Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Assessment and management of respiratory function in patients with Duchenne muscular dystrophy: current and emerging options
Authors LoMauro A, D'Angelo MG, Aliverti A
Received 20 June 2015
Accepted for publication 22 July 2015
Published 28 September 2015 Volume 2015:11 Pages 1475—1488
DOI https://doi.org/10.2147/TCRM.S55889
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Antonella LoMauro,1 Maria Grazia D’Angelo,2 Andrea Aliverti1
1Department of Electronics, Information and Bioengineering, Politecnico di Milano, Milan, 2IRCCS Eugenio Medea, Bosisio Parini, Italy
Abstract: Duchenne muscular dystrophy (DMD) is an X-linked myopathy resulting in progressive weakness and wasting of all the striated muscles including the respiratory muscles. The consequences are loss of ambulation before teen ages, cardiac involvement and breathing difficulties, the main cause of death. A cure for DMD is not currently available. In the last decades the survival of patients with DMD has improved because the natural history of the disease can be changed thanks to a more comprehensive therapeutic approach. This comprises interventions targeted to the manifestations and complications of the disease, particularly in the respiratory care. These include: 1) pharmacological intervention, namely corticosteroids and idebenone that significantly reduce the decline of spirometric parameters; 2) rehabilitative intervention, namely lung volume recruitment techniques that help prevent atelectasis and slows the rate of decline of pulmonary function; 3) scoliosis treatment, namely steroid therapy that is used to reduce muscle inflammation/degeneration and prolong ambulation in order to delay the onset of scoliosis, being an additional contribution to the restrictive lung pattern; 4) cough assisted devices that improve airway clearance thus reducing the risk of pulmonary infections; and 5) non-invasive mechanical ventilation that is essential to treat nocturnal hypoventilation, sleep disordered breathing, and ultimately respiratory failure. Without any intervention death occurs within the first 2 decades, however, thanks to this multidisciplinary therapeutic approach life expectancy of a newborn with DMD nowadays can be significantly prolonged up to his fourth decade. This review is aimed at providing state-of-the-art methods and techniques for the assessment and management of respiratory function in DMD patients.
Keywords: DMD, spirometry, respiratory muscles, cough device, NIV, steroids, idebenone, respiratory function, lung volume recruitment, scoliosis, spinal fusion, mdx, GRMD
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked myopathy that affects one out of 3,600–6,000 male births.1–3 It is caused by mutations in the dystrophin gene leading to a defect in the protein dystrophin which results in progressive weakness and wasting of all the striated muscles. At the beginning, the proximal lower limb muscles are affected followed by the shoulder muscles, by the distal limb muscle, and ultimately by the respiratory muscles. The consequences are progressive impairment of motor skills, loss of ambulation before teen ages, orthopedic complications, cardiac involvement, and breathing difficulties. Without any intervention, death occurs around the age of 19 due to cardiac and respiratory failure.4–7 At the moment therapy is not available, but in the last decades the survival of these patients improved and a newborn with DMD now has a life expectancy into the fourth decade.3,8–10 The natural history of the disease can now be changed thanks to a more comprehensive therapeutic approach comprising interventions targeted to the manifestations and complications of the disease, particularly in respiratory care.7,11–14 ACE-inhibitions are used to prevent or improve cardiomyopathy. Corticosteroids are used to reduce muscle inflammation, minimize later complications, and prolong ambulation.3,15,16 When the patient becomes non-ambulatory, treatments aim to reduce the risk of progressive scoliosis and stabilizing pulmonary function. According to the internationally recognized guidelines, a structured approach to respiratory management of DMD during the progression of the disease comprises: 1) the monitoring of respiratory function to deal with respiratory complications timely; 2) lung volume recruitment (LVR) techniques to open the lung and reduce atelectasis; 3) the use of a cough assisted device to improve airway clearance and to reduce the risk of pulmonary infections; and 4) the use of non-invasive ventilation (NIV) to deal with nocturnal hypoventilation, sleep disordered breathing, and ultimately respiratory failure.8,11,13,14
The genetic therapy represents hope for the future to develop a definitive cure for DMD. Many promising researches on skeletal muscles are currently under development although not specifically addressed to respiratory muscles.17,18
The present review article is thought to provide a state-of-the-art methods and techniques for the assessment and management of respiratory function in DMD patients. In particular, the techniques currently used for respiratory functional evaluation are illustrated, indicating limits, advantages, and usefulness for assessing the effects of different possible kinds of intervention.
Assessment of respiratory function
The causes of death in DMD patients are both respiratory and cardiac, with the former being the main cause until the 1980s to 1990s. More recently, the structured approach to respiratory problems and, in particular, the institution of nocturnal NIV, had a major effect on survival. Since respiratory failure, however, is still among the main causes of death in patients affected by DMD,3,5–7,12 it is extremely important to measure lung and respiratory muscle function in order to monitor the progression of the disease, identify early signs of respiratory failure, and therefore plan optimal interventions to improve the quality of life and longevity in these patients. The assessment of respiratory function is also important to quantify the possible effects of innovative therapeutic trials.
According to their invasiveness and to the collaboration required to the patients, lung and respiratory muscle function tests are classified as non-invasive/non-volitional, non-invasive/volitional, invasive/non-volitional, and invasive/volitional. These tests can be “specific” or “global” according to whether they allow to distinguish between the contribution and the relative involvement of the different respiratory muscles, namely the diaphragm, the intercostal muscles, the abdominal muscles, and the accessory inspiratory muscles. These tests can assess: i) the level of muscle activation by measuring the electrical activity of the muscle (electromyography [EMG]); ii) the force developed by the muscles by measuring the pressure developed; iii) the resulting action of the contraction of the muscles by measuring flow and/or volume; or iv) the morphology of the muscles with different imaging techniques.19
Non-invasive/non-volitional tests
The electrical activity of each respiratory muscle can be non-invasively measured by transcutaneous EMG using surface electrodes. Unfortunately, at rest the signal/noise ratio is very poor, particularly in weak muscles, because of the “cross-talk” of other muscles, including cardiac activity.20,21
The ventilatory pattern during quiet breathing at rest can be measured at the mouth on a breath-by-breath base by a pneumotachograph preferable using a mask rather than a mouthpiece in order to avoid possible leakage due to weakness of cheek muscle and macroglossia, which are common characteristics in the advanced stages of the disease. The face mask connection to the lung function device is a valid alternative; however, it is important to choose the appropriate prediction equations, depending on subject-instrument interface.22 From the integration of the flow signal, tidal volume (VT) and breathing frequency (fR) are measured and minute ventilation (VE = VT*fR) and rapid and shallow breathing index (RSBi = fR/VT) derived.23 These are global respiratory indexes that can be performed at any age and in different postures: standing, seated, and supine.
Measurements of thoraco-abdominal motion do not require the use of mouthpiece or mask and can be divided into three groups:
- those recording the anteroposterior variations of chest wall diameters, such as the magnetometers (ie, coils placed anteriorly and posteriorly on the ribcage and the abdomen);24
- those measuring variations of ribcage and abdominal cross-sectional areas, such as respiratory inductive plethysmography (ie, coils sewn in belts or wearable garments placed below the axillary line and above the umbilicus);19,25
- those measuring total and compartmental chest wall volumes, such as opto-electronic plethysmography that detects the 3D displacement of a large set of reflective markers placed on the thoraco-abdominal surface of the subject.26–29
Although the shape of the diaphragm can be reconstructed with an excellent resolution by using magnetic resonance imaging (MRI), its clinical use is still limited because of the high cost. Moreover, patients are not always able to maintain the supine posture for the prolonged time required for the acquisition.30
On the other hand, ultrasound (US) imaging is increasingly used since it allows to non-invasively study the diaphragm at rest from three different views: 1) the displacement of the diaphragm dome by positioning the US probe on the anterior abdominal wall; 2) the length of apposition of the diaphragm by positioning the US probe on the lateral ribcage; and 3) the thickness of the diaphragm by positioning the US probe in the intercostal space around the eighth rib.31–33
Non-invasive/volitional tests
All non-invasive/volitional tests require repetition of maximal efforts to get reliable results that can fatigue the weak muscles of DMD patients. They also require full cooperation and are difficult for children to perform. Cooperative children over the age of 7 are considered able to perform reproducible and therefore reliable maneuvers.
Maximal static inspiratory (MIP) and expiratory (MEP) pressures measured at the mouth provide a global index of inspiratory and expiratory muscle strength, respectively. Being MIP and MEP influenced by respiratory muscle length that in turn depends on lung volume, it is crucially important to control the lung volume at which the maneuver is performed. MIP has to be performed at residual volume (RV) and MEP at total lung capacity (TLC), respectively when inspiratory and expiratory muscles are maximally stretched.19,34
Sniff nasal inspiratory pressure (SNIP) measured at the nostril is generally easier to perform, since the requested maneuvers is more natural. If performed correctly (ie, with an outward movement of the belly), it can be considered a specific index of the strength of the diaphragm, although in presence of nasal obstruction it underestimates the real pressure.19,35,36
Inspiratory muscle fatigue can be assessed by the tension time index (TTI) that is calculated as P0.1/MIP· duty cycle, where P0.1 is the pressure measured during the first 100 milliseconds of occlusion while breathing quietly and the duty cycle is the ratio between inspiratory time and the total time duration of the breath. High values of TTI indicate that inspiratory muscles are at risk of fatigue.37–40
Peak expiratory flow (PEF) and cough peak flow (CPF) are measured at the mouth respectively during a maximal exhalation and cough. They provide information on the expiratory muscles.41
Traditional spirometric parameters such as forced vital capacity (FVC) and forced expiratory volume in 1 second (FEV1) are global indexes of the respiratory function. Values above 80% of their ratio (FEV1/FVC), the Tiffenau index, can indicate the presence of restrictive lung disease. The diagnosis has to be confirmed by reduced values of TLC. A drop in FVC (>20%), when the patient goes from sitting to supine position, is a more specific sign of diaphragmatic weakness.42
TLC, functional residual capacity, and RV are absolute lung volumes that cannot be measured by simple spirometry. They require either nitrogen washout technique or body plethysmography. Wheel-chair bound DMD patients can meet great difficulties in entering a body plethysmograph.19,43–46
Invasive/non-volitional tests
Invasive/non-volitional tests imply the use of esophageal and/or gastric unpleasant balloon-catheter systems. They provide specific indexes of the different respiratory muscles. The use of the catheters is not recommended in very severe DMD patients with difficulties in swallowing to avoid regurgitation and aspiration.19 An additional caution of pressure measurements by esophageal and gastric balloons is represented by the possible bradycardia mediated by the stimulation of the vagal nerve. They can be used during quiet breathing at rest without requiring any manouvers.47
The measurement of esophageal pressure (POES), based on transducer-catheter-balloon system placed in the lower third of the esophagus, is considered a useful surrogate of pleural pressure while abdominal gastric pressure (PGA) is measured by placing the transducer-catheter-balloon system in the stomach. The trans-diaphragmatic pressure (PDI) is calculated as PGA–POES and represents the force generated by the diaphragm.19 The ratio between swings of PGA and PDI (ΔPGA/ΔPDI) during spontaneous quiet breathing is able to provide an assessment of paradoxical breathing.48
Peak PDI during magnetic stimulation of the phrenic nerve tests the strength of the diaphragm in non-cooperative patients.49
Volumetric computed tomography imaging can be used to obtain 3D reconstruction of the diaphragm with an excellent resolution, but the radiation exposure limits the clinical use of this technique in DMD patients.50
Invasive/volitional tests
Invasive/volitional tests include the measurements of POES, PGA, and PDI during maximal maneuvers like sniff and cough.51,52 Maximal POES and PDI during sniff maneuvers represent reliable indexes of the strength of global inspiratory muscles and of the diaphragm, respectively;36,53,54 while PGA during maximal cough is an index of the strength of expiratory muscles.51,55 Because cough efficacy depends on the inspiratory volume preceding the maneuver, lung volume is a source of variability and should be controlled.56
Table 1 summarizes the different techniques, categorized into invasive/non-invasive, and volitional/non-volitional, which are used to evaluate different ventilatory functional parameters in DMD patients, as reported in the literature.
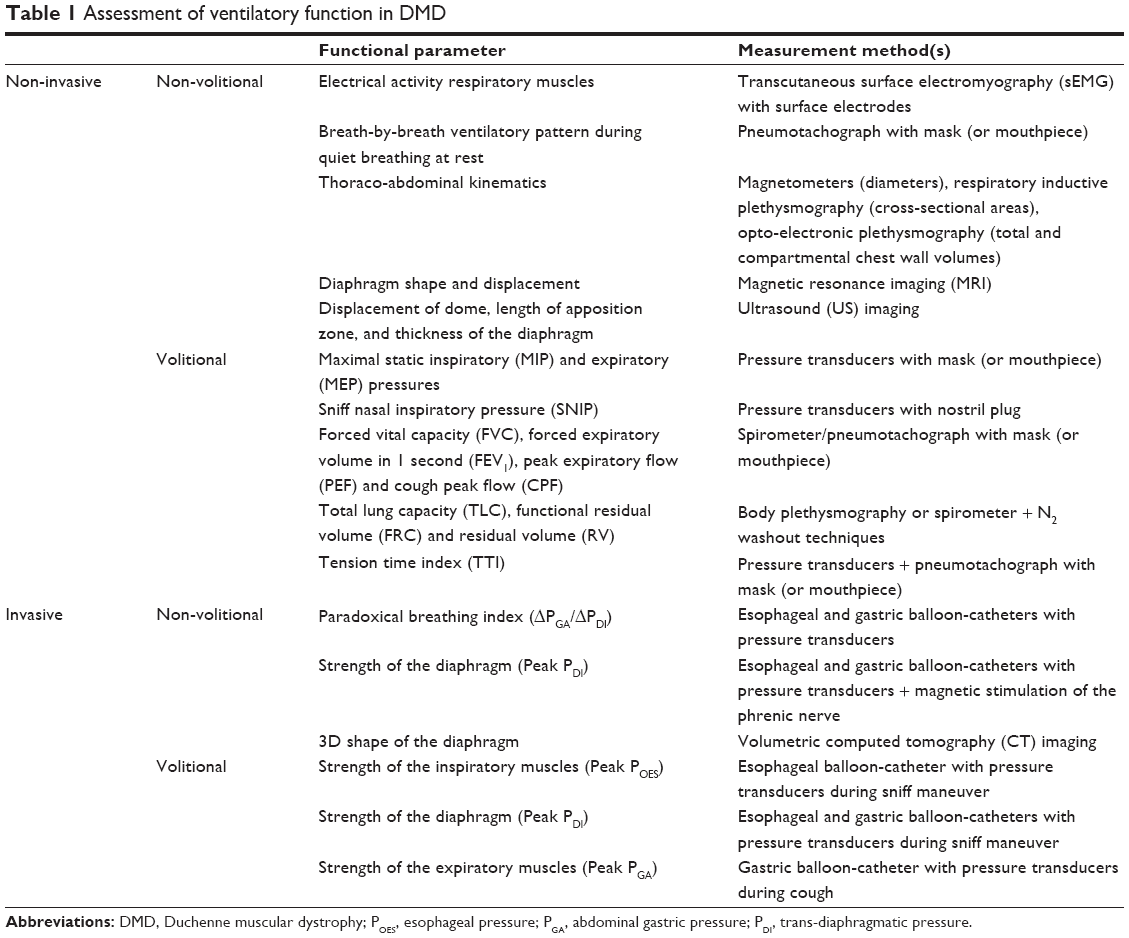 | Table 1 Assessment of ventilatory function in DMD |
Respiratory function in DMD
Respiratory function assessed by non-invasive non-volitional tests
During spontaneous quiet breathing at rest in supine position, VE decreases because of the reduction of VT with preserved respiratory rate, particularly in older DMD patients.57 In seated position VE does not change significantly because respiratory rate increases to compensate for the fall of VT.37,58,59
The abdominal contribution to VT in supine position during inspiration is an index of the action of the diaphragm. It progressively decreases with age and it is an important predictor of nocturnal hypoexemia57,60 and inefficient cough.61
Quantitative muscle US is a non-invasive child-friendly tool able to measure DMD muscle changes in terms of both echo intensity and muscle thickness.62 DMD patients are characterized by poor spirometry, reduced inspiratory muscle force (lower MIP), and higher diaphragmatic thickness, measured at its appositional zone by B-mode ultrasonography, compared to healthy controls. This may indicate pseudo-hypertrophy of the diaphragm secondary to infiltration of connective tissue and fat deposition as occurs in limb muscles.63
Recent studies investigating MRI measurements of T2 relaxation time (MRI-T2) on lower limb muscles in patients and on canine model confirm the feasibility of MRI to objectively and non-invasively measure muscle pathology in DMD.64–67 MRI-T2 of the sternocleidomastoid muscle of DMD patients are significantly lower than an age-matched control group indicating altered tissue composition.68 MRI, therefore, represents a potentially promising technique for early detection of disease progression not only of limb muscles but also of the diaphragm and other respiratory muscles in DMD. It can also be used to assess the efficacy of therapeutic interventions on the muscles.
Respiratory function assessed by non-invasive volitional tests
The absolute value of FVC follows a pathognomonic pattern characterized by an ascending phase, a peak approximately at 12–14 years, and a plateau followed by a descending phase.59,69–74 The absolute value reached at the plateau correlates with life expectancy in these patients.75 Values of FVC lower than 2.1 L are associated to lower CPF and represent important thresholds to start with techniques that improve airway clearance.76 FVC below 1 L is a suitable indicator to consider starting more intensive patient support,69 being a good predictor of severe oxygen desaturation, uncontrolled hypercapnia, and impending death within 12 months.69,77,78 The decline in FVC has recently been shown to be associated with ventilation inhomogeneity assessed by lung clearance index measured with the multiple-breath washout technique.58 The maximal percentage postural difference of FVC in DMD patients is approximately 9%. It tends to decrease with age like MIP values. This can indicate that a global weakness of both inspiratory and expiratory muscles is not able to compensate for the diaphragmatic impairment.79
In DMD, FVC expressed as percentage of the predicted values (FVC%) starts approximately 80% at the age of 10 and then it linearly declines with age as shown by different authors indicating a progressive respiratory muscles weakness.59,69,70,72–74,80–83 FVC% and its decline are prognostic markers in DMD.69
Inspiratory flow reserve volume is defined as the ratio between the maximal inspiratory flow during tidal breathing and during FVC. This ratio is higher in DMD compared to controls indicating a reduction in inspiratory flow reserve in DMD.84
When listed as a function of age, the absolute values of FEV1, functional residual capacity, TLC, RV, inspiratory and expiratory reserve volumes reveal ascending, plateau, and descending phases like FVC.70,73,74 Between the ages of 8 and 12 the restrictive pattern is mainly due to reduced expiratory reserve volumes and increased RV. This can indicate an early deterioration of expiratory muscles. After the age of 16, the decline of lung volumes is associated with reduced inspiratory reserve volumes and TLC and therefore with inspiratory muscles’ impairment.70
MIP and MEP in Duchenne patients are reduced compared to normal values and they decrease with age, with MEP being globally lower than MIP. Expiratory muscles, therefore, seem to be more compromised while inspiratory muscles seem to be relatively preserved.59,70,81 MIP predicts the extent of hypercapnia70 and starts to decline from the age of 10 years onward. Because FVC decay starts a bit later, MIP maneuver seems to be affected earlier than spirometry.72 The mean decline with age of MIP varies from 4% to 6.9%.59,70,72
MEP is associated with cough efficiency: if MEP is higher than 60 cm H2O, DMD patient is expected to have an adequate cough; while MEP below 45 cm H2O is an indicator to start cough-assisting techniques.8,85,86
SNIP increases until the age of 10.5 and decreases thereafter with a mean decline of 4.8 cm H2O (5.2% predicted) per year. Because it declines earlier than FVC, it can be considered an early sign of inspiratory muscle strength. Reliable values of SNIP can be obtained from the age of 5 and therefore its measure should be initiated early in the course of the disease.59,71
With the progression of DMD, fatigue index (TTI) increases37,87 and consequently, without any intervention, older patients are likely to approach respiratory muscle fatigue.
Respiratory function assessed by invasive non-volitional tests
As indicated earlier, ΔPGA/ΔPDI ratio measured at rest during spontaneous quiet breathing is a non-volitional marker that reflects the relative contribution of the diaphragm to VT. In teenagers with DMD, ΔPGA/ΔPDI is negative indicating that the diaphragm is weaker than the other inspiratory muscles and it paradoxically moves upward. PDI secondary to magnetic stimulation of the phrenic nerve is lower in DMD patients than patients with spinal muscle atrophy and congenital myopathy indicating that Duchenne patients are characterized by the weakest diaphragm.51
Respiratory function assessed by invasive volitional tests
Trans-diaphragmatic and POES during sniff are low and progressively decrease after the age of 10 years. The evolution of gastric pressure during cough follows a similar decrement pattern, being significantly reduced by the age of 8 years.51,59
Figure 1 shows a schematic diagram summarizing the natural course of the different respiratory functional parameters over time according to what has been reported in the up-to-date literature.
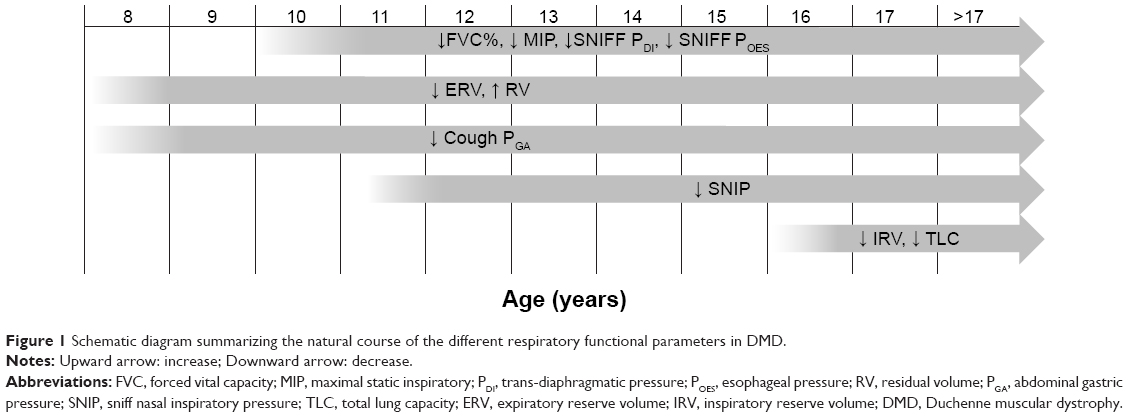 | Figure 1 Schematic diagram summarizing the natural course of the different respiratory functional parameters in DMD. |
Pharmacological intervention
There is no current curative treatment for DMD and the only medications available and used are glucocorticoid steroids.3,88 The main effects of corticosteroids on skeletal muscles are inflammation reduction, mass increment and deterioration delay. Steroid therapy delays the motor impairment of DMD children by improving muscular strength leading to prolonged ambulation for up to 2–3 years longer.16,89–95 At the beginning, steroid therapy was prescribed to improve motor function. Successively some authors suggested prolonging the treatment also after patients are wheelchair bound for its potential effect in preventing scoliosis and respiratory deterioration. There are studies showing no effect of steroid therapy on the respiratory function in treated DMD patients.80,83 A stabilization of FVC and FEV1 after 2 years of treatment has been described96 while another study reports a significant increase of MEP and cough peak flow (CPF) after treatment without FVC improvement/stabilization.97 Many authors have found the effectiveness of steroid treatment in improving spirometry in their cohort of DMD patients.98–100 Two reasons can explain such a discrepancy in the role of steroid therapy on lung function in DMD: the lack of standardization in steroid administration and/or the binary subdivision (steroid vs non-steroid) according to the use of therapy. The non-steroid group includes not only young patients, but also old patients who never took the therapy (because its introduction in the clinical management of DMD is relatively new) together with patients who chronically took steroids but in a discontinuous way. As already pointed out by Mayer et al80 the authors believe that the heterogeneity of the two groups can introduce a bias effect on the results since old and therefore more compromised patients can worsen the overall lung function of the group, while the discontinuous-users can contribute in a positive way thanks to the possible beneficial effect of the therapy. The cooperative international neuromuscular research group Duchenne natural history study15,16 groups 340 DMD patients according to age and to the use of steroids as follows: 1) naïve patients: never used steroids or treated <1 month total and not currently under medication; 2) current recipients; and 3) past users: treated in the past for >1 month and not currently under medication. An overall improved lung function in terms of FVC, FEV1, PEF, MIP, and MEP has been found in the current steroid users across the age groups 10–12 and 13–15 years old.15,16 This demonstrates how crucial it is to properly subdivide patients while testing the efficacy of new therapy in order to avoid possible bias due to the patients’ heterogeneity in their clinical conditions because of the progressive nature of the disease.
Since the use of corticosteroids has well-known side effects, such as weight gain, cataract, osteopenia, behavioral changes, increased insulin resistance, and delayed puberty, there is a high medical need for new effective, safe, and well-tolerated pharmacological therapy able to slow the impairment or even ameliorate the respiratory function in DMD. In a preclinical placebo-controlled study in the mdx mouse idebenone, a potent antioxidant shows cardio-protective effect and improvement in exercise performance.101 A 12-month Phase 2 randomized placebo-controlled trial on a small group of DMD patients shows a significant treatment effect not only on cardiac markers, but also on PEF, as absolute values and as percentage of the predicted values. The beneficial effect of idebenone on PEF is more pronounced in naïve steroid patients rather than in current users, suggesting an inhibitory effect of glucocorticoid steroids.98 Finally, the trial reaches Phase 3 by studying the efficacy of idebenone on respiratory function in DMD patients not on concomitant steroid therapy. Idebenone significantly reduces the decline of different parameters of the respiratory function, namely PEF, FVC, and FEV1, and results have shown it to be safe and well tolerated by patients therefore representing a new therapeutic approach for DMD.102
The goal for future pharmacological therapy is to have at least equal efficacy than glucocorticosteroids but with lower side effect profile. Multiple new treatment approaches have emerged ranging from molecular based therapies aimed to reduce muscle fibrosis or to promote muscle blood flow. It seems more likely that a combination of different approaches, targeting more than one pathway, may be more satisfactory.17,18 Alternative approaches including mutation-specific therapy (gene replacement or repair and stem cell therapy) represent the future possibility to find a definite cure for DMD. There have been genetic-based treatment experiments in the last few years in clinical trials, ie, ataluren for non-sense mutation in the dystrophin gene and antisense oligonucleotide induced exon skipping.103,104 The promising results of some of these studies still have to face the lack of accurate clinical and biological outcome measures able to discriminate between and describe modifications in the disease history.
The ongoing clinical trials are not specifically addressed to study new therapeutic effects on respiratory muscles and consequently do not have respiratory function measures as primary outcomes. The following trials consider respiratory variables as secondary outcome in addition to other clinical and functional measures: a) evaluating the long-term safety of ACE-031 in subjects with DMD;105 b) long-term safety trial of ataluren to treat Becker and Duchenne muscular dystrophies;105 c) NCT02286947;106 d) NCT00264888;106 e) NCT00759876;106 f) NCT01009294;106 g) NCT01239758;106 h) NCT01099761;106 and i) NCT01462292.106
Respiratory muscle training (RMT) and physiotherapy intervention
Respiratory muscles are morphologically and functionally skeletal muscles and therefore RMT can be a possible treatment to improve their strength and endurance and to try to counteract the progressive weakness induced by DMD. The role of RMT in neuromuscular patients is still controversial being potentially dangerous since it may accelerate muscle fatigue by overwork. Beneficial effects of RMT have been shown in DMD. These comprise increased strength of expiratory muscle,107 improved muscle endurance,108 and augmented MIP.109 Another study demonstrates that inspiratory muscle training (IMT) through resistive breathing and/or efforts against a variable inspiratory resistance allows diaphragmatic training in terms of strength and endurance. Peak esophageal and PDI during sniff maneuvers have been measured before and after 6 months of training. In ten out of 15 patients, respiratory muscle function parameters improved and the effect has remained even 6 months after the end of training. RMT is not effective in patients with FVC lower than 25% predicted and/or PaCO2 higher than 45 mmHg. It seems that RMT is not applicable in the most compromised patients, maybe because they do not have enough functional muscle mass left or because their respiratory muscles are so impaired and thus more susceptible to damage.110
IMT is strongly recommended preoperatively in case of spinal surgery in order to prevent pulmonary complication secondary to anesthesia.111 IMT improves the functionality of respiratory muscles by increasing FVC to 26.2% before surgery and by reducing postoperative risks.112
RMT can be simply implemented within video games with the possibility to control all the training processes. Improvements in respiratory performance correlate positively with the actual amount of training and negatively with baseline lung function test. This implies that RMT seems to be more effective in DMD patients who have moderate respiratory impairment.113
The main limitations of these studies are the reduced number of patients and the lack of standardized protocols. RMT therapeutic effects rely on two factors: the amount of training and the relatively good condition of the patient.13
LVR, also known as “breath stacking”, is a method to inflate and recruit the lung. It consists of an inspiratory pressure applied to the airways, the hold of the delivered volume of air by glottic closure, followed by a spontaneous or assisted forced expiratory maneuver. In DMD patients, the progressive impairment of the ventilatory pump reduces the range of motion of the chest wall and therefore the ribcage becomes stiff with reduced compliance of the chest wall and of the lung. The main consequence is the formation of areas of atelectasis. These lead to reduced oxygenation of the blood and to impaired CO2 elimination due to the imbalance between alveolar ventilation and blood perfusion of the capillary. LVR helps to prevent atelectasis, to limit contractures of the thoracic muscles, to maintain the motion of the chest wall particularly of the costo-vertebral joints, and to improve lung compliance. It can be used also in uncooperative and in very weak patients who cannot hold a mouthpiece and therefore it is applicable to DMD patients.114,115 LVR has been shown to slow the rate of decline of pulmonary function compared to the decline before the start. The values of FVC and peak cough flow improve after LVR.114,116 Because LVR can reach higher pressure compared to in-exsufflator, great care should be taken to avoid barotrauma.114 Failure to successfully stack the breath may occur in very sick patients whose VT is lower than the dead space of the mask and whose inspiratory effort is not enough to trigger the valve. In such a case, it is recommendable to use a supported breath stacking device in which a resuscitator bag helps to augment VT and to open the valve.115
Glossopharyngeal breathing (GPB) represents an autonomous lung-inflation maneuver that these patients should be taught, practiced and encouraged particularly in the last stage of the disease. GPB has been proven to increase lung volume by inflating one gulp at a time and to delay the onset of daytime ventilator use. It can be performed in the event of ventilator failure and during the inspiratory cough phase, since cough flow correlates with pre-cough volume.116
Recently, a 10-month program of yoga breathing exercises has been shown to ameliorate the pulmonary function in DMD patients in terms of significant improvement of FVC and FEV1.117
Finally, very severe DMD patients use a spontaneous movement called the “row-a-boat phenomenon”, ie, swing of the torso back and front rhythmically synchronized with fR, to compensate for the atrophied respiratory muscles. By simply allowing the row-a-boat movement, FVC increases up to 50.8% compared to the values reached when the movement is prevented.118
Management of scoliosis and spinal fusion
In the natural history of DMD, the development of scoliosis is a frequent complication that impacts on respiratory function, seating, and comfort. Ninety percent of untreated DMD boys develop scoliosis, secondary to muscle weakness, pelvic imbalance,119 and after becoming wheelchair bound full-time.120 For this reason, prolonging ambulation in DMD patients is one of the main strategies to delay the onset of scoliosis.121 Other strategies include physiotherapy (passive stretching) to maintain lower limb range of motion, night splints or serial casting.3,122 Moreover, surgery can release contractures of lower limbs and then rehabilitation with “KAFO” (knee ankle foot orthoses) may prolong standing and walking for 2 years.3,119,122–125 There is level III-2 evidence that the use of corticosteroids delays the onset of scoliosis by extending independent ambulation. Steroid therapy has been shown to reduce the need for surgical treatment to reduce scoliosis in DMD from 90% to 15%.88,100,119,126,127 Long-term corticosteroid treatment, however, carries important side effects including worsened bone fragility and the risk of vertebral fractures.
When the scoliosis curvature exceeds the 40° of Cobb angle, scoliosis rapidly progresses and may impair breathing by reducing lung capacity on both the convex and the concave sides.128 Since spinal orthoses seem to have minimal effects on preventing, delaying or minimally correcting the curve progression, surgical correction becomes the indication.129 Because of the cardiopulmonary impairment of DMD, spinal surgery is better when performed during early adolescence in order to avoid the high risk of developing postoperative pneumonia, prolonging ventilatory dependence, cardiac failure, and eventually death.130 In the literature, there are conflicting reports about the effect of surgical spine stabilization on pulmonary function. They all consider the evolution of FVC. There are studies reporting positive effects of spinal fusion in terms of stabilization of FVC131,132 or slightly slowing FVC deterioration with respect to a control non-operative group or to pre-operative patients.128,133–135 It should be noted that in these papers there was a continuous reduction in FVC after surgery, although to a lesser extent. Other studies have found no ameliorative effects on the decline of pulmonary function.83,136–141
The decision to have surgical fixation of the spine therefore has to be considered mainly to improve the general well-being of the patient (maintenance of comfortable sitting balance, release of pain due to muscle contractures, and improvement of quality of life) rather than to improve his respiratory function which will continue to decline anyway. Nevertheless, in DMD patients the combination of spinal fusion with nocturnal ventilation prolongs median survival to 30 years.141
Cough assisted devices
The main causes that require intubation for DMD patients are episodes of pneumonia and acute respiratory failure secondary to benign upper-respiratory infection. This is a consequence of retained secretion due to inability to cough effectively.142 In DMD, cough is considered adequate when CPF >270 L/min and ineffective if CPF <160 L/min and therefore cough augmentation is required. When CPF is between 160 and 270 L/min cough is considered effective when the patient is in good and stable condition, but it becomes inadequate in case of viral illness when secretions increase.111,143,144 Once assisted coughing is recommended, it should be used not only when the patient is ill, but once or twice-per-day as maintenance therapy to improve airway clearance and reduce atelectasis. Since CPF strongly depends on the volume of air inspired pre-coughing,56 the first goal of cough assisted techniques is to maximally inflate the lungs. This can be achieved autonomously by GPB, manually through the use of an inflating Ambu bag or mechanically assisted. The expulsive cough phase can be spontaneous, when the patient is still able to generate sufficient intrathoracic pressures, manually assisted by abdominal compression or mechanically forced by a cough assisted device. The combination of inspiratory volume support with manual assisted expiratory cough phase is more efficacious than the single intervention per se.145,146 The use of mechanical in-exsufflator is effective in preventing pulmonary complications like pneumonia and respiratory failure and in decreasing the number of hospitalizations due to respiratory illness.4 The addition of intrapulmonary percussive ventilation to assist mucus clearance techniques is a safe method to augment the efficacy of these methods to keep the airways of tracheotomized DMD patients clear.147 Care must be taken to adequately pre-set the timing and the levels of pressure in order to be well tolerated by the patient, to maximize cough effectiveness, and to avoid possible complications like pneumothorax, gastroesophageal reflux, and pulmonary hemorrhage.
CPF correlates with MIP and MEP97,146 and all the three parameters improve after long-term steroid therapy. Therefore the steroid-induced preservation of both inspiratory and expiratory muscle strength translates to better cough efficiency.97
The combination of cough assisted devices with NIV before surgery helps in easy extubation after surgery without postoperative respiratory complications.148
NIV
The onset or chronic respiratory insufficiency is a fatal inevitable complication of DMD. Patients not treated with mechanical ventilation die by the end of the second decade of life.7,37,75,77 The use of NIV combined with adequate airway clearance prolongs survival up to the third decade of life in DMD patients.7,9,10,77 In these patients, nocturnal NIV helps to reduce the fatigue of respiratory muscles by lowering TTI. As a result dyspnea reduces, as indicated by lower Borg scale values. After nocturnal NIV, vital capacity slightly increases while MIP does not change. Two additional hours of diurnal NIV improves the respiratory condition of severe DMD patients who present with nocturnal hypoventilation symptoms like breathlessness in the evening. In these patients, the benefits of nocturnal NIV last 6 hours. The same levels of lower TTI and reduced dyspnea are restored and maintained after the 2 additional hours of diurnal NIV that represents a good tool to reduce the load on the respiratory muscles rising from morning to evening.37 This is an important therapeutic intervention for DMD patients who are dyspneic during diurnal unassisted breathing.
Moreover, intermittent positive-pressure breathing (IPPB) shows positive effects on regional lung ventilation. A session of 30 consecutive breaths up to 30 cm H2O performed in different positions (supine, left and right lateral position) increases the regional ventilation of the lungs. Such increment is maintained also 3 hours after the end of IPPB if the patient wears an abdominal belt. The supine position is characterized by the greatest improvement in lung ventilation particularly in the anterior regions. In order to reduce risks of hyperinflation in only one part of the lung, it is recommended to use several positions for the IPPB protocol.149
With the progression of the clinical course of the disease, DMD patients develop respiratory failure characterized by daytime hypercapnia, hypoxia, and impairment in swallowing. The traditional approach to deal with this last stage of the disease would be to introduce tracheostomy ventilation.150 Twenty-four hours’ NIV represents a safe and non-invasive alternative to tracheostomy.9,143,151 The advantage of 24-hour NIV compared to invasive ventilation is the possibility to engage normal verbal communication, to eat, to breath-stack as desired, and to avoid the clinical complications of tracheotomized patients. Dysphagia also benefits from 24 hours of NIV maybe because oropharyngeal muscles improve their strength and endurance associated with the reduced work of breathing. The swallowing mechanism may improve and therefore it can support better nutrition.152
Tracheostomy ventilation becomes necessary in case of severe aspiration risk, severe respiratory infection, and after onset of severe bulbar weakness that is rarely developed in patients with DMD. The combined use of NIV and mechanically assisted cough allows the decannulation and extubation even for unweanable DMD patients.9,153
Animal studies
DMD animal models, both mice and canine, are important reliable models for a physiological study of the disease and/or to test new pharmacological treatment efficacy, particularly for the diaphragm, the most important respiratory muscle. Exposure to episodic hypoxia mocks the sleep disorder breathing of DMD patients and it results in a 30% reduction in diaphragmatic strength in mdx mice.154 Fourteen weeks’ treatment with phosphodiesterase 5 inhibitors reduced the dysfunction of the diaphragm in mdx mice by enhancing the contractility of the diaphragm, by slowing muscle fibrosis, and by normalizing the expression of pro-inflammatory cytokine.155 Improvement in diaphragmatic health, in terms of muscle mass normalization and reduced fiber necrosis, can be obtained in neonatal mdx mice with the combination of antisense therapy with dystrophin restoration and myostatin inhibition.156 Reduction of the oxidative damage and of the inflammatory response in the mdx mouse diaphragm can be achieved using deferoxamine, a potent iron chelating agent.157 It is very important to restore diaphragmatic function not only from a respiratory point of view but also since the rescue of the diaphragm in mdx mice, by increasing its UTRN or dystrophin expression, prevented heart dysfunction. The decline in lung function secondary to respiratory muscles’ impairment increases pulmonary vascular pressure therefore leading to pulmonary hypertension.158 In DMD patients, pulmonary hypertension increases right atrial pressure with possible right ventricular failure.159 An early component of the hypercapnic ventilatory reflex has been revealed in mdx mice, demonstrating a malfunction in the peripheral chemosensory drive. This is supposed to be a further contributor to the respiratory failure in mdx mice. Therefore the monitoring of peripheral chemosensory drive function may be useful to better manage DMD patients with respiratory failure.160 The DMD mdx mouse model with haploinsufficiency of the UTRN gene is characterized by more severe diaphragmatic fibrosis and may improve when testing antifibrotic therapies, especially in the most severe stage.161
Golden Retriever muscular dystrophy (GRMD) is a canine model of DMD. GRMD dogs guarantee an adequate level of ventilation at rest by increasing their respiratory rate in order to compensate for a lower VT compared to normal dogs. GRMD does not show abdominal paradox. When the respiratory drive is stimulated by doxapram, abdominal expiratory muscles are recruited early to compensate for the loss of diaphragmatic function. The diaphragm in GRMD is weaker, shorter, thicker, and severely fibrotic compared to the other respiratory and locomotor muscles.162
Conclusion
Until now clinical trials have not considered respiratory function variables as primary outcomes. This, however, would be highly recommended, as respiratory problems are among the main causes of death in DMD patients. The non-invasive and non-volitional methods and techniques for respiratory functional assessment which are nowadays available should be considered as a tool not only for experimental trials, but also for clinical monitoring starting during the very early stages of the disease.
Disclosure
The authors have no conflict of interest to declare.
References
Emery AE. Population frequencies of inherited neuromuscular diseases – a world survey. Neuromuscul Disord. 1991;1(1):19–29. | ||
Griggs RC, Bushby K. Continued need for caution in the diagnosis of Duchenne muscular dystrophy. Neurology. 2005;64(9):1498–1499. | ||
Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93. | ||
Gomez-Merino E, Bach JR. Duchenne muscular dystrophy: prolongation of life by noninvasive ventilation and mechanically assisted coughing. Am J Phys Med Rehabil. 2002;81(6):411–415. | ||
Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002;12(10):926–929. | ||
Saito T, Tatara K. Database of Wards for Patients with Muscular Dystrophy in Japan; 2012. Available from: http://cdn.intechopen.com/pdfs/36737/InTech-Database_of_wards_for_patients_with_muscular_dystrophy_in_japan.pdf. Accessed July 16, 2015. | ||
Passamano L, Taglia A, Palladino A, et al. Improvement of survival in Duchenne Muscular Dystrophy: Retrospective analysis of 835 patients. Acta Myol. 2012;31(2):121–125. | ||
Finder JD, Birnkrant D, Carl J, et al. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med. 2004;170(4):456–465. | ||
Bach JR, Martinez D. Duchenne muscular dystrophy: continuous noninvasive ventilatory support prolongs survival. Respir Care. 2011;56(6):744–750. | ||
Ishikawa Y, Miura T, Ishikawa Y, et al. Duchenne muscular dystrophy: survival by cardio-respiratory interventions. Neuromuscul Disord. 2011;21(1):47–51. | ||
Birnkrant DJ, Bushby KM, Amin RS, et al. The respiratory management of patients with Duchenne muscular dystrophy: A DMD care considerations working group specialty article. Pediatr Pulmonol. 2010;45(8):739–748. | ||
Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9(2):177–189. | ||
Finsterer J. Cardiopulmonary support in Duchenne muscular dystrophy. Lung. 2006;184(4):205–215. | ||
Wagner KR, Lechtzin N, Judge DP. Current treatment of adult Duchenne muscular dystrophy. Biochim Biophys Acta. 2007;1772(2):229–237. | ||
McDonald CM, Henricson EK, Abresch RT, et al. The cooperative international neuromuscular research group Duchenne natural history study – a longitudinal investigation in the era of glucocorticoid therapy: design of protocol and the methods used. Muscle Nerve. 2013;48(1):32–54. | ||
Henricson EK, Abresch RT, Cnaan A, et al. The cooperative international neuromuscular research group Duchenne natural history study: glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and othe. Muscle Nerve. 2013;48(1):55–67. | ||
Malik V, Rodino-Klapac LR, Mendell JR. Emerging drugs for Duchenne muscular dystrophy. Expert Opin Emerg Drugs. 2012;17(2):261–277. | ||
Beytía Mde L, Vry J, Kirschner J. Drug treatment of Duchenne muscular dystrophy: available evidence and perspectives. Acta Myol. 2012;31(1):4–8. | ||
American Thoracic Society/European Respiratory Society. ATS/ERS Statement on respiratory muscle testing. Am J Respir Crit Care Med. 2002;166(4):518–624. | ||
Duiverman ML, van Eykern LA, Vennik PW, Koëter GH, Maarsingh EJ, Wijkstra PJ. Reproducibility and responsiveness of a noninvasive EMG technique of the respiratory muscles in COPD patients and in healthy subjects. J Appl Physiol (1985). 2004;96(5):1723–1729. | ||
Beck J, Sinderby C, Weinberg J, Grassino A. Effects of muscle-to-electrode distance on the human diaphragm electromyogram. J Appl Physiol (1985). 1995;79(3):975–985. | ||
Wohlgemuth M, van der Kooi EL, Hendriks JC, Padberg GW, Folgering HT. Face mask spirometry and respiratory pressures in normal subjects. Eur Respir J. 2003;22(6):1001–1006. | ||
Tobin MJ, Perez W, Guenther SM, et al. The pattern of breathing during successful and unsuccessful trials of weaning from mechanical ventilation. Am Rev Respir Dis. 1986;134(6):1111–1118. | ||
Mead J, Peterson N, Grimby G, Mead J. Pulmonary ventilation measured from body surface movements. Science. 1967;156(3780):1383–1384. | ||
Chadha TS, Watson H, Birch S, et al. Validation of respiratory inductive plethysmography using different calibration procedures. Am Rev Respir Dis. 1982;125(6):644–649. | ||
Cala SJ, Kenyon CM, Ferrigno G, et al. Chest wall and lung volume estimation by optical reflectance motion analysis. J Appl Physiol (1985). 1996;81(6):2680–2689. | ||
Aliverti A, Dellacà R, Pelosi P, Chiumello D, Gatihnoni L, Pedoti A. Compartmental analysis of breathing in the supine and prone positions by optoelectronic plethysmography. Ann Biomed Eng. 2001;29(1):60–70. | ||
Romei M, Mauro A Lo, D’Angelo MG, et al. Effects of gender and posture on thoraco-abdominal kinematics during quiet breathing in healthy adults. Respir Physiol Neurobiol. 2010;172(3):184–191. | ||
Boudarham J, Pradon D, Prigent H, et al. Optoelectronic plethysmography as an alternative method for the diagnosis of unilateral diaphragmatic weakness. Chest. 2013;144(3):887–895. | ||
Cluzel P, Similowski T, Chartrand-Lefebvre C, Zelter M, Derenne JP, Grenier PA. Diaphragm and chest wall: assessment of the inspiratory pump with MR imaging-preliminary observations. Radiology. 2000;215(2):574–583. | ||
Gauthier AP, Verbanck S, Estenne M, Segebarth C, Macklem PT, Paiva M. Three-dimensional reconstruction of the in vivo human diaphragm shape at different lung volumes. J Appl Physiol (1985). 1994;76(2):495–506. | ||
Ueki J, De Bruin PF, Pride NB. In vivo assessment of diaphragm contraction by ultrasound in normal subjects. Thorax. 1995;50(11):1157–1161. | ||
Testa A, Soldati G, Giannuzzi R, et al. Ultrasound M-mode assessment of diaphragmatic kinetics by anterior transverse scanning in healthy subjects. Ultrasound Med Biol. 2011;37(1):44–52. | ||
Evans JA, Whitelaw WA. The assessment of maximal respiratory mouth pressures in adults. Respir Care. 2009;54(10):1348–1359. | ||
Stefanutti D, Fitting JW. Sniff nasal inspiratory pressure. Reference values in Caucasian children. Am J Respir Crit Care Med. 1999;159(1):107–111. | ||
Fauroux B, Aubertin G, Cohen E, Clément A, Lofaso F. Sniff nasal inspiratory pressure in children with muscular, chest wall or lung disease. Eur Respir J. 2009;33(1):113–117. | ||
Toussaint M, Soudon P, Kinnear W. Effect of non-invasive ventilation on respiratory muscle loading and endurance in patients with Duchenne muscular dystrophy. Thorax. 2008;63(5):430–434. | ||
Hayot M, Ramonatxo M, Matecki S, Milic-Emili J, Prefaut C. Noninvasive assessment of inspiratory muscle function during exercise. Am J Respir Crit Care Med. 2000;162(6):2201–2207. | ||
Hussain SN, Pardy RL. Inspiratory muscle function with restrictive chest wall loading during exercise in normal humans. J Appl Physiol (1985). 1985;58(6):2027–2032. | ||
Mulreany LT, Weiner DJ, McDonough JM, Panitch HB, Allen JL. Noninvasive measurement of the tension-time index in children with neuromuscular disease. J Appl Physiol (1985). 2003;95(3):931–937. | ||
Bianchi C, Baiardi P. Cough peak flows: standard values for children and adolescents. Am J Phys Med Rehabil. 2008;87(6):461–467. | ||
Fromageot C, Lofaso F, Annane D, et al. Supine fall in lung volumes in the assessment of diaphragmatic weakness in neuromuscular disorders. Arch Phys Med Rehabil. 2001;82(1):123–128. | ||
Miller MR, Crapo R, Hankinson J, et al. General considerations for lung function testing. Eur Respir J. 2005;26(1):153–161. | ||
Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. | ||
Wanger J, Clausen JL, Coates A, et al. Standardisation of the measurement of lung volumes. Eur Respir J. 2005;26(3):511–522. | ||
Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J. 2005;26(5):948–968. | ||
Hillman DR, Finucane KE. Respiratory pressure partitioning during quiet inspiration in unilateral and bilateral diaphragmatic weakness. Am Rev Respir Dis. 1988;137(6):1401–1405. | ||
Gilbert R, Auchincloss JH, Peppi D. Relationship of rib cage and abdomen motion to diaphragm function during quiet breathing. Chest. 1981;80(5):607–612. | ||
Mills GH, Kyroussis D, Hamnegard CH, Polkey MI, Green M, Moxham J. Bilateral magnetic stimulation of the phrenic nerves from an anterolateral approach. Am J Respir Crit Care Med. 1996;154(4 Pt 1):1099–1105. | ||
Pettiaux N, Cassart M, Paiva M, Estenne M. Three-dimensional reconstruction of human diaphragm with the use of spiral computed tomography. J Appl Physiol (1985). 1997;82(3):998–1002. | ||
Nicot F, Hart N, Forin V, et al. Respiratory muscle testing: a valuable tool for children with neuromuscular disorders. Am J Respir Crit Care Med. 2006;174(1):67–74. | ||
Fauroux B, Quijano-Roy S, Desguerre I, Khirani S. The value of respiratory muscle testing in children with neuromuscular disease. Chest. 2015;147(2):552–559. | ||
Héritier F, Rahm F, Pasche P, Fitting JW. Sniff nasal inspiratory pressure. A noninvasive assessment of inspiratory muscle strength. Am J Respir Crit Care Med. 1994;150(6 Pt 1):1678–1683. | ||
Laroche CM, Mier AK, Moxham J, Green M. The value of sniff esophageal pressures in the assessment of global inspiratory muscle strength. Am Rev Respir Dis. 1988;138(3):598–603. | ||
Man WD, Kyroussis D, Fleming TA, et al. Cough gastric pressure and maximum expiratory mouth pressure in humans. Am J Respir Crit Care Med. 2003;168(6):714–717. | ||
Smith JA, Aliverti A, Quaranta M, et al. Chest wall dynamics during voluntary and induced cough in healthy volunteers. J Physiol. 2012;590(Pt 3):563–574. | ||
Lo Mauro A, D’Angelo MG, Romei M, et al. Abdominal volume contribution to tidal volume as an early indicator of respiratory impairment in Duchenne muscular dystrophy. Eur Respir J. 2010;35(5):1118–1125. | ||
Stehling F, Dohna-Schwake C, Mellies U, Grosse-Onnebrink J. Decline in Lung Volume With Duchenne Muscular Dystrophy Is Associated With Ventilation Inhomogeneity. Respir Care. Epub 2015 May 5. | ||
Khirani S, Ramirez A, Aubertin G, et al. Respiratory muscle decline in Duchenne muscular dystrophy. Pediatr Pulmonol. 2014;49(5):473–481. | ||
Romei M, D’Angelo MG, LoMauro A, et al. Low abdominal contribution to breathing as daytime predictor of nocturnal desaturation in adolescents and young adults with Duchenne Muscular Dystrophy. Respir Med. 2012;106(2):276–283. | ||
Lomauro A, Romei M, D’Angelo MG, Aliverti A. Determinants of cough efficiency in Duchenne muscular dystrophy. Pediatr Pulmonol. 2014;49(4):357–365. | ||
Jansen M, van Alfen N, Nijhuis van der Sanden MW, van Dijk JP, Pillen S, de Groot IJ. Quantitative muscle ultrasound is a promising longitudinal follow-up tool in Duchenne muscular dystrophy. Neuromuscul Disord. 2012;22(4):306–317. | ||
De Bruin PF, Ueki J, Bush A, Khan Y, Watson A, Pride NB. Diaphragm thickness and inspiratory strength in patients with Duchenne muscular dystrophy. Thorax. 1997;52(5):472–475. | ||
Wang J, Fan Z, Vandenborne K, et al. A computerized MRI biomarker quantification scheme for a canine model of Duchenne muscular dystrophy. Int J Comput Assist Radiol Surg. 2013;8(5):763–774. | ||
Hollingsworth KG, Garrood P, Eagle M, Bushby K, Straub V. Magnetic resonance imaging in Duchenne muscular dystrophy: longitudinal assessment of natural history over 18 months. Muscle Nerve. 2013;48(4):586–588. | ||
Senesac CR, Lott DJ, Forbes SC, et al. Longitudinal Evaluation of Muscle Composition Using Magnetic Resonance in 4 Boys With Duchenne Muscular Dystrophy: Case Series. Phys Ther. 2015;95(7):978–988. | ||
Willcocks RJ, Arpan IA, Forbes SC, et al. Longitudinal measurements of MRI-T2 in boys with Duchenne muscular dystrophy: effects of age and disease progression. Neuromuscul Disord. 2014;24(5):393–401. | ||
Mavrogeni S, Tzelepis GE, Athanasopoulos G, et al. Cardiac and sternocleidomastoid muscle involvement in Duchenne muscular dystrophy: an MRI study. Chest. 2005;127(1):143–148. | ||
Phillips MF, Quinlivan RC, Edwards RH, Calverley PM. Changes in spirometry over time as a prognostic marker in patients with duchenne muscular dystrophy. Am J Respir Crit Care Med. 2002;164(12):2191–2194. | ||
Hahn A, Bach JR, Delaubier A, Renardel-Irani A, Guillou C, Rideau Y. Clinical implications of maximal respiratory pressure determinations for individuals with Duchenne muscular dystrophy. Arch Phys Med Rehabil. 1997;78(1):1–6. | ||
Nève V, Cuisset JM, Edmé JL, et al. Sniff nasal inspiratory pressure in the longitudinal assessment of young Duchenne muscular dystrophy children. Eur Respir J. 2013;42(3):671–680. | ||
Gayraud J, Ramonatxo M, Rivier F, Humberclaude V, Petrof B, Matecki S. Ventilatory parameters and maximal respiratory pressure changes with age in Duchenne muscular dystrophy patients. Pediatr Pulmonol. 2010;45(6):552–559. | ||
Tangsrud S, Petersen IL, Lødrup Carlsen KC, Carlsen KH. Lung function in children with Duchenne’s muscular dystrophy. Respir Med. 2001;95(11):898–903. | ||
Humbertclaude V, Hamroun D, Bezzou K, et al. Motor and respiratory heterogeneity in Duchenne patients: implication for clinical trials. Eur J Paediatr Neurol. 2012;16(2):149–160. | ||
Rideau Y, Jankowski LW, Grellet J. Respiratory function in the muscular dystrophies. Muscle Nerve. 1981;4(2):155–164. | ||
Gauld LM, Boynton A. Relationship between peak cough flow and spirometry in Duchenne muscular dystrophy. Pediatr Pulmonol. 2005;39(5):457–460. | ||
Toussaint M, Chatwin M, Soudon P. Mechanical ventilation in Duchenne patients with chronic respiratory insufficiency: clinical implications of 20 years published experience. Chron Respir Dis. 2007;4(3):167–177. | ||
Phillips MF, Smith PE, Carroll N, Edwards RH, Calverley PM. Nocturnal oxygenation and prognosis in Duchenne muscular dystrophy. Am J Respir Crit Care Med. 1999;160(1):198–202. | ||
Won YH, Choi WA, Kim DH, Kang SW. Postural Vital Capacity Difference with Aging in Duchenne Muscular Dystrophy. Muscle Nerve. Epub 2015 Feb 27. | ||
Mayer OH, Finkel RS, Rummey C, et al. Characterization of pulmonary function in Duchenne Muscular Dystrophy. Pediatr Pulmonol. 2015;50(5):487–494. | ||
McDonald CM, Abresch RT, Carter GT, et al. Profiles of neuromuscular diseases. Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1995;74(5 Suppl):S70–S92. | ||
Steffensen BF, Lyager S, Werge B, Rahbek J, Mattsson E. Physical capacity in non-ambulatory people with Duchenne muscular dystrophy or spinal muscular atrophy: a longitudinal study. Dev Med Child Neurol. 2002;44(9):623–632. | ||
Roberto R, Fritz A, Hagar Y, et al. The natural history of cardiac and pulmonary function decline in patients with duchenne muscular dystrophy. Spine (Phila Pa 1976). 2011;36(15):E1009–E1017. | ||
De Bruin PF, Ueki J, Bush A, Y Manzur A, Watson A, Pride NB. Inspiratory flow reserve in boys with Duchenne muscular dystrophy. Pediatr Pulmonol. 2001;31(6):451–457. | ||
Boitano LJ. Management of airway clearance in neuromuscular disease. Respir Care. 2006;51(8):913–922. | ||
Panitch HB. Respiratory issues in the management of children with neuromuscular disease. Respir Care. 2006;51(8):885–893. | ||
Matecki S, Topin N, Hayot M, et al. A standardized method for the evaluation of respiratory muscle endurance in patients with Duchenne muscular dystrophy. Neuromuscul Disord. 2001;11(2):171–177. | ||
Bushby K, Muntoni F, Urtizberea A, Hughes R, Griggs R. Report on the 124th ENMC International Workshop. Treatment of Duchenne muscular dystrophy; defining the gold standards of management in the use of corticosteroids. 2–4 April 2004, Naarden, The Netherlands. Neuromuscul Disord. 2004;14(8–9):526–534. | ||
Yilmaz O, Karaduman A, Topaloğlu H. Prednisolone therapy in Duchenne muscular dystrophy prolongs ambulation and prevents scoliosis. Eur J Neurol. 2004;11(8):541–544. | ||
Mendell JR, Moxley RT, Griggs RC, et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320(24):1592–1597. | ||
Fenichel GM, Florence JM, Pestronk A, et al. Long-term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurology. 1991;41(12):1874–1877. | ||
Fenichel GM, Mendell JR, Moxley RT, et al. A comparison of daily and alternate-day prednisone therapy in the treatment of Duchenne muscular dystrophy. Arch Neurol. 1991;48(6):575–579. | ||
Griggs RC, Moxley RT 3rd, Mendell JR, et al. Prednisone in Duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical Investigation of Duchenne Dystrophy Group. Arch Neurol. 1991;48(4):383–388. | ||
Connolly AM, Schierbecker J, Renna R, Florence J. High dose weekly oral prednisone improves strength in boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2002;12(10):917–925. | ||
Escolar DM, Hache LP, Clemens PR, et al. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology. 2011;77(5):444–452. | ||
Machado DL, Silva EC, Resende MB, Carvalho CR, Zanoteli E, Reed UC. Lung function monitoring in patients with duchenne muscular dystrophy on steroid therapy. BMC Res Notes. 2012;5(1):435. | ||
Daftary AS, Crisanti M, Kalra M, Wong B, Amin R. Effect of long-term steroids on cough efficiency and respiratory muscle strength in patients with Duchenne muscular dystrophy. Pediatrics. 2007;119(2):e320–e324. | ||
Buyse GM, Goemans N, Van Den Hauwe M, Meier T. Effects of glucocorticoids and idebenone on respiratory function in patients with duchenne muscular dystrophy. Pediatr Pulmonol. 2013;48(9): 912–920. | ||
Biggar WD, Gingras M, Fehlings DL, Harris VA, Steele CA. Deflazacort treatment of Duchenne muscular dystrophy. J Pediatr. 2001;138(1):45–50. | ||
Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. 2006;16(4):249–255. | ||
Buyse GM, Van der Mieren G, Erb M, et al. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance. Eur Heart J. 2009;30(1):116–124. | ||
Buyse GM, Voit T, Schara U, et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double-blind randomised placebo-controlled phase 3 trial. Lancet. 2015;385:1748–1757. | ||
Wilton SD, Veedu RN, Fletcher S. The emperor’s new dystrophin: finding sense in the noise. Trends Mol Med. 2015;21(7):417–426. | ||
Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50(4):477–487. | ||
Muscular Dystrophy UK [homepage on the Internet]. Find a clinical trial. Available from: http://www.musculardystrophyuk.org/progress-in-research/patient-registries-and-clinical-trials/clinical-trials/find-a-clinical-trial/page/2/?mdcore_clinicaltrial_region_tag=-1&mdcore_clinicaltrial_condition=duchenne-muscular-dystrophy. Accessed July 20, 2015. | ||
ClinicalTrials.gov. Duchenne search results. Available from: https://clinicaltrials.gov/ct2/results?term=duchenne&pg=5. Accessed July 20, 2015. | ||
Gozal D, Thiriet P. Respiratory muscle training in neuromuscular disease: long-term effects on strength and load perception. Med Sci Sports Exerc. 1999;31(11):1522–1527. | ||
Topin N, Matecki S, Le Bris S, et al. Dose-dependent effect of individualized respiratory muscle training in children with Duchenne muscular dystrophy. Neuromuscul Disord. 2002;12(6):576–583. | ||
Winkler G, Zifko U, Nader A, et al. Dose-dependent effects of inspiratory muscle training in neuromuscular disorders. Muscle Nerve. 2000;23(8):1257–1260. | ||
Wanke T, Toifl K, Merkle M, Formanek D, Lahrmann H, Zwick H. Inspiratory muscle training in patients with Duchenne muscular dystrophy. Chest. 1994;105(2):475–482. | ||
Birnkrant DJ, Panitch HB, Benditt JO, et al. American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Chest. 2007;132(6):1977–1986. | ||
Takaso M, Nakazawa T, Okada T, et al. Surgical management of severe scoliosis with high risk pulmonary dysfunction in Duchenne muscular dystrophy: Patient function, quality of life and satisfaction. Int Orthop. 2010;34(5):695–702. | ||
Vilozni D, Bar-Yishay E, Gur I, Shapira Y, Meyer S, Godfrey S. Computerized Respiratory Muscle Training in Children with Duchenne Muscular Dystrophy. Neuromuscul Disord. 1994;4(3):249–255. | ||
McKim DA, Katz SL, Barrowman N, Ni A, LeBlanc C. Lung volume recruitment slows pulmonary function decline in duchenne muscular dystrophy. Arch Phys Med Rehabil. 2012;93(7):1117–1122. | ||
Jenkins HM, Stocki A, Kriellaars D, Pasterkamp H. Breath stacking in children with neuromuscular disorders. Pediatr Pulmonol. 2014;49(6):544–553. | ||
Bach JR, Bianchi C, Vidigal-Lopes M, Turi S, Felisari G. Lung inflation by glossopharyngeal breathing and “air stacking” in Duchenne muscular dystrophy. Am J Phys Med Rehabil. 2007;86(4):295–300. | ||
Rodrigues MR, Carvalho CR, Santaella DF, Lorenzi-Filho G, Marie SK. Effects of yoga breathing exercises on pulmonary function in patients with Duchenne muscular dystrophy: an exploratory analysis. J Bras Pneumol. 2014;40(2):128–133. | ||
Yasuma F, Kato T, Matsuoka Y, Konagaya M. Row-a-boat phenomenon: Respiratory compensation in advanced Duchenne muscular dystrophy. Chest. 2001;119(6):1836–1839. | ||
Hsu JD, Quinlivan R. Scoliosis in Duchenne muscular dystrophy (DMD). Neuromuscul Disord. 2013;23(8):611–617. | ||
Shapiro F, Zurakowski D, Bui T, Darras BT. Progression of spinal deformity in wheelchair-dependent patients with Duchenne muscular dystrophy who are not treated with steroids: coronal plane (scoliosis) and sagittal plane (kyphosis, lordosis) deformity. Bone Joint J. 2014;96–B(1):100–105. | ||
Hsu JD. The development of current approaches to the management of spinal deformity for patients with neuromuscular disease. Semin Neurol. 1995;15(1):24–28. | ||
Scott OM, Hyde SA, Goddard C, Dubowitz V. Prevention of deformity in Duchenne muscular dystrophy. A prospective study of passive stretching and splintage. Physiotherapy. 1981;67(6):177–180. | ||
Kinali M, Main M, Eliahoo J, et al. Predictive factors for the development of scoliosis in Duchenne muscular dystrophy. Eur J Paediatr Neurol. 2007;11(3):160–166. | ||
Hyde SA, Scott OM, Goddard CM, Dubowitz V. Prolongation of ambulation in Duchenne muscular dystrophy by appropriate orthoses. Physiotherapy. 1982;68(4):105–108. | ||
Rodillo EB, Fernandez-Bermejo E, Heckmatt JZ, Dubowitz V. Prevention of rapidly progressive scoliosis in Duchenne muscular dystrophy by prolongation of walking with orthoses. J Child Neurol. 1988;3(4):269–274. | ||
Lebel DE, Corston JA, McAdam LC, Biggar WD, Alman BA. Glucocorticoid treatment for the prevention of scoliosis in children with Duchenne muscular dystrophy: long-term follow-up. J Bone Joint Surg Am. 2013;95(12):1057–1061. | ||
Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;(1):CD003725. | ||
Kurz LT, Mubarak SJ, Schultz P, Park SM, Leach J. Correlation of scoliosis and pulmonary function in Duchenne muscular dystrophy. J Pediatr Orthop. 1983;3(3):347–353. | ||
Harvey A, Baker L, Williams K. Non-surgical prevention and management of scoliosis for children with Duchenne muscular dystrophy: what is the evidence? J Paediatr Child Health. 2014;50(10):E3–E9. | ||
Duckworth AD, Mitchell MJ, Tsirikos AI. Incidence and risk factors for post-operative complications after scoliosis surgery in patients with Duchenne muscular dystrophy : a comparison with other neuromuscular conditions. Bone Joint J. 2014;96–B(7):943–949. | ||
Velasco MV, Colin AA, Zurakowski D, Darras BT, Shapiro F. Posterior spinal fusion for scoliosis in duchenne muscular dystrophy diminishes the rate of respiratory decline. Spine (Phila Pa 1976). 2007;32(4):459–465. | ||
Galasko CS, Delaney C, Morris P. Spinal stabilisation in Duchenne muscular dystrophy. J Bone Joint Surg Br. 1992;74(2):210–214. | ||
Takaso M, Nakazawa T, Imura T, et al. Surgical management of severe scoliosis with high-risk pulmonary dysfunction in Duchenne muscular dystrophy. Int Orthop. 2010;34(3):401–406. | ||
Cervellati S, Bettini N, Moscato M, Gusella A, Dema E, Maresi R. Surgical treatment of spinal deformities in Duchenne muscular dystrophy: a long term follow-up study. Eur Spine J. 2004;13(5):441–448. | ||
Galasko CS, Williamson JB, Delaney CM. Lung function in Duchenne muscular dystrophy. Eur Spine J. 1995;4(5):263–267. | ||
Alexander WM, Smith M, Freeman BJC, Sutherland LM, Kennedy JD, Cundy PJ. The effect of posterior spinal fusion on respiratory function in Duchenne muscular dystrophy. Eur Spine J. 2013;22(2):411–416. | ||
Jenkins JG, Bohn D, Edmonds JF, Levison H, Barker GA. Evaluation of pulmonary function in muscular dystrophy patients requiring spinal surgery. Crit Care Med. 1982;10(10):645–649. | ||
Kennedy JD, Staples AJ, Brook PD, et al. Effect of spinal surgery on lung function in Duchenne muscular dystrophy. Thorax. 1995;50(11):1173–1178. | ||
Miller RG, Chalmers AC, Dao H, Filler-Katz A, Holman D, Bost F. The effect of spine fusion on respiratory function in Duchenne muscular dystrophy. Neurology. 1991;41(1):38–40. | ||
Van Opstal N, Verlinden C, Myncke J, Goemans N, Moens P. The effect of Luque-Galveston fusion on curve, respiratory function and quality of life in Duchenne muscular dystrophy. Acta Orthop Belg. 2011;77(5):659–665. | ||
Eagle M, Bourke J, Bullock R, et al. Managing Duchenne muscular dystrophy – the additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul Disord. 2007;17(6):470–475. | ||
Bach JR, Rajaraman R, Ballanger F, et al. Neuromuscular ventilatory insufficiency: effect of home mechanical ventilator use v oxygen therapy on pneumonia and hospitalization rates. Am J Phys Med Rehabil. 1998;77(1):8–19. | ||
Bach JR, Ishikawa Y, Kim H. Prevention of pulmonary morbidity for patients with Duchenne muscular dystrophy. Chest. 1997;112(4):1024–1028. | ||
Bach JR, Saporito LR. Criteria for extubation and tracheostomy tube removal for patients with ventilatory failure. A different approach to weaning. Chest. 1996;110(6):1566–1571. | ||
Ishikawa Y, Bach JR, Komaroff E, Miura T, Jackson-Parekh R. Cough Augmentation in Duchenne Muscular Dystrophy. Am J Phys Med Rehabil. 2008;87(9):726–730. | ||
Kang SW, Kang YS, Sohn HS, Park JH, Moon JH. Respiratory muscle strength and cough capacity in patients with Duchenne muscular dystrophy. Yonsei Med J. 2006;47(2):184–190. | ||
Toussaint M, De Win H, Steens M, Soudon P. Effect of intrapulmonary percussive ventilation on mucus clearance in duchenne muscular dystrophy patients: a preliminary report. Respir Care. 2003;48(10):940–947. | ||
Mills B, Bach JR, Zhao C, Saporito L, Sabharwal S. Posterior spinal fusion in children with flaccid neuromuscular scoliosis: the role of noninvasive positive pressure ventilatory support. J Pediatr Orthop. 2013;33(5):488–493. | ||
Guérin C, Vincent B, Petitjean T, et al. The short-term effects of intermittent positive pressure breathing treatments on ventilation in patients with neuromuscular disease. Respir Care. 2010;55(7):866–872. | ||
Lofaso F, Orlikowski D, Raphael JC. Ventilatory assistance in patients with Duchenne muscular dystrophy. Eur Respir J. 2006;28(3):468–469. | ||
Bach JR, Gonçalves MR, Hon A, et al. Changing trends in the management of end-stage neuromuscular respiratory muscle failure: recommendations of an international consensus. Am J Phys Med Rehabil. 2013;92(3):267–277. | ||
McKim DA, Griller N, LeBlanc C, Woolnough A, King J. Twenty-four hour noninvasive ventilation in Duchenne muscular dystrophy: A safe alternative to tracheostomy. Can Respir J. 2013;20(1):e5–e9. | ||
Bach JR, Gonçalves MR, Hamdani I, Winck JC. Extubation of patients with neuromuscular weakness: a new management paradigm. Chest. 2010;137(5):1033–1039. | ||
Farkas GA, McCormick KM, Gosselin LE. Episodic hypoxia exacerbates respiratory muscle dysfunction in DMD(mdx) mice. Muscle Nerve. 2007;36(5):708–710. | ||
Percival JM, Whitehead NP, Adams ME, Adamo CM, Beavo JA, Froehner SC. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J Pathol. 2012;228(1):77–87. | ||
Lu-Nguyen NB, Jarmin SA, Saleh AF, Popplewell L, Gait MJ, Dickson G. Combination antisense treatment for destructive exon skipping of myostatin and open reading frame rescue of dystrophin in neonatal mdx mice. Mol Ther. 2015;23(8):1341–1348. | ||
Moraes LH, de Burgos RR, Macedo AB, de Almeida Hermes T, de Faria FM, Minatel E. Reduction of Oxidative Damage and Inflammatory Response in the Diaphragm Muscle of mdx Mice Using Iron Chelator Deferoxamine. Biol Trace Elem Res. 2015;167(1):115–120. | ||
Crisp A, Yin H, Goyenvalle A, et al. Diaphragm rescue alone prevents heart dysfunction in dystrophic mice in vitro. Hum Mol Genet. 2011;20(3):413–421. | ||
Yotsukura M, Miyagawa M, Tsuya T, Ishihara T, Ishikawa K. Pulmonary hypertension in progressive muscular dystrophy of the Duchenne type. Jpn Circ J. 1988;52(4):321–326. | ||
Mosqueira M, Baby SM, Lahiri S, Khurana TS. Ventilatory chemosensory drive is blunted in the mdx mouse model of Duchenne Muscular Dystrophy (DMD). PLoS One. 2013;8(7):e69567. | ||
Huang P, Cheng G, Lu H, Aronica M, Ransohoff RM, Zhou L. Impaired respiratory function in mdx and mdx/utrn(+/−) mice. Muscle Nerve. 2011;43(2):263–267. | ||
Mead AF, Petrov M, Malik AS, et al. Diaphragm Remodeling and Compensatory Respiratory Mechanics in a Canine Model of Duchenne Muscular Dystrophy. J Appl Physiol (1985). 2014;116(7):807–815. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.