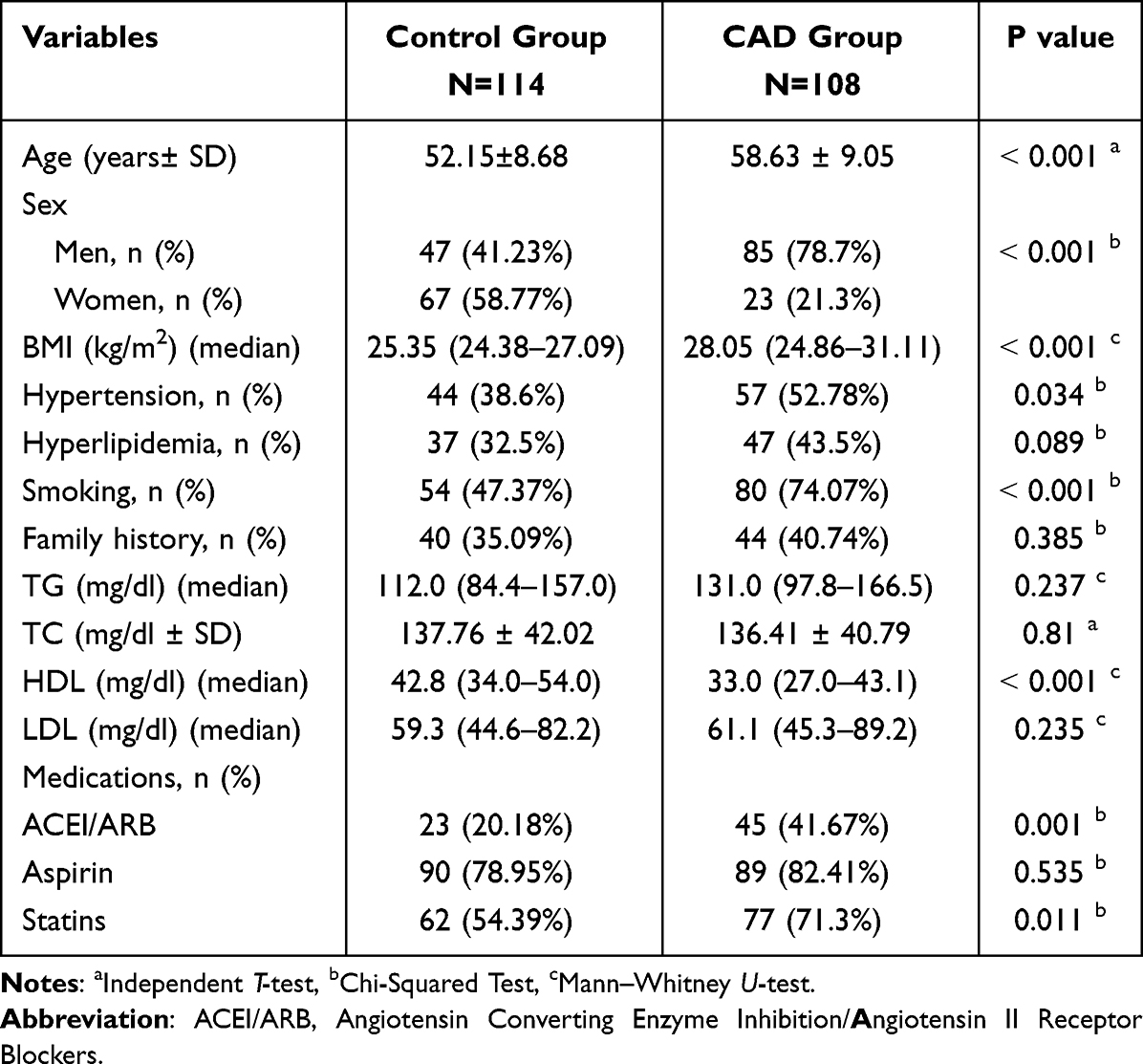 |
Table 1 Demographic, Clinical, and Biochemical Characteristics of the Study Groups |
In terms of lipid profiles, no statistically significant differences were observed between the two groups in terms of median TG, mean TC, and median LDL-C levels (p > 0.05). However, median HDL-C levels were significantly lower in the CAD group [33.0 (27.0–43.1) mg/dl] compared to the control group [42.8 (34.0–54.0) mg/dl] (p < 0.001). Furthermore, significant differences were observed between the groups in terms of gender, smoking status, and hypertension, which are established risk factors for CAD.29,30 Conversely, no significant differences were found between the groups in terms of hyperlipidemia and family history (p > 0.05). These findings are consistent with the notion that CAD patients exhibit a higher prevalence of common cardiac risk factors compared to healthy controls.31
Plasma Apelin Concentrations and Coronary Artery Disease
Our investigation revealed a significant disparity in plasma apelin levels between patients diagnosed with CAD and healthy controls. Specifically, the median plasma apelin concentration was substantially lower in the CAD group (Median= 2707.49, IQR=1584.25 pg/mL) compared to the control group (Median= 3727.39, IQR=2598.52 pg/mL), with a highly statistically significant difference (p < 0.001) (Figure 1). This finding suggests a potential correlation between apelin levels and CAD risk.
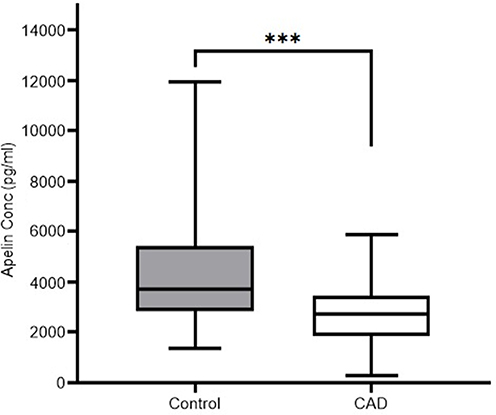 |
Figure 1 Plasma apelin concentrations in controls and CAD patients. ***p ≤ 0.001 vs control group. |
To evaluate whether plasma apelin levels differ according to the clinical severity of CAD, the patient cohorts were classified into two groups based on clinical presentation: Unstable angina (USA, n = 66) and Stable Angina (SA, n = 42). The statistical analysis revealed a highly significant difference in plasma apelin concentrations based on CAD severity (Mann–Whitney U = 843, p <0.001). Notably, we have reported significantly lower plasma apelin levels in patients with unstable CAD (Median = 2394 pg/mL, IQR = 1511 pg/mL) compared to those diagnosed with SA (Stable Angina; Median = 3151 pg/mL, IQR = 2561 pg/mL). These findings indicate that decreased circulating apelin concentrations are strongly associated with more severe, unstable clinical presentations of CAD.
Further analysis of the data revealed no significant differences in plasma apelin concentrations between male and female subjects within both the CAD (Median=2578.53, IQR=1639.47 vs Median=2851.37, IQR=1127.31) and control (Median=3815.31, IQR=2391.25 vs Median=3710.38, IQR=2381.48) groups. These results imply that apelin levels are not influenced by gender in the context of CAD.
Association Between Plasma Apelin Levels and Cardiovascular Risk Factors
Our analysis, utilizing Spearman’s rank correlation, revealed that plasma apelin levels generally did not exhibit significant correlations (p>0.05) with most studied parameters in both the control and patient groups (Table 2). Specifically, Spearman’s rho (ρ) values for age, TG, TC, HDL, and LDL ranged from −0.136 to −0.003 in the control group and −0.071 to 0.150 in the CAD group.
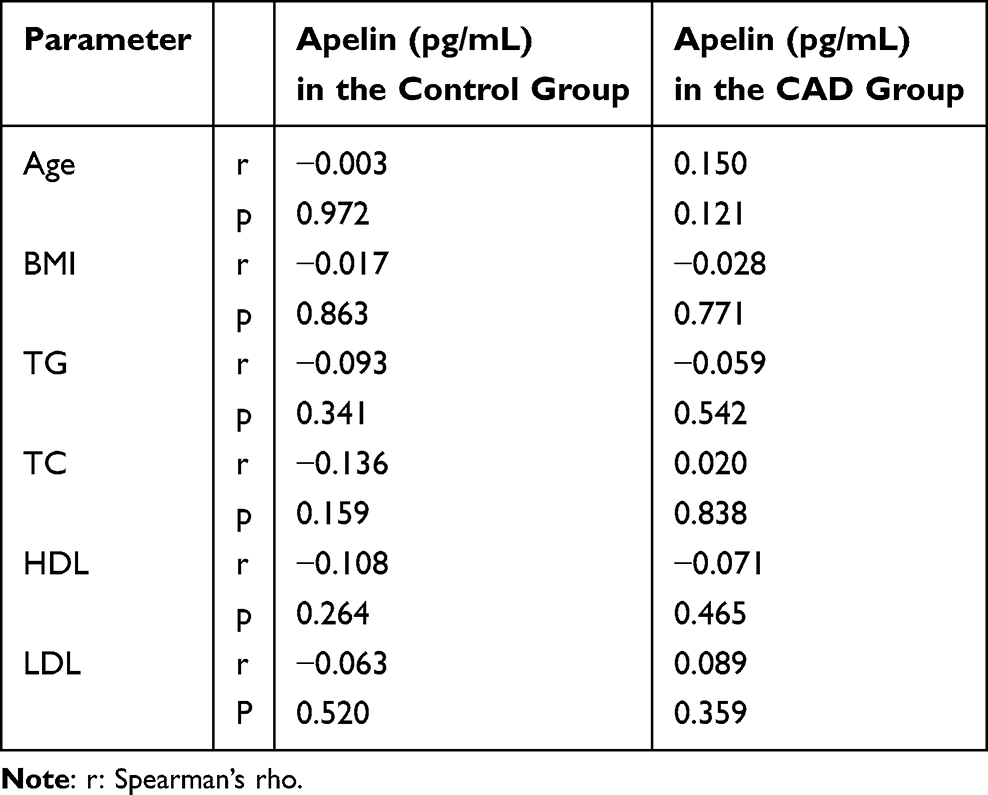 |
Table 2 The Correlation of Apelin with Other Studied Parameters in the Study Subject |
To further explore the interplay between plasma apelin levels and cardiovascular risk factors, we conducted a multivariable linear regression analysis incorporating all studied risk factors, as well as the presence of CAD (Table 3). The variables were entered into the model based on their clinical relevance. Notably, the results showed that CAD was independently associated with plasma apelin levels (β = −0.421; 95% CI −2186.61 to −975.47; p < 0.001). While traditional risk factors, including age, sex, lipids, and smoking, did not exhibit significant associations. These findings suggest that plasma apelin levels are primarily influenced by the presence of CAD, rather than other cardiovascular risk factors, in the Syrian cohort studied. This conclusion is consistent with prior research highlighting the potential role of apelin in cardiovascular disease pathophysiology.32–34
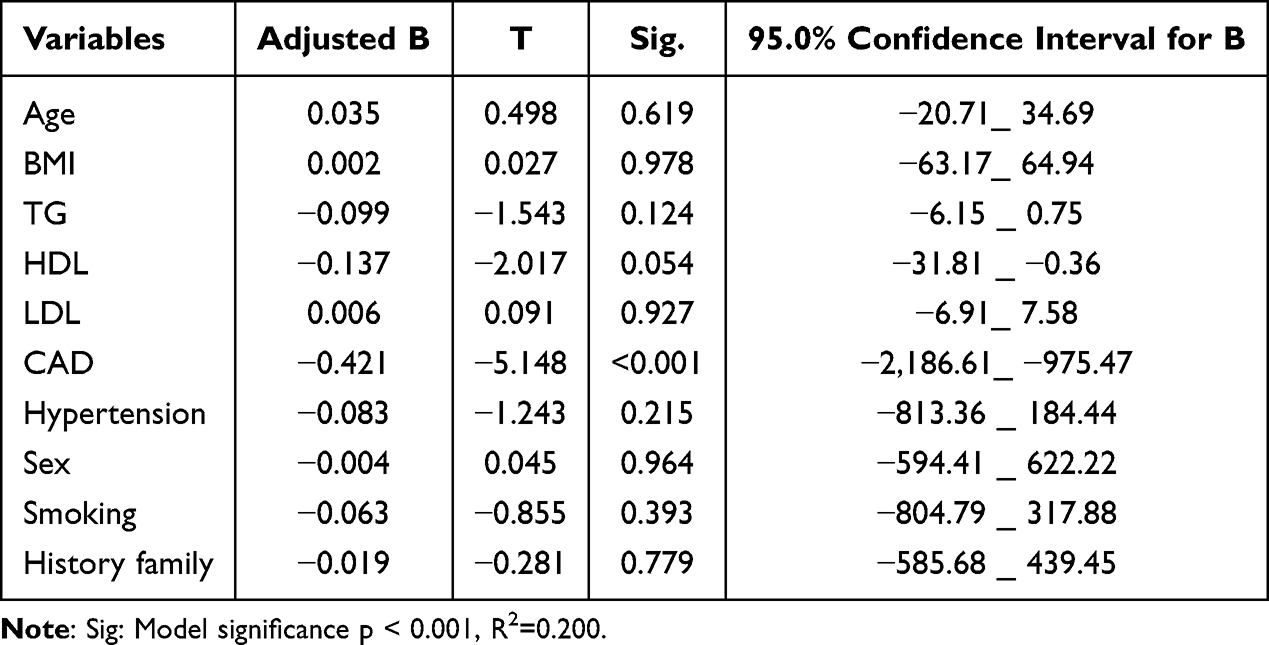 |
Table 3 Multivariable Regression Analysis for Plasma Apelin Levels and the Other Factors in the Studied Subjects |
Genotypic Distribution and Hardy-Weinberg Equilibrium of the Apelin Gene −1860T>C Polymorphism
The XhoI restriction enzyme digestion reaction was employed to genotype the Apelin gene −1860T>C polymorphism, and the resulting fragment patterns are illustrated in Figure 2. The amplified product (732 bp) was subjected to electrophoresis on a 2.5% agarose gel, yielding distinct bands corresponding to the three possible genotypes: TT, TC, and CC (Figures S1 and S2).
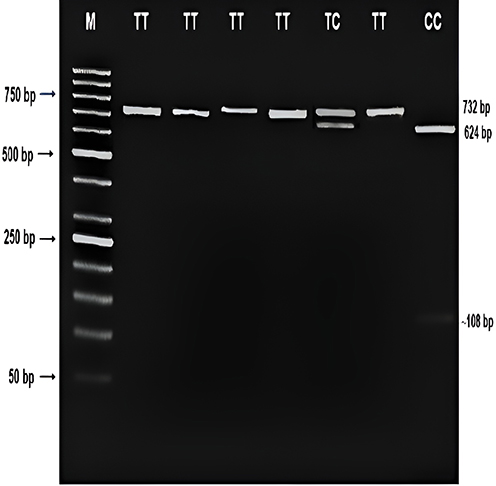 |
Figure 2 Products of the digestion reaction using the XhoI restriction enzyme at the 1860T/C site and the corresponding genotypes of the −1860T>C polymorphism in the Apelin gene. |
Given the X-chromosomal location of the Apelin gene, Hardy-Weinberg equilibrium (HWE) testing was performed exclusively in females. The observed genotype frequencies of the −1860T>C polymorphism in both the patient and control groups were consistent with HWE (p > 0.05). This finding suggests that the sampled population is representative, randomly distributed, and genetically analyzable.
These results imply that the study sample is genetically representative of the Syrian population, thereby providing a suitable basis for assessing the correlation between the Apelin gene −1860T>C polymorphism and coronary artery disease risk.
The distribution of the −1860T>C polymorphism genotypes frequencies in female patients with CAD and healthy controls is presented in Table 4. Because males have only one X chromosome, genotype information is not available.
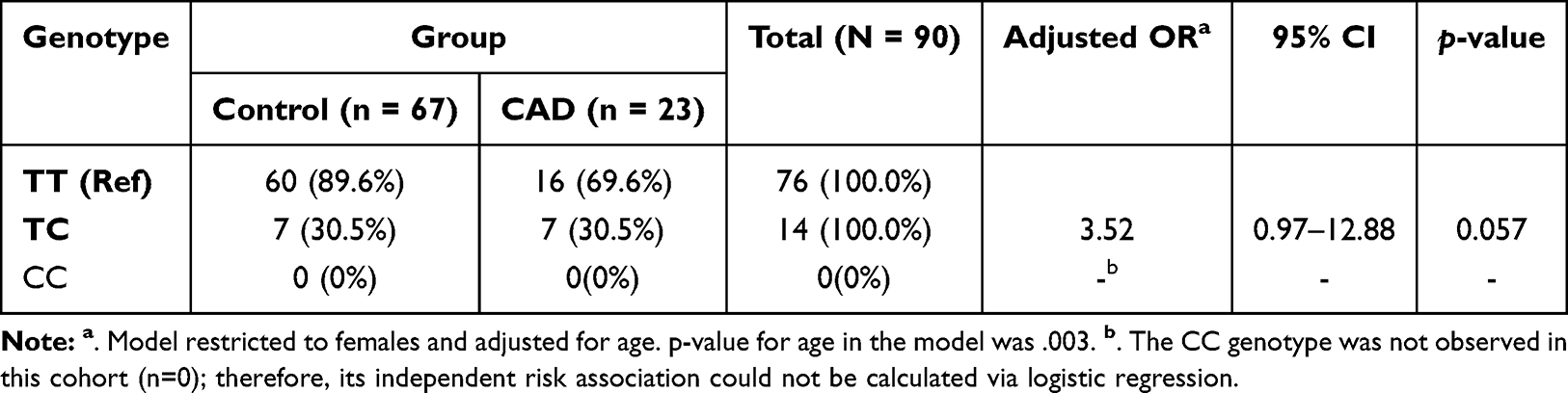 |
Table 4 Age-Adjusted Association of Genotype −1860T>C Polymorphism with Coronary Artery Disease Risk in Females |
Our results indicate that the genotype distribution differed significantly between the CAD and control groups in females (Table 4). Specifically, the heterozygous TC genotype frequency was substantially higher in female CAD patients compared to controls within their groups (30.4% vs 10.4%, respectively). After adjusting for age, multivariable logistic regression analysis demonstrated that carriers of the TC genotype exhibit a more than 3.5-fold increased risk of developing CAD compared to those carrying the homozygous wild-type TT genotype, reaching borderline statistical significance (OR = 3.52, 95% CI: 0.97–12.88, p = 0.057) (Figure 3). These findings suggest that the APLN −1860T>C heterozygous genotype displays a strong trend toward an independent association with CAD susceptibility in the studied female Syrian population.
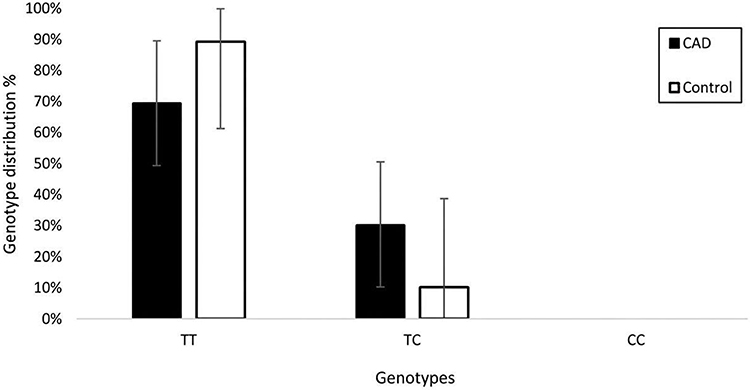 |
Figure 3 Genotype distribution for females in the studied groups. |
Allele Frequencies and Risk Association Analysis
Notable disparities in allelic frequencies were observed between CAD patients and control individuals, with significant differences detected in female subjects (Table 5). Specifically, in the female cohort, the C allele frequency was higher in CAD patients compared to controls (15.2% vs 5.2%, respectively). After adjusting for age, this disparity corresponds to a nearly threefold increased risk of developing CAD in female carriers of the C allele compared to those carrying the T allele (OR = 2.99, 95% CI: 0.89–10.06, p = 0.077). In the male cohort, while the frequency of the mutant C allele was higher in CAD patients than in controls (12.9% vs 4.2%), the difference reached borderline significance after adjusting for age (OR = 4.90, 95% CI: 0.97–24.78, p = 0.054).
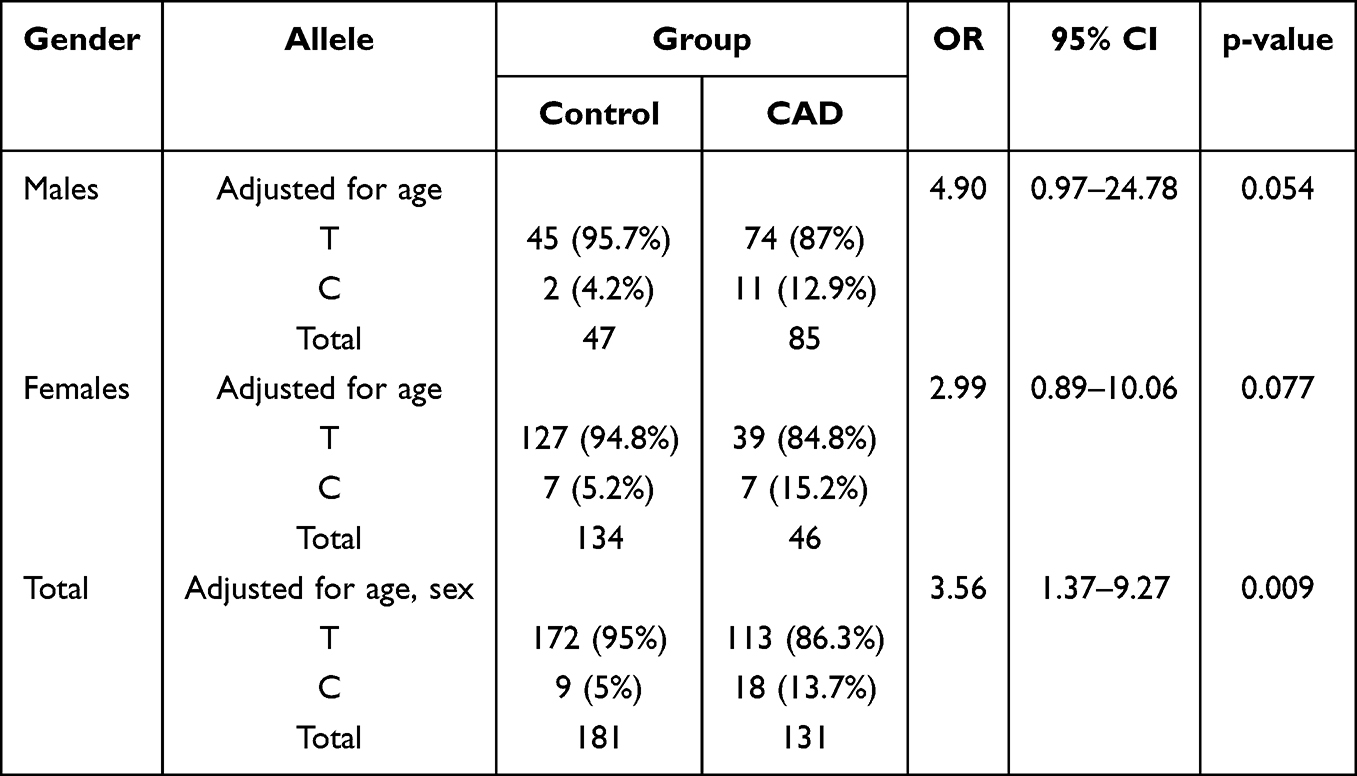 |
Table 5 Allele Frequencies of the −1860T>C Polymorphism in the Study Groups Stratified by Sex |
However, when considering the total study population, the allelic distribution differed significantly between CAD patients and controls, with a higher frequency of the C allele in the patient group (13.7% vs 5% in controls). Multivariable logistic regression analysis, adjusted for age and sex, confirmed that the C allele is independently associated with a 3.5-fold increased risk of CAD (OR = 3.56, 95% CI: 1.37–9.27, p = 0.009).
These findings suggest that the APLN −1860T>C polymorphism is a significant genetic risk factor for CAD in the studied Syrian population, independent of traditional demographic variables.
Biochemical Markers and APLN −1860 T>C Association Analysis
To investigate the potential impact of the −1860 T>C polymorphism on lipid metabolism, we analyzed the association between genotype/allele frequencies and various biochemical markers (TG, TC, HDL-C, and LDL-C), stratified by sex (Table 6). No statistically significant associations were observed between the −1860 T>C variant and lipid profiles in either the female (n=90) or male (n=132) cohorts. Of note, the CC genotype was not detected among the female participants, precluding further comparative analysis for that group. Similarly, comparing the T and C alleles in males revealed no significant differences in any measured biochemical parameters. These results suggest that while the −1860 T>C polymorphism is significantly associated with CAD risk and plasma apelin levels as previously described, it does not directly influence circulating lipid profiles in this Syrian cohort. This implies that the contribution of the APLN gene to CAD pathogenesis may occur through pathways independent of traditional lipid metabolism.
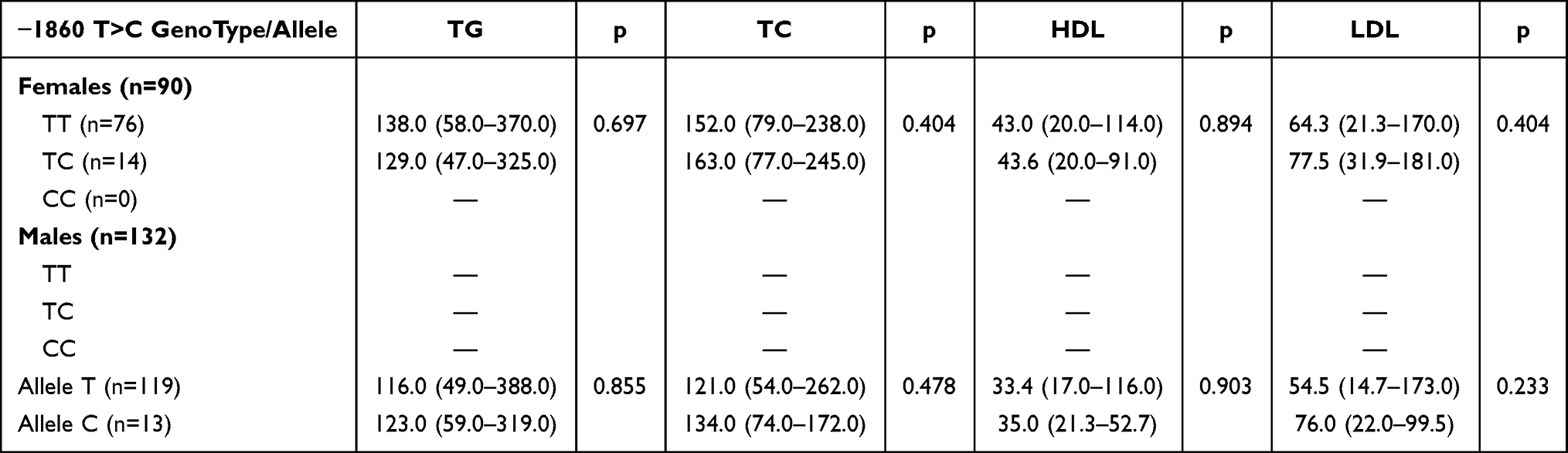 |
Table 6 Association Between APLN −1860T>C Genotype and Biochemical Markers Stratified by Sex |
Influence of −1860T>C Polymorphism on Plasma Apelin Levels
To elucidate the relationship between the −1860T>C polymorphism and plasma apelin levels, we compared the concentrations of apelin in individuals with different genotypes. In the female cohort, plasma apelin levels were slightly lower in individuals with the TC genotype [median: 2896 pg/mL (range: 422–5580 pg/mL)] compared to those with the TT genotype [median: 3286 pg/mL (range: 467–11950 pg/mL)], although this difference did not reach statistical significance (p = 0.305). It is important to note that the CC genotype was not detected in the female study population, precluding further comparative analysis for this group. Notably, a statistically significant difference in plasma apelin levels was observed between carriers of the C allele and those carrying the T allele in the male cohort [median: 1502 pg/mL (range: 714–7853 pg/mL) vs. 3216 pg/mL (range: 293–10703 pg/mL), respectively, p = 0.002].
Discussion
Correlation Between Apelin Gene −1860T>C Polymorphism and Coronary Artery Disease Risk: Implications and Clinical Significance
The principal findings of this study demonstrate a significant association between the apelin gene −1860T>C polymorphism and susceptibility to CAD in a Syrian cohort. Notably, patients with CAD exhibited lower plasma apelin levels compared to individuals with angiographically normal coronary arteries. This association suggests that the apelin levels may be associated with CAD risk independent of other clinical variables. As the first study of its kind in Syria, our investigation provides valuable insights into the correlation between apelin and CAD.
CAD is a major contributor to the global burden of morbidity and mortality, often in conjunction with comorbidities such as hypertension and myocardial infarction.5 The interplay of environmental and genetic factors is well established in the development of CAD and the occurrence of acute coronary syndrome.35 The rising incidence of myocardial infarction in young individuals, frequently in the absence of traditional risk factors, underscores the need to identify novel risk factors, including genetic polymorphisms, that may confer susceptibility to CAD in these patients.
Disparities in apelin levels emerge when comparing our findings to those reported in existing literature. Our study revealed a significant decline in plasma apelin levels in patients with CAD compared to controls (p < 0.001). A substantial number of investigations establish a significant reduction in plasma apelin concentrations among CAD patients. For instance, a meta-analysis of observational studies in China showed that, overall, circulating apelin concentration was significantly lower in CAD patients than in controls.36 As Hazbar and Sahab34 demonstrated, apelin levels were lower in patients with CAD compared to healthy individuals. The Kozani study,37 which shared similarities with our study in terms of sample size, also reported a significant decrease in apelin levels among CAD patients. Similarly, Akcılar et al23 found a significant decrease in plasma apelin levels in Turkish patients with CAD compared to controls. The results of Rachwalik et al14 indicate that the levels of apelinergic peptides (AP-13, AP-17, ELA, and APJ) are altered in patients with CAD, which may be a potential diagnostic indicator.
The current investigation has consistently demonstrated a significant correlation between diminished plasma apelin levels and the presence, severity, and clinical manifestations of cardiovascular disease, including stable and unstable angina. An inverse association has been observed between plasma apelin levels and the severity of CAD, underscoring the intricate relationship between these factors. Notably, existing clinical evidence indicates that circulating apelinergic peptides vary across specific acute cardiovascular conditions, with a more pronounced depletion of the protective apelin/APJ axis pathway occurring during acute vascular stress compared to chronic stable ischemic phenotypes. The cumulative evidence from these experiments suggests a robust correlation between decreased plasma apelin levels and an increased risk of CAD. These findings have important implications for the diagnosis and management of CAD, highlighting the potential utility of plasma apelin levels as a biomarker for disease severity and risk stratification. The observed correlation may be attributed to the potential role of apelin in modulating vascular tone, blood pressure, and inflammation, all of which are critical factors in the development and progression of CAD, supporting its role as a protective factor against CAD progression.38
In contrast, Motawi et al39 demonstrated a statistically significant increase in serum apelin levels in CAD patients (2.15±0.15 ng/mL) relative to controls (0.96±0.48 ng/mL). Variations in reported apelin levels across studies underscore the complex and multifactorial nature of its role in CAD.
The inconsistencies in apelin level profiles across studies highlight the complexity of establishing a standardized normal level for apelin. This variability is likely a consequence of differences in study populations, sample size, experimental designs, and laboratory assays employed. As such, it is essential to acknowledge these limitations when interpreting the results of apelin-based CAD risk assessment studies. Further research is warranted to elucidate the sources of these discrepancies and to develop more accurate and reliable methods for apelin level determination.
Our study’s correlation analysis revealed that plasma apelin levels generally did not exhibit significant correlations (p > 0.05) with most studied biochemical parameters in both groups. Various results have been demonstrated in different studies. Hussein et al,40 showed a slight variation in apelin concentrations among groups categorized by BMI according to the World Health Organization (WHO) criteria. Additionally, Namazi et al1 found a significant positive correlation between serum apelin and HDL levels by multivariable regression analysis. However, our results are consistent with the study by Hazbar et al34 which also failed to detect significant correlations between apelin and other biochemical parameters.
Our investigation reveals a notable lack of correlation between apelin levels and other examined parameters, suggesting that the association between decreased plasma apelin concentrations and the incidence of CAD may be independent of conventional cardiovascular risk factors. Crucially, this lack of correlation with traditional circulating lipids (as well as the baseline similarity in total cholesterol and LDL-C levels between our study groups) is heavily influenced by clinical intervention. In our cohort, 71.3% of the CAD patients were actively receiving intensive statin therapy (Table 1), which naturally modifies and normalizes circulating lipid parameters. This clinical reality strongly implies that this observation highlights the intricate relationship between apelin and CAD, warranting further research to elucidate the underlying mechanisms. The discrepancies in study findings may be attributed to variations in study populations, methodologies, or analytical techniques employed. Nonetheless, our results emphasize the importance of considering the complexity of apelin’s involvement in CAD, which may not be fully captured by traditional risk factor assessments.
The apelin −1860T>C SNP located in the promoter region of the apelin gene has recently been identified.41 Notably, the current results reveal a significant association between the apelin −1860T>C polymorphism and CAD susceptibility (p = 0.041). Multivariable logistic regression demonstrated that female carriers of the heterozygous TC genotype displayed a more than 3.5-fold increased risk of developing CAD compared to the homozygous wild-type TT genotype, reaching borderline statistical significance (OR = 3.52, 95% CI: 0.97–12.88, p = 0.057; Table 5). This finding is consistent with the observation that the CC genotype was absent in our study population, a phenomenon also reported by Martinez et al42 At the allelic level within the female stratum, the mutant C allele frequency was higher in CAD patients than in controls (15.2% vs 5.2%), similarly demonstrating a strong borderline trend toward risk after age adjustment (OR = 2.99, 95% CI: 0.89–10.06, p = 0.077; Table 4). Interestingly, we did not observe a statistically significant difference in allele frequency among males in both patient and control groups, although a highly pronounced borderline risk trend was noted after age adjustment (OR = 4.90, 95% CI: 0.97–24.78, p = 0.054). A key strength of our study is accounting for the X-chromosomal location of the APLN gene. Because males are hemizygous, a single copy of the mutant C allele fully dictates their genetic exposure. Interestingly, while the C allele showed a borderline significant trend toward CAD risk in both males (OR=4.90, p=0.054) and females (OR=2.99, p=0.077), analyzing the combined total allele pool revealed a highly significant, independent 3.5-fold increase in CAD susceptibility (OR=3.56, p=0.009). This underscores the necessity of evaluating total allelic burden in X-linked cardiovascular genetic studies, as stratified sample sizes may lack the statistical power to capture the full impact of hemizygous and heterozygous variant distributions independently. Our post-hoc power analysis confirmed that the female stratum was slightly underpowered at 77% for genotype comparisons. However, when pooling the entire study population together (N=222), the increased sample size yielded robust statistical power. In this combined model, adjusted for both age and sex, the mutant C allele was confirmed to be a highly significant, independent genetic risk factor, conferring a 3.5-fold increased susceptibility to CAD (OR = 3.56, 95% CI: 1.37–9.27, p = 0.009).
The Minor Allele Frequency (MAF) and the association between the −1860 T>C polymorphism and CAD varied among different studies. Kidoya et al43 reported a higher MAF of 0.347 for the −1860T>C SNP in a Chinese population compared with the Syrian population. Ackilar et al23 demonstrated that the distribution of apelin −1860T>C genotypes and alleles differed significantly between CAD patients and controls, with the CC and CT genotypes, as well as the C allele, being associated with an increased risk of CAD.
Conspicuously, our findings are not consistent with the study of Jin et al44 which failed to detect a statistically significant difference in the genotype and allele distributions of the rs56204867 polymorphism between CAD patients and controls. Furthermore, our results align with those of Ackilar et al,23 who reported lower plasma apelin levels in CAD patients with CC and TC genotypes compared to TT genotypes, although the difference was not statistically significant. This reduction may reflect depletion of the protective apelin/APJ axis pathway due to chronic vascular stress, or a consequence of established atherosclerosis, rather than a purely primary genetic effect. Clinical evidence indicates that circulating apelinergic peptides vary across cardiovascular conditions, with increased levels observed in acute coronary syndromes and decreased levels in chronic ischemic or hypertension.7 Several studies have reported that circulating apelin levels increase following surgical revascularization, providing indirect support for a causal or at least bidirectional relationship between apelin levels and CAD severity.9 Nevertheless, due to the cross-sectional, case–control design of the present study, a definitive causal inference cannot be established.
It is worth noting that the current study’s results are not directly comparable to existing literature, as there is a dearth of information on the relationship between variants in the apelin −1860T>C gene and plasma apelin levels. Intriguingly, plasma apelin was significantly downregulated in mutant C allele carriers compared to wild-type T allele carriers exclusively within the CAD cohort (p = 0.002), a difference absent in healthy controls. Because the −1860T>C variant resides in the APLN promoter, this disease-specific effect is physiologically plausible. Promoter activity is heavily driven by cellular stress, hypoxia, and vascular inflammation characteristic of active CAD. In healthy controls, the promoter remains quiescent, keeping the variant functionally silent. Under the ischemic stress of CAD, the mutant C allele likely disrupts a critical transcription factor binding site, crippling the compensatory upregulation of protective apelin. This indicates that the polymorphism’s functional impact is unmasked by active disease pathology.
The cumulative evidence from these studies suggests a robust correlation between the −1860T>C polymorphism and susceptibility to CAD. However, inconsistencies in the results of some studies highlight the need for more comprehensive research to clarify the role of the apelin −1860T>C polymorphism in the risk of developing CAD. To elucidate the relationship between the apelin −1860T>C polymorphism and CAD risk, forthcoming studies should prioritize recruitment of larger, more diverse cohorts, complemented by robust statistical analyses. This approach will facilitate a more comprehensive understanding of the polymorphism’s impact on CAD susceptibility.
Study Limitations
One important limitation of this study is the relatively small number of female CAD patients included in the subgroup analysis. This imbalance reflects the underlying epidemiology of CAD, where women—particularly in the studied population—tend to present at older ages and are less frequently referred for invasive diagnostic procedures such as coronary angiography. Additionally, sociocultural and healthcare access factors may contribute to the underrepresentation of female patients in clinical settings. Although post-hoc power analysis indicated moderate to adequate power for certain comparisons, the reduced sample size in the female subgroup may limit the ability to detect modest genetic effects and increase the risk of type II error. Therefore, the sex-specific findings should be interpreted with caution and considered exploratory. Nevertheless, given that the APLN gene is located on the X chromosome, sex-stratified analysis remains biologically relevant and may provide important insights into potential sex-specific genetic mechanisms in CAD.
Conclusion
This pioneering case-control study establishes a significant correlation between the apelin gene −1860T>C polymorphism and coronary artery disease (CAD) risk in a Syrian cohort. Our findings demonstrate a notable increase in the frequency of the TC genotype among CAD patients compared to controls, implying that this genotype may be associated with an increased risk of CAD. Furthermore, our data suggest that C allele carriers are more likely to develop CAD than T allele individuals, supporting the hypothesis that apelin gene variants contribute to CAD susceptibility.
Particularly, our results show that plasma apelin levels are lower in CAD patients compared to controls. These levels were lower in individuals with the TC genotype compared to those with the TT genotype, which may have detrimental effects on CAD pathophysiology. Low apelin levels were independently associated with coronary artery disease, irrespective of the traditional cardiovascular risk factors included in the study. These findings collectively implicate the −1860T>C variation in apelin as a genetic risk factor for CAD, highlighting its potential as a genetic biomarker for early identification of CAD predisposition. While our study provides valuable insights into the apelin-CAD association, further research is necessary to fully elucidate the precise role of apelin in CAD and to explore the therapeutic implications of this finding.
Abbreviations
ACEI/ARB, Angiotensin Converting Enzyme Inhibition/Angiotensin II Receptor Blockers; BMI, Body Mass Index; CAD, Coronary Artery Disease; 95% CI, 95% Confidence interval; ELISA, Enzyme-linked immunosorbent assay; HDL-C, High-density lipoprotein cholesterol; HWE, Hardy-Weinberg equilibrium; LDL-C, Low-density lipoprotein cholesterol; OR, Odds Ratio; PCR-RFLP, Polymerase chain reaction-restriction fragment length polymorphism; SNPs, Single-nucleotide polymorphisms; TC, Total cholesterol; TG, Triglycerides; UV, Ultra Violet; XhoI, Xanthomonas holcicola.
Data Sharing Statement
The datasets generated and/or analyzed during the current study are not publicly available because they contain sensitive clinical and genetic information derived from a relatively small and geographically localized cohort, which may increase the potential risk of participant re-identification despite data de-identification procedures. In addition, the informed consent obtained from participants and the ethical approval granted by the Ethics Committee of Damascus University of Medical Sciences did not include provisions for unrestricted public data deposition or open-access genomic data sharing. Besides, deidentified participant-level data underlying the findings reported in this article, including selected demographic, clinical, biochemical, plasma apelin concentration, and genotyping data relevant to the published analyses, may be considered for limited academic review by qualified researchers upon reasonable scientific request directed to the corresponding author. Requests will be evaluated on a case-by-case basis in accordance with institutional ethical regulations, participant confidentiality obligations, and applicable data protection requirements. The study protocol and statistical analysis methodology may also be made available upon reasonable request for the purpose of academic verification and scientific transparency. Data, if approved, will be accessible through secure institutional communication with the corresponding author beginning from the date of publication and for a period of five years thereafter. No publicly accessible repository will be used for data distribution to preserve participant privacy and comply with ethical restrictions governing human genetic research.
Ethics Approval and Consent to Participate
Informed consent was obtained from all participants involved in this study. Participants provided both oral and written informed consent forms before their inclusion in the research. Approval was obtained from the ethics committee of Damascus University of Medical Sciences (Approval No. 9/A.Kh, dated 23/7/2019). The study was conducted in compliance with the Helsinki Declaration. Before data collection, the researchers provided a comprehensive explanation of the study’s objectives, potential benefits, and the selection criteria to the participants. They were explicitly informed of their right to withdraw from the research at any point without facing any consequences. Throughout the data collection process, strict adherence to confidentiality measures was ensured. All data collection procedures were conducted anonymously and in line with ethical standards.
Acknowledgments
We would like to express our gratitude to Prof. Loai Aljerf (ORCID: 0000-0002-1132-9659) for his meticulous review and editing of this article, ensuring the highest standards of academic excellence.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors reported no funding received for this study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Namazi G, Salami R, Pourfarzam M, et al. Association of the serum apelin, but not ghrelin, with the presence and severity of coronary artery disease. Indian Heart J. 2021;73(2):214–16. doi:10.1016/j.ihj.2021.01.013
2. Jebari-Benslaiman S, Galicia-García U, Larrea-Sebal A, et al. Pathophysiology of atherosclerosis. Int J Mol Sci. 2022;23(6):3346. doi:10.3390/ijms23063346
3. Diakowska D, Wyderka R, Krzystek-Korpacka M, et al. Plasma levels of apelinergic system components in patients with chronic and acute coronary syndromes—A pilot study. J Clin Med. 2021;10(19):4420. doi:10.3390/jcm10194420
4. Aherrahrou R, Guo L, Nagraj VP, et al. Genetic regulation of atherosclerosis-relevant phenotypes in human vascular smooth muscle cells. Circ Res. 2020;127(12):1552–1565. doi:10.1161/CIRCRESAHA.120.317415
5. Nowzari Z, Masoumi M, Nazari-Robati M, et al. Association of polymorphisms of leptin, leptin receptor and apelin receptor genes with susceptibility to coronary artery disease and hypertension. Life Sci. 2018;207:166–171. doi:10.1016/j.lfs.2018.06.007
6. Krasniqi X, Vincelj J, Kocinaj D, et al. Evaluative value of Apelin-12 in acute myocardial infarction. Int Cardiovasc Res J. 2023;17(2):33–38.icrj.139796.
7. Wyderka R, Osuch Ł, Ołpińska B, et al. The impact of the apelinergic system on the cardiovascular system. Int J Mol Sci. 2025;26(20):10087. doi:10.3390/ijms262010087
8. Wysocka MB, Pietraszek-Gremplewicz K, Nowak D. The role of apelin in cardiovascular diseases, obesity and cancer. Front Physiol. 2018;9(9):557. doi:10.3389/fphys.2018.00557
9. Liu W, Yan J, Pan W, et al. Apelin/Elabela-APJ: a novel therapeutic target in the cardiovascular system. Ann Transl Med. 2020;8(5):243. doi:10.21037/atm.2020.02.07
10. Murza A, Trân K, Bruneau-Cossette L, et al. Apelins, ELABELA, and their derivatives: peptidic regulators of the cardiovascular system and beyond. Peptide Science. 2019;111(1):e24064. doi:10.1002/pep2.24064
11. Rikitake Y. The apelin/APJ system in the regulation of vascular tone: friend or foe? J Biochem. 2021;169(4):383–386. doi:10.1093/jb/mvaa129
12. Chapman FA, Maguire JJ, Newby DE, et al. Targeting the apelin system for the treatment of cardiovascular diseases. Cardiovasc Res. 2023;119(17):2683–2696. doi:10.1093/cvr/cvad171
13. Babapour B, Doustkami H, Avesta L, et al. Negative association of apelin plasma levels with epicardial fat thickness in patients with stable angina and acute myocardial infarction: a case–control study. J Res Med Sci. 2024;11(29):26. doi:10.4103/jrms.jrms_478_22
14. Rachwalik M, Leśków A, Matusiewicz M, et al. Assessment of levels of apelinergic system peptides in serum and epicardial adipose tissue in patients with multivessel coronary artery disease who underwent myocardial revascularisation. Biomedicines. 2025;13(4):809. doi:10.3390/biomedicines13040809
15. Askin L, Askin HS, Tanrıverdi O, et al. Serum apelin levels and cardiovascular diseases. Northern Clin Istanbul. 2022;9(3):290–294. doi:10.14744/nci.2021.33427
16. Zhou Y, Wang Y, Qiao S, et al. Effects of Apelin on cardiovascular aging. Front Physiol. 2017;8:1035. doi:10.3389/fphys.2017.01035
17. Cardoso Dos Santos LM, Azar P, Brun C, et al. Apelin is expressed in intimal smooth muscle cells and promotes their phenotypic transition. Sci Rep. 2023;13(1):18736. doi:10.1038/s41598-023-45470-z
18. He L, Zhou Q, Huang Z, et al. PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPKα and exacerbates atherosclerotic lesions. J Cell Physiol. 2019;234(6):8668–8682. doi:10.1002/jcp.27527
19. Chun HJ, Ali ZA, Kojima Y, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008;118(10):3343–3354. doi:10.1172/JCI34871
20. Niu W, Wu S, Zhang Y, et al. Validation of genetic association in apelin–AGTRL1 system with hypertension in a larger Han Chinese population. J Hypertens. 2010;28(9):1854–1861. doi:10.1097/HJH.0b013e32833b1fad
21. Li J, Feng M, Wang Y, et al. The relationship between three X-linked genes and the risk for hypertension among northeastern Han Chinese. J Renin Angiotensin Aldosterone Syst. 2015;16(4):1321–1328. doi:10.1177/1470320314534510
22. Wang T, Liu C, Jia L, et al. The association between apelin polymorphisms and hypertension in China: a meta-analysis. J Renin Angiotensin Aldosterone Syst. 2019;20(1):1470320319827204. doi:10.1177/1470320319827204
23. Akcılar R, Yümün G, Bayat Z, et al. Characterization of the apelin −1860T>C polymorphism in Turkish coronary artery disease patients and healthy individuals. Int J Physiol Pathophysiol Pharmacol. 2015;7(4):165–171.
24. Suriyaprom K, Pheungruang B, Tungtrongchitr R, et al. Relationships of apelin concentration and APLN T-1860C polymorphism with obesity in Thai children. BMC Pediatr. 2020;20(1):455. doi:10.1186/s12887-020-02350-z
25. World Health Organization. Everyday actions for better health – WHO recommendations. 2013. Available from: https://www.who.int/europe/news-room/fact-sheets/item/everyday-actions-for-better-health-who-recommendations.
26. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Hypertension. 2018;71(6):1269–1324. doi:10.1161/HYP.0000000000000066
27. Mach F, Baigent C, Catapano AL, et al. ESC/EAS Guidelines for the management of dyslipidemia: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111–188. doi:10.1093/eurheartj/ehz455
28. Karkhaneh A, Bagherieh M, Sadeghi S, et al. Evaluation of eight formulas for LDL-C estimation in Iranian subjects with different metabolic health statuses. Lipids Health Dis. 2019;18(1):231. doi:10.1186/s12944-019-1178-1
29. Brown JC, Gerhardt TE, Kwon E. risk factors for coronary artery disease. In: StatPearls. Treasure Island (FL); StatPearls Publishing. 2023. https://www.ncbi.nlm.nih.gov/books/NBK554410/.
30. Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. 2019;140(11):e596–646. doi:10.1161/CIR.0000000000000678
31. Gabulova R, Marzà-Florensa A, Rahimov U, et al. Risk factors in cardiovascular patients: challenges and opportunities to improve secondary prevention. World J Cardiol. 2023;15(7):342. doi:10.4330/wjc.v15.i7.342
32. Zhong J-C, Zhang -Z-Z, Wang W, et al. Targeting the apelin pathway as a novel therapeutic approach for cardiovascular diseases. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):1942–1950. doi:10.1016/j.bbadis.2016.11.007
33. Kuba K, Sato T, Imai Y, et al. Apelin and Elabela/Toddler; double ligands for APJ/Apelin receptor in heart development, physiology, and pathology. Peptides. 2019;111:62–70. doi:10.1016/j.peptides.2018.04.011
34. Hazbar AM, Sahab KS. The use of plasma apelin alteration in diagnosis of atherosclerosis. J Biochem Tech. 2018;9(3):23–26.
35. Güven R, Akyol KC, Bayar N, et al. The association between apelin gene polymorphism and coronary artery disease in young patients with acute obstructive coronary syndrome. Turk Kardiyoloji Dernegi arsivi. 2017;45(6):520–526. doi:10.5543/tkda.2017.95849
36. Chen T, Wu B, Lin R. Association of apelin and apelin receptor with the risk of coronary artery disease: a meta-analysis of observational studies. Oncotarget. 2017;8(34):57345–57355. doi:10.18632/oncotarget.17360
37. Kadoglou NPE, Lampropoulos S, Kapelouzou A, et al. Serum levels of apelin and ghrelin in patients with acute coronary syndromes and established coronary artery disease—KOZANI STUDY. Translational Res. 2010;155(5):238–246. doi:10.1016/j.trsl.2010.01.004
38. Zaki MZ, Elshrkawy EA, Ghobrial AG, et al. Role and significance of serum Apelin in coronary artery diseases. MJMR. 2024;35(4):167–181. doi:10.21608/MJMR.2024.311419.1779
39. Motawi TMK, Mahdy SG, El-Sawalhi MM, et al. Serum levels of chemerin, apelin, vaspin, and omentin-1 in obese type 2 diabetic Egyptian patients with coronary artery stenosis. Can J Physiol Pharmacol. 2018;96(1):38–44. doi:10.1139/cjpp-2017-0272
40. Hussein GM. Assessment of serum chemerin, omentin-1 and apelin concentration in normal weight, overweight and obese men in Al-Hilla city. J Univ Babylon Pure Appl Sci. 2017;25(3):1219–1226.
41. Heinonen MV, Purhonen AK, Miettinen P, et al. Apelin, orexin-A and leptin plasma levels in morbid obesity and effect of gastric banding. Regul Pept. 2005;130(1–2):7–13. doi:10.1016/j.regpep.2005.05.003
42. Esteban-Martínez RL, Pérez-Razo JC, Vargas-Alarcón G, et al. Polymorphisms of APLN-APLNR system are associated with essential hypertension in Mexican-Mestizo individuals. Exp Mol Pathol. 2016;101(1):105–109. doi:10.1016/j.yexmp.2016.07.007
43. Kidoya H, Ueno M, Yamada Y, et al. Spatial and temporal role of the apelin/APJ system in the caliber size regulation of blood vessels during angiogenesis. EMBO J. 2008;27(3):522–534. doi:10.1038/sj.emboj.7601982
44. Jin W, Su X, Xu M, et al. Interactive association of five candidate polymorphisms in Apelin/APJ pathway with coronary artery disease among chinese hypertensive patients. PLoS One. 2012;7(12):e51123. doi:10.1371/journal.pone.0051123
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.