Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Assessment of Weighted Gene Co-Expression Network Analysis to Explore Key Pathways and Novel Biomarkers in Muscular Dystrophy
Authors Xu X, Hao Y, Wu J, Zhao J, Xiong S
Received 12 January 2021
Accepted for publication 23 March 2021
Published 13 April 2021 Volume 2021:14 Pages 431—444
DOI https://doi.org/10.2147/PGPM.S301098
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Xiaoxue Xu,1 Yuehan Hao,1 Jiao Wu,2 Jing Zhao,1 Shuang Xiong3
1Department of Neurology, The First Hospital of China Medical University, Shenyang, People’s Republic of China; 2Department of Neurology, The People’s Hospital of Liaoning Province, Shenyang, People’s Republic of China; 3Liaoning Academy of Analytic Science, Construction Engineering Center of Important Technology Innovation and Research and Development Base in Liaoning Province, Shenyang, People’s Republic of China
Correspondence: Xiaoxue Xu
Department of Neurology, The First Hospital of China Medical University, No. 155, Nanjing Street, Heping District, Shenyang, Liaoning, People’s Republic of China
Email [email protected]
Purpose: This study aimed to explore the key molecular pathways involved in Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) and thereby identify hub genes to be potentially used as novel biomarkers using a bioinformatics approach.
Methods: Raw GSE109178 data were collected from the Gene Expression Omnibus (GEO) database. Weighted gene co-expression network analysis (WGCNA) was conducted on the top 50% of altered genes. The key modules associated with the clinical features of DMD and BMD were identified. Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed using the DAVID website. A protein-protein interaction (PPI) network was constructed using the STRING website. MCODE, together with the Cytohubba plug-ins of Cytoscape, screened out the potential hub genes, which were subsequently verified via receiver operating characteristic (ROC) curves in other datasets.
Results: Among the 11 modules obtained, the black module was predominantly associated with pathology and DMD, whereas the light-green module was primarily related to age and BMD. Functional enrichment assessments indicated that the genes in the black module were primarily clustered in “immune response” and “phagosome,” whereas the ones in the light-green module were chiefly enriched in “protein polyubiquitination”. Eleven essential genes were eventually identified, including VCAM1, TYROBP, CD44, ITGB2, CSF1R, LCP2, C3AR1, CCL2, and ITGAM for DMD, along with UBA5 and UBR2 for BMD.
Conclusion: Overall, our findings may be useful for investigating the mechanisms underlying DMD and BMD. In addition, the hub genes discovered might serve as novel molecular markers correlated with dystrophinopathies.
Keywords: Duchenne muscular dystrophy, Becker muscular dystrophy, WGCNA, biomarker, pathway
Introduction
Muscular dystrophies (MDs) are a group of genetic neuromuscular disorders caused by gene mutations. To date, more than 40 genes have been recognized to participate in the phenotypic variations of MDs, suggesting a complicated pathogenesis. Allelic mutations in several genes can affect onset either before or after the acquisition of walking, thereby discriminating congenital muscular dystrophies from limb girdle muscular dystrophies (LGMD). The distribution of the impaired encoding protein also contributes to a variety of MD subtypes.1
The most common forms of MD, Duchenne Muscular Dystrophy (DMD) and Becker muscular dystrophy (BMD), are both X‐linked disorders characterized by progressive muscle weakness.2 They are caused by mutations in dystrophin, which encodes the dystrophin protein, an important component of the plasma membrane cytoskeleton. The absence of functional dystrophin leads to a severe form of DMD, in which patients lose the ability to walk in childhood and subsequently die from respiratory insufficiency or cardiomyopathy in early adulthood. Other organs, such as the brain, are compromised over the course of the disease causing delayed cognitive development in some patients.3–5 BMD is milder than DMD due to only partial dystrophin deficiency, but also displays diverse symptoms, including gradual ambulatory disability.6 Unfortunately, although several treatment strategies have been applied to slow the progression and improve the quality of life of patients with DMD/BMD, curative methods for some MDs have not yet been developed. Gene-based therapies are usually considered promising, but are limited by poor targeting and low efficiency issues.7 Therefore, a comprehensive understanding of the molecular pathways and hub genes involved is of utmost importance to discover new therapeutic targets, explore the underlying mechanism, and to identify biomarkers for prognostic evaluation.
Rapid advancements in high-throughput sequencing technology have reduced the cost of gene expression profiling, providing a favorable tool with high sensitivity and accuracy for large-scale genomic screening. This technique has been increasingly utilized in a number of studies to identify differentially expressed genes in disease occurrences, including DMD.8,9 However, compared to prior research focusing on single gene-gene relationship, research focusing on systematic expression patterns is still limited, leaving a huge knowledge gap on the highly correlated genes responsible for the specific clinical features of interest. Weighted gene co-expression network analysis (WGCNA) is a powerful bioinformatics application for describing the connections among various genes by constructing a co–expression network. It offers straightforward interpretations of gene modules associated with clinical traits for further exploration of biological functions and regulatory mechanisms.10 WGCNA has been successfully employed to investigate the key pathways and identify hub genes as novel biomarkers in many human disorders, ranging from cancers to neurological diseases.11–14
In the present study, the expression profile of patients with MD was acquired to create co-expression modules using the WGCNA algorithm. The crucial modules that were significantly linked to clinical manifestations were highlighted. Functional assessments were carried out to gain more insight into the development of DMD or BMD at the molecular level. Protein-protein interaction (PPI) network analysis revealed essential genes as candidates of interest. We aimed to uncover the generic organization principles of functional cellular networks, and understand the mechanisms for identification of new biomarkers for DMD and BMD.
Materials and Methods
Raw Data Collection
Three gene expression profile datasets (GSE109178, GSE6011, and GSE13608) were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/gds/). GSE109178 platform is “GPL570, [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array”, which was used as the training dataset. This dataset contained muscle biopsies from patients with DMD (n = 17), BMD (n = 11), LGMD2I (LGMD type 2I) (n = 7), LGMD2B (LGMD type 2B) (n = 8), as well as from normal controls (n = 6). All other detailed information is summarized in Table 1 and Supplementary Table 1. GSE6011 and GSE13608 were used to verify the hub genes of DMD and BMD. The GSE6011 and GSE13608 platforms were “GPL96, [HG-U133A] Affymetrix Human Genome U133A Array” and “GPL570, [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array,” respectively. GSE6011 contained skeletal muscle biopsies obtained from 22 patients with DMD and 14 healthy donors. GSE13608 included skeletal muscle biopsies taken from five patients with BMD and six normal adults. The series matrix file and the annotation SOFT table of the platform were downloaded. Figure 1 shows the workflow of the present project.
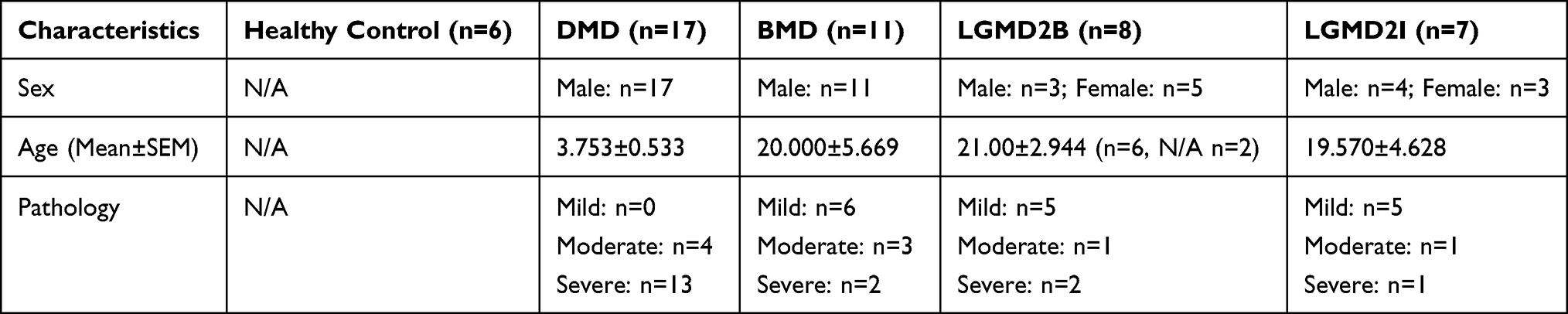 |
Table 1 Demographic and Clinical Characteristics of Subjects in GSE109178 |
 |
Figure 1 Schematic chart of the work flow indicates the steps of the current work. |
Data Pre-Processing
For all three datasets, the probe names were converted into gene symbols via the Perl program and normalized by quantiles. Subsequently, the data were pre-processed with the “Impute” package in “R” and the values of multiple probes for the same gene were averaged. After log2-transformation, 21,756 genes in GSE109178 were further analyzed using the “Limma” package in “R,” with the 10,800 genes exhibiting the most (top 50%) variation in expression levels being screened out for the subsequent WGCNA.15
Co-Expression Network Analysis Utilizing WGCNA
The WGCNA analysis was carried out via the “WGCNA” package in “R”, to generate a co-expression network and to disclose the key modules which were highly associated with clinical features of muscular dystrophy.10 First, a soft threshold ranging from 1 to 30 was determined. This was the lowest value for obtaining a relatively high scale-free network without batch effects when the degree of independence exceeded 0.8. Furthermore, the weighted adjacency matrix was transformed into a topological overlap measure (TOM) matrix to measure the connectivity property of a gene in the network. Finally, based on the TOM dissimilarity, the genes with similar expression profiles were then classified into different modules to construct a clustering dendrogram through linkage hierarchical clustering. The minimal number of genes for each module was set to 30, and the threshold for merging similar modules was set to 0.2.
Selecting Significant Modules of Clinical Features
To determine module–trait relationships, we calculated the gene significance (GS), which was defined as the log10 transformation of the P-value (GS = lgP) in the linear regression between gene expression and clinical information, as well as module significance (MS), which was defined as the average absolute GS of all the genes involved in the module. Generally, the module with the highest MS score among all selected modules was recognized as closely related to the clinical trait.10,16 Moreover, we only selected the positively correlated modules with R2 > 0.4, and P < 0.01, for the subsequent analysis.
GO Enrichment and KEGG Pathway Analyses for Key Modules
Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of genes in key modules were performed using the Database for Annotation, Visualization and Integration Discovery (DAVID, https://david.ncifcrf.gov). A P-value of < 0.01 was set as the cut-off criterion in each category. The top 15 records of each category of GO analysis (Biological Process, Cellular Component and Molecular Function) and KEGG pathways were visualized using the “Ggplot2” package.
Construction of a PPI Network and Topological Analysis
The PPI network of the genes in key modules was evaluated using the online search tool STRING (Search Tool for the Retrieval of Interacting Genes, https://string-db.org). The plug-in Molecular Complex Detection (MCODE) of Cytoscape17 was employed to check the clusters and visualize the protein interactions. The criteria for selection were as follows: MOCDE score > 5, degree cutoff = 2, max. Depth = 100, k-core = 2, and node score cutoff = 0.2). The potential key genes were explored using the CytoHubba plug-in, and the top 20 nodes ranked by degree were collected for subsequent analysis.18,19
Identification of Candidate Hub Genes
Genes with higher relevance were screened out using module connectivity, measured by module membership (MM) ≥ median and clinical trait relationships, measured by GS ≥ median.20 These genes, along with 20 potential key genes acquired from CytoHubba previously, underwent Venn diagram analysis (http://bioinformatics.psb.ugent.be/webtools/Venn/) to identify candidate hub genes. The outputs were of great importance in the PPI network for key modules and were tightly linked to clinical features.
Selection of Real Hub Genes Through Expression Level and (Receiver Operating Characteristic) ROC Curve
We further utilized two independent datasets, GSE6011 and GSE13608, to verify the real hub genes in DMD and BMD, respectively. After preprocessing, the raw data were extracted and unpaired Student’s t-test (two-tailed) was used to evaluate significant differences between the patients with MD and healthy subjects. The diagnostic accuracy of these genes was evaluated using the area under the curve (AUC) of the ROC in Prism 5.0. The significantly altered genes (P < 0.05) with AUC > 0.80 (P < 0.05) were regarded as true hub genes.
Results
Construction of a Co-Expression Network via WGCNA
As the training dataset, GSE109178 possessed complete clinical information. A total of 10,725 genes in GSE109178 were first employed to construct co-expression networks with WGCNA, after removing genes without information. The hierarchical clustering dendrogram showed that all 49 samples clustered well, without outliers, and were mainly divided into two groups. All samples from healthy donors (GSM2934813–GSM2934818) were enrolled in one cluster (Figure 2A). Subsequently, a power of β = 13 was selected as the soft-threshold to generate a scale-free network with an independence degree of up to 0.9, as shown in Figure 2B. Figure 2C indicates that 11 co-expression modules were exported using the average linkage hierarchical clustering algorithm. There were 1763 genes in the black module, 4594 in the dark-grey module, 1079 genes in the dark-olive-green module, 72 genes in the dark-orange module, 100 genes in the dark-red module, 345 genes in the green-yellow module, 398 genes in the gray60 module, 314 genes in the light-green module, 455 genes in the purple module, 176 genes in the royal-blue module, and 64 genes in the sky-blue module. The remaining 1365 genes that did not belong to any module went to the gray module, which was ruled out for the following analysis.
 |
Figure 2 WGCNA analysis. (A) The clustering dendrograms of samples. The samples were well divided into two group with no outlier; (B) determination of the soft-thresholding power (β) in WGCNA. The left panel shows the influence of soft-threshold power on the scale-free fit index and the right panel shows the impact of soft-threshold power on the mean connectivity; (C) gene clustering tree built by hierarchical clustering of adjacency-based dissimilarity to detect 11 co-expression clusters with corresponding color assignments. Each color represents a module and the gray module indicates none co-expression among the genes; (D) the module-trait relationships. Each row correlates to a module eigengene, column to a trait. Each cell includes the corresponding correlation and P - value. Positive correlation is in red and negative correlation is in blue; (E) correlated hierarchical clustering of the adjacency modules. Branches of the dendrogram group are correlated; (F) correlated heatmap of eigengene adjacency. Light-blue represents low adjacency, while red represents high adjacency. |
Key Modules Associated with Clinical Features
The module-trait relationships shown in Figure 2D depict the correlation between the available clinical messages (sex, age, pathology, DMD, BMD, LGMD2B, and LGMD2I) and each module in GSE109178 by calculating the value of MS. It is noteworthy that the black module had a strong correlation with pathology (R2 = 0.66, P = 2E-07) and DMD (R2 = 0.63, P = 1E-06). The genes in the light-green module were significantly affected by age (R2 = 0.41, P = 0.004) and BMD (R2 = 0.43, P = 0.002). In addition, the sky-blue module was most significantly associated with LGMD2I (R2 = 0.64, P = 6E-07). Figure 2E shows the interaction-based relationships for the 11 modules, suggesting that all the modules were chiefly divided into two clusters, based on their ME correlation. The heatmap in Figure 2F shows the adjacencies in the eigengene network, which also indicates a high level of independence among the modules. Considering the higher occurrences of DMD and BMD, we focused on the black and light-green modules for further analysis.
Functional Enrichment Analyses of Genes in Key Modules
The scatter plots in Figures 3A and B and 4A and B demonstrate the correlation between module membership and gene significance in the clinical features of the black and light-green modules, respectively. We conducted GO and KEGG analyses via the DAVID website to expand our understanding of the function of the genes in the key modules. Figure 3C–E demonstrates that the genes in the black module were mainly gathered in BP, CC, and MF, including in the “extracellular matrix organization” (GO:0030198, P = 7.68E-12), “extracellular exosome” (GO:0070062, P = 3.07E-15), and “extracellular matrix structural constituent” (GO:0005201, P = 1.15E-06) pathways. In addition, the genes also referred to “inflammatory response” (GO:0006954, P = 5.66E-10) and “immune response” (GO:0006955, P = 1.77E-05). Figure 3F presents the enriched KEGG pathway of the black module genes, including “Staphylococcus aureus infection” (hsa05150, P = 2.87E-08), “Phagosome” (hsa04145, P = 6.77E-08), and “Tuberculosis” (hsa05152, P = 6.43E-07). As for the light-green module, as demonstrated in Figure 4C–E, the genes were mainly clustered in BP, CC, and MF, including “antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-independent” (GO:0002480, P = 7.84E-06), “ER to Golgi transport vesicle membrane” (GO:0012507, P = 1.49E-05), and “protein binding” (GO:0005515, P = 1.02E-05). In addition, they also referred to “protein polyubiquitination” (GO:0000209, P = 8.29E-04) and “ubiquitin-protein transferase activity” (GO:0004842, P = 2.47E-05). Figure 4F shows the enriched KEGG pathway of the light-green module genes, which include “Phagosome” (hsa04145, P=3.28E-04), “Antigen processing and presentation” (hsa04612, P=3.87E-04), and “Graft-versus-host disease” (hsa05332, P=0.002666998). The results of GO and KEGG analyses for black and light-green modules are listed in Supplementary Tables 2–5.
 |
Figure 3 GO enrichment and KEGG analyses for black module. (A) Scatterplot of gene significance for pathology vs module membership in black module; (B) scatterplot of gene significance for DMD vs module membership in black module; (C–E) the top 15 terms of GO categories of biological process (BP), cellular component (CC) and molecular function MF, respectively; (F) the top 15 terms of KEGG analysis. P-value < 0.01 was considered significantly. |
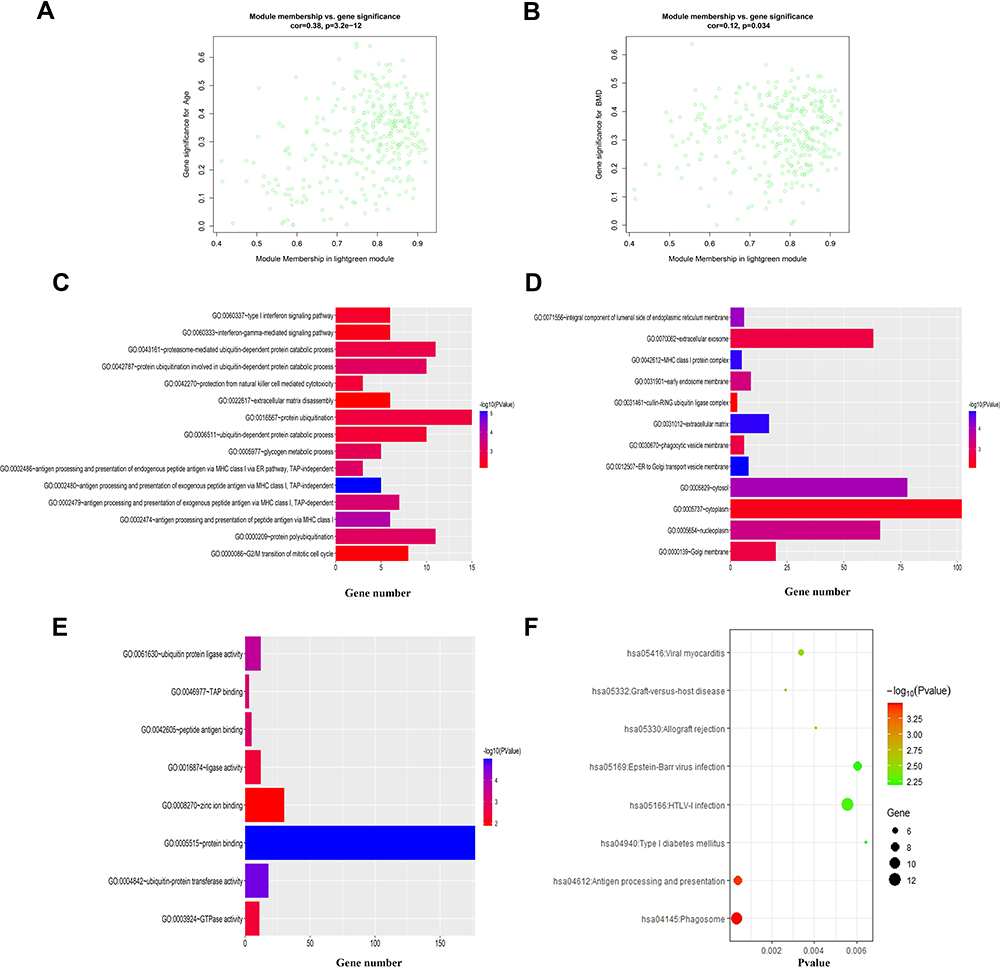 |
Figure 4 GO enrichment and KEGG analyses for light-green module. (A) Scatterplot of gene significance for age vs module membership in light-green module; (B) scatterplot of gene significance for BMD vs module membership in light-green module; (C–E) the top 15 terms of GO categories of biological process (BP), cellular component (CC) and molecular function (MF), respectively; (F) KEGG analysis. P-value < 0.01 was considered significantly. |
PPI Network Analysis and Candidate Hub Genes
To further probe the hub genes in key modules, we generated a protein-protein network and performed a topological assessment. Figure 5A shows the protein-protein cluster with the highest score (33.238) by MCODE in the black module, which was composed of 43 nodes and 698 interactive edges. As shown in Figure 5B, the top 20 genes ranked by degree, calculated using CytoHubba also formed a sub-network. Detailed information on these genes is provided in Supplementary Table 6. Similarly, 17 nodes and 136 edges constituted the highest score (17) cluster in the light-green module, as shown in Figure 5C, while the top 20 genes from CytoHubba are listed in Supplementary Table 7, and the sub-network is shown in Figure 5D. Since higher MM and GS should indicate a closer association of the gene with the clinical feature, we then filtered the genes in key modules under the condition of MM ≥ median (MM Black ≥ 0.6321; MM light-green ≥ 0.7077) and GS ≥ median (black, GSDMD ≥ 0.2908; GSBMD ≥ 0.2867). A total of 690 genes in the black module and 106 genes in the light-green module were screened out. After cross-matching with the top 20 genes using CytoHubba, 12 genes in the black module and 8 genes in the light-green module were selected as potential hub genes (Figure 6A and B).
 |
Figure 5 Diagram of PPI networks. The balls represent the gene nodes, the connecting lines represent the interactions. The size of the ball is corresponding to its node degree, indicating that the bigger the ball is, the higher node degree is. (A) The cluster with highest score analyzed by MCODE in black module; (B) the top 20 hub gene in black module by Cytohubba; (C) the cluster with highest score analyzed by MCODE in light-green module; (D) the top 20 hub gene candidates in light-green module. |
 |
Figure 6 The validation for hub genes. (A) Venn diagram for selecting hub candidates in black module; (B) Venn diagram for selecting hub candidates in light-green module; (C) the ROC curves for true hub genes in black module; (D) the ROC curves for true hub genes in light-green module. |
Identification of Real Hub Genes
ROC curves and AUCs are useful tools for evaluating test accuracy and identifying biomarkers to distinguish between patients with and without a health outcome of interest.21,22 Therefore, in order to identify the true hub genes as biomarkers of DMD and BMD, after testing the expression level for all the candidates, ROC curves were generated and the AUC (95% CIs) was calculated by capitalizing the independent expression profiles (Table 2, Figure 6C and D). According to our criteria, we discovered that nine abnormally expressed genes exhibited a high predictive accuracy for DMD, including VCAM1, TYROBP, CD44, ITGB2, CSF1R, LCP2, C3AR1, CCL2, and ITGAM, as well as two genes for BMD, including UBA5 and UBR2.
 |
Table 2 The Validation of the Potential Hub Genes of DMD and BMD |
Discussion
The purpose of the current work was: 1) to determine the key molecular pathways involved in DMD and BMD, and 2) to identify the potential hub genes that could be used as biomarkers for monitoring disease progression or predicting prognosis. Therefore, we selected GSE109178 with sufficient clinical messages and applied WGCNA for correlated gene mining. This bioinformatics approach transforms the gene expression profile into a co-expression network, offering informative insights on a systematic level.23 In this study, we detected 11 modules in the GSE109178 dataset. In patients with DMD, the genes in the key (black) module were mainly located in the “extracellular exosome” and “immune response” pathways. For BMD samples, the genes in the key (light-green) module were chiefly enriched in “protein polyubiquitination,” which has not previously been demonstrated. Combined with PPI analysis, nine and two genes were eventually filtered out as the hub genes for DMD and BMD, respectively. These critical genes may have a diagnostic value in the pathogenesis of MD. To the best of our knowledge, this is the first report of WGCNA concerning MDs, particularly BMD, for exploring trait-module relationships and potential biomarkers.
As a common form of MD, DMD affects approximately 1 in 3500 live male births worldwide.24,25 Mutations in dystrophin lead to the loss of dystrophin protein, which is vital for maintaining muscle integrity. Dystrophic muscles undergo cycles of necrosis and muscle repair, along with chronic inflammation. Ultimately, the impaired muscle is replaced by adipose and scar tissue, resulting in fibrosis. A number of complex secondary damages and altered expressions of multiple genes played a role in this procedure.26 According to our results, both pathology and DMD were tightly linked to the black module. Indeed, the symptoms of DMD are much more serious than those of other MDs,27 implying that manipulation of these genes might be beneficial in improving muscle function. Moreover, “extracellular exosome” was referred to in the functional terms of the black module. Exosomes are extracellular vesicles with diameters of 40–150 nm. As carriers of multiple signaling factors, exosomes are thought to be involved in numerous physiological and pathological activities.28 In terms of muscle regeneration, exosomes can be secreted by differentiating myoblasts to deliver essential molecules, including proteins, inflammatory cytokines, miRNAs, and lipids.29,30 In particular, the enhanced serum levels of non-coding RNAs found in patients with DMD were coordinated with the expression of CD63, an exosomal surface marker, indicating that the impaired muscle likely released exosomes to “rescue” neighboring cells from injury or death.31 Conversely, growing evidence suggests that exosomes might exert pathogenic roles in DMD by promoting phenotypic transformation from normal fibroblasts to myofibroblasts.32 Accordingly, the precise role of exosomes remains a matter of debate. Meanwhile, in light of our functional assessments of black module genes, defective downstream cascades, including immunological and inflammatory processes, as well as autophagy, might result in identification of muscle pathology in DMD.33 Normal skeletal muscles have a low ability to generate localized immune responses, whereas muscles with dystrophin deficiency can sustain a proinflammatory microenvironment activating innate immune pathways, due to a missing immune privileged status.33 Steroid administration mitigated symptoms in patients with DMD by suppressing muscle-specific T cell accumulation.34 Similarly, as an immunosuppressor, rapamycin ameliorated dystrophic phenotypes in mdx mice,35 suggesting that reducing the immunity burden might be a promising therapy. Moreover, the key genes revealed in this study included VCAM1 (vascular cell adhesion protein 1), TYROBP (transmembrane immune signaling adaptor TYROBP), CD44, ITGB2 (integrin subunit beta 2), CSF1R (colony stimulating factor 1 receptor), LCP2 (lymphocyte cytosolic protein 2), C3AR1 (complement C3a receptor 1), CCL2 (C-C motif chemokine ligand 2), and ITGAM (integrin subunit alpha M). With the exception of VCAM1, CD44, and CCL2, none of these genes have been investigated in DMD–related experimental studies.36–38 Likewise, most of these genes were listed in “response to wounding”-related modules in dystrophic co-expression networks constructed by other teams using different DMD datasets, which was in line with the outcome of this study.39 In view of the diagnostic value of these genes, there is an urgent need to evaluate the likelihood of these genes as novel biomarkers to monitor disease progression.
BMD is significantly less severe than DMD, and therefore, a number of “preclinical” or “asymptomatic” cases have been identified.40 Intriguingly, our results indicated that the key BMD module was associated with age. This relationship probably explains the delayed onset and much longer normal life expectancy reported in patients with BMD.41,42 In addition, given that the same pathogenic gene (dystrophin) is implicated in both conditions, we initially speculated that BMD might share a majority of molecular pathways or hub genes with DMD. Notably, light-green module genes clustered in distinct GO and KEGG categories, in which “protein ubiquitination” was highlighted. As one of the protein post-translational modifications, ubiquitylation commences with the transfer of ubiquitin to proteins, subsequently resulting in the formation of a ubiquitin chain on the targets. This orchestrated procedure is strictly managed by several enzymes, including ATP-dependent activating enzyme (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin protein ligase (E3). Ubiquitination is thought to control protein degradation because it favors multiple biological functions, such as selective autophagy. Hence, dysfunction of the ubiquitin proteasome machinery could contribute to muscle pathologies.43,44 Nevertheless, most lines of evidence are directly linked to DMD rather than BMD. Altered levels of proteasome and ubiquitin were discovered in the cytoplasm of necrotic fibers from patients with DMD.45 Restoration of the dystrophin protein by blocking the proteasome was observed in golden retriever muscular dystrophy dog models.46 Four E3 ubiquitin ligases (Zfand5, FBXO33, Amn1, and Trim75) likely regulated the mutated dystrophin levels in a new DMD mouse model, affecting disease progression.47 Additionally, despite the insufficient evidence in BMD, the two hub genes we found here are closely associated with ubiquitination. The encoding product of UBA5 (ubiquitin-like modifier activating enzyme 5) is a member of the E1-like ubiquitin-activating enzyme family. UBR2 encodes the ubiquitin protein ligase E3 component, n-recognin 2. Further research is required to demonstrate the exact function of ubiquitination-associated molecules in the context of BMD. On the other hand, we noticed that “phagosome” appeared in KEGG enrichments of the light-green module. As a regular physiological activity, autophagosomes engulf cellular components and organelles to fuse with lysosomes, which activates the process of degradation. Importantly, in a dystrophic animal model, inhibition of autophagic signaling likely led to increased autophagosome escape, implying that impaired autophagic trafficking might favor dystrophic progression.48,49 Combined with several immune system-related terms in GO/KEGG enrichments in light-green modules (such as GO:0002480-antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-independent; GO:0002474-antigen processing and presentation of peptide antigen via MHC class I antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-dependent; hsa04612-Antigen processing and presentation, etc), we hypothesized that chronic inflammation due to autophagosome escape might be involved in BMD occurrence.
Despite these important findings, we acknowledge that our study has several limitations. First, the potential molecular pathways were mainly recognized based on data mining through bioinformatic methods and therefore still need to be experimentally validated. Second, the number of samples in the raw dataset was relatively small; therefore, more work focused on the potential biomarkers is necessary by using a larger sample size, especially for those that have not yet been reported in DMD or BMD. Third, functional studies are warranted to identify drug candidate targets and develop potential therapies. In addition, important findings of other types of MD (such as LGMD2I), which were neglected in the current study, should also be further studied.
In summary, the current study concentrated on the co-occurrence network generated through WGCNA using the expression profiles from MDs. The analysis of the key modules revealed the involvement of “exocellular exosomes” and “immune response” in DMD as well as “ubiquitination” in BMD, indicating the distinct pathways in the two muscular dystrophies. We also identified the hub genes, including VCAM1, TYROBP, CD44, ITGB2, CSF1R, LCP2, C3AR1, CCL2, and ITGAM for DMD, along with UBA5 and UBR2 for BMD. Our results not only offer a reference for a more comprehensive understanding of dystrophinopathies, but also provide new perspectives on the underlying mechanism. Additionally, the key molecules disclosed here might become novel biomarkers for the diagnosis, treatment, and prognosis of DMD and BMD.
Acknowledgments
This work was kindly supported by National Natural Science Foundation of China (81601129) and Science Foundation of Shenyang (F16-205-1-48).
Disclosure
The authors have no conflict of interests.
References
1. Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. The Lancet. 2019;394:2025–2038. doi:10.1016/S0140-6736(19)32910-1
2. Wein N, Alfano L, Flanigan KM. Genetics and emerging treatments for Duchenne and Becker muscular dystrophy. Pediatr Clin North Am. 2015;62(3):723–742. doi:10.1016/j.pcl.2015.03.008
3. Sun C, Serra C, Lee G, et al. Stem cell-based therapies for Duchenne muscular dystrophy. Exp Neurol. 2020;323:113086. doi:10.1016/j.expneurol.2019.113086
4. Anand A, Tyagi R, Mohanty M, et al. Dystrophin induced cognitive impairment: mechanisms, models and therapeutic strategies. Ann Neurosci. 2015;22:108–118. doi:10.5214/ans.0972.7531.221210
5. Annexstad EJ, Lund-Petersen I, Rasmussen M. Duchenne muscular dystrophy. Tidsskr nor Laegeforen. 2014;134:1361–1364. doi:10.4045/tidsskr.13.0836
6. Waldrop MA, Flanigan KM. Update in Duchenne and Becker muscular dystrophy. Curr Opin Neurol. 2019;32:722–727. doi:10.1097/WCO.0000000000000739
7. Cordova G, Negroni E, Cabello-Verrugio C, et al. Combined therapies for Duchenne muscular dystrophy to optimize treatment efficacy. Front Genet. 2018;9:114. doi:10.3389/fgene.2018.00114
8. Zhang R, Lv L, Ban W, et al. Identification of hub genes in Duchenne muscular dystrophy: evidence from bioinformatic analysis. J Comput Biol. 2020;27:1–8. doi:10.1089/cmb.2019.0167
9. Xu X, Hao Y, Xiong S, et al. Comprehensive analysis of long non-coding RNA-associated competing endogenous RNA network in Duchenne muscular dystrophy. Interdiscip Sci. 2020;12:447–460. doi:10.1007/s12539-020-00388-2
10. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi:10.1186/1471-2105-9-559
11. Niemira M, Collin F, Szalkowska A, et al. Molecular signature of subtypes of non-small-cell lung cancer by large-scale transcriptional profiling: identification of key modules and genes by weighted gene co-expression network analysis (WGCNA). Cancers. 2019;12:37. doi:10.3390/cancers12010037
12. Yin L, Cai Z, Zhu B, et al. Identification of key pathways and genes in the dynamic progression of HCC based on WGCNA. Genes. 2018;9:92. doi:10.3390/genes9020092
13. Soleimani Zakeri NS, Pashazadeh S, MotieGhader H. Gene biomarker discovery at different stages of Alzheimer using gene co-expression network approach. Sci Rep. 2020;10:12210. doi:10.1038/s41598-020-69249-8
14. Bhargava P, Fitzgerald KC, Calabresi PA, et al. Metabolic alterations in multiple sclerosis and the impact of vitamin D supplementation. JCI Insight. 2017;2:e95302. doi:10.1172/jci.insight.95302
15. Niu X, Zhang J, Zhang L, et al. Weighted gene co-expression network analysis identifies critical genes in the development of heart failure after acute myocardial infarction. Front Genet. 2019;10:1214. doi:10.3389/fgene.2019.01214
16. Wright RM, Aglyamova GV, Meyer E, et al. Gene expression associated with white syndromes in a reef building coral, Acropora hyacinthus. BMC Genomics. 2015;16:371. doi:10.1186/s12864-015-1540-2
17. Otasek D, Morris JH, Bouças J, et al. Cytoscape automation: empowering workflow-based network analysis. Genome Biol. 2019;20:185. doi:10.1186/s13059-019-1758-4
18. Wang W, Lou W, Ding B, et al. A novel mRNA-miRNA-lncRNA competing endogenous RNA triple sub-network associated with prognosis of pancreatic cancer. Aging. 2019;11:2610–2627. doi:10.18632/aging.101933
19. Chin CH, Chen SH, Wu HH, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. doi:10.1186/1752-0509-8-S4-S11
20. Lunnon K, Ibrahim Z, Proitsi P, et al. Mitochondrial dysfunction and immune activation are detectable in early Alzheimer’s disease blood. J Alzheimers Dis. 2012;30:685–710. doi:10.3233/JAD-2012-111592
21. Heller G, Seshan VE, Moskowitz CS, et al. Inference for the difference in the area under the ROC curve derived from nested binary regression models. Biostatistics. 2017;18:260–274. doi:10.1093/biostatistics/kxw045
22. Soreide K, Korner H, Soreide JA. Diagnostic accuracy and receiver-operating characteristics curve analysis in surgical research and decision making. Ann Surg. 2011;253:27–34. doi:10.1097/SLA.0b013e318204a892
23. Wang M, Wang L, Pu L, et al. LncRNAs related key pathways and genes in ischemic stroke by weighted gene co-expression network analysis (WGCNA). Genomics. 2020;112:2302–2308. doi:10.1016/j.ygeno.2020.01.001
24. Hrach HC, Mangone M. miRNA profiling for early detection and treatment of Duchenne muscular dystrophy. Int J Mol Sci. 2019;20:4638. doi:10.3390/ijms20184638
25. Mehdizadeh A, Barzegar M, Negargar S, et al. The current and emerging therapeutic approaches in drug-resistant epilepsy management. Acta Neurol Belg. 2019;119:155–162. doi:10.1007/s13760-019-01120-8
26. Haslett JN, Sanoudou D, Kho AT, et al. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc Natl Acad Sci U S A. 2002;99:15000–15005. doi:10.1073/pnas.192571199
27. Heydemann A. Skeletal muscle metabolism in Duchenne and Becker muscular dystrophy-implications for therapies. Nutrients. 2018;10:796. doi:10.3390/nu10060796
28. Murphy C, Withrow J, Hunter M, et al. Emerging role of extracellular vesicles in musculoskeletal diseases. Mol Aspects Med. 2018;60:123–128. doi:10.1016/j.mam.2017.09.006
29. Yáñez-Mó M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066. doi:10.3402/jev.v4.27066
30. Lam NT, Gartz M, Thomas L, et al. Influence of microRNAs and exosomes in muscle health and diseases. J Muscle Res Cell Motil. 2020;41:269–284. doi:10.1007/s10974-019-09555-5
31. Aoi W. Frontier impact of microRNAs in skeletal muscle research: a future perspective. Front Physiol. 2014;5:495.
32. Zanotti S, Gibertini S, Blasevich F, et al. Exosomes and exosomal miRNAs from muscle-derived fibroblasts promote skeletal muscle fibrosis. Matrix Biol. 2018;74:77–100. doi:10.1016/j.matbio.2018.07.003
33. Rosenberg AS, Puig M, Nagaraju K, et al. Immune-mediated pathology in Duchenne muscular dystrophy. Sci Transl Med. 2015;7:299rv294. doi:10.1126/scitranslmed.aaa7322
34. Flanigan KM, Campbell K, Viollet L, et al. Anti-dystrophin T cell responses in Duchenne muscular dystrophy: prevalence and a glucocorticoid treatment effect. Hum Gene Ther. 2013;24:797–806. doi:10.1089/hum.2013.092
35. Eghtesad S, Jhunjhunwala S, Little SR, et al. Rapamycin ameliorates dystrophic phenotype in mdx mouse skeletal muscle. Mol Med. 2011;17:917–924. doi:10.2119/molmed.2010.00256
36. Malecova B, Gatto S, Etxaniz U, et al. Dynamics of cellular states of fibro-adipogenic progenitors during myogenesis and muscular dystrophy. Nat Commun. 2018;9:3670. doi:10.1038/s41467-018-06068-6
37. Kong WH, Sung DK, Kim H, et al. Self-adjuvanted hyaluronate–antigenic peptide conjugate for transdermal treatment of muscular dystrophy. Biomaterials. 2016;81:93–103. doi:10.1016/j.biomaterials.2015.12.007
38. Bronisz-Budzyńska I, Chwalenia K, Mucha O, et al. miR-146a deficiency does not aggravate muscular dystrophy in mdx mice. Skelet Muscle. 2019;9:22. doi:10.1186/s13395-019-0207-0
39. Mukund K, Subramaniam S. Dysregulated mechanisms underlying Duchenne muscular dystrophy from co-expression network preservation analysis. BMC Res Notes. 2015;8:182. doi:10.1186/s13104-015-1141-9
40. Angelini C, Marozzo R, Pegoraro V. Current and emerging therapies in Becker muscular dystrophy (BMD). Acta Myol. 2019;38:172–179.
41. Mori-Yoshimura M, Mizuno Y, Yoshida S, et al. Psychiatric and neurodevelopmental aspects of Becker muscular dystrophy. Neuromuscul Disord. 2019;29:930–939. doi:10.1016/j.nmd.2019.09.006
42. Flanigan KM. Duchenne and Becker muscular dystrophies. Neurol Clin. 2014;32:671–688. doi:10.1016/j.ncl.2014.05.002
43. Bachiller S, Alonso-Bellido IM, Real LM, et al. The ubiquitin proteasome system in neuromuscular disorders: moving beyond movement. Int J Mol Sci. 2020;21:6429. doi:10.3390/ijms21176429
44. Sandri M, Coletto L, Grumati P, et al. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J Cell Sci. 2013;126:5325–5333. doi:10.1242/jcs.114041
45. Kumamoto T, Fujimoto S, Ito T, et al. Proteasome expression in the skeletal muscles of patients with muscular dystrophy. Acta Neuropathol. 2000;100:595–602. doi:10.1007/s004010000229
46. Gazzerro E, Assereto S, Bonetto A, et al. Therapeutic potential of proteasome inhibition in Duchenne and Becker muscular dystrophies. Am J Pathol. 2010;176:1863–1877. doi:10.2353/ajpath.2010.090468
47. McCourt JL, Talsness DM, Lindsay A, et al. Mouse models of two missense mutations in actin-binding domain 1 of dystrophin associated with Duchenne or Becker muscular dystrophy. Hum Mol Genet. 2018;27:451–462. doi:10.1093/hmg/ddx414
48. Spaulding HR, Kelly EM, Quindry JC, et al. Autophagic dysfunction and autophagosome escape in the mdx mus musculus model of Duchenne muscular dystrophy. Acta Physiol. 2018;222:222. doi:10.1111/apha.12944
49. Fiacco E, Castagnetti F, Bianconi V, et al. Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death Differ. 2016;23:1839–1849. doi:10.1038/cdd.2016.70
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.