Back to Journals » Journal of Inflammation Research » Volume 15
SK-Channel Activation Alters Peripheral Metabolic Pathways in Mice, but Not Lipopolysaccharide-Induced Fever or Inflammation
Authors Bredehöft J, Dolga AM ![]() , Honrath B, Wache S, Mazurek S, Culmsee C
, Honrath B, Wache S, Mazurek S, Culmsee C ![]() , Schoemaker RG, Gerstberger R, Roth J, Rummel C
, Schoemaker RG, Gerstberger R, Roth J, Rummel C ![]()
Received 14 September 2021
Accepted for publication 25 November 2021
Published 23 January 2022 Volume 2022:15 Pages 509—531
DOI https://doi.org/10.2147/JIR.S338812
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Ning Quan
Janne Bredehöft,1 Amalia M Dolga,2 Birgit Honrath,2,3 Sybille Wache,1 Sybille Mazurek,1 Carsten Culmsee,3,4 Regien G Schoemaker,5 Rüdiger Gerstberger,1 Joachim Roth,1,4 Christoph Rummel1,4
1Institute of Veterinary Physiology and Biochemistry, Justus Liebig University Giessen, Giessen, Germany; 2Department of Molecular Pharmacology, Groningen Research Institute of Pharmacy, University of Groningen, Groningen, Netherlands; 3Institute of Pharmacology and Clinical Pharmacy, Philipps University of Marburg, Marburg, Germany; 4Center for Mind, Brain and Behavior-CMBB, Giessen and Marburg, Germany; 5Department of Neurobiology, GELIFES, University of Groningen, Groningen, Netherlands
Correspondence: Christoph Rummel
Institute of Veterinary Physiology and Biochemistry, Justus Liebig University Giessen, Frankfurter Strasse 100, Giessen D-35392, Germany
Tel +49 641 99 38155
Fax +49 641 99 38159
Email [email protected]
Purpose: Previously, we have shown that CyPPA (cyclohexyl-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-pyrimidin-4-yl]-amine), a pharmacological small-conductance calcium-activated potassium (SK)–channel positive modulator, antagonizes lipopolysaccharide (LPS)-induced cytokine expression in microglial cells. Here, we aimed to test its therapeutic potential for brain-controlled sickness symptoms, brain inflammatory response during LPS-induced systemic inflammation, and peripheral metabolic pathways in mice.
Methods: Mice were pretreated with CyPPA (15 mg/kg IP) 24 hours before and simultaneously with LPS stimulation (2.5 mg/kg IP), and the sickness response was recorded by a telemetric system for 24 hours. A second cohort of mice were euthanized 2 hours after CyPPA or solvent treatment to assess underlying CyPPA-induced mechanisms. Brain, blood, and liver samples were analyzed for inflammatory mediators or nucleotide concentrations using immunohistochemistry, real-time PCR and Western blot, or HPLC. Moreover, we investigated CyPPA-induced changes of UCP1 expression in brown adipose tissue (BAT)–explant cultures.
Results: CyPPA treatment did not affect LPS-induced fever, anorexia, adipsia, or expression profiles of inflammatory mediators in the hypothalamus or plasma or microglial reactivity to LPS (CD11b staining and CD68 mRNA expression). However, CyPPA alone induced a rise in core body temperature linked to heat production via altered metabolic pathways like reduced levels of adenosine, increased protein content, and increased UCP1 expression in BAT-explant cultures, but no alteration in ATP/ADP concentrations in the liver. CyPPA treatment was accompanied by altered pathways, including NFκB signaling, in the hypothalamus and cortex, while circulating cytokines remained unaltered.
Conclusion: Overall, while CyPPA has promise as a treatment strategy, in particular according to results from in vitro experiments, we did not reveal anti-inflammatory effects during severe LPS-induced systemic inflammation. Interestingly, we found that CyPPA alters metabolic pathways inducing short hyperthermia, most likely due to increased energy turnover in the liver and heat production in BAT.
Keywords: neuroinflammation, fever, inflammatory markers, small-conductance calcium-activated potassium channels, locomotor activity, brown adipose tissue
Graphical Abstract:

Introduction
Systemic inflammation is accompanied by an inflammatory response in the central nervous system. Experimentally, this response can be induced by administration of lipopolysaccharide (LPS), a component of Gram-negative bacteria cell walls. Injection of LPS induces severe illness symptoms, including fever, anorexia, and reduced activity.1–4 The proinflammatory cytokine IL6 is induced by LPS and considered one of the most important circulating cytokines in the context of fever,5 and its plasma concentration directly correlates with the magnitude of the febrile response.6 In the brain, sickness symptoms are accompanied by signs of brain inflammation like characteristic spatiotemporal expression pattern of various inflammatory mediators including transcription factors, cytokines, and inducible forms of enzymes (eg, COX2 and, microsomal prostaglandin E synthase [mPGES]) responsible for a pronounced brain-intrinsic formation of PGE2. Moreover, IL6-induced STAT3 translocation in brain endothelial cells7 or the vascular organ of lamina terminalis (OVLT),8 expression of suppressor of cytokine signaling (SOCS)3, a negative regulator and activity marker of the Janus kinase (JAK)–STAT3 pathway,3 and LPS-induced IκBα, an inhibitor and activity marker of NFκB3,9 contribute to brain inflammatory signaling during systemic inflammation.3
Microglia are the most important immune-system cells in the brain.10,11 Their involvement in neuroinflammatory processes has been well described and broadly discussed, eg, using models of microglia depletion in organotypic brain slice cultures or in vivo.12–15 Increased microglial reactivity is regarded as an important mechanism in the pathogenesis of different neurological diseases, such as multiple sclerosis and stroke.15,16 Upon LPS stimulation, activated microglial cells have elevated levels of intracellular Ca2+ concentrations17 and produce and release increased amounts of inflammatory mediators, including TNFα, IL1β, and IL6.2,18 Expression levels of NF-IL6, CD68, or CD11b can be applied as activity markers of microglia and macrophages.19,20 Moreover, immunoreactivity of CD11b, the complement receptor type 3, has been used as an indicator for microglial activity or in methods applied targeting CD11b for microglia isolation from the brain.21–23
Small-conductance calcium-activated potassium channels (SK) are expressed as subtypes, namely SK1, SK2, and SK3, and are present in various areas of the brain,24 including the hippocampus and hypothalamus.25 These SK channels have protective effects within the brain by maintaining Ca2+ homeostasis in neurons and microglial cells.26 In neurons, SK-channel activation prevents excessive stimulation of NMDA receptors, which is otherwise accompanied by Ca2+ influx into the cells. As such, SK-channel activation has been proposed as an efficient strategy to inhibit excitotoxic stimulation of NMDA receptors.27–29 We have recently shown protection from oxidative stress–induced neuronal death and promotion of neuronal resilience by metabolic mechanisms for SK-channel activation.30,31 Furthermore, the LPS-induced inflammatory response of microglia is reduced through the opening or activation of SK channels.32
CyPPA (cyclohexyl-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-pyrimidin-4-yl]-amine) is a well-established pharmacological activator of SK channels, acting on SK2- and SK3-channel subtypes.33 SK3-channel mRNA is found in murine microglia32,34 and significantly increased after LPS stimulation in rat primary microglial cell cultures.35 Administration of positive modulators of SK channels reduces ischemic neuronal death in model systems of focal cerebral ischemia and global ischemia after cardiac arrest.27,29 Activation of SK channels in vitro has been shown to modulate inflammatory processes: SK activation by CyPPA reduced LPS-induced reactivity of cultured microglia in a dose-dependent manner,32,36 and the LPS-mediated inflammatory response of murine primary microglial cells was attenuated by CyPPA and resulted in reduced production and release of TNFα, IL6, and nitric oxide. Furthermore, LPS-induced morphological alterations of microglial cells were attenuated by CyPPA.32 Therefore, SK-channel modulation could represent a promising target for therapeutic intervention in vivo in pathological conditions involving neuroinflammation.
In vivo brain inflammatory responses can be mimicked by stimulating rodents with LPS.37,38 In the present study, we aimed at translating previously described in vitro anti-neuroinflammatory properties of CyPPA32,36 to such an in vivo setup. For this purpose, C57Bl/6J mice were treated with CyPPA and with a LPS dose of 2.5 mg/kg, which induces robust fever and other sickness components like anorexia and reduced locomotor activity but no lethality.1,39 In order to investigate whether protective effects of this drug were exerted within the brain or were rather attributed to modification of the strength of peripherally derived afferent pro-inflammatory signaling, we evaluated the effects of CyPPA on sickness response including fever, reduced activity and anorexia; as well as expression profiles of inflammatory mediators in circulation and in the hypothalamus, a pivotal brain structure involved in regulating food intake, and body temperature. CD11b staining was utilized to assess microglial reactivity in the fimbria of hippocampus, a brain region which has previously shown strong immune-to-brain signaling during systemic inflammation.1
To investigate underlying mechanisms by which CyPPA treatment alone unexpectedly increase core body temperature (Tb), we carried out an additional experiment to further assess CyPPA action versus solvent in vivo, ie, if this response was fever or hyperthermia, two distinct pathophysiological responses. Fever is an active brain-controlled response to fight infection and inflammation and hyperthermia, a situation where heat-dissipation mechanisms are overwhelmed by heat production or heat exposure.40 We characterized a potential inflammatory response in plasma and the hypothalamus region of the brain by CyPPA alone. It was important to evaluate the effect on the hypothalamus, since it plays a crucial role in thermoregulation and also includes the OVLT, a circumventricular organ with a leaky blood–brain barrier that is a pivotal brain structure for fever-induction pathways and immune-to-brain communication.37,41,42 For a broader view, the cortex was also assessed for the inflammatory potential of CyPPA. Additionally, samples from the liver, which fulfills a large number of important metabolic tasks and also expresses SK2 and SK3 channels, were used to analyze nucleotides as markers for changes in energy turnover related to increased heat generation.43 Indeed, recent evidence has shown that CyPPA can induce metabolic changes.30 Moreover, additional experiments with brown adipose tissue (BAT)–explant cultures were used to assess the induction of UCP1, another important metabolic heat-generating mechanism.44
Methods
Animals
Male C57Bl/6J mice aged 7–8 weeks were purchased from Charles River (Sulzfeld, Germany) and used for the experiments. Animals were housed individually in a climatic chamber (Typ 10’US/+5 to + 40 DU; Weiss Umwelttechnik, DU, Germany) in controlled environmental conditions of 50% humidity, 30°C, and a 12-hour day/night cycle. Powdered standard laboratory chow (Ssniff Spezialdiäten) and water were available ad libitum. Eight days before the experiment, an intra-abdominal radio transmitter was implanted and took telemetric recordings of locomotor activity and Tb every 5 minutes, with data displayed in 15-minute increments for better visualization (MiniMitter). At 4–5 days after surgery, animals were moved to special experimental cages equipped with tunnels to access the powdered laboratory chow and rodent sipper valves for water access. The powdered laboratory chow and water bottles were placed on scales outside the experimental cages to enable accurate measurement of food and water intake over 24 hours without the risk of spillage.
Injections started 5 days after moving the mice to the experimental cages for habituation. Surgery was conducted under general anesthesia induced by intraperitoneal (IP) administration of xylazine (about 5–12 mg/kg body weight [BW]; CP-Pharma, Burgdorf, Germany) and ketamine (about 100 mg/kg BW; Medistar Arzneimittelvertrieb). Mice received analgesic treatment by oral administration of meloxicam (about 1 mg/kg BW; Boehringer Ingelheim Vetmedica) starting 1 day before surgery and lasting up to 2 days postsurgery. For habituation to experimental procedures and confinement, mice were handled once a day from at least 5 days before injection, involving weighing, inspection of the abdominal cicatrix, and the whole procedure for injection, but without actually inserting a needle. All animal procedures were performed in strict accordance with relevant guidelines and regulations for the care and use of animals, ie, EU Directive 2010/63/EU on the protection of animals used for scientific purposes and were authorized (ethics approval) by the local government authorities Regierungspräsidium Giessen (RP Giessen) after review by the animal-welfare officers of Justus Liebig University Giessen, RP Giessen and the Animal Protection Committee Giessen (Tierschutzkommission Giessen, project GI 18/2 Nr 28/2013). The animal-welfare officers of Justus Liebig University Giessen approved the harvesting of BAT for ex vivo explant cultures (project AZ 679_M).
Treatment and Experimental Protocols
CyPPA 5 mg (C5493-5MG; Sigma-Aldrich) was dissolved in 175 µL ethanol (Sigma-Aldrich) and 83 µL Kolliphor EL (synonym Cremophor EL, C5135-500G; Sigma-Aldrich) and brought to a final volume of 1.67 mL by adding 0.9% sterile and pyrogen-free saline up to 1 hour before injection. The final solution contained 5% Kolliphor and 10% ethanol, similar to what has been previously reported (10% Cremophor and 4% ethanol).45 To increase drug content, ethanol content can be up to 10% (according to the manufacturer’s instructions). For in vivo injection of 125 µL in a mouse with BW of 25 g (5 mL/kg) and the mouse having an approximate total blood volume of 2 mL (80 mL/kg), the initial ethanol concentration in plasma may reach 0.0625%. While the solvent was applied in both PBS and LPS-treated groups, effects of very low ethanol concentrations cannot be completely excluded. Indeed, although hard to compare to in vivo application, previous studies have shown that concentration of ethanol twice as high on brain-slice cultures evoked modulation of responsiveness of neurons involved in thermoregulation.46 Solvent was stored in aliquots at –20°C and defrosted before administration. Mice were treated IP with 5 mL/kg BW CyPPA solution or its solvent twice within 24 hours (at −24 and 0 hours). The final dose of 15 mg/kg BW CyPPA was well tolerated, while higher doses (30 mg/kg CyPPA) significantly reduced basic explorative activity in the mice.47 A third IP injection containing 2.5 mg/kg BW LPS (E. coli, serotype O111:B4; Sigma-Aldrich) at a dose previously reported to induce robust fever, changes in behavior, and brain inflammatory response in mice, but not death,1,39 or Dulbecco’s PBS (both at equal volumes of 5 mL/kg BW) was administered together with the second application of 15 mg/kg CyPPA or its solvent. All injections were performed between 9:00 and 10:40 am.
Experiments
A time line is presented in Supplementary Figure 1.
Experiment I
Five animals per treatment group (solvent + PBS, solvent + LPS, CyPPA + PBS, and CyPPA + LPS) were given terminal anesthesia with sodium pentobarbital (Merial, Hallbergmoos, Germany) at a dose of 100 mg/kg BW 24 hours after the last treatment. The time point of terminal anesthesia (24 hours) was chosen based on previous experiments investigating increased microglial reactivity induced by LPS and evidenced by changes in microglia morphology and cytokine production after LPS stimulation in vitro, which was blocked by CyPPA treatment.36 To investigate brain-controlled sickness symptoms, such as fever and anorexia, we focused on the hypothalamus for screening mRNA expression of inflammatory marker proteins. To assess LPS-induced morphological changes indicative of microglial reactivity, CD11b immunoreactivity was investigated in the fimbria of hippocampus, showing strong CD11b staining. Indeed, our previous findings revealed strong inflammatory signaling to the fimbria by neutrophil recruitment during LPS-induced systemic inflammation, supporting its role in immune-to-brain communication.1
Experiment II
Six animals per group (CyPPA + PBS and solvent + PBS) were euthanized 2 hours after the last injection, which was the peak of CyPPA-induced increase of Tb. Blood was collected via cardiac puncture with a heparinized sterile syringe, and all 32 animals were transcardially perfused with about 4–6 mL ice-cold 0.9% sodium chloride solution. Brains and livers were immediately removed, shock-frozen on powdered dry ice, and stored until further analysis at –54°C. Here, we investigated hypothalamic (control centers of thermoregulation and regulation of BW/food intake) and cortical mRNA expression of inflammatory marker proteins to assess a potential overall inflammatory response by CyPPA. Moreover, we focused on the OVLT, with its pivotal role in fever-induction pathways,37,41,42 to further assess if the CyPPA effect was a fever or hyperthermia. As we did not detect evidence of a fever-inducing effect in the brain, we further analyzed the potential activation of peripheral effector organs to contribute to a hyperthermic increase in Tb. For this purpose, we investigated nucleotides as indicators of increased energy turnover in the liver. In addition, we added a separate series of experiments using ex vivo fat-explant cultures after organ withdrawal, namely BAT, as it is an important thermoeffector organ in rodents.44
Tissue Processing
Brain sections were identified according to a neuroanatomical atlas48 and sliced at a thickness of 20 µm. The OVLT (bregma 0.62 to 0.38), a pivotal brain structure for fever induction and immune-to-brain communication pathways,41,49 and the fimbria of hippocampus (bregma −0.34 to −1.22), a brain area that shows increased numbers of microglia in mouse models of immunodeficiency50 and strong staining for CD11b, were prepared as described previously on poly-l-lysine (Biochrom)–coated glass slides1 and stored at –54°C until immunohistochemistry could be performed. Additional 80 µm sections between bregma 0.38 and −0.34 were stacked and the hypothalamus and the cortex identified and dissected out. The hypothalamus and cortex were then divided into the left and right hemisphere. Samples were stored at –54°C until Western blot or PCR processing.
Immunohistochemistry
Brain sections were air-dried and immersion-fixed for 10 minutes in 2% paraformaldehyde (Merck), followed by washing three times in PBS for 5 minutes. Slices were incubated for 1 hour in blocking solution containing 0.3% Triton X-100 (Sigma-Aldrich) and 10% normal donkey serum (Biozol). Diluted in blocking solution, sections were incubated with primary antibodies (rat anti-CD11b, diluted 1:500, 101201, BioLegend; rabbit anti-STAT3, diluted 1:4,000, sc-482, Santa Cruz Biotechnology; antisheep von Willebrand factor diluted 1:3,000, SARTW-IG; Affinity Biologicals) for 20–22 hours at 4°C. After three more washing steps, additional incubations for the STAT3-antibody protocol were performed using an avidin–biotin blocking kit (Sp-2001, Vector Laboratories) according to the manufacturer’s instructions, followed by a biotinylated antirabbit antibody (diluted 1:200, BA-1000, Vector Laboratories). For all other primary antibodies, secondary antibodies were incubated for 2 hours (diluted 1:500, Alexa 488–conjugated antisheep, A11015; Life Technologies; Cy3-conjugated antirat, 712–165-150; Jackson ImmunoResearch). For visualization of STAT3 signals, 90 minutes’ incubation with Cy3-conjugated streptavidin (diluted 1:600, 800–367-5296; Jackson ImmunoResearch) was carried out. Sections were counterstained for 10 minutes with DAPI diluted in PBS 1:8,000, (Mobitec), followed by a final three washing steps. Applied primary antibodies have previously been characterized for specificity of staining in the mouse brain.1,51–53 All sections were coverslipped with Citifluor and stored at 4°C until microscopic analysis. Unless stated otherwise, all procedures were carried out at room temperature.
Microscopic Analysis
Images were taken with a black-and-white Spot Insight camera (Diagnostic Instruments) attached to a light/fluorescent Olympus BX50 microscope. Using the software MetaMorph 7.7.5.0 (Molecular Devices), images for each staining were taken with equal exposure and individual photographs merged to RGB images. Brightness, contrast, and color balance were adjusted equally for comparison among treatment groups with Photoshop 5.5 (Adobe Systems). Digital mouse-brain maps were adapted to corresponding brain levels as per a brain atlas.48 At least two brain sections of the OVLT of each animal (perfused at the 2-hour time point, experiment II) were semiquantitatively evaluated with the help of a five-point scale based on intensity and quantity of signals, as previously described.7 In pictures taken of the fimbria of hippocampus (four to eight per animal, 20× magnification, perfused at the 24-hour time point, experiment I), which showed the strongest staining after screening for CD11b immunoreactivity, an area of interest, was taken covering the complete fimbria of hippocampus, but leaving the surrounding membranous tissue out. The area covered by CD11b-positive tissue was obtained using color cube–based image analyses (Image Pro Plus) and expressed as a percentage of the area of interest. Quantitative analysis was performed based on previously published procedures.54–56
Protein Extraction and Western Blot Analysis
For Western blot analysis, cortical tissue samples (10–25 mg) were suspended in 150 µL lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100) supplemented with protease- and phosphatase-inhibitor cocktails (Roche) using a 26 G syringe. After centrifugation for 10 minutes at 10,000 revolutions per minute, cell supernatants were collected and the protein content determined using the Pierce BCA protein assay kit (Thermo Fisher Scientific). Protein (25 µg) from each sample was separated on 10% SDS gel, followed by transfer onto a PVDF membrane (Millipore). After blocking with 5% skim milk in TBST (Tween–Tris base buffer), membranes were incubated with primary antibodies overnight at 4°C: anti-p65 (1:1,000, rabbit, sc-372; Santa Cruz Biotechnology), anti–phospho p65 Ser535 (1:1,000, rabbit, sc-33020; Santa Cruz Biotechnology), anti-IκBα (1:1,000, mouse, 4814; Cell Signaling Technology), and anti–α-tubulin (1:10.000, mouse; Sigma-Aldrich). Membranes were incubated with antimouse and antirabbit peroxidase-conjugated secondary antibodies (1:1,000; Vector Laboratories) for at least 1 hour at room temperature. Images were acquired with the ChemiDoc XRS Imaging System (BioRad Laboratories) and quantified according to the optical density of the bands (p65 and phospho p65 65 kDa, IκBα 30 kDa, α-tubulin 50 kDa) in relation to α-tubulin (loading control, 50 kDa) using ImageLab software (BioRad Laboratories).
Real-Time PCR
RNA extraction of 7–27 mg hypothalamic and ~40 mg BAT tissue samples and reverse transcription were conducted as previously described.1,57 Applied Biosystems assay IDs for the genes analyzed are: CD68 Mm03047340_m1, COX2 Mm00478374_m1, GAPDH 4352339E-1009032, IL6 Mm00446190_m1, mPGES Mm00452105_m1, NF-IL6 Mm00843434_m1, IκBα Mm00477798_m1, SOCS3 Mm00545913_s1, UCP1 Mm01244861_m1, and β-actin Mm02619580_g1. GAPDH was chosen as the housekeeping gene from 12 commonly used mouse genes using a Perfect Probe GeNorm mouse 12-gene kit and GeNorm software (ge-PP-12.mo; PrimerDesign) on hypothalamic mouse samples, and β-actin was chosen for BAT samples because of its higher expression stability between groups of BAT samples. Therefore, GAPDH and β-actin were used as a reference and to normalize cDNA amounts between different reactions. Sample values are depicted as x-fold differences from a control sample. The 2–∆∆Ct method was used to calculate relative expression as reported previously.58,59
Plasma IL6 and TNFα Measurement
IL6 and TNFα levels were measured in blood-plasma samples using bioassays. Detection was based on an IL6-dependent growth effect on B9 hybridoma cells and a cytotoxic effect of TNFα on WEHI cells. The detection limit was 3 IU/mL for IL6 and 6 pg/mL for TNFα. Both bioassays have been previously described.60
HPLC Analysis of Nucleotides
About 300 mg liver-tissue samples of animals perfused 2 hours after treatment (experiment II, CyPPA + PBS, solvent + PBS) were immediately homogenized in 3 mL ice-cold 0.6 N HClO4 (Sigma-Aldrich) per gram wet weight using a mixer mill (2×15 seconds, 30 Hz) and cooled grinding balls (Retsch). After centrifugation (20 minutes, 40,000 g, 4°C), supernatants were adjusted with ice-cold 0.6 N KOH (Merck) to pH 5–6. To remove KClO4, a second centrifugation was conducted. The supernatants were lyophilized (0.3 mbar, −55°C, 20 hours; Martin Christ Gefriertrocknungsanlagen) and stored at −80°C until the measurements. Unless otherwise noted, all operations were carried out on ice. Nucleotides were measured by reverse-phase ion-pair liquid-chromatography using a Nucleodur C18 gravity column (Macherey-Nagel). In the mobile phase, a buffer gradient of two base solutions (buffer A 50 mM KH2PO4 [AppliChem] and 2.5 mM tetra-butyl-ammonium hydrogen sulfate [TBA-HS]; Sigma Aldrich; pH 6); buffer B 70 mM KH2PO4, 3.5 mM TBA-HS, and 30% v/v methanol (Rotisolv (Roth) was used to separate nucleotides. Quantification of the nucleotides was carried out by integration of the peak areas of sample and standard nucleotides using Clarity Chromatography software (Data Apex). Nucleotide concentrations were normalized to the wet weight of the liver samples. In order to compare protein concentrations in liver samples of control and CyPPA-treated mice, the cell pellets of the first centrifugation step were suspended in PBS, and protein concentrations were determined using the Bradford protein assay (Sigma-Aldrich) according to the manufacturer’s specifications.
Brown Adipose Tissue–Explant Culture Experiments
Interscapular BAT was dissected after skin disinfection (70% ethanol; Stockmeiern) and explant cultures had been performed as recently described with small modifications to account for the high energy needs of this highly metabolic tissue.61 In brief, for each independent experiment, BAT from eight mice was washed with cold PBS (Capricorn Scientific), cut into ~40 mg pieces to ensure nutrient supply, and stored on ice in oxygenated Earle’s balanced salt solution (Thermo Fisher Scientific) supplemented with penicillin–streptomycin (100 U/mL, Thermo Fisher Scientific), and 20 mM HEPES (Sigma-Aldrich). For each experiment, eight wells of BAT were cultured in 12-well plates with DMEM supplemented with high glucose (GlutaMax Supplement, Thermo Fisher Scientific) containing 10% FBS (Capricorn), penicillin–streptomycin (100 U/mL), and 40 mM HEPES for 2 hours at 37°C (95% humidity, 5% CO2, and 95% air). Afterward, BAT was washed (FBS-free medium) and incubated for 2 hours with 10 µg/mL LPS (E. coli serotype 0111:B4, lot 043M4104; Sigma-Aldrich) or PBS dissolved in DMEM (FBS-free) and cotreated with CyPPA (25 µM according to previous in vitro assessments30) or solvent (see above). BAT-explant cultures have been used to investigate cytokine release into supernatants.61 After collection of the supernatants, BAT was frozen on dry ice and stored at −80°C for further analyses of UCP1 expression.
Data Analysis and Statistics
Data were analyzed separately for the 24- and 2-hour time points (experiments I and II) using paired Student’s t-tests or two-way ANOVA for treatment 1 (CyPPA or solvent) and treatment 2 (LPS or PBS) followed by Bonferroni post hoc tests (Prism 5, GraphPad). One outlier each on UCP1 and SOCS3 expression was detected with Grubbs’s test and excluded from statistical analyses. The thermal index was calculated for each animal by integration of the area under the curve using Microsoft Excel based on average temperature 2 hours before injection (baseline). The thermal index/fever index has frequently been used to characterize febrile responses.8,62–65 For statistical analyses, Tb (at 15-minute intervals) was separated into 2-hour intervals for experiments with an end point after 24 hours (experiment I) and divided into 0.5-hour intervals for experiments with an end point after 2 hours (experiment II) and analyzed using three-way repeated-measure ANOVA. Between-subject factors were treatment 1 (CyPPA or solvent) and 2 (LPS or PBS), the within-subject factor was time. Bonferroni correction for multiple-comparison analysis was carried out, followed by Tukey’s post hoc test in cases of positive interactions (Statistica 12, StatSoft). All data are shown as means ± SEM. P<0.05 was considered statistically significant.
Results
Experiment I: CyPPA-Treated Mice Showed Increased Core Body Temperature and Motor Activity Without an Effect on LPS-Induced Febrile Response
To investigate whether CyPPA influenced LPS-induced febrile response or Tb, animals were injected with CyPPA or the vehicle (solvent) in the presence of LPS or PBS (Figure 1A). Animals treated with solvent and PBS exhibited a normal circadian day/night rhythm, altered only by a stress-induced Tb rise as a consequence of handling and injection. Stimulation with 2.5 mg/kg LPS induced a short febrile response (1–3 hours, P=0.012) followed by a dramatic decrease in Tb during the night (7–21 hours, P<0.001) compared to counterpart controls (solvent + PBS and CyPPA + PBS). Interestingly, another observation was made alongside the LPS-mediated effect: CyPPA-treated animals (CyPPA + PBS/LPS) showed a significant increase in Tb up to 3 hours after injection compared to control mice (solvent + PBS/LPS, P<0.001). Mice stimulated with CyPPA alone (CyPPA + PBS) showed an increase in Tb comparable to mice treated with LPS only (solvent + LPS), while animals treated with CyPPA + LPS (apparently) exhibited the strongest increase in Tb; however, this effect did not reach significance. Mice treated with CyPPA + PBS had significantly higher Tb than solvent + PBS–treated animals (Figure 1B, post hoc P<0.05). However, after 24 hours, this CyPPA-induced Tb effect was no longer detectable (Figure 1B). The decrease in Tb in solvent + PBS–treated mice up to 2 hours after injection represents a normal temperature drop in the circadian rhythm occurring in the morning after lights have been turned on.
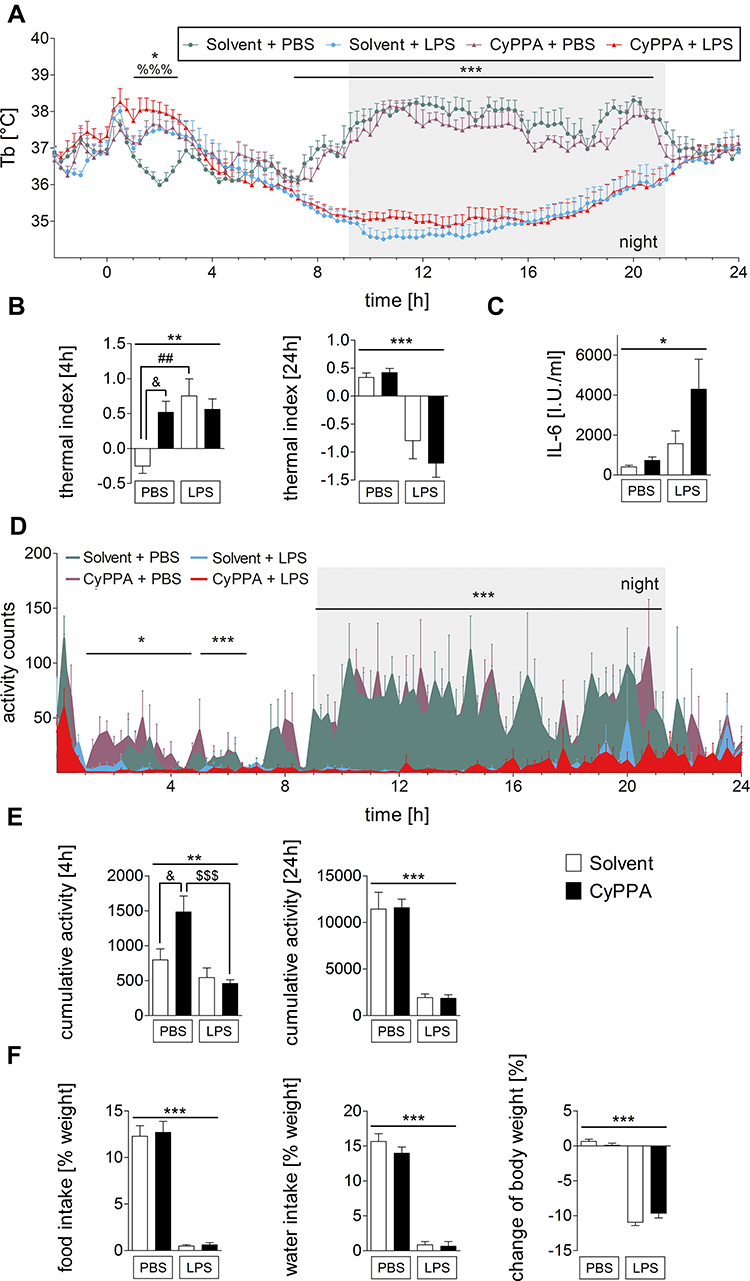 |
Figure 1 In vivo data and circulating IL6 levels in experimental and control groups of mice after injection of PBS or 2.5 mg/kg LPS and 15 mg/kg CyPPA or solvent. Core body temperature (Tb) (A), thermal index over 4 and 24 hours (B), IL6 plasma levels (C), locomotor activity over time (D), cumulative motor activity (E), body weight, food and water intake (F). Injection at time point 0. (A-–C) six animals per group; (D-,E) n=4 for solvent + LPS, 5 for the other groups; (F) five per group. %=Main effect of treatment 1 (solvent vs CyPPA), *=main effect of treatment 2 (PBS vs LPS), #=solvent + PBS vs solvent + LPS, &=CyPPA + PBS vs solvent + PBS, $=CyPPA + PBS vs CyPPA + LPS; %%%P<0.001; *P<0.05; **P<0.01; ***P<0.001; ##P<0.01; &P<0.05; $$$P<0.001. |
To clarify the influence of CyPPA on circulating mediators involved in immune-to-brain communication, we analyzed IL6 plasma concentrations. At 24 hours after LPS/PBS treatment, IL6 showed basal levels in control groups (CyPPA/solvent + PBS). After LPS stimulation, IL6 levels increased (Figure 1C, P=0.0108), while TNFα was not detectable in any plasma samples. Locomotor activity of PBS-treated mice showed a circadian rhythm with increased activity during the night. LPS in particular reduced nocturnal activity (Figure 1D, P<0.001) as previously found.1 Comparable to the Tb data, CyPPA + PBS–treated mice showed increased locomotor activity compared to the control group (solvent + PBS) for 4 hours following the drug injection, as revealed by cumulative analysis of the first 4 hours of the activity profile (Figure 1E, post hoc P<0.05). LPS-induced anorexia was not influenced by CyPPA treatment, and neither was food or water intake (Figure 1F, treatment 1 [CyPPA], BW P=0.4497, food intake P=0.7453, water intake P=0.2727; treatment 2 [LPS], P<0.001).
CyPPA Did Not Alter LPS-Induced Hypothalamic mRNA Expression of Inflammatory Markers
To obtain data on central inflammatory mediators, hypothalamic mRNA expression of several cytokines, transcription factors, and other inflammatory genes were analyzed with RT-PCR. As expected, 24 hours after 2.5 mg/kg LPS injection, mRNA expression of SOCS3 (P<0.001), COX2 (P<0.001), mPGES (P<0.001), and IκBα (P<0.01) had increased compared to PBS-treated mice. mRNA expression of NF-IL6, CD68 (an activity marker of microglia and macrophages19,20), and IL6 remained unaltered. However, 24 hours following CyPPA or solvent administration in the presence or absence of LPS or PBS, we did not detect any alterations that could be attributed to CyPPA alone or in combination with the LPS-induced hypothalamic expression of the inflammatory markers (Figure 2).
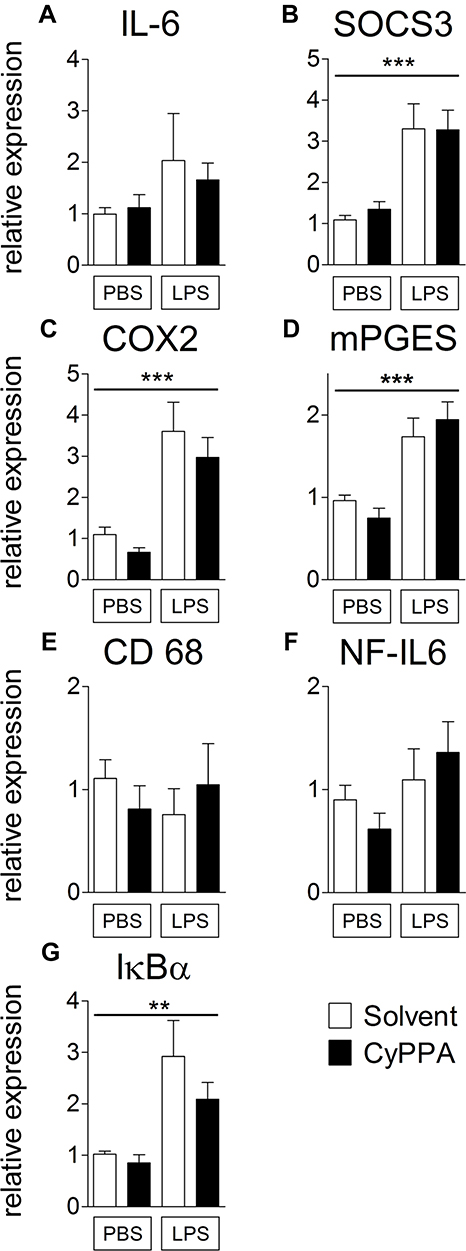 |
Figure 2 Comparison of hypothalamic mRNA expression of inflammatory marker proteins 24 hours after injection of PBS or 2.5 mg/kg LPS and 15 mg/kg CyPPA or solvent. The pro-inflammatory cytokine interleukin (IL)6 (A), the negative regulator and marker for signal transducer and activator of transcription 3 activation, suppressor of cytokine signaling (SOCS)3 (B), the rate limiting enzymes of prostaglandin synthesis, namely, cyclooxygenase (COX)2 (C), and microsomal prostaglandin synthase (mPGES,D) are displayed. Moreover, CD68 a marker of microglia activity (E), the transcription factor NF-IL6 (F) and the marker of nuclear factor κB activation, inhibitors (I)κBα (G) are shown. No difference was observed in CyPPA- (15 mg/kg) treated animals compared to solvent treated ones. n=5 for all groups. *Main effect of treatment 2 (PBS vs LPS); **P<0.01; ***P<0.001. |
CyPPA Did Not Influence CD11b Signals in the Fimbria of Hippocampus
Recently, CyPPA has been shown to antagonize LPS-induced reactivity of cultured microglial cells in a dose-dependent manner.32 For more detailed characterization of the in vivo effect of CyPPA on increased microglial reactivity induced by LPS, immunohistochemistry for the detection of CD11b was performed 24 hours following LPS stimulation. Qualitative analyses of CD11b signals in the fimbria of hippocampus showed larger cell bodies and less complex processes than controls, as previously shown for glial morphology after LPS stimulation (Figure 3A–D and high magnifications in a-d).66 However, no obvious morphological difference was observed following CyPPA treatment. Quantitative analysis of CD11b staining did not yield differences when all four groups were compared (Figure 3E).
 |
Figure 3 Assessment of microglial CD11b staining (red) in the fimbria of hippocampus 24 hours after treatment with PBS, or 2.5 mg/kg LPS and solvent, or CyPPA (15 mg/kg). CD11b immunoreactivity (red) in the fimbria of hippocampus (overview); nuclear DAPI staining (blue). Representative microphotographs of all four treatment groups (A–D solvent vs CyPPA and PBS vs LPS) and representative high-magnification insets, which were also derived from the fimbria of hippocampus, are depicted in A–D. The area covered by CD11b-positive tissue (red) was obtained using color cube-based image analyses (Image Pro Plus) and expressed as percentage of the area of interest (E). Five animals per group, four to eight microphotographs per animal analyzed; scale bar in D represents 100 µm and applies to all overview microphotographs; 10 µm in d for all inserts. |
Further Investigations to Analyze Effects of CyPPA on Physiological Parameters
In vivo CyPPA treatment resulted in an increase of both abdominal T and locomotor activity in the first 3–4 hours following administration in PBS-control animals. To further investigate whether this was a specific effect of CyPPA and to reveal potential mechanisms involved, another set of animals were treated with either CyPPA + PBS or solvent + PBS and euthanized 2 hours after treatment (experiment II).
Experiment II: Tb of Mice Had Increased Significantly 2 Hours after CyPPA Treatment Alone
After 1.75 hours, animals that had received CyPPA + PBS showed a statistically significant rise in Tb (Figure 4A, P=0.016), showing the same overall reaction pattern as in Figure 1 (see Supplementary Figure 2 for comparison). Thermal index analyses (2 hours, Figure 4B) did not reach statistical significance. It is important to note that the room was entered several times during the 2 hours to inject and perfuse animals, thus disturbing all animals present. This has to be taken into account as a confounding factor when comparing the Tb response between experiments I and II. IL6 plasma concentrations remained at basal levels in both treatment groups at the 2-hour time point (Figure 4C), while bioactive TNFα was not detectable in any sample. In contrast to Tb measurements, CyPPA did not elevate locomotor activity (Figure 4D and E). Therefore, this experiment suggests that the temperature increase was not due to increased activity detected in experiment I (Figure 1D and E).
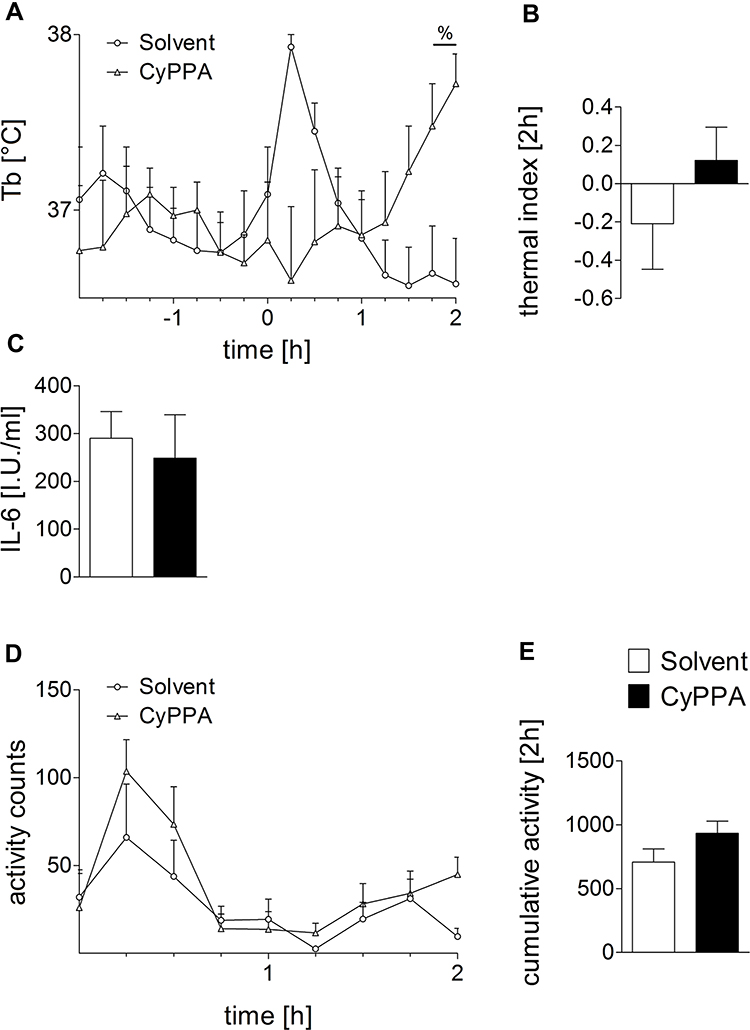 |
Figure 4 Early CyPPA (15 mg/kg) versus solvent action in vivo 2 hours after treatment. Core body temperature (Tb [°C]) (A), the thermal index (B), IL6 plasma levels (C), as well as locomotor activity over time (D) and cumulated (E) are displayed. Six animals per group. |
Hypothalamic and Cortical mRNA Expression of Different Inflammatory Markers Had Altered 2 Hours after CyPPA Stimulation
mRNA expression of inflammatory markers was analyzed 2 hours after treatment with CyPPA or its solvent and PBS. Interestingly, expression of NF-IL6 (Figure 5F, P=0.0494) and IκBα (Figure 5G, P=0.0011) had increased in the hypothalamus in CyPPA-stimulated mice compared to controls. Expression of the negative regulator of STAT3 signaling SOCS3, however, had significantly reduced (Figure 5B, P=0.0144) in the hypothalamus. As an endogenous regulator, SOCS3 inhibits JAK67 and thereby regulates the activity of STAT3. IL6, COX2, mPGES, and CD68 were unaffected by the treatment (Figure 5A P=0.8343, C P=0.8926, D P=0.9034, E P=0.1258). Cortical and hypothalamic expression of IκBα was significantly increased by CyPPA (Figure 5N, P=0.0045). In contrast, NF-IL6 expression was only significantly higher in the hypothalamus, but not the cortex (Figure 5M, P=0.1946). Moreover, the reduction in SOCS3 expression seen in the hypothalamus was not detectable in the cortex (Figure 5I, P=0.8771). Likewise, there were no differences between treatment groups for IL6, COX2, mPGES, or CD68 expression in the cortex (Figure 5H P=0.5864, J P=0.4861, K P=0.4663, L P=0.5345).
 |
Figure 5 Assessment of early CyPPA action on hypothalamic and cortical mRNA expression of inflammatory marker proteins 2 hours after CyPPA (15 mg/kg) treatment compared to control animals (solvent) (A–N). The proinflammatory cytokine IL6 (A and H), the negative regulator and marker for STAT3 activation, SOCS3 (B and I), the rate-limiting enzymes of prostaglandin synthesis, namely COX2 (C and J), and microsomal prostaglandin synthase (mPGES; D, K) are displayed. CD68 a marker of microglial activity (E and L), the transcription factor NF-IL6 (F and M) and marker of NFκB-activation inhibitors (I) κBα (G and N) are shown. Six animals per group; one outlier discarded for SOCS3 in the CyPPA group (n=5). *P<0.05 and **P<0.01 for solvent vs CyPPA. |
Cortical Expression Patterns of p65 Protein Altered by CyPPA
The elevated mRNA expression of IκBα in the hypothalamus and cortex 2 hours after CyPPA treatment of mice was indicative of modulatory effects of the SK-channel agonist on the NFκB signaling pathway. To further test this hypothesis, analyses of various specific markers of the NFκB signaling pathway were conducted by Western blot (Figure 6A; see Supplementary Figure 3 for corresponding entire Western blot images). It is known that phosphorylation of p65 (pp65) coincides with the degradation of IκB and finally leads to activation of p65.68 In the present study, IκBα and pp65 levels were not affected by CyPPA treatment (Figure 6C P=0.4566, D P=0.5406). In contrast, p65 levels were significantly reduced by CyPPA treatment compared to controls (Figure 6B, P=0.0257). However, the proportion of phosphorylated to unphosphorylated p65 did not differ between treatment groups (Figure 6E P=0.2680, F P=0.4373).
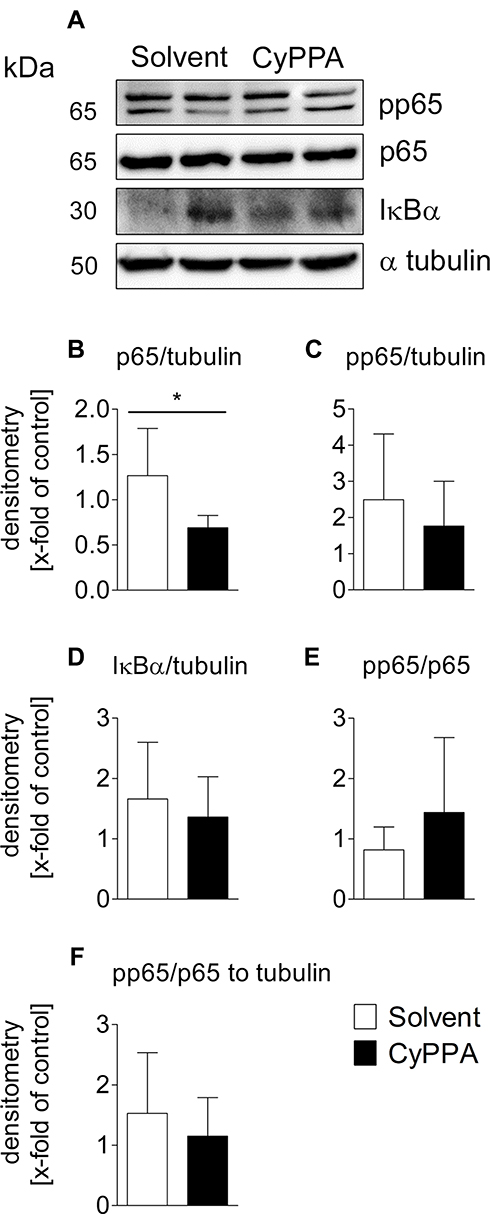 |
Figure 6 Western blot analyses of early CyPPA action on cortical NFκB signaling 2 hours after CyPPA (15 mg/kg) or solvent treatment. Exemplary protein bands are shown out of 6 samples P65 (A), pp65 (phospho p65) (B), IκBα (C), and pp65/p65 quotients without (D and E) and with normalization to tubulin (F) are displayed. Quantification was based on tubulin expression levels. Six per group. *P<0.05 for solvent vs CyPPA. |
CyPPA Did Not Induce STAT3 Immunofluorescence in the Hypothalamus
PCR results indicated reduced expression of SOCS3 2 hours after CyPPA treatment. To further analyze possible changes in STAT3 activation at the protein level, immunohistochemical detection of STAT3, which is known to be activated at 1.5 hours with a peak around 3–4 hours after induction of systemic inflammation, was performed.3,7 Sections from both treatment groups (CyPPA and solvent, Figure 7C and D) revealed few and weak cytoplasmic STAT3 signals (red) in the region of the OVLT (overview, Figure 7A), a pivotal brain structure for fever induction.41,49 von Willebrand factor (green), a marker protein for endothelial cells, was used to depict potential endothelial STAT3 activation, which is known to occur during systemic inflammatory insults.7 An LPS-treated animal was stained as positive control for nuclear STAT3 translocation (activation), and is shown in Figure 7B with specific staining as shown in several previous studies.3,51,52 Semiquantitative evaluation of STAT3 immunoreactivity revealed no visible differences between the two treatment groups (Figure 7E, P=0.5223).
 |
Figure 7 Analyses of CyPPA-mediated induction of hypothalamic nuclear STAT3 translocation 2 hours after treatment with either solvent or CyPPA (15 mg/kg). Immunohistochemical detection of STAT3 (red) in the vascular organ of the lamina terminalis (OVLT; schematic neuroanatomical overview in A) depicted as nuclear signals (pink) in an LPS-treated animal as positive control with typical distribution (B). Representative microphotographs of solvent (C) and CyPPA treatment (D) with high-magnification insets of areas highlighted with dotted squares. For better orientation, von Willebrand factor (green) was used to label endothelial cells. Semiquantitative evaluation of the immunohistochemistry between solvent and CyPPA groups (at least two brain sections per animal, six animals per group) is shown in E. Bars in B apply to all overview microphotography and represent 100 µm, 10 µm for inserts. |
CyPPA Treatment Changed Hepatic Adenosine Concentrations and Increased UCP1 Expression in BAT-Explant Cultures
In order to investigate whether the increased Tb in CyPPA-treated mice was possibly due to an increased metabolic rate. Various nucleotides involved in energy metabolism, were measured (Figure 8A–F). In livers of CyPPA-treated animals, a significant reduction in adenosine (Figure 8A, P=0.0273) and a slight but not significant decrease in AMP (Figure 8B, P=0.1245) were observed. Concentrations of neither ATP (Figure 8D, P=0.1245) nor GTP (Figure 8F, P=0.5106), nor the ATP:ADP ratio (Figure 8G, P=0.1455), significantly increased (Figure 8G and H, P=0.1455, P=0.7132) in the livers of CyPPA-treated mice compared to untreated controls. Interestingly, we detected a significant elevation in protein content in liver samples from CyPPA-treated mice compared to controls (Figure 8I, P<0.0001). Activation of UCP1 in BAT induces short circuits in the respiratory chain, which represents another efficient metabolic heat-producing pathway.44 As such, we used ex vivo BAT-explant cultures in a new set of experiments and found increased UCP1 expression 2 hours after CyPPA treatment compared to controls (Figure 9, P<0.005). LPS had not significantly altered UCP1 mRNA expression at this time point. These results suggest a contribution of increased BAT activity to CyPPA-induced hyperthermia in our in vivo experiments.
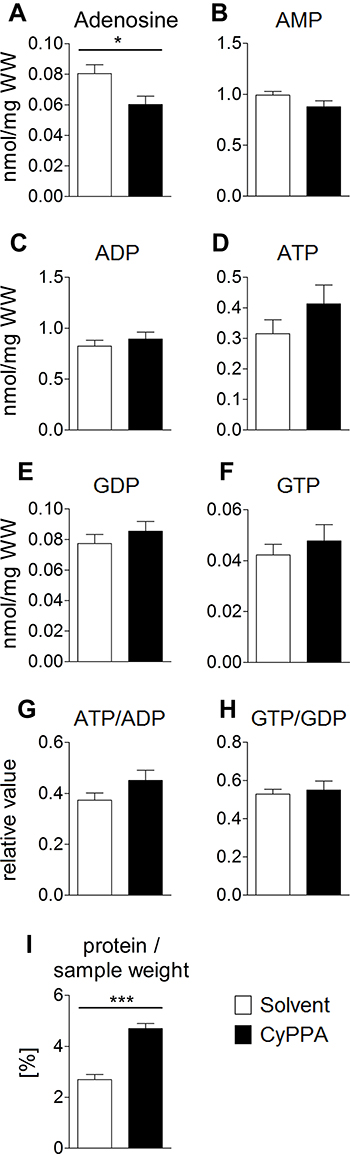 |
Figure 8 HPLC analysis of nucleotides in the liver as markers for changes in energy turnover induced by CyPPA 2 hours after CyPPA (15 mg/kg) or solvent application (A–I). Amount of intracellular adenosine (A), nmol/mg wet weight (WW), AMP B), ADP (C), ATP (D), GDP (E), GTP (F) concentrations and ATP:ADP (G) and GTP:GDP ratios. Cellular protein levels are displayed in I. n=6 animals per group. *P<0.05; ***P<0.001 for solvent vs CyPPA. |
 |
Figure 9 Comparison of brown adipose tissue (BAT)–explant cultures in experimental and control groups. Mouse BAT was stimulated with LPS or PBS and cotreated with solvent or CyPPA (25 µM) for 2 hours and mRNA expression of UCP1 was assessed. Results out of four independent experiments, with every well consisting of one mouse (biological replicate). One well was discarded due to bacterial contamination, one after outlier testing. PBS eight and LPS seven wells. **P<0.005 for solvent vs CyPPA. |
Discussion
LPS-treated mice showed adipsia, anorexia, and fever, followed by hypothermia. These changes were associated with elevated mRNA levels of several inflammatory markers. However, no statistically significant effects of the SK-channel opener CyPPA on LPS-induced sickness response, brain inflammation, or plasma IL6 were observed. Expression of all SK channels (SK1–3) by microglial cells has been demonstrated,35,36,69,70 and among these SK3 is the most important for the reactivity of microglia.36 Pharmacological inhibition of SK3 reduces the neurotoxic effect of activated microglia in rat primary cell cultures.35,36 CyPPA is not only a potent inhibitor of cytokine secretion by microglial cells but also affects LPS-induced morphological changes in microglia. Simultaneous application of LPS and CyPPA in cultured murine microglial cells leads to a reduction in LPS-induced increases in microglia activation, as assessed at the level of morphological alterations and cytokine production.32
In the present study, we detected a robust LPS-induced qualitative morphological change in microglia in the fimbria of hippocampus at 24 hours (experiment I, CD11b). CD11b immunoreactivity has previously been confirmed to stain increased microglia reactivity.21 Moreover, CD11b deficiency reduced microglial proinflammatory activation in a mouse model of Parkinson’s disease.71 However, CD11b-staining coverage was not significantly altered by LPS or CyPPA. Similarly, mRNA expression analyses of the microglial reactivity marker CD68 did not reveal significant changes. As such, effects of CyPPA on increased LPS-induced microglia reactivity that we previously observed in vitro could not be demonstrated in in vivo experiments where CyPPA (15 mg/kg) was administered together with LPS. Potential reasons for this discrepancy might pertain to the timing of CyPPA treatment and the magnitude of the robust systemic inflammatory response elicited by LPS in vivo. In addition, insufficient distribution of CyPPA in the brain parenchyma could also have contributed to the differences, but good penetration of CyPPA into brain tissue was confirmed by the manufacturer (personal communication by Saniona to A Dolga) and has been reported in a pilot study.72 Nonetheless, we have to acknowledge that higher doses of CyPPA, which may induce stronger side effects, could potentially alter brain microglial reactivity in vivo when applied systemically. Additional microglial reactivity markers should be used in future studies to assess modulatory effects. We did not find any obvious changes from CyPPA administration in brain or peripheral inflammatory markers during robust LPS-induced systemic inflammation. However, in a recent study, IP injection with NS309, another positive modulator of SK (KCa3.1/KCa2) channels, reduced levels of the proinflammatory cytokines IL1β, IL6, and TNFα and chemokines MCP1, MIP2, and RANTES following traumatic brain injury. RT-PCR and Western blot revealed that NS309 mediated an increase in TSG6 expression and an inhibition of NFκB activation.43 In SOD1G93A high-copy mice, a mouse model for amyotrophic lateral sclerosis, early treatment with CyPPA prolonged the survival and motor function of SOD mice. Most interestingly, long-term beneficial effects of CyPPA were associated with modulation of neuroinflammation and mitochondrial function.73
Application of novel depletion paradigms for microglia in rats and mice by another lab after our experiments had been completed revealed that LPS-induced sickness was not altered in the absence of microglia.74 While the authors did not show data on LPS-induced fever, they observed an enhanced inflammatory response with depletion of macrophage-like cells in rats and mice. Therefore, even if other markers of increased microglia reactivity have been altered by CyPPA in vivo, this may not result in changes in the sickness response. Moreover, our experiments demonstrated that the inflammatory response was not exaggerated systemically and only very moderately altered in the brain by CyPPA. Interestingly, an unexpected intrinsic effect of CyPPA was seen in vivo. Animals showed increased locomotor activity 2–4 hours after CyPPA application and elevated Tb up to ~4 hours compared to controls. To clarify underlying central or peripheral mechanisms, signaling pathways and brain areas involved, two additional experimental groups (experiment II — CyPPA + PBS and solvent + PBS) were analyzed at the peak of this response 2 hours after injection of CyPPA. In addition, BAT-explant cultures were analyzed to reveal potential stimulating effects of CyPPA on the known heat-generating function of this highly metabolically active tissue by increased expression of UCP1 and uncoupling of the respiratory chain.75,76
SOCS3 Expression in the Brain 2 Hours after CyPPA Injections in Mice
We demonstrated an influence of CyPPA on the hypothalamic expression of SOCS3, an activity marker of the transcription factor STAT3. However, neither STAT3 immunoreactivity in the OVLT nor SOCS3 expression was altered in the cortex of these animals. Therefore, CyPPA does not seem to affect SOCS3 expression by inhibition of STAT3 activity. In general, SOCS3 cannot only be influenced by the transcription factor STAT377 but also by NF-IL6. Binding of NF-IL6 on the SOCS3 promoter has been shown for several cell types.78 In the present study, increased NF-IL6 expression 2 hours after CyPPA treatment was accompanied by reduced SOCS3 expression in the hypothalamus, suggesting that NF-IL6 was not directly involved here. Overall, our data reveal mixed results for CyPPA modulating STAT3/SOCS3 signaling. An effect of SK2- or SK3-channel activation on JAK–STAT3 signaling has not been reported so far. Future studies need to confirm a potential modulating effect, as STAT3/SOCS3 has been shown to play an important role in neuroinflammatory processes.79 Whether modulatory effects on SOCS3 expression in vivo may be involved in previously described neuroprotective and anti-inflammatory properties of CyPPA in vitro28,80 remains an open question.
Brain NFκB Signaling 2 Hours after CyPPA Injections in Mice
We found increased IκBα mRNA expression in the cortex and hypothalamus, but reduced p65 protein concentrations in the cortex. This observation most likely indicates inhibition of the whole NFκB signaling pathway due to an enhanced inhibitory effect of IκBα on NFκB activation linked to reduced NFκB protein levels. However, some caution needs to be taken when using IκBα as an activity marker for NFκB signaling, as these molecules do not strictly reflect each other’s activity.81 We found an in vivo influence of CyPPA on the NFκB signaling pathway under noninflammatory conditions at protein and mRNA levels. The NFκB signaling pathway is well known to be involved in neuroinflammatory and neurodegenerative processes.82–84 Therefore, the anti-inflammatory potential of CyPPA as a potent inhibitor of the NFκB pathway is of much interest for the treatment of such conditions and might thereby contribute to protective effects.
Peripheral Cytokines and Enzymes Essential for Prostaglandin Synthesis 2 Hours after CyPPA Injection in Mice
Although we have previously shown that CyPPA reduces TNFα release from microglial cells in vitro, most likely via a Ca2+-independent mechanism,32 a significant effect on TNFα expression was not detected in vivo in the present study, as TNFα levels remained below the detection limit of 6 pg/mL. Similarly, CyPPA successfully reduced the LPS-induced increase in IL6 in microglial cell cultures.32 Again, this phenomenon was not observed under in vivo conditions. Neither plasma concentration nor hypothalamic mRNA expression of IL6 was altered by CyPPA compared to controls. The lack of systemic and central effects on expression and release of IL6 and TNFα by CyPPA in vivo might be attributed to cell type–specific effects of CyPPA and pharmacokinetic properties, respectively.
CyPPA Increases Tb under Noninflammatory Conditions 2 Hours after Injection in Mice
Interestingly, we were able to show that administration of CyPPA induced several changes in mice, even without any apparent inflammatory stimulation. Tb of CyPPA-treated animals increased about 1 hour after injection and reached a statistically significant level at the end of the second hour post injection. This increase in Tb was accompanied by enhanced locomotor activity of these animals, suggesting that the rise in Tb might be related to some activity-inducing effects of CyPPA. The second cohort of animals, which were euthanized 2 hours after CyPPA stimulation (experiment II), also showed a significant rise in Tb, but locomotor activity was not significantly increased. This observation suggests that increased locomotor activity might not represent the cause of the increase in Tb after CyPPA treatment.
In general, CyPPA and other SK-channel modulators are known to improve locomotor function in mouse models. For example, in a model of spinocerebellar ataxia type 3 IP administration of 10 mg/kg of SK-channel agonist SKA31 improved motor coordination and balance in transgenic mice.85 Further, the SK-channel agonist NS13001 and CyPPA enhanced motor performance on a RotaRod and beamwalk in a transgenic mouse model for spinocerebellar ataxia type 2 after oral administration of 30 mg/kg NS13001 or 10 mg/kg CyPPA per day for 3 consecutive weeks.72 In another mouse model of spinocerebellar ataxia type 2, the performance of animals receiving a chronic cerebellar perfusion of 80 ng/hour 1EBIO, another SK-channel agonist, significantly improved on the RotaRod compared to controls.86 SK-channel activation prevents degeneration of Purkinje cells.72 Administration of 15 mg/kg CyPPA leads to significantly reduced distances covered in open-field tests87 as well as to reduced methylphenidate-induced hyperactivity and stereotypical behavior in a concentration-dependent manner (3–30 mg/kg CyPPA).47 Contrary to these previous studies, we observed induction of locomotor activity in CyPPA-treated animals in the first experiment, but this effect was not confirmed in the second study. Previous studies have even described the development of seizures induced by SK channel–inhibitor overdoses.88 In our study, no signs of cramping or tremor were observed in any animal. An overdose of the SK-channel agonist CyPPA and a resulting paradoxical pharmaceutical reaction can thus be excluded, which in fact only occurs when applying much higher doses of CyPPA (eg, 30 mg/kg47) than used in the present study (15 mg/kg).
In order to investigate the potential cause of increased Tb by CyPPA and to distinguish if this response represents fever or hyperthermia, further analyses were conducted. Both IL6 plasma concentrations and hypothalamic IL6 mRNA expression (both time points, experiment I and II) were unaltered, subsequent IL6-induced STAT3 activation was not detectable, and neither COX2 nor mPGES expression was increased. In general, STAT3 activation is mainly mediated by circulating IL6, which is one of the most important endogenous mediators for manifestation of fever via induction of the rate-limiting enzymes (COX2 and mPGES) of PGE2 synthesis.51,89,90 Therefore, our results do not support the induction of a classic fever by CyPPA. SOCS3 mRNA expression was even reduced 2 hours after CyPPA treatment compared to controls. As such, the observed Tb increase most likely represents non–pyrogen-induced hyperthermia, rather than pyrogen-induced PGE2-dependent fever. For instance, locomotor activity did not seem to contribute to such hyperthermic responses in the present study. Aside from our study, other groups have previously shown that locomotor activity does not contribute to stress-induced hyperthermia during cage switch.91,92
Interestingly, NF-IL6 expression was significantly increased in the hypothalamus, but not in the cortex 2 hours after CyPPA administration under noninflammatory condition (experiment II). NF-IL6 is a ubiquitous inflammatory transcription factor that can be used as activation marker for a variety of brain-cell phenotypes.1,3,37,93 We have shown that NF-IL6 deficiency in mice reduces motor activity under basal conditions potentially related to altered endogenous corticosterone and proopiomelanocortin levels.1 It remains to be confirmed whether CyPPA influences the aforementioned systems by induction of NF-IL6 contributing to changes in locomotor activity.
Hepatic Energy Levels 2 Hours after Injections in Mice
One potential heat-generating mechanism that could further contribute to the observed hyperthermic response to CyPPA might pertain to increased metabolism. Measurement of intracellular nucleotide levels in the liver of CyPPA-treated mice revealed a significant decrease in adenosine levels within 2 hours after CyPPA application (15 mg/kg). ATP levels and the ATP:ADP ratio slightly but not significantly increased in the livers of CyPPA-treated mice. Notably, measurements of the intracellular protein content pointed to an increased rate of ATP consumption in protein synthesis. These results indicate that in mice treated for 2 hours with 15 mg/kg CyPPA, the metabolism of the liver is able to provide enough ATP to compensate for ATP-consumption processes. Adding 25 µM CyPPA in vitro to a culture medium of neuronal HT22 cells induces a slight decrease in basal mitochondrial respiration and an increase in glycolysis, thereby preserving ATP levels.30,94,95
UCP1 Expression 3 Hours after CyPPA Stimulation in BAT-Explant Cultures
BAT-explant experiments support a contribution of UCP1-mediated BAT-induced thermogenesis to CyPPA-induced hyperthermia in vivo.44 Interestingly, while UCP1-induced thermogenesis in BAT contributes to cold-induced heat generation,44 recent evidence suggests that this process does not contribute to stress-induced hyperthermia or fever.96,97 Overall, we report that CyPPA alters metabolic pathways in the liver and BAT, which adds to previous reports showing CyPPA action on cellular metabolism.30
Limitations
Differences among model systems should be considered when comparing the present observations on CyPPA action in vivo to previous in vitro data.32 Very likely, initiation of fever is a peripheral response to LPS,98 but central effects are mediated by cytokines and other humoral/cellular pathways independent of LPS, which — besides endothelial cells and circumventricular organs3,4,99 — do not directly affect microglia/neural cell reactivity in the brain. The effects detected in the brain are then likely indirect and not necessarily attributable to LPS treatment. Nonetheless, others have reported that the maintenance of the inflammatory response to endotoxemia is mediated by TLR4 in the brain.100
The use of solvents (DMSO in vitro versus ethanol/Kolliphor in vivo) should be taken into account when addressing these discrepancies. This difference is due to the fact that DMSO is not suitable for in vivo applications because of its potent modulatory effects on inflammation.101 While hard to compare to in vivo applications, previous studies have shown that double the concentration of ethanol than applied here evoked modulation of responsiveness of neurons involved in thermoregulation on brain-slice cultures.46 While some effects of the solvent cannot be excluded, we observed significant effects of CyPPA compared to the solvent. Moreover, CyPPA may need higher concentrations in vivo to reach sufficient concentration at the target cells in the brain to inhibit increased microglial reactivity. With the reproducible effects we observed in the present study, ie, increased Tb and other parameters reducing basic exploratory activity at higher doses of CyPPA,47 the promise of this drug to modulate neuroinflammation may need more consideration. In addition, while other microglial markers than CD11b or CD68 like Iba1102,103 may have been altered in our in vivo model, recent studies have found heightened inflammation after microglia depletion in rodents and no abrogation of inflammation-induced sickness.74 We have to acknowledge that changes in metabolism in the brain and in particular hypothalamic brain regions induced by CyPPA can be taken into account. However, since we did not find any of the postulated alterations that are characteristic of responses to inflammatory stimuli and fever regulation in the hypothalamus, our present study focused on peripheral tissues, ie, BAT as an important thermoregulatory effector organ in rodents, and the liver, with its known function in metabolism.
Conclusion
We observed mixed effects of the SK activator CyPPA in reducing JAK–STAT and increasing NFκB and NF-IL6 signaling pathways under noninflammatory conditions. This may be related to modulatory potential in neurological pathologies associated with neuroinflammation and neurodegeneration. In fact, the inflammatory transcription factors involved are known to contribute to brain inflammation or regulation of serotonin–kynurenine pathway balance.3 No effects of CyPPA on increases in microglial reactivity induced by LPS and on parameters of sickness response like fever and anorexia were observed in vivo after application of a strong LPS stimulus. Overall, while CyPPA might be suitable to modulate and treat less severe neuroinflammatory processes than investigated here, the observed CyPPA-induced hyperthermia was most likely related to decreased adenosine concentrations and increased protein contents in the liver, as well as to increased UCP1-mediated BAT thermogenesis.
Abbreviations
ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; BAT, brown adipose tissue; BW, body weight; CyPPA, N-cyclohexyl-N-[2-(3,5-dimethyl-pyrazol-1-yl)]-6-methyl-4-pyrimidinamine; GDP, guanosine diphosphate; GTP, guanosine triphosphate;; IP, intraperitoneal[ly]; JAK, Janus kinase; LPS, lipopolysaccharide; mPGES, microsomal prostaglandin E synthase; OVLT, vascular organ of the lamina terminalis; SK, small-conductance calcium-activated potassium channels; SOCS, suppressor of cytokine signaling; Tb, core body temperature; TBA-HS, tetra-butyl-ammonium hydrogen sulfate.
Data Sharing
Please contact C Rummel for data requests.
Ethics Approval
All animal procedures were authorized (ethics approval) by the local government authority, ie, Regierungspräsidium Giessen (RP Giessen) after review by the animal-welfare officers of Justus Liebig University Giessen, RP Giessen and the Animal Protection Committee Giessen, ie, Tierschutzkommission Giessen (project GI 18/2 Nr 28/2013). The animal welfare officers of Justus Liebig University Giessen approved the harvesting of brown adipose tissue for ex vivo explant cultures (project AZ 679_M).
Acknowledgments
We would like to thank Ms S Michels, Ms D Marks, Ms J Murgott, and Ms D Ott for excellent technical assistance, as well as Mr A Eva for his help performing HPLC experiments and Ms J Hernandez for English proofreading.
Author Contributions
All authors made a significant contribution to the work reported, whether in conception, study design, execution, acquisition of data, analysis and interpretation, or all these areas, took part in drafting, revising, or critically reviewing the article, gave final approval to the version to be published, have agreed on the journal to which the article has been submitted, and agree to be accountable for all aspects of the work.
Funding
Part of this work was supported by Deutsche Forschungsgemeinschaft (DFG FOR2107/CU 43/9-1 to CC, DO 1525/3-1 to AMD) and by funding of the “Lung-brain axis in health and disease” initiative by the Research Campus Mid-Hessen (FCMH) to CC and CR. AMD is the recipient of a Rosalind Franklin Fellowship cofunded by European Union and University of Groningen.
Disclosure
Professor Dr Christoph Rummel reports grants from DFG FOR2107/CU 43/9-1 awarded to CC and DFG DO 1525/3-1 and personal fees from Rosalind Franklin Fellowship awarded to AMD and support by funding of the “Lung-brain axis in health and disease” initiative by the Research Campus Mid-Hessen (FCMH) awarded to CC and CR during the conduct of the study. The authors declare that they have no competing interests.
References
1. Schneiders J, Fuchs F, Damm J, et al. The transcription factor nuclear factor interleukin 6 mediates pro- and anti-inflammatory responses during LPS-induced systemic inflammation in mice. Brain Behav Immun. 2015;48:147–164. doi:10.1016/j.bbi.2015.03.008
2. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. doi:10.1038/nrn2297
3. Rummel C. Inflammatory transcription factors as activation markers and functional readouts in immune-to-brain communication. Brain Behav Immun. 2016;54:1–14. doi:10.1016/j.bbi.2015.09.003
4. Peek V, Harden LM, Damm J, et al. LPS primes brain responsiveness to high mobility group box-1 protein. Pharmaceuticals. 2021;14(6). doi:10.3390/ph14060558.
5. Cartmell T, Poole S, Turnbull AV, Rothwell NJ, Luheshi GN. Circulating interleukin-6 mediates the febrile response to localised inflammation in rats. J Physiol. 2000;526(Pt 3):653–661. doi:10.1111/j.1469-7793.2000.00653.x
6. LeMay LG, Vander AJ, Kluger MJ. Role of interleukin 6 in fever in rats. Am J Physiol. 1990;258(3 Pt 2):R798–803. doi:10.1152/ajpregu.1990.258.3.R798
7. Rummel C, Voss T, Matsumura K, et al. Nuclear STAT3 translocation in Guinea pig and rat brain endothelium during systemic challenge with lipopolysaccharide and interleukin-6. J Comp Neurol. 2005;491(1):1–14. doi:10.1002/cne.20653
8. Rummel C, Hubschle T, Gerstberger R, Roth J. Nuclear translocation of the transcription factor STAT3 in the Guinea pig brain during systemic or localized inflammation. J Physiol. 2004;557(Pt 2):671–687. doi:10.1113/jphysiol.2003.058834
9. Laflamme N, Rivest S. Effects of systemic immunogenic insults and circulating proinflammatory cytokines on the transcription of the inhibitory factor kappaB alpha within specific cellular populations of the rat brain. J Neurochem. 1999;73(1):309–321. doi:10.1046/j.1471-4159.1999.0730309.x
10. Benarroch EE. Microglia: multiple roles in surveillance, circuit shaping, and response to injury. Neurology. 2013;81(12):1079–1088. doi:10.1212/WNL.0b013e3182a4a577
11. Chan WY, Kohsaka S, Rezaie P. The origin and cell lineage of microglia: new concepts. Brain Res Rev. 2007;53(2):344–354. doi:10.1016/j.brainresrev.2006.11.002
12. Berve K, West BL, Martini R, Groh J. Sex- and region-biased depletion of microglia/macrophages attenuates CLN1 disease in mice. J Neuroinflammation. 2020;17(1):323. doi:10.1186/s12974-020-01996-x
13. Masuch A, van der Pijl R, Funer L, et al. Microglia replenished OHSC: a culture system to study in vivo like adult microglia. Glia. 2016;64(8):1285–1297. doi:10.1002/glia.23002
14. Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014;9(6):2124–2138. doi:10.1016/j.celrep.2014.11.018
15. Nissen JC, Thompson KK, West BL, Tsirka SE. Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp Neurol. 2018;307:24–36. doi:10.1016/j.expneurol.2018.05.021
16. Teeling JL, Perry VH. Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: underlying mechanisms. Neuroscience. 2009;158(3):1062–1073. doi:10.1016/j.neuroscience.2008.07.031
17. Hoffmann A, Kann O, Ohlemeyer C, Hanisch UK, Kettenmann H. Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J Neurosci. 2003;23(11):4410–4419. doi:10.1523/JNEUROSCI.23-11-04410.2003
18. Wuchert F, Ott D, Murgott J, et al. Rat area postrema microglial cells act as sensors for the toll-like receptor-4 agonist lipopolysaccharide. J Neuroimmunol. 2008;204(1–2):66–74. doi:10.1016/j.jneuroim.2008.07.017
19. Bauer J, Sminia T, Wouterlood FG, Dijkstra CD. Phagocytic activity of macrophages and microglial cells during the course of acute and chronic relapsing experimental autoimmune encephalomyelitis. J Neurosci Res. 1994;38(4):365–375. doi:10.1002/jnr.490380402
20. Damoiseaux JG, Dopp EA, Calame W, Chao D, MacPherson GG, Dijkstra CD. Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology. 1994;83(1):140–147.
21. Goings GE, Kozlowski DA, Szele FG. Differential activation of microglia in neurogenic versus non-neurogenic regions of the forebrain. Glia. 2006;54(4):329–342. doi:10.1002/glia.20381
22. Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302(5651):1760–1765. doi:10.1126/science.1088417
23. Srinivasan K, Friedman BA, Larson JL, et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat Commun. 2016;7:11295. doi:10.1038/ncomms11295
24. Stocker M, Pedarzani P. Differential distribution of three Ca(2+)-activated K(+) channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol Cell Neurosci. 2000;15(5):476–493. doi:10.1006/mcne.2000.0842
25. Sailer CA, Kaufmann WA, Marksteiner J, Knaus HG. Comparative immunohistochemical distribution of three small-conductance Ca2+-activated potassium channel subunits, SK1, SK2, and SK3 in mouse brain. Mol Cell Neurosci. 2004;26(3):458–469. doi:10.1016/j.mcn.2004.03.002
26. Kuiper EFE, Nelemans A, Luiten P, Nijholt I, Dolga A, Eisel U. KCa2 and KCa3 Channels in Learning and Memory Processes, and Neurodegeneration. Front Pharmacol. 2012;3. doi:10.3389/fphar.2012.00003
27. Allen D, Nakayama S, Kuroiwa M, et al. SK2 channels are neuroprotective for ischemia-induced neuronal cell death. J Cereb Blood Flow Metab. 2011;31(12):2302–2312. doi:10.1038/jcbfm.2011.90
28. Dolga AM, Granic I, Blank T, et al. TNF-α mediates neuroprotection against glutamate-induced excitotoxicity via NF-κB-dependent up-regulation of KCa2.2 channels. J Neurochem. 2008. doi:10.1111/j.1471-4159.2008.05701.x
29. Dolga AM, Terpolilli N, Kepura F, et al. KCa2 channels activation prevents [Ca2+]i deregulation and reduces neuronal death following glutamate toxicity and cerebral ischemia. Cell Death Dis. 2011;2(4):e147. doi:10.1038/cddis.2011.30
30. Krabbendam IE, Honrath B, Dilberger B, et al. SK channel-mediated metabolic escape to glycolysis inhibits ferroptosis and supports stress resistance in C. elegans. Cell Death Dis. 2020;11(4):263. doi:10.1038/s41419-020-2458-4
31. Honrath B, Matschke L, Meyer T, et al. SK2 channels regulate mitochondrial respiration and mitochondrial Ca(2+) uptake. Cell Death Differ. 2017;24(5):761–773. doi:10.1038/cdd.2017.2
32. Dolga AM, Letsche T, Gold M, et al. Activation of KCNN3/SK3/K(Ca)2.3 channels attenuates enhanced calcium influx and inflammatory cytokine production in activated microglia. Glia. 2012;60(12):2050–2064. doi:10.1002/glia.22419
33. Hougaard C, Eriksen BL, Jørgensen S, et al. Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+channels. Br J Pharmacol. 2007;151(5):655–665. doi:10.1038/sj.bjp.0707281
34. Khanna R, Roy L, Zhu X, Schlichter LC. K+ channels and the microglial respiratory burst. Am J Physiol Cell Physiol. 2001;280(4):C796–806. doi:10.1152/ajpcell.2001.280.4.C796
35. Schlichter LC, Kaushal V, Moxon-Emre I, Sivagnanam V, Vincent C. The Ca2+ activated SK3 channel is expressed in microglia in the rat striatum and contributes to microglia-mediated neurotoxicity in vitro. J Neuroinflammation. 2010;7(1):4. doi:10.1186/1742-2094-7-4
36. Dolga AM, Culmsee C. Protective Roles for Potassium SK/K(Ca)2 Channels in Microglia and Neurons. Front Pharmacol. 2012;3:196. doi:10.3389/fphar.2012.00196
37. Damm J, Luheshi GN, Gerstberger R, Roth J, Rummel C. Spatiotemporal nuclear factor interleukin-6 expression in the rat brain during lipopolysaccharide-induced fever is linked to sustained hypothalamic inflammatory target gene induction. J Comp Neurol. 2011;519(3):480–505. doi:10.1002/cne.22529
38. Gautron L, Lafon P, Chaigniau M, Tramu G, Laye S. Spatiotemporal analysis of signal transducer and activator of transcription 3 activation in rat brain astrocytes and pituitary following peripheral immune challenge. Neuroscience. 2002;112(3):717–729. doi:10.1016/S0306-4522(02)00115-X
39. Aguilar-Valles A, Kim J, Jung S, Woodside B, Luheshi GN. Role of brain transmigrating neutrophils in depression-like behavior during systemic infection. Mol Psychiatry. 2014;19(5):599–606. doi:10.1038/mp.2013.137
40. Roth J, Blatteis CM. Mechanisms of fever production and lysis: lessons from experimental LPS fever. Compr Physiol. 2014;4(4):1563–1604.
41. Bredehoft J, Bhandari DR, Pflieger FJ, et al. Visualizing and Profiling Lipids in the OVLT of Fat-1 and Wild Type Mouse Brains during LPS-Induced Systemic Inflammation Using AP-SMALDI MSI. ACS Chem Neurosci. 2019;10(10):4394–4406. doi:10.1021/acschemneuro.9b00435
42. Roth J, Harre EM, Rummel C, Gerstberger R, Hubschle T. Signaling the brain in systemic inflammation: role of sensory circumventricular organs. Front Biosci. 2004;9:290–300. doi:10.2741/1241
43. Chen MX, Gorman SA, Benson B, et al. Small and intermediate conductance Ca(2+)-activated K+ channels confer distinctive patterns of distribution in human tissues and differential cellular localisation in the colon and corpus cavernosum. Naunyn Schmiedebergs Arch Pharmacol. 2004;369(6):602–615. doi:10.1007/s00210-004-0934-5
44. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84(1):277–359. doi:10.1152/physrev.00015.2003
45. Mourre C, Manrique C, Camon J, et al. Changes in SK channel expression in the basal ganglia after partial nigrostriatal dopamine lesions in rats: functional consequences. Neuropharmacology. 2017;113(Pt A):519–532. doi:10.1016/j.neuropharm.2016.11.003
46. Wollweber BT, Schneider H, Voigt K. Ethanol effects on temperature-sensitive hypothalamic neurons in rat brain slices. J Therm Biol. 2004;29:345–350. doi:10.1016/j.jtherbio.2004.08.083
47. Herrik KF, Redrobe JP, Holst D, et al. CyPPA, a Positive SK3/SK2 Modulator, Reduces Activity of Dopaminergic Neurons, Inhibits Dopamine Release, and Counteracts Hyperdopaminergic Behaviors Induced by Methylphenidate1. Front Pharmacol. 2012;3:3.
48. Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. San Diego, California, USA: Academic Press; 2001.
49. Simm B, Ott D, Pollatzek E, et al. Effects of prostaglandin E2 on cells cultured from the rat organum vasculosum laminae terminalis and median preoptic nucleus. Neuroscience. 2016;313:23–35. doi:10.1016/j.neuroscience.2015.11.034
50. Lorke DE, Ip CW, Schumacher U. Increased number of microglia in the brain of severe combined immunodeficient (SCID) mice. Histochem Cell Biol. 2008;130(4):693–697. doi:10.1007/s00418-008-0463-2
51. Rummel C, Sachot C, Poole S, Luheshi GN. Circulating interleukin-6 induces fever through a STAT3-linked activation of COX-2 in the brain. Am J Physiol Regul Integr Comp Physiol. 2006;291(5):R1316–1326. doi:10.1152/ajpregu.00301.2006
52. Rummel C, Inoue W, Sachot C, Poole S, Hubschle T, Luheshi GN. Selective contribution of interleukin-6 and leptin to brain inflammatory signals induced by systemic LPS injection in mice. J Comp Neurol. 2008;511(3):373–395. doi:10.1002/cne.21850
53. Torika N, Asraf K, Apte RN, Fleisher-Berkovich S. Candesartan ameliorates brain inflammation associated with Alzheimer’s disease. CNS Neurosci Ther. 2018;24(3):231–242. doi:10.1111/cns.12802
54. Latta CH, Sudduth TL, Weekman EM, et al. Determining the role of IL-4 induced neuroinflammation in microglial activity and amyloid-β using BV2 microglial cells and APP/PS1 transgenic mice. J Neuroinflammation. 2015;12(1):41. doi:10.1186/s12974-015-0243-6
55. Perego C, Fumagalli S, De Simoni M-G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation. 2011;8(1):174. doi:10.1186/1742-2094-8-174
56. Hovens IB, Nyakas C, Schoemaker RG. A novel method for evaluating microglial activation using ionized calcium-binding adaptor protein-1 staining: cell body to cell size ratio. Neuroimmunol Neuroinflammation. 2014;1(2):82–88. doi:10.4103/2347-8659.139719
57. Damm J, Harden LM, Gerstberger R, Roth J, Rummel C. The putative JAK-STAT inhibitor AG490 exacerbates LPS-fever, reduces sickness behavior, and alters the expression of pro- and anti-inflammatory genes in the rat brain. Neuropharmacology. 2013;71:98–111. doi:10.1016/j.neuropharm.2013.03.014
58. Dangarembizi R, Erlwanger KH, Rummel C, Roth J, Madziva MT, Harden LM. Brewer’s yeast is a potent inducer of fever, sickness behavior and inflammation within the brain. Brain Behav Immun. 2018;68:211–223. doi:10.1016/j.bbi.2017.10.019
59. Koenig S, Luheshi GN, Wenz T, Gerstberger R, Roth J, Rummel C. Leptin is involved in age-dependent changes in response to systemic inflammation in the rat. Brain Behav Immun. 2014;36:128–138. doi:10.1016/j.bbi.2013.10.019
60. Welsch J, Hubschle T, Murgott J, et al. Fever induction by systemic stimulation with macrophage-activating lipopeptide-2 depends upon TLR2 but not CD36. Innate Immun. 2012;18(3):541–559. doi:10.1177/1753425911426892
61. Peek V, Neumann E, Inoue T, et al. Age-Dependent Changes of Adipokine and Cytokine Secretion From Rat Adipose Tissue by Endogenous and Exogenous Toll-Like Receptor Agonists. Front Immunol. 2020;11:1800. doi:10.3389/fimmu.2020.01800
62. Roth J, McClellan JL, Kluger MJ, Zeisberger E. Attenuation of fever and release of cytokines after repeated injections of lipopolysaccharide in Guinea-pigs. J Physiol. 1994;477(Pt 1):177–185. doi:10.1113/jphysiol.1994.sp020182
63. Voss T, Rummel C, Gerstberger R, Hubschle T, Roth J. Fever and circulating cytokines induced by double-stranded RNA in Guinea pigs: dependence on the route of administration and effects of repeated injections. Acta Physiol. 2006;187(3):379–389. doi:10.1111/j.1748-1716.2006.01587.x
64. Simons CT, Kulchitsky VA, Sugimoto N, Homer LD, Szekely M, Romanovsky AA. Signaling the brain in systemic inflammation: which vagal branch is involved in fever genesis? Am J Physiol. 1998;275(1):R63–68. doi:10.1152/ajpregu.1998.275.1.R63
65. Zeisberger E, Roth J. Tolerance to pyrogens. Ann N Y Acad Sci. 1998;856:116–131. doi:10.1111/j.1749-6632.1998.tb08320.x
66. Savage JC, St-Pierre MK, Hui CW, Tremblay ME. Microglial Ultrastructure in the Hippocampus of a Lipopolysaccharide-Induced Sickness Mouse Model. Front Neurosci. 2019;13:1340. doi:10.3389/fnins.2019.01340
67. Sasaki A, Yasukawa H, Suzuki A, et al. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells. 1999;4(6):339–351. doi:10.1046/j.1365-2443.1999.00263.x
68. Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25(51):6717–6730. doi:10.1038/sj.onc.1209937
69. Hayashi Y, Kawaji K, Sun L, et al. Microglial Ca(2+)-activated K(+) channels are possible molecular targets for the analgesic effects of S-ketamine on neuropathic pain. J Neurosci. 2011;31(48):17370–17382. doi:10.1523/JNEUROSCI.4152-11.2011
70. Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci. 2007;27(1):234–244. doi:10.1523/JNEUROSCI.3593-06.2007
71. Hou L, Qu X, Qiu X, Huang R, Zhao X, Wang Q. Integrin CD11b mediates locus coeruleus noradrenergic neurodegeneration in a mouse Parkinson’s disease model. J Neuroinflammation. 2020;17(1):148. doi:10.1186/s12974-020-01823-3
72. Kasumu Adebimpe W, Hougaard C, Rode F, et al. Selective Positive Modulator of Calcium-Activated Potassium Channels Exerts Beneficial Effects in a Mouse Model of Spinocerebellar Ataxia Type 2. Chem Biol. 2012;19(10):1340–1353. doi:10.1016/j.chembiol.2012.07.013
73. Draper CSI. ALS-Induced Excitability Changes in Individual Motorneurons and the Spinal Motorneuron Network in SOD1-G93A Mice at Symptom Onset OhioLINK Electronic Theses and Dissertations Center. Wright State University; 2021.
74. Vichaya EG, Malik S, Sominsky L, Ford BG, Spencer SJ, Dantzer R. Microglia depletion fails to abrogate inflammation-induced sickness in mice and rats. J Neuroinflammation. 2020;17(1):172. doi:10.1186/s12974-020-01832-2
75. Golozoubova V, Cannon B, Nedergaard J. UCP1 is essential for adaptive adrenergic nonshivering thermogenesis. Am J Physiol Endocrinol Metab. 2006;291(2):E350–357. doi:10.1152/ajpendo.00387.2005
76. Nicholls DG, Locke RM. Thermogenic mechanisms in brown fat. Physiol Rev. 1984;64(1):1–64. doi:10.1152/physrev.1984.64.1.1
77. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374(Pt 1):1–20. doi:10.1042/bj20030407
78. Yarwood SJ, Borland G, Sands WA, Palmer TM. Identification of CCAAT/enhancer-binding proteins as exchange protein activated by cAMP-activated transcription factors that mediate the induction of the SOCS-3 gene. J Biol Chem. 2008;283(11):6843–6853. doi:10.1074/jbc.M710342200
79. Qin H, Yeh WI, De Sarno P, et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A. 2012;109(13):5004–5009. doi:10.1073/pnas.1117218109
80. Dolga AM, Netter MF, Perocchi F, et al. Mitochondrial Small Conductance SK2 Channels Prevent Glutamate-induced Oxytosis and Mitochondrial Dysfunction. J Biol Chem. 2013;288(15):10792–10804. doi:10.1074/jbc.M113.453522
81. Nadjar A, Combe C, Laye S, et al. Nuclear factor kappaB nuclear translocation as a crucial marker of brain response to interleukin-1. A study in rat and interleukin-1 type I deficient mouse. J Neurochem. 2003;87(4):1024–1036. doi:10.1046/j.1471-4159.2003.02097.x
82. Liu DL, Zhao LX, Zhang S, Du JR. Peroxiredoxin 1-mediated activation of TLR4/NF-kappaB pathway contributes to neuroinflammatory injury in intracerebral hemorrhage. Int Immunopharmacol. 2016;41:82–89. doi:10.1016/j.intimp.2016.10.025
83. Srinivasan M, Bayon B, Chopra N, Lahiri DK. Novel Nuclear Factor-KappaB Targeting Peptide Suppresses beta-Amyloid Induced Inflammatory and Apoptotic Responses in Neuronal Cells. PLoS One. 2016;11(10):e0160314. doi:10.1371/journal.pone.0160314
84. Zhang Y, Gao L, Cheng Z, et al. Kukoamine A Prevents Radiation-Induced Neuroinflammation and Preserves Hippocampal Neurogenesis in Rats by Inhibiting Activation of NF-kappaB and AP-1. Neurotox Res. 2016;31(2):259–268. doi:10.1007/s12640-016-9679-4
85. Shakkottai VG, Do Carmo Costa M, Dell’Orco JM, Sankaranarayanan A, Wulff H, Paulson HL. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci. 2011;31(36):13002–13014. doi:10.1523/JNEUROSCI.2789-11.2011
86. Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9(3):389–397. doi:10.1038/nn1648
87. Vick KA, Guidi M, Stackman RW. In vivo pharmacological manipulation of small conductance Ca2+-activated K+ channels influences motor behavior, object memory and fear conditioning. Neuropharmacology. 2010;58(3):650–659. doi:10.1016/j.neuropharm.2009.11.008
88. Lam J, Coleman N, Garing AL, Wulff H. The therapeutic potential of small-conductance KCa2 channels in neurodegenerative and psychiatric diseases. Expert Opin Ther Targets. 2013;17(10):1203–1220. doi:10.1517/14728222.2013.823161
89. Eskilsson A, Mirrasekhian E, Dufour S, Schwaninger M, Engblom D, Blomqvist A. Immune-induced fever is mediated by IL-6 receptors on brain endothelial cells coupled to STAT3-dependent induction of brain endothelial prostaglandin synthesis. J Neurosci. 2014;34(48):15957–15961. doi:10.1523/JNEUROSCI.3520-14.2014
90. Rummel C, Matsumura K, Luheshi GN. Circulating IL-6 contributes to peripheral LPS-induced mPGES-1 expression in the rat brain. Brain Res Bull. 2011;86(5–6):319–325. doi:10.1016/j.brainresbull.2011.09.006
91. Long NC, Vander AJ, Kunkel SL, Kluger MJ. Antiserum against tumor necrosis factor increases stress hyperthermia in rats. Am J Physiol. 1990;258(3 Pt 2):R591–595. doi:10.1152/ajpregu.1990.258.3.R591
92. Koenig S, Bredehoft J, Perniss A, Fuchs F, Roth J, Rummel C. Age Dependent Hypothalamic and Pituitary Responses to Novel Environment Stress or Lipopolysaccharide in Rats. Front Behav Neurosci. 2018;12:55. doi:10.3389/fnbeh.2018.00055
93. Fuchs F, Damm J, Gerstberger R, Roth J, Rummel C. Activation of the inflammatory transcription factor nuclear factor interleukin-6 during inflammatory and psychological stress in the brain. J Neuroinflammation. 2013;10:140. doi:10.1186/1742-2094-10-140
94. Richter M, Nickel C, Apel L, et al. SK channel activation modulates mitochondrial respiration and attenuates neuronal HT-22 cell damage induced by H2O2. Neurochem Int. 2015;81:63–75. doi:10.1016/j.neuint.2014.12.007
95. Richter M, Vidovic N, Honrath B, et al. Activation of SK2 channels preserves ER Ca(2+) homeostasis and protects against ER stress-induced cell death. Cell Death Differ. 2016;23(5):814–827. doi:10.1038/cdd.2015.146
96. Eskilsson A, Shionoya K, Enerback S, Engblom D, Blomqvist A. The generation of immune-induced fever and emotional stress-induced hyperthermia in mice does not involve brown adipose tissue thermogenesis. FASEB J. 2020;34(4):5863–5876. doi:10.1096/fj.201902945R
97. Dittner C, Lindsund E, Cannon B, Nedergaard J. At thermoneutrality, acute thyroxine-induced thermogenesis and pyrexia are independent of UCP1. Mol Metab. 2019;25:20–34. doi:10.1016/j.molmet.2019.05.005
98. Steiner AA, Chakravarty S, Rudaya AY, Herkenham M, Romanovsky AA. Bacterial lipopolysaccharide fever is initiated via Toll-like receptor 4 on hematopoietic cells. Blood. 2006;107(10):4000–4002. doi:10.1182/blood-2005-11-4743
99. Ott D, Murgott J, Rafalzik S, et al. Neurons and glial cells of the rat organum vasculosum laminae terminalis directly respond to lipopolysaccharide and pyrogenic cytokines. Brain Res. 2010;1363:93–106. doi:10.1016/j.brainres.2010.09.083
100. Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci. 2005;25(7):1788–1796. doi:10.1523/JNEUROSCI.4268-04.2005
101. Santos NC, Figueira-Coelho J, Martins-Silva J, Saldanha C. Multidisciplinary utilization of dimethyl sulfoxide: pharmacological, cellular, and molecular aspects. Biochem Pharmacol. 2003;65(7):1035–1041. doi:10.1016/S0006-2952(03)00002-9
102. Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation. 2015;12:114. doi:10.1186/s12974-015-0332-6
103. Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia. 2016;64(2):300–316. doi:10.1002/glia.22930
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.