Back to Archived Journals » Hypoxia » Volume 7
Aryl hydrocarbon receptor acts as a tumor suppressor in a syngeneic MC38 colon carcinoma tumor model
Authors Yakkundi P, Gonsalves E, Galou-Lameyer M, Selby MJ, Chan WK
Received 28 November 2018
Accepted for publication 18 February 2019
Published 10 April 2019 Volume 2019:7 Pages 1—16
DOI https://doi.org/10.2147/HP.S196301
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Poonam Yakkundi,1 Eleanor Gonsalves,1 Maria Galou-Lameyer,1 Mark J Selby,2 William K Chan3
1Animal Biology Group, Bristol-Myers Squibb Company, Redwood City, CA, 94063, USA; 2Immuno-Oncology Group, Bristol-Myers Squibb Company, Redwood City, CA, 94063, USA; 3Department of Pharmaceutics and Medicinal Chemistry, Thomas J Long School of Pharmacy & Health Sciences, University of the Pacific, Stockton, CA, 95211, USA
Background: Aryl hydrocarbon receptor (AHR), commonly known as an environmental sensor involved in the metabolism and elimination of xenobiotic substances, is also an important modulator in the development and functioning of the immune system. AHR expression is varied in the T cell subsets with the highest expression in T-helper 17 and T regulatory cells. It has been reported that AHR can act as a tumor promoter or a tumor suppressor, depending on the tumor type.
Methods: In an effort to understand the role played by AHR in tumor growth, the MC38 syngeneic colon carcinoma tumor model was used on C57BL/6 or ahr knockout (KO, -/-) mice with or without AHR antagonist (CH223191) treatment. Tumor sizes were measured, and biomarkers were quantified in tumor microenvironment and draining lymph nodes using flow cytometry. Enzyme-linked immunosorbent assay was used to determine the amount of cytokines in tumors.
Results: In ahr deficient mice, MC38 tumors progress more rapidly than in wild-type mice, accompanied by an increase in tumor-associated macrophages and M2 macrophages and a decrease in CD8a positive cytotoxic lymphocytes. Analysis of cytokines in the tumor microenvironment reveals a pro-inflammatory phenotype. Similar changes were observed by pharmacologic blockade of the receptor using CH223191.
Conclusion: AHR acts as a tumor suppressor in mice implanted with MC38 colon carcinoma cells as evidenced by either a blockade or deficiency of AHR.
Keywords: aryl hydrocarbon receptor, MC38, colon carcinoma, syngeneic, tumor suppressor, AHR deficiency
Introduction
Aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor which resides in the cytoplasm in its latent state.1 Mouse AHR, which was cloned in the early 90’s by two groups,2,3 is a member of the basic-helix-loop-helix/Per-ARNT-Sim family of proteins. When AHR engages with a ligand in the cytoplasm, it transits into the nucleus where it forms a complex with the AHR nuclear translocator protein (ARNT). The AHR/ARNT heterodimer binds to specific dioxin response element on the genome and activates the transcription of AHR target genes, most notably the cytochrome P450 1A1 gene (cyp1a1).4 AHR has long been known to be an environmental sensor involved in the metabolism of environmental toxins like 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and other planar chlorinated aromatic hydrocarbons.5 In the last decade or so, researchers became aware of its role in mediating/regulating the development and function of cells of the innate and adaptive immune system.6 The latter function of AHR was mediated by its response to endogenous ligands generated from diet, microbiota, or physiological process of the host cells. AHR can be understood as an important sensor acting as a physiological mediator of immune function by providing cues from the environment.
AHR is involved in the terminal differentiation of hematopoietic stem cells7 and many immune cells. In the cells of the innate and adaptive immune system, AHR is expressed in dendritic cells (DCs),8 macrophages,9 natural killer (NK) cells,10 and intraepithelial lymphocytes.11 Among T cells, AHR is silent in naïve, Th1, and Th2 cells, but its expression is high in Th17, Tr1, and Treg cells.12,13 It has been shown that AHR affects Treg and Th17 cell differentiation in a ligand-dependent manner – treatment with TCDD in mice increased Treg cells and ameliorated autoimmune disease EAE while treatment with 6-formylindolo[3,2-b]carbazole led to increased Th17 cells and exacerbated EAE.14 Treatment with 2-(1H-indol-3-ylcarbonyl)-4-thiazolecarboxylic acid methyl ester, an endogenous AHR ligand, promoted tolerogenic DCs, resulting in increased Foxp3+ Treg cells and reduced EAE symptoms.15 In addition, AHR can either be a tumor promoter or suppressor depending on the tumor type. Its role in epithelial–mesenchymal transition and progression is also controversial. Differential AHR expression has been observed in tumor versus normal tissues; it can be considered either a negative or a positive prognostic factor.16 The reason for the dual role of AHR in cancer is not known. Many AHR agonists such as TCDD are known carcinogens.17 Kynurenine, an AHR endogenous ligand generated through the indoleamine 2,3-dioxygenase (IDO)-mediated tryptophan metabolism, is associated with cancer progression by increasing Treg cells and suppressing T effector cells.18 Some AHR activators such as β-naphthoflavone, indole-3-carbinol (I3C, from cruciferous vegetables), and its dimer 3,3ʹ-diindolylmethane exhibit promising anti-tumor activities.16 There is evidence that AHR may curb tumor growth: for example, AHR appears to lower the risk of liver carcinogenesis,19 colon cancer,20 and ovarian cancer.21 In lung cancer, an increase in AHR expression decreases autophagy which leads to tumor suppression; in addition, it inhibits migration of lung cancer cell lines.22 On the contrary, AHR agonists drive the growth of gliomas23 and breast cancer.24 Based on the conflicting role of AHR in cancer, we wanted to better understand its role in tumor growth using a syngeneic mouse colon carcinoma model. MC38 is a syngeneic mouse colon adenocarcinoma tumor model on C57BL/6 background. Here we provide evidence supporting that AHR can act as a tumor suppressor. Changes in tumor burden after activation of the receptor using TCDD, blockade of the receptor using the antagonist CH223191, and removal of the receptor function using ahr deficient mice were examined. The tumor microenvironment was interrogated by examining the tumor-infiltrating leukocytes (TILs) along with cytokine and chemokine profiling in an effort to understand changes in tumor burden. Analysis of cytokines in the tumor microenvironment was performed to determine the inflammatory status.
Material and methods
Animals
For all tumor burden studies, the wild-type (WT) C57BL/6 mice were purchased from Jackson Laboratories. The mice were housed in the Bristol-Myers Squibb animal facility per the company’s guidelines. All animal studies were approved by the Institutional Animal Care and Use Committee of the company. The animal facility at Bristol Myers Squibb in Redwood City, California, is an AAALAC accredited facility. The mice used in the study were all female and 8 weeks of age at the time of tumor implants. For the studies involving ahr WT and knockout (KO, -/-) mice, these mice were obtained from matings of male and female ahr heterozygous (+/-) littermates that were purchased from Jackson Laboratories. The ahr deficient mice were originally generated by the Bradfield’s group.25 Some of WT and KO mice were also obtained from ahr (-/-) X ahr (-/-) and ahr (+/+) X ahr (+/+) matings, respectively. The KO mice from both sets of matings did not exhibit any differences in their tumor burden. These mice were housed and bred at the Bristol-Myers Squibb animal facility. Female mice between 8 and 12 weeks of age at the time of tumor implants were used in the study. All WT and KO mice that were put on a tumor study had an acclimatization period of 5 days in the protocol room.
Genotyping
Genotyping was done by polymerase chain reaction (PCR) to distinguish between WT and KO mice. DNA was extracted using Extract N Amp Tissue extraction kit. PCR was set up using 1X PCR buffer from Sigma using the primers 5ʹ-GGATTTGACTTAATTCCTTCAGCGG-3ʹ (wild type reverse), 5ʹ-TCTTGGGCTCGATCTTGTGTCAGGAACAGG-3ʹ (common) and 5ʹ-TGGATGTGGAATGTGTGCGAG-3ʹ (mutant reverse). The PCR sequences were obtained from Jackson Laboratory genotyping database/repository.
Culture of MC38, B16F10, and Hepa1c1c7 cells
Murine colon carcinoma MC38 cells were grown in RPMI, murine melanoma B16F10 cells in DMEM, and murine hepatoma Hepa1c1c7 cells in MEM supplemented with 10% fetal bovine serum and were cultured at 37°C with 5% CO2. Hepa1c1c7 and B16F10 cell lines were purchased from ATCC (Manassas, VA). MC38 cell line was a gift from Dr Jim Allison (MD Anderson Cancer Center, University of Texas). The use of MC38 and B16F10 cells for syngeneic tumor studies was approved by the institutional review board of the company.
Tumor model
8- to 12-week-old C57BL/6 or ahr KO (-/-) mice were injected subcutaneously on the right flank with either 2×106 MC38 cells or 1×106 B16F10 cells. TCDD dissolved in DMSO (at 50 µg/mL concentration) was purchased from Cambridge Isotope Laboratories. TCDD was mixed with NF grade corn oil (CO136-25ML, Spectrum Chemicals) and was administered intraperitoneally at 1 µg per mouse, one day prior to MC38 cell implant to ensure that AHR was activated before implantation. CH223191 (C8124-5MG) and DMSO were purchased from Sigma-Aldrich, mixed in corn oil and administered at 10 mg/kg body weight orally, once a day starting on day 7. On day 7, the mice were randomized based on the tumor burden and divided into the control and the treatment group such that both groups had the same average tumor burden. The weight and tumor burden were measured twice a week using Studylog database (from Studylog Systems Inc.). The height, width, and length of the tumors were measured using a caliper, and the tumor burden was calculated using the formula LxWxH/2. The tumor study involving ahr WT and KO mice did not receive any treatment. The health of the mice and husbandry was monitored on a daily basis per the guidelines of Bristol-Myers Squibb animal facility. For the immuno-phenotyping experiment, the tumors and tumor-draining lymph nodes constituting the axillary, brachial, and inguinal lymph nodes on the tumor-bearing side were excised on day 17 for TIL analysis by flow cytometry.
Flow cytometry
T cells, DCs, neutrophils, NK cells, and macrophages were measured in tumors (TILs) and in tumor-draining lymph nodes. Tissues were ground using GentleMACS™ dissociator and stained with fluorescent antibodies from Biolegend, BD Biosciences, and eBioscience. A 15 color antibody panel was run on BD Fortessa and analyzed using FlowJo. The following clones were used: F4/80 (BM8), CD80 (16-10A1), CD11c (N418), CD86 (GL-1), Ly6G (1A8), CD11b (M1/70), Ly6c (HK1.4), CD45 (30-F11), CD4 (GK1.5), CD8a (53-6.7), Foxp3 (FJK-16s), and NK1.1. In order to release leucocytes from the fibroblasts, the ground tumors and lymph nodes were digested in the presence of collagenase IV (Worthington) and DNase I (Sigma Aldrich) for 30 mins at 37°C.
Cytokine/chemokine analysis of the tumor microenvironment
The tumors from ahr WT, ahr KO, and vehicle- and CH223191-treated mice were harvested and ground in RPMI medium using gentleMACSTM tissue grinder. The samples were filtered using a 100 µM filter and spun at 400 g for 5 mins at 4°C; the supernatant was used for analysis. The cytokine and chemokine analysis was done using Millipore’s bead-based Luminex assays and run on Magpix. TGF-β enzyme-linked immunosorbent assay (ELISA) was done using ELISA kit from R n D Systems.
RT-PCR
RNA from MC38 and Hepa1c1c7 cells was extracted using Direct-zol Total RNA extraction kit (Zymo Research). RNA quantitation was done using a QIAxpert (Qiagen). 1 µg of RNA per reaction was used to set up the cDNA reaction. cDNA was prepared immediately from RNA using the SuperScript III first strand synthesis super mix for qRT-PCR (Thermo Fisher Scientific) following manufacturer’s instructions. The prepared cDNA was immediately used to set up the RT-PCR reaction. 2 µl of cDNA was used per sample to set the RT-PCR reaction with three technical replicates. Real-time PCR was done using Sso advanced SYBR green master mix (Bio-Rad). The samples were run on the LC480 cycler (Roche). The housekeeping gene gapdh was used as the standard. Fold induction was calculated using the 2−∆∆CT method.26 All the primers for the assay were ordered from Integrated DNA Technologies. The primer sequences are shown in Table 1.
 | Table 1 PCR primer sequences used for SYBR green qPCR analysis |
Statistical analysis
Statistics were performed using GraphPad PRISM 7. Error bars represent standard deviation. For differences in tumor burden between groups, one-way and two-way ANOVA (with Bonferroni post test) were used. Differences between ahr WT versus KO mice and vehicle versus CH223191-treated mice among different lymphocyte subsets were determined by the Student’s t-test (unpaired) comparing the means of the two study groups. A p-value of <0.05 was considered as a significant difference.
Results
MC38 tumor growth was promoted in ahr deficient mice
We examined whether AHR would have any role in MC38 tumor growth. MC38 tumor experiments using ahr deficient (KO) and wild-type (WT) C57BL/6 mice were performed as described (Figure 1A). Each group consisted of 8–10 mice. On day 7, mice were not randomized, but the average tumor burdens in the WT and KO mice were similar. The tumors between the two groups started showing differences on day 17 post-implantation and became significant by day 21 (Figure 1B). The tumor burden in the ahr KO mice was almost double the volume to their WT counterparts on day 21.
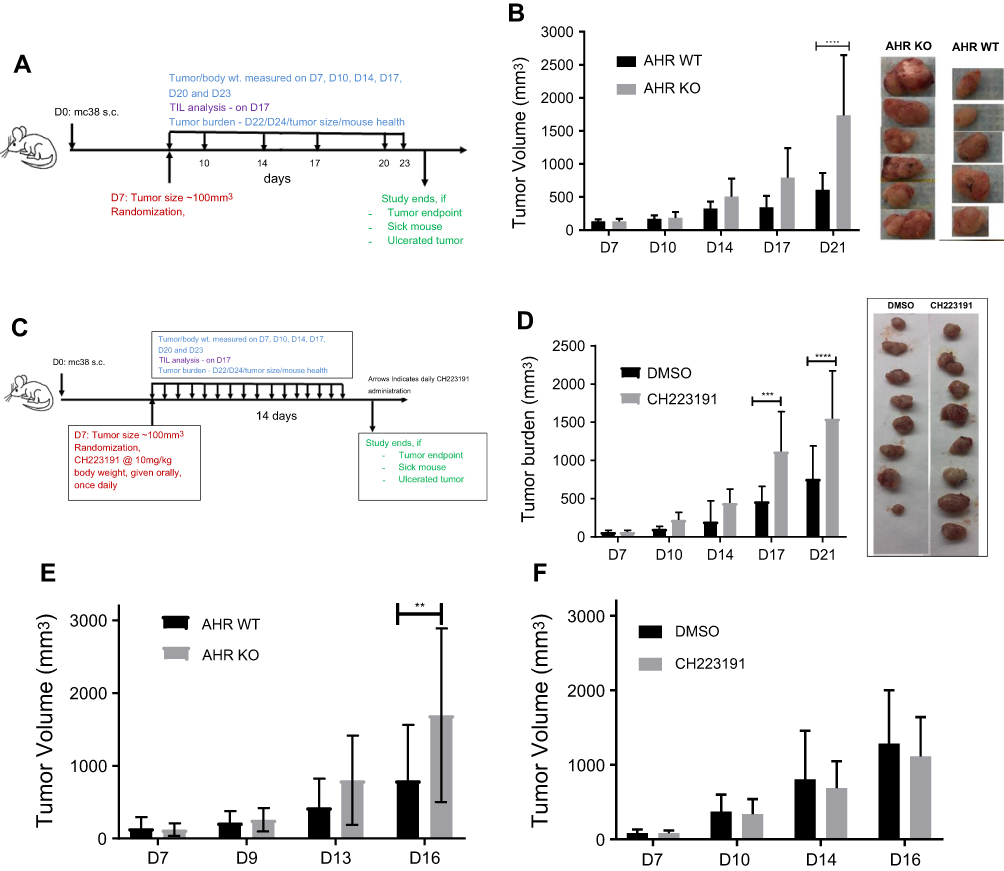 | Figure 1 (A) Schematic of tumor study. (B) MC38 tumor burden was increased in ahr deficient mice (KO) in comparison to ahr wild-type (WT) C57BL/6 mice. Tumor pictures were obtained at day 21. (C) Schematic of tumor study with CH223191 administration. (D) Pharmacologic blockade of AHR by CH223191 increased MC38 tumor burden in C57BL/6 mice. Tumor pictures were obtained at day 21. (E) B16F10 tumor burden was increased in ahr deficient (KO) mice in comparison to the wild-type (WT) C57BL/6 mice. (F) B16F10 tumor burden in C57BL/6 mice in the presence of CH223191 did not result in any significant changes in tumor volume. Error bars represent means±SD, n=5–7 (A); n=7–10 (D); n=10–11 (E); n=10–12 (F); **p<0.005; ***p<0.0005; ****p<0.0001. Abbreviations: wt, weight; D, day; TIL, tumor-infiltrating lymphocytes; s.c., subcutaneous. |
AHR antagonist promoted MC38 tumor growth
Next, we examined whether inhibition of AHR function would elicit similar tumor burden as observed in the ahr deficient mice. CH223191, a highly specific AHR antagonist,27 was administered at a daily dose of 10 mg/kg, starting day 7 until the end of the tumor study (Figure 1C). Mice in the study were in good health, and the administration of the compound did not appear to cause any adverse effects. Mice that developed tumor ulcerations were removed from the study; more specifically, tumors on three mice (out of 10) were found ulcerated on day 17 and were removed from the experiment. We began to observe significant changes in the tumor burden starting on day 17 (Figure 1D). The study was terminated at day 21 as most of the mice in the study had reached the maximum tumor volume of 2,000 mm3 or exhibited ulcerated tumors. We observed that the MC38 tumor burden was increased when AHR was inhibited by an AHR-specific antagonist.
B16F10 tumor grew faster in ahr deficient mice
Next, we examined whether AHR would suppress another type of tumor growth in C57BL/6 mice. One million B16F10 melanoma cells were implanted in co-housed ahr WT (+/+) and ahr KO (AHR-/-) mice and remained untreated for the duration of the experiment. Same protocol as described in Figure 1A was followed. The tumor measurements started on day 7 with a starting number of 10–11 mice per group. There were significant changes in the tumor burden on day 16 post implantations. Similar to MC38 tumors, B16F10 tumors grew faster in the absence of AHR in comparison to the wild-type counterparts (Figure 1E). Unlike the MC38 tumor burden study, this study could not be continued beyond day 16 as mice developed ulcerated tumors and their health status, monitored by weight and health, began to decline.
B16F10 tumor did not exhibit differences in the tumor burden with or without CH223191 treatment
Next, we examined whether inhibition of AHR function would increase the B16F10 tumor growth. A B16F10 tumor study was done as described in Figure 1C, except that 1 million B16F10 cells were subcutaneously implanted. On day 7, the tumors were measured, the mice randomized based on tumor burden, then divided into the treatment and control groups. The antagonist CH223191 or vehicle was administered every day orally starting from day 7 and continued until the end of the tumor study. There were no significant changes in the tumor burden up to day 16 after the subcutaneous implants (Figure 1F). The study had to be brought down because many mice developed ulcerated tumors, and their health began to decline.
Multiple dosing of TCDD was required to suppress MC38 tumor growth
Next, we examined whether activation of AHR function by an exogenous ligand would suppress the MC38 tumor growth. We performed tumor burden experiment with a single dose of TCDD (Figure 2A) and observed that there was no significant change in the tumor burden when mice were treated with 1 µg of TCDD (Figure 2B). However, when mice were treated with three 1 µg doses of TCDD (day 1, 7, and 10), the tumor growth was significantly suppressed on day 21 and 24 (Figure 2C). There were no gross changes in body weight or the health status of the mice with multiple doses of TCDD.
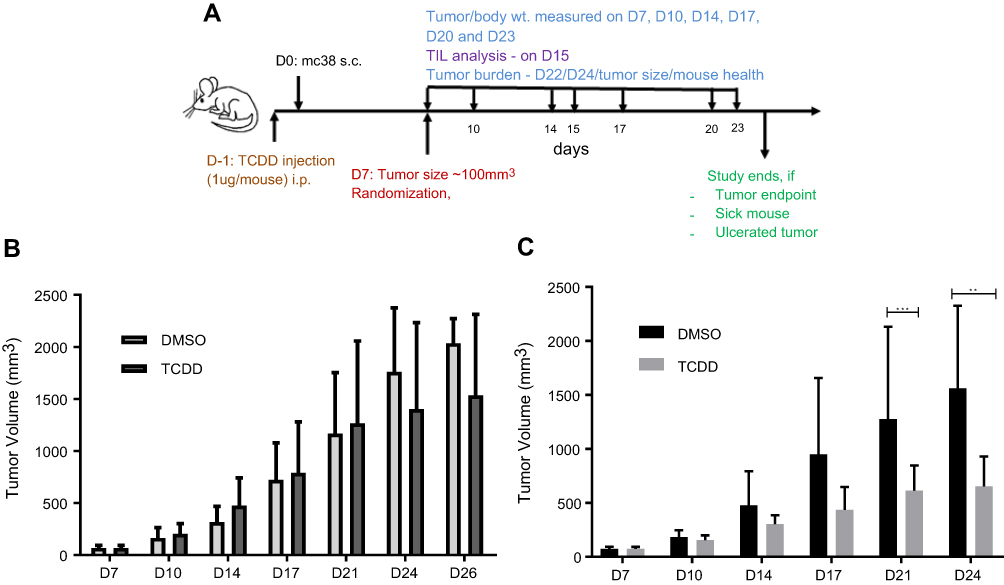 | Figure 2 (A) Schematic of MC38 tumor study on C57BL/6 mice with TCDD treatment. (B) MC38 tumor burden with one dose of TCDD (1 µg/mouse). (C) MC38 tumor burden with three doses of TCDD (1 µg per dose). The mice in the study were measured using a balance and recorded in the STUDYLOG database. The tumors were measured twice a week. Error bars represent means±SD, n=6–12 (B); n=8–10 (C); **p<0.005; ***p<0.0005.Abbreviations: D, day; wt, weight; TIL, tumor-infiltrating lymphocytes; s.c., subcutaneous. |
MC38 cells did not appear to possess appreciative AHR function
To determine if MC38 cells respond to AHR ligands, expression of a TCDD-driven AHR target gene in these cells was evaluated by quantitative PCR. The widely cited AHR responsive Hepa1c1c7 mouse hepatoma cells were used as the control for comparison. Expression levels of cyp1a1 in Hepa1c1c7 in comparison to that of MC38 were measured at 6 and 24 hrs in the presence or absence of 1nM TCDD. The induced expression levels of cyp1a1 in Hepa1c1c7 was much higher than that of MC38 cells – 78-fold versus 1.2-fold after 24 hrs (Figure 3A). To determine the consequences of TCDD incubation of MC38 and B16F10 cells, these cells were exposed to 1nM TCDD and the expression of cyp1a1 was measured at 3, 6, 18, and 24 hrs after TCDD treatment. There was very modest induction of the cyp1a1 message in MC38 cells; it was highest 3 hrs after TCDD treatment (2.7-fold) and decreased thereafter to 24 hrs (Figure 3B). Contrarily, the cyp1a1 message levels of B16F10 were quite steady over the 24-hr period and were significantly higher than that of MC38 (17- to 19-fold) (Figure 3C). Moreover, in MC38 cells treated with 1nM TCDD, there was no detectable induction among six AHR target genes – ugt1a1, ahrr, nqo, cyp1a1, cyp1a2, and cyp1b1 (Figure 3D), suggesting that AHR may not elicit normal function in MC38 cells. Perhaps AHR expression in MC38 cells may be irrelevant; thus, any effects on tumor burden should be mediated by the host AHR.
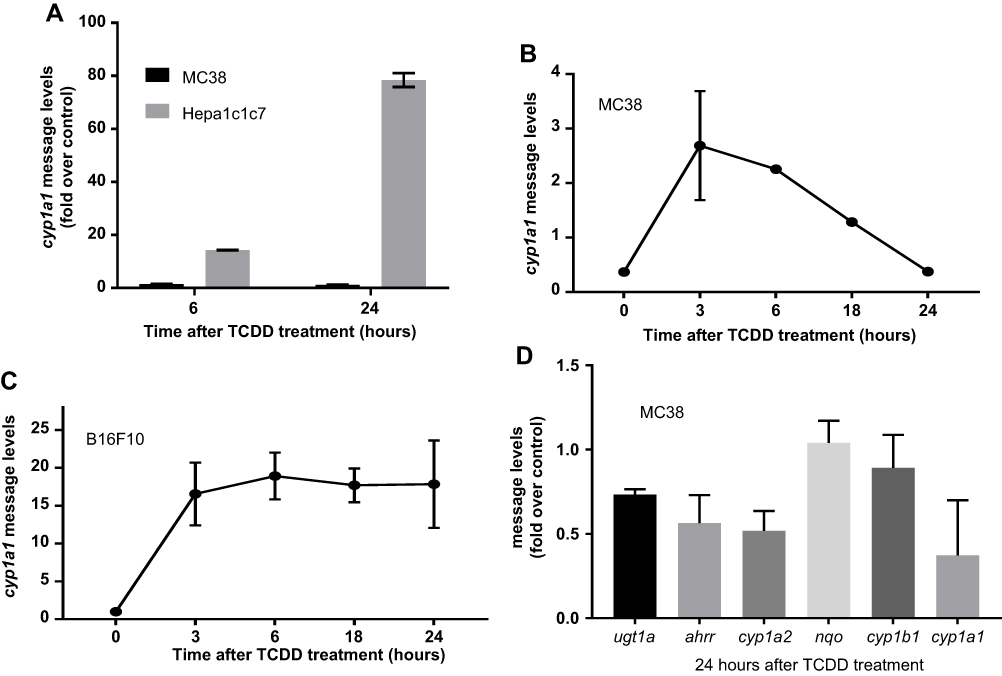 | Figure 3 (A) mRNA levels were measured in MC38 and Hepa1c1c7 cell lines 6 and 24 hrs after 1 nM TCDD treatment. (B) cyp1a1 mRNA levels were measured in MC38 cell line at 3, 6, 18, and 24 hrs after 1 nM TCDD treatment. (C) cyp1a1 mRNA levels were measured in B16F10 cell line at 3, 6, 18, and 24 hrs after 1 nM TCDD treatment. (D) AHR gene battery mRNA levels were measured in MC38 cells at 24 hrs after 1nM TCDD treatment. Error bars represent means±SD, n=3 except zero timepoint in (B) with n=2. All qPCR data were normalized to the gapdh control.Abbreviations: cyp1a1, cytochrome P450 1A1; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin. |
A decrease of the CD8/Treg ratio and NK cells and an increase of tumor-associated macrophages (TAMs) and M2 macrophages were observed in the tumor microenvironment of ahr deficient mice and CH223191-treated wild-type mice
Immune monitoring of TILs was undertaken to identify changes that correlated with differences in tumor burdens in either ahr deficient or wild-type mice treated with antagonist CH223191. Tumor burden differences were manifested at day 17 – same day when tumors were harvested for analysis. Fluorescence-activated cell sorting (FACS) analysis of TILs in WT versus ahr KO mice (Figure 4) and WT mice±CH223191 treatment (Figure 5) revealed that an increase in MC38 tumor burden resulted in a small, but not significant, increase in Foxp3+ Treg cells in the tumor microenvironment (Figures 4A and 5A). However, there was a significant decrease in the CD8a+ T cells with increased tumor burden (Figures 4B and 5B). These changes became more apparent when we observed a clear, significant decrease in the CD8a to Foxp3 Treg ratio, consistent with the increase in tumor burden when there was less or no AHR function (Figures 4C and 5C). The tumor-draining lymph nodes were harvested on day 17 from same mice as noted earlier. FACS analysis did not show any differences among wild-type, ahr deficient, and CH223191-treated wild-type mice (Figures 4K and L, 5K and L). NK cell marker NK1.1 was used to interrogate TILs. Reduced NK cells appeared to correlate with increased tumor burden in the ahr KO mice (Figure 4J) and CH223191-treated mice (Figure 5J) compared to controls. There were no changes in the infiltrating monocytes (Ly6C+) or neutrophils (Figures 4D and E, 5D and E) among myeloid cells in TILs. No changes were observed in DC subsets represented by CD103 (Figures 4F and 5F) and CD11b (Figures 4G and 5G). Importantly, there was a significant increase in the F4/80 positive tumor-associated macrophages (TAMs) and the CD206+ M2 macrophages with increased tumor burden (Figures 4H and I, 5H and I), although there was no change in the surface expression of the MHCII.
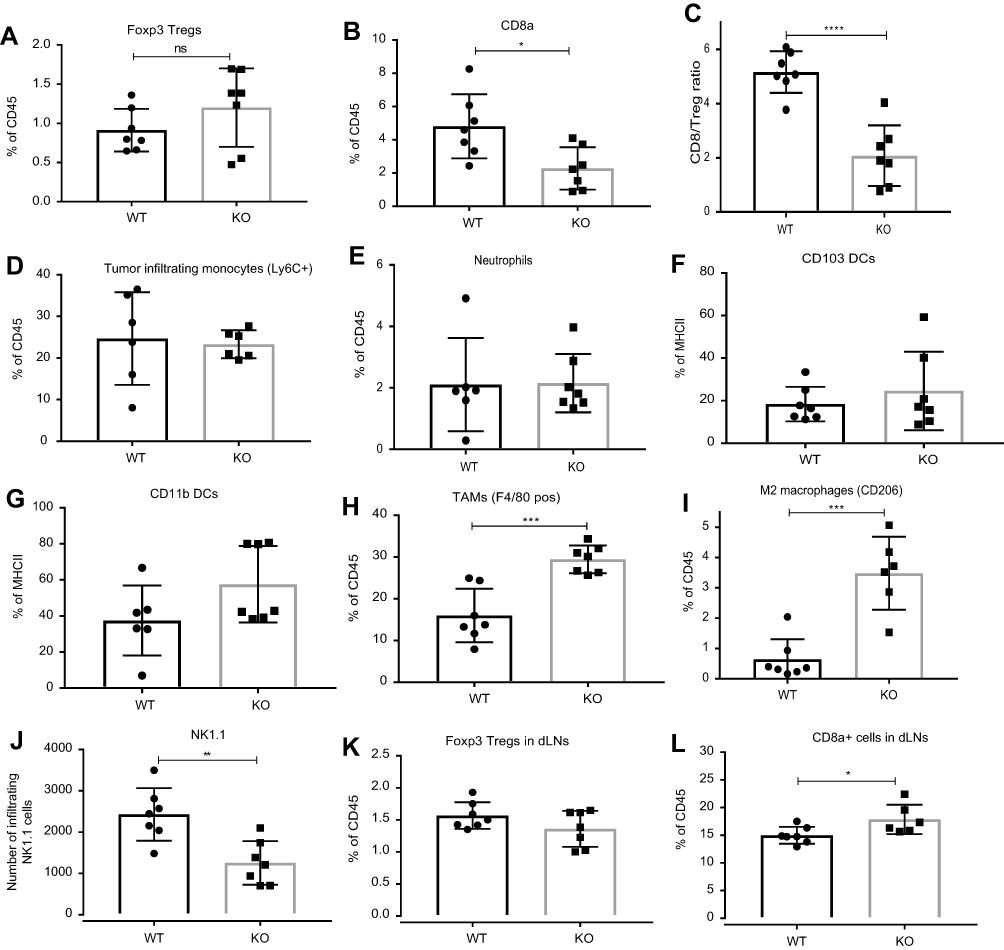 | Figure 4 FACS analysis of tumor microenvironment. Increase in MC38 tumor burden in ahr knockout (KO) when compared to the wild-type (WT) mice resulted in (A) no change in Foxp3+ Treg cells; (B) reduced CD8a+ T cell infiltration; (C) reduced CD8a/Treg ratio; (D) no change in Ly6C+ monocyte infiltration; (E) no change in neutrophil infiltration; (F) and (G) no change in CD103+ and CD11b+dendritic cells (DC); (H) increased F4/80+ TAM; (I) increased CD206 + M2 macrophages; (J) decreased NK1.1 cells; (K) and (L) increased number of CD8a+ T cells but no change in Foxp3+ Treg cells in draining lymph nodes (dLNs). These data are a representative of two experiments. Error bars represent means±SD, n=6–7; *p<0.05; **p<0.005; ***p<0.0005; ****p<0.0001.Abbreviations: DC, dendritic cells; ns, not significant. |
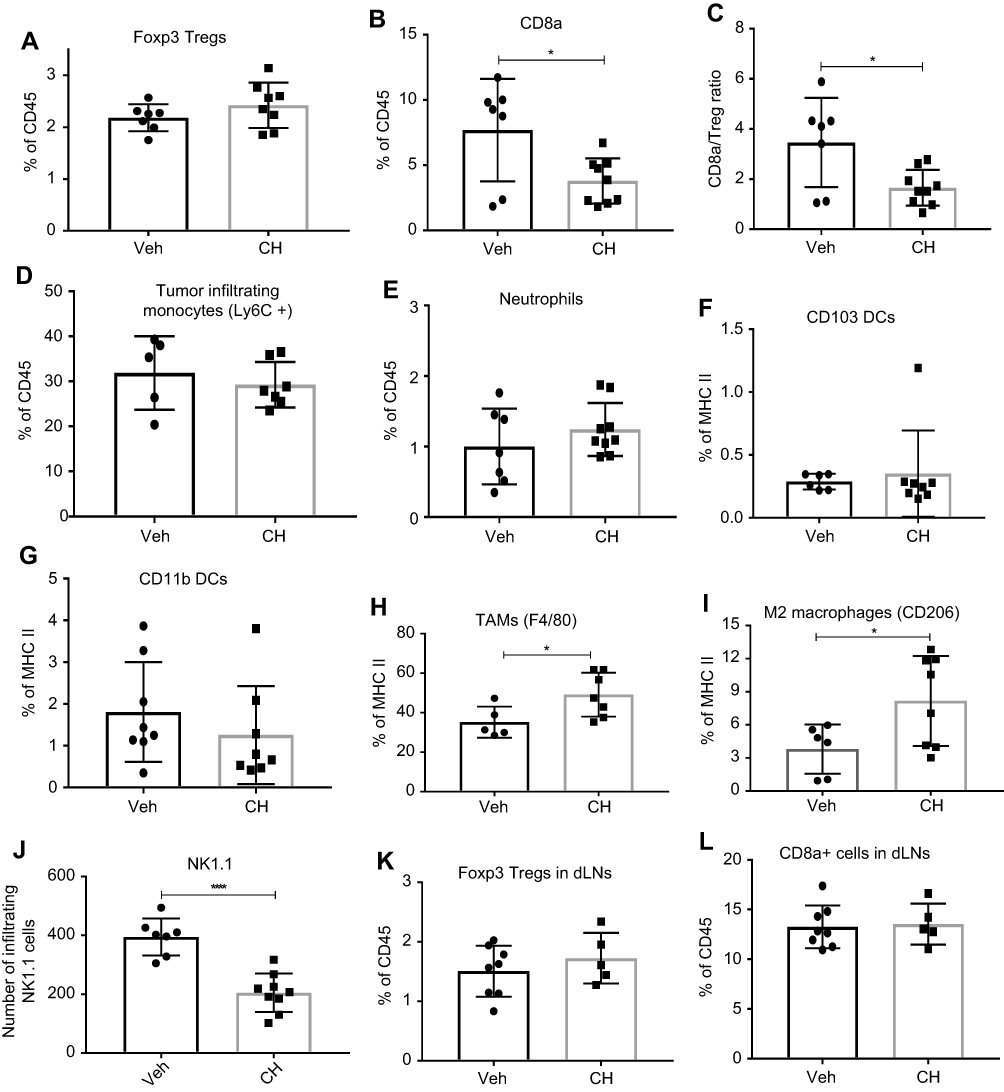 | Figure 5 FACS analysis of tumor microenvironment. Increase in MC38 tumor burden in CH223191 (CH)-treated mice when compared to the DMSO vehicle (Veh)-treated wild-type mice resulted in (A) no change in Foxp3+ Treg cells; (B) reduced CD8a +T cell infiltration; (C) reduced CD8a/Treg ratio; (D) no change in Ly6C+ monocyte infiltration; (E) no change in neutrophil infiltration; (F) and (G) no change in CD103+ and CD11b+ dendritic cells (DC); (H) increased F4/80+ TAMs (I) increased CD206 + M2 macrophages; (J) decreased NK cells; (K) and (L) no change in number of Foxp3+ Treg and CD8a+ T cells in draining lymph nodes (dLNs). These data are a representative of 4 experiments. Error bars represent means±SD, n=5–8; *p<0.05; ****p<0.0001.Abbreviations: Treg, T regulatory cells; CH, CH223191; DC, dendritic cells; NK, natural killer cells. |
Expression of cytokines and chemokines in the tumor microenvironment of the ahr deficient and CH223191-treated wild-type mice revealed a pro-inflammatory status
Next, we examined the inflammation status of the tumor microenvironment by measuring the amount of various cytokines and chemokines. Eotaxin levels, a chemokine secreted by Th2 and other cell types in response to IL-4 and IL-13 to mobilize eosinophils from bone marrow to blood circulation, were significantly lower in tumor supernatants from ahr KO mice compared to WT mice (Figure 6B), although there was no difference in tumors of mice treated with vehicle and antagonist (Figure 7A). Notably, the levels of IFN-γ were significantly reduced with an increase in tumor burden (Figures 6C and 7B). There was no change in the levels of IL-1a, IL-2, IL-13, IL-10, MIG (CXCL9), and VEGF; however, there was a significant increase in the pro-inflammatory cytokine IL-6 and a decrease in IL-1b and TNF-α levels in the ahr KO and antagonist-treated mice as tumor burden increased (Figures 6 and 7, Table 2). CCL5/RANTES levels were also reduced as the tumor burden increased.
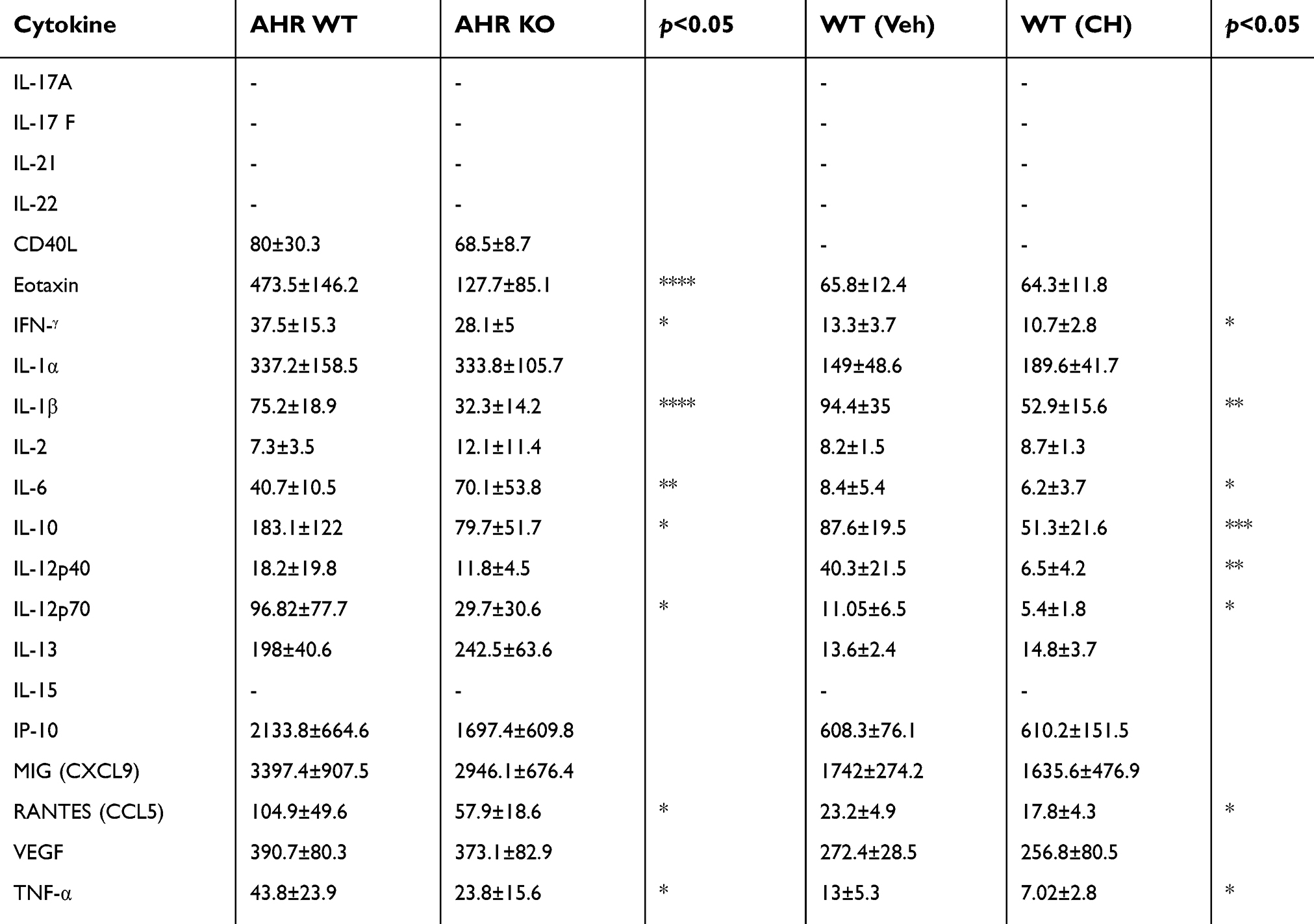 | Table 2 The amount of cytokines/chemokines (in pg/mL) in the tumor microenvironment of wild-type (WT) versus ahr knockout (KO) mice and vehicle- versus CH223191 (CH)-treated wild-type mice. Statistical analysis was done using unpaired Student’s t-test |
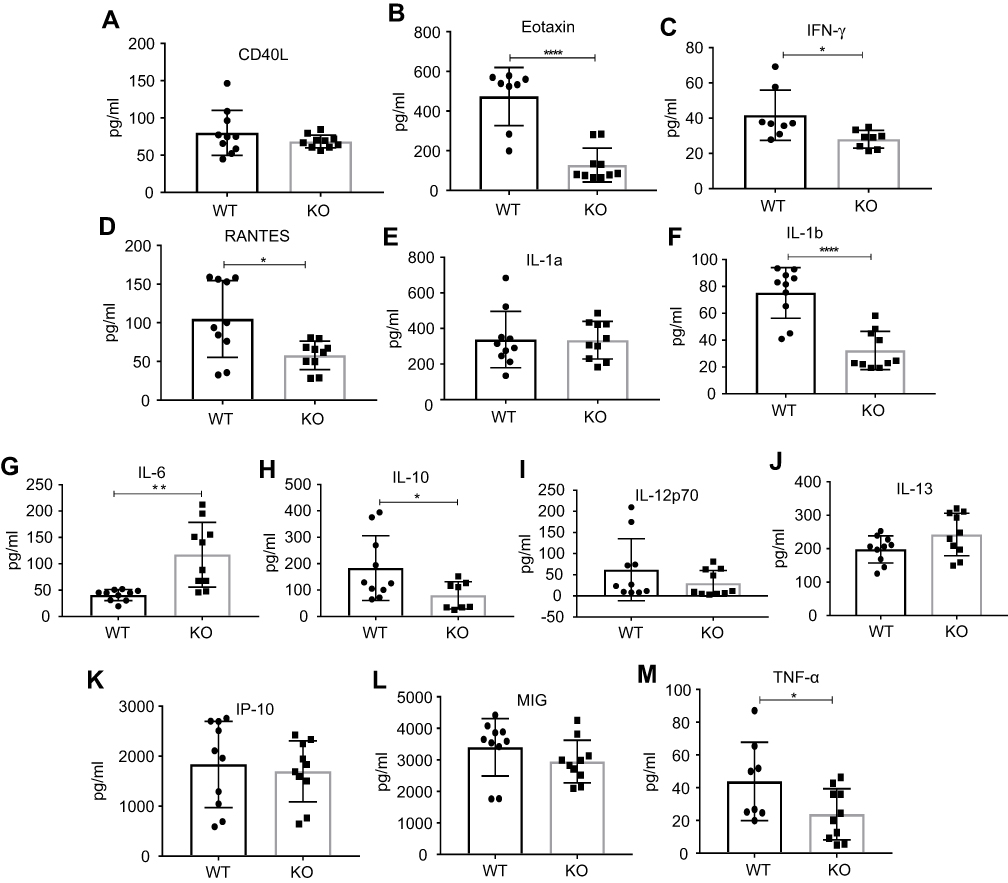 | Figure 6 Amount of cytokines and chemokines (A–M) in tumor microenvironment of wild-type (WT) vs ahr knockout (KO) C57BL/6 mice. Eotaxin, IFNγ, RANTES, IL-1b, IL-10, and TNF-α were reduced whereas IL-6 was increased in KO mice when compared to WT. Statistical analysis was done using unpaired Student’s t-test. Error bars represent means±SD, n=8–10; *p<0.05; **p<0.005; ****p<0.0001. |
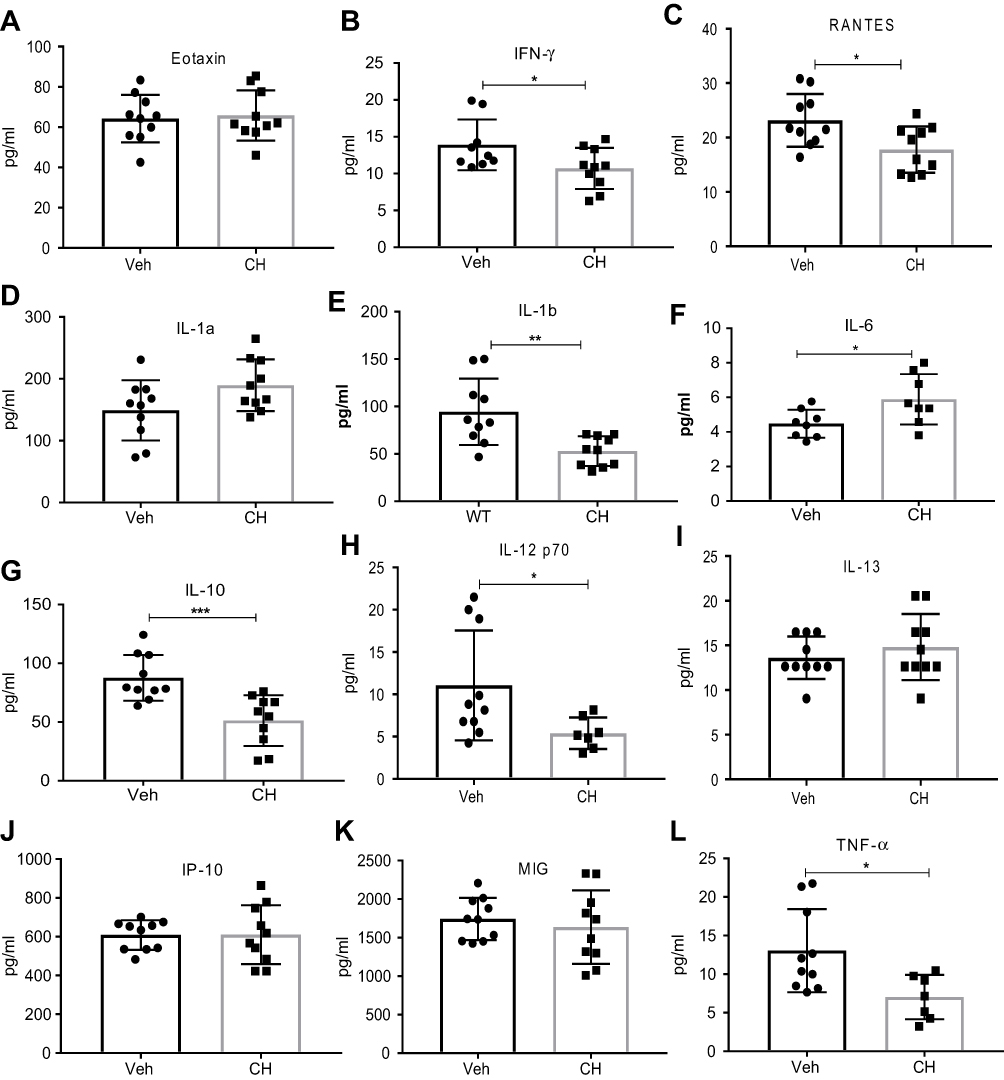 | Figure 7 Amount of cytokines/chemokines in tumor microenvironment when compared between DMSO vehicle (Veh)- and CH223191 (CH)-treated C57BL/6 mice. IFNγ, RANTES, IL-1b, IL-10, IL-12p70, and TNF-α were reduced whereas IL-6 was increased with CH223191 treatment when compared to DMSO treatment. Statistical analysis was done using unpaired Student’s t-test. Error bars represent means±SD, n=8–10; *p<0.05; **p<0.005; ***p<0.0005. |
Discussion
We have used a mouse colon carcinoma syngeneic model to elucidate the role of AHR in cancer. Syngeneic models have an intact immune system and are very useful in understanding the role of immune system in modulating cancer growth.28 Some of the commonly used syngeneic tumor models in cancer research are CT26 and MC38 colon carcinoma, B16F10 melanoma, and 4T1 breast cancer models. Preclinical data using surrogate antibodies supported clinical investigation, yielding to ultimate approval of human-specific PD-1 and CTLA-4 antibodies. In our studies, we have used MC38 and B16F10 syngeneic models to explore the AHR role in MC38 and B16F10 tumor progression.
Tumor burden was increased in the ahr deficient and the CH223191-treated mice, suggesting that a ligand-dependent activation of AHR function is responsible for the suppression of tumor growth. Although a single TCDD dose did not alter tumor burden, it promoted peripheral suppression of immune response as determined by FACS data on cells from tumors and spleen (data not shown). A single dose of TCDD was immunosuppressive, as confirmed by the suppression of LPS-elicited humoral responses in dioxin-treated mice (Supplementary materials and Figure S1), suggesting that AHR is already active that further activation by a single dose of TCDD does not elicit any more AHR function. However, multiple doses of TCDD did suppress the MC38 tumor growth, validating that activation of the AHR function can curb tumor growth.
Our B16F10 tumor study data showed increased tumor burden in ahr deficient mice, similar finding as reported by Sunwoo et al,10 although no changes were seen when the receptor was blocked by CH223191. We observed that B16F10 tumors were much more aggressive and less consistent than the MC38 tumors, making it difficult to discern the AHR effect on tumor growth. In addition, we cannot rule out the possibility that our treatment of CH223191 was insufficient to block the overall AHR function since the B16F10 tumors possessed substantially higher AHR function, as compared to the MC38 tumors. Although both Sunwoo’s and our groups observed increased tumor growth in ahr deficient mice when compared to the wild-type control, function of AHR in the stroma cells has been shown to drive the tumor growth.29 Since these researchers used AHR-knockdown B16F10 cells for tumor study, it could be the off-target effect of the manipulated B16F10 cells that may contribute to tumor growth. Interestingly, our finding appears to contradict with a recent report stating that AHR can drive PD-1 expression in CD8+ T lymphocytes and in turn can promote mouse melanoma tumor growth.30 Our observation that recruitment of CD8+ T cells into the tumor microenvironment was significantly reduced in mice with compromised AHR function may nullify the AHR-mediated PD-1 effect in these cells. It is interesting that we observed tumor promotion with AHR antagonism whereas some researchers observed otherwise, although differences in experimental design and environment are known to influence the outcome of a tumor study. Unlike the highly tumorigenic tumor-repopulating cells, IDO expression in MC38 cells was detected only after IFNγ treatment (data not shown). Realizing that the IFNγ levels were repressed in the MC38 tumor microenvironment in ahr deficient and CH223191-treated mice, we argue that synthesis of kynurenine by MC38 tumors should be modest, if any, to affect the tumor burden.
MC38 cells showed very low TCDD-dependent cyp1a1 expression when compared to Hepa1c1c7 cells, suggesting that the receptor, which is detectable by Western analysis (data not shown), might not be active. Thus, the AHR-mediated increase in tumor burden is most likely mediated by stromal cells and TILs rather than MC38 cells. We performed a comprehensive FACS analysis of TILs composed of NK cells, T cell subsets, and myeloid cells in search of evidence which could explain our tumor burden studies. There was a reduction in total NK cell numbers and activated NK cells with increased tumor burden. It has been demonstrated that AHR is essential for the cytolytic activity of NK cells in RMA-S cells implanted in ahr deficient mice.10 Unlike RMA-S cells, however, MC38 cells express MHC class I which may negatively affect NK cell function; whether NK cells play a role in controlling MC38 cells is remained to be determined. Analysis of the myeloid populations demonstrated that there were no changes in the tumor-infiltrating Ly6C+ monocytes or neutrophils. Among DCs, which are the primary antigen presenting cells,31 no changes were observed in the infiltrating DCs, the classical DCs (CD11b), cross-presenting DCs (CD103), and activated DCs (CD80 and CD86, data not shown) with increased tumor burden. However, a significant reduction in F4/80+ TAMs and CD206+ macrophages was observed in the wild-type mice. TAMs are frequently detected in the tumor microenvironment.32 They are differentiated myeloid populations with immune suppressive properties that are thought to block anti-tumor immunity and promote angiogenesis.33 The CD206+ macrophages or M2 macrophages are also thought to be immune suppressive and promote tumor growth.34 It appeared that inhibition of TAM and M2 macrophages into the tumor microenvironment is partly responsible for the AHR function in suppressing tumor growth.
Flow cytometry analysis of tumor lymphocytes revealed a significant decrease in cytotoxic T lymphocytes (CD8a+) and CD8/Treg ratio in ahr deficient and antagonist-treated mice when compared to controls. This is in line with our finding that AHR suppresses the MC38 tumor growth by causing more cytotoxic T cell recruitment into the tumor microenvironment. Paradoxically, it has been reported that activation of AHR by kynurenine would trigger precisely the opposite effect on Treg and CD8a+ T cells.18 It is conceivable that different endogenous ligands are at play in this MC38 tumor microenvironment.14 There were no changes in Ki67 staining in Foxp3+ Treg and CD8a+ T cells in the ahr deficient and antagonist-treated mice when compared to the wild-type counterparts (data not shown), suggesting that the reduced cytotoxic T lymphocytes was due to reduced infiltration of these cells into the tumor environment but not reduced proliferation.
Intratumoral cytokine analysis of the tumor microenvironment revealed an increase in the pro-inflammatory cytokine IL-6 and the reduction of IFN-γ, TNF-α, and IL-1b with increased tumor burden in the ahr deficient and antagonist-treated wild-type mice when compared to controls. There was a reduction in the amount of IL-10, an anti-inflammatory cytokine, in the ahr deficient and antagonist-treated mice. IL-12 is a cytokine that is involved in the differentiation of Th1 cells, mainly secreted by DCs, which activates effector T and NK cells and is involved in IFN-γ production and activation of anti-tumor immunity.34 The IL-12p40 and p70 subunits of IL-12 cytokine were reduced in both the antagonist treated group and in the KO mice, consistent with the weaker anti-tumor T cell response. Likewise, reductions in RANTES/CCL5, a chemoattractant for T cells, basophils, and other leucocytes,35 maybe consistent with lower levels of CD8a T cell infiltration into tumors from antagonist-treated or KO mice. Based on the cytokine/chemokine analysis of the tumor milieu, there is a shift towards a pro-inflammatory environment by AHR and a chronic pro-inflammatory tumor environment is thought to result in tumor initiation and/or progression.
Conclusion
Collectively, we conclude that AHR plays a crucial role in the MC38 tumor development in C57BL/6 mice, the absence of which leads to tumor progression by reducing the infiltration of anti-tumor cytotoxic T lymphocytes, promoting the recruitment of F4/80+ TAMs and CD206+ M2 macrophages, and promoting a proinflammatory tumor microenvironment.
Acknowledgments
Funding for this study and article was provided by Bristol-Myers Squibb Company. All experiments were done at a Bristol-Myers Squibb Company facility in Redwood City, CA, USA. A portion of this paper was presented in 2016 at the Aryl Hydrocarbon Receptor Conference as a poster presentation with interim findings.
Disclosure
PY, EG, MGL, and MJS are employed by Bristol-Myers Squibb Company. The authors report no other conflicts of interest in this work.
References
1. Poland A, Glover E, Kende AS. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. J Biol Chem. 1976;251:4936–4946.
2. Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci USA. 1992;89:8185–8189.
3. Ema M, Sogawa K, Watanabe N, et al. cDNA cloning and structure of mouse putative Ah receptor. Biochem Biophys Res Comm. 1992;184:246–253.
4. Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci. 2011;124:1–22. doi:10.1093/toxsci/kfr218
5. Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519–561. doi:10.1146/annurev.pharmtox.40.1.519
6. Gutierrez-Vazquez C, Quintana FJ. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity. 2018;48:19–33. doi:10.1016/j.immuni.2017.12.012
7. Boitano AE, Wang J, Romeo R, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–1348. doi:10.1126/science.1191536
8. Nguyen NT, Kimura A, Nakahama T, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci USA. 2010;107:19961–19966. doi:10.1073/pnas.1014465107
9. Kimura A, Naka T, Nakahama T, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med. 2009;206:2027–2035. doi:10.1084/jem.20090560
10. Shin JH, Zhang L, Murillo-Sauca O, et al. Modulation of natural killer cell antitumor activity by the aryl hydrocarbon receptor. Proc Natl Acad Sci USA. 2013;110:12391–12396. doi:10.1073/pnas.1302856110
11. Li Y, Innocentin S, Withers DR, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147:629–640. doi:10.1016/j.cell.2011.09.025
12. Apetoh L, Quintana FJ, Pot C, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol. 2010;11:854–861. doi:10.1038/ni.1912
13. Korn T. How T cells take developmental decisions by using the aryl hydrocarbon receptor to sense the environment. Proc Natl Acad Sci USA. 2010;107:20597–20598. doi:10.1073/pnas.1015420107
14. Quintana FJ, Basso AS, Iglesias AH, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi:10.1038/nature06880
15. Quintana FJ, Murugaiyan G, Farez MF, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2010;107:20768–20773. doi:10.1073/pnas.1009201107
16. Safe S, Cheng Y, Jin UH. The aryl hydrocarbon receptor (AhR) as a drug target for cancer chemotherapy. Curr Opin Toxicol. 2017;2:24–29. doi:10.1016/j.cotox.2017.01.012
17. Poland A, Knutson JC. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Ann Rev Pharmacol Toxicol. 1982;22:517–554. doi:10.1146/annurev.pa.22.040182.002505
18. Platten M, von Knebel Doeberitz N, Oezen I, et al. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol. 2014;5:673.
19. Fan Y, Boivin GP, Knudsen ES, et al. The aryl hydrocarbon receptor functions as a tumor suppressor of liver carcinogenesis. Cancer Res. 2010;70:212–220. doi:10.1158/0008-5472.CAN-09-3090
20. Kawajiri K, Kobayashi Y, Ohtake F, et al. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+mice with natural ligands. Proc Natl Acad Sci USA. 2009;106:13481–13486. doi:10.1073/pnas.0902132106
21. Wang K, Li Y, Jiang YZ, et al. An endogenous aryl hydrocarbon receptor ligand inhibits proliferation and migration of human ovarian cancer cells. Cancer Lett. 2013;340:63–71. doi:10.1016/j.canlet.2013.06.026
22. Tsai CH, Li CH, Cheng YW, et al. The inhibition of lung cancer cell migration by AhR-regulated autophagy. Sci Rep. 2017;7:41927. doi:10.1038/srep41927
23. Platten M, Opitz C, Wick W. The aryl hydrocarbon receptor as a promoter of malignant glioma. Cell Cycle. 2012;11:643–644. doi:10.4161/cc.11.4.19357
24. Stanford EA, Wang Z, Novikov O, et al. The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol. 2016;14:20. doi:10.1186/s12915-016-0240-y
25. Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci USA. 1996;93:6731–6736.
26. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi:10.1006/meth.2001.1262
27. Kim SH, Henry EC, Kim DK, et al. Novel compound 2-methyl-2H-pyrazole-3-carboxylic acid (2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) prevents 2,3,7,8-TCDD-induced toxicity by antagonizing the aryl hydrocarbon receptor. Mol Pharmacol. 2006;69:1871–1878. doi:10.1124/mol.105.021832
28. Landgraf M, McGovern JA, Friedl P, et al. Rational design of mouse models for cancer research. Trends Biotech. 2018;36:242–251. doi:10.1016/j.tibtech.2017.12.001
29. Contador-Troca MA-BA, Barrasa E, Rico-Leo EM, et al. The dioxin receptor has tumor suppressor activity in melanoma growth and metastasis. Carcinogenesis. 2013;34:2683–2693. doi:10.1093/carcin/bgt248
30. Liu Y, Liang X, Dong W, et al. Tumor-repopulating cells induce PD-1 expression in CD8(+) T cells by transferring kynurenine and AhR activation. Cancer Cell. 2018;33:480–494. doi:10.1016/j.ccell.2018.02.005
31. Longhi MP, Trumpfheller C, Idoyaga J, et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med. 2009;206:1589–1602. doi:10.1084/jem.20090247
32. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–220. doi:10.1016/j.it.2016.01.004
33. Takeya M, Komohara Y. Role of tumor-associated macrophages in human malignancies: friend or foe? Path Int. 2016;66:491–505. doi:10.1111/pin.12440
34. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi:10.1016/j.cell.2010.01.025
35. Matter CM, Handschin C. RANTES (regulated on activation, normal T cell expressed and secreted), inflammation, obesity, and the metabolic syndrome. Circulation. 2007;115:946–948. doi:10.1161/CIRCULATIONAHA.106.685230
Supplementary materials
Effect of TCDD on LPS-elicited humoral challenge
LPS (Lipopolysaccharide) is a component of the bacterial cell wall and an endotoxin which elicits an innate immune response via TLR4 pathway and also a humoral immune response mediated by CD4+ T cells. This experiment was based on the paper from Kaminski et al.1 The idea behind using this model was to see if the TCDD dosage of 1 µg per mouse suppresses the activation of humoral response initiated by 25 µg of LPS.
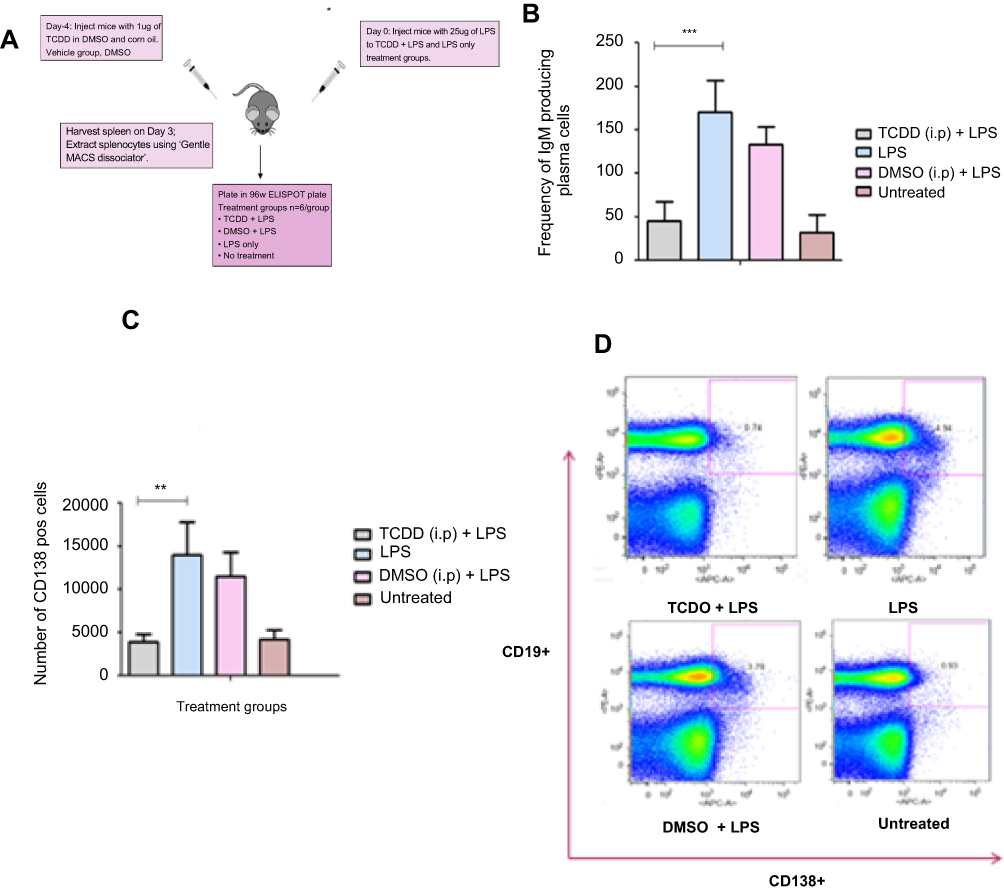 | Figure S1 (A) Schematic of the TCDD-mediated suppression of the humoral response due to administration of 25 µg of LPS in C57BL/6 mice. (B) Humoral response measured by the frequency of IgM producing plasma cells elicited on day 3 after LPS challenge and day 4 after TCDD administration. (C) Reduced CD138+ plasma cell number after TCDD and LPS challenge. (D) FACS plot showing CD19+ CD138+ plasma cells in splenocytes from mice administered with TCDD and LPS challenge. Abbreviations: 2,3,7,8-tetra-chlorodibenzo-p-dioxin ; LPS |
The experiment was done as described in Figure S1. Mouse splenocytes were plated into a 96-well ELISPOT assay plate, and the frequency of the IgM-producing plasma cells was measured. There was an increase in the number of IgM-producing plasma cells upon LPS challenge. This response was reduced by almost three-fold in mice that were exposed to TCDD prior to LPS challenge. In the DMSO+LPS group that received no TCDD, the humoral response was similar to the LPS alone group. The untreated group did not elicit any humoral response.
Next, CD138+ plasma cells were enumerated by FACS. Plasma cells are terminally differentiated B cells that produce specific antibodies for the antigen they have encountered. CD138 (Syndecan-1) is a specific plasma cell surface marker which B cells acquire after antigen-driven differentiation. CD19 was also used as a B-lymphocyte marker.
In supplementary Figure S1C, LPS challenge in the LPS and DMSO+LPS groups resulted in increased numbers of CD138hi plasma cells. In supplementary Figure S1D, plasma cell populations identified as CD19hiCD138hi cells were measured and a 3-fold reduction was observed in the TCDD-treated group. Therefore, TCDD at 1 µg/mouse suppressed the humoral response elicited by LPS challenge as measured by CD19hiCD138hi cells.
With the FACS analysis of B cells from the LPS challenge and the T cell subsets (from spleen and tumor), 1 µg/mouse TCDD dose is sufficient to suppress an LPS-elicited humoral response, the effector CD4+ T cells in the spleen and the CD8+ T cells in the tumor. However, this dosage has no effect on tumor volume. Therefore, it was decided to increase the TCDD dosing frequency, but below the lethal dose of TCDD (LD50), namely 160 µg/kg for C57BL/6.2
References
1. North CM, Crawford RB, Lu H, et al. Simultaneous in vivo time course and dose response evaluation for TCDD-induced impairment of the LPS-stimulated primary IgM response. Toxicol Sci. 2009;112:123–132.
2. Hogaboam JP, Moore AJ, Lawrence BP. The aryl hydrocarbon receptor affects distinct tissue compartments during ontogeny of the immune system. Toxicol Sci. 2008;102:160–170.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.