")
Back to Journals » Medical Devices: Evidence and Research » Volume 14
Analysis of the Regulatory, Legal, and Medical Conditions for the Prescription of Mobile Health Applications in the United States, The European Union, and France
Authors Hassanaly P , Dufour JC
Received 24 July 2021
Accepted for publication 14 October 2021
Published 24 November 2021 Volume 2021:14 Pages 389—409
DOI https://doi.org/10.2147/MDER.S328996
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Parina Hassanaly,1 Jean Charles Dufour2
1Aix Marseille Université, Inserm, IRD, SESSTIM, Sciences Economiques & Sociales de la Santé & Traitement de l’Information Médicale, ISSPAM, Marseille, France; 2Aix Marseille Université, APHM, Inserm, IRD, SESSTIM, Sciences Economiques & Sociales de la Santé & Traitement de l’Information Médicale, ISSPAM, Hop Timone, BioSTIC, Biostatistique et Technologies de l’Information et de la Communication, Marseille, France
Correspondence: Parina Hassanaly
Aix Marseille Université, Faculté de Médecine, 27 Boulevard Jean Moulin, Marseille, 13005, France
Tel +33 491 324772
Email [email protected]
Introduction: Mobile health (mHealth) is now considered an important approach to extend traditional health services and to meet the growing medical needs. The prescribability of mHealth applications is a complex problem because it depends on a large number of factors and concerns a wide range of disciplines and actors in the industrial, health, normative, and regulatory domains.
Objective: Our study correlated data from the scientific literature with data on regulatory developments in the United States, the European Union, and France with the aim of identifying the conditions for the prescription of mHealth applications.
Methods: The search method adopted was the systematic literature review process by Brereton et al. All empirical evidence from the relevant fields of study was gathered and then evaluated to answer our predefined research questions. The WoS and PubMed databases were queried for the period between 1 January 1975 and 30 November 2020. A total of 165 articles (15 with a direct focus and 150 with an indirect focus on mHealth prescribing) were analyzed/cross-referenced. The ScienceDirect database was consulted to complement the collected data when needed. Data published by international and national regulatory bodies were analyzed in light of the scientific data obtained from the WoS, PubMed, and ScienceDirect databases.
Results: The International Medical Device Regulators Forum has ensured the international structuring of the regulatory field in collaboration with participating countries. The creation and updating of databases have allowed the tracking of medical device versions/upgrades and incidents. The regulatory organizations of the United States, the European Union, and France are currently consulting healthcare personnel, manufacturers, and patients to establish evaluation criteria for usability and quality of instructions for use that take into consideration patients’ level of literacy. These organizations are also providing support to manufacturers who wish to file marketing applications. Marketing, privacy, and cybersecurity measures are evolving with developments in technology and state cooperation policies. The prescription of mHealth applications will gain social acceptance only if consistency and coordination are ensured at all stages of the process: from pre-design, through verification of medical effectiveness, to ethical consideration during data collection and use, and on to marketing.
Conclusion: The conditions for mHealth prescribability include the adaptation of international regulation by the different states, the state provision of marketing support, and the evaluation of mHealth applications. For mHealth to gain social acceptance, increased collaboration among physicians, manufacturers, and “information technology stakeholders” is needed. Once this is achieved, MHealth can become the cornerstone of successful health care reform.
Keywords: national and international laws and regulations, medical device, evaluation of technologies, mHealth, prescription feasibility
Introduction
Mobile health (mHealth) is the use of applications and/or mobile connected devices for the purpose of supporting medical and public health practices.1,2 Mobile health applications consist of two types. The first is software as a medical device (SaMD), which performs medical functions through software installation on generic devices such as tablets or smartphones. The second is software in a medical device (SiMD), which requires both hardware and software components to operate – for instance, an mHealth application that interfaces and interacts with a material medical device (MD).3 Insofar as they are qualified as MDs, both SaMDs and SiMDs must comply with the regulatory frameworks established by national and international authorities for marketing, quality, safety of use, usability, and data security. Material MDs, and their embedded SiMDs if any, have long been required to undergo certification by national and international regulatory systems. By contrast, the requirement for certification of SaMDs, particularly mHealth applications, was implemented only recently.
Mobile health can play a positive role in various domains that affect the health status of individuals (wellbeing, prevention, care, monitoring or surveillance of diagnosed diseases, etc.) while also benefiting the health care system as a whole.4 It is now considered an important approach to extend traditional health services and a means to meet the growing medical needs.
In their review of the literature, Gagnon et al5 identified several factors that might contribute to the adoption of mHealth: perceived usefulness and ease of use, design and technical concerns, cost, time, privacy and safety issues, familiarity with the technology, risk-benefit assessment, and interaction with others (colleagues, patients, and management). Several studies6–9 have noted that physicians can play a major role in mHealth adoption by recommending mHealth applications to their patients. Yet, despite the recent publication of policy recommendations promoting the use of mHealth applications in routine clinical practice, these technologies are rarely recommended and even less prescribed by physicians.10
The prescribability of mHealth applications is a complex problem because it depends on a large number of factors and concerns a wide range of disciplines and actors in the industrial, health, normative, and regulatory domains. Moreover, patient acceptance of these technologies is not self-evident. Patients must be assured that their data will be protected and must be provided information via explanatory notices on how to use these applications.11
Indeed, while the prescription of drugs is a well-known practice in the medical field, the prescription of software is not. For mHealth prescription to gain social acceptance, prescribers and patients must be protected through regulating this novel practice and requiring the certification of mHealth applications. In this regard, J. Forsstrom12 highlighted as early as 1997 the urgency of implementing the certification of medical software to ensure their widespread adoption in clinical practice. In addition, the performance outcomes of mHealth applications must be evaluated in clinical trials, such that prescribers’ actions are supported by evidence and possibly covered by insurance.11 There is indeed a striking gap between the large number of mHealth applications available in app stores and the small number of applications evaluated by researchers and other actors.13
In 2015, the Institute for Healthcare Informatics (IMS)8 published a report proposing a series of actions to build a maturity model for the prescription of mHealth applications. The IMS noted that the prescription process had to be designed by all actors via the creation of technological standards and controls related to security and privacy. It also highlighted that mHealth applications had to be clinically evaluated prior to their medical use and integration into health information systems (HIS). The majority of IMS proposals have since been cited by researchers interested in the use of mHealth in clinical research.14–17 In their study examining the possibility of prescribing mHealth applications in oncology, Berkowitz17 et al recalled the importance of obtaining marketing authorization from the US Food and Drug Administration (FDA). Fox BI18 argued that the prescription of certain types of mHealth applications could contribute to reducing hospital readmissions provided that the following conditions were met: provision of support to patients in their use of these applications; prioritization of applications adapted to patients’ literacy level; verification of effective use; and allowance of dedicated nursing time. In their literature review of mHealth prescribing for the period 2005–2015, Daifi et al19 proposed the development of stores with applications evaluated based on clinical data and approved for medical use by the FDA. Byambasuren et al20,21 identified the following two barriers to prescription of mHealth applications by general practitioners: lack of knowledge about effective applications and lack of reliable sources (eg app stores) to access them. They also observed that it is often impossible to affirm the effectiveness of mHealth applications based on the available literature, and highlighted the lack of robustness of existing randomized clinical trials (RCTs).
The 2017 report of the IQVIA (formerly IMS)22 assessed the degree of maturity of the four criteria listed in the 2015 IMS report8 to help promote the prescription of mHealth applications – ie patient usability; accuracy, effectiveness, and safety; data privacy and security assurance; mitigation of malpractice risk – and added two important criteria – ie acceptable financial incentives; acceptable clinical workflow and usability. All or part of these criteria have been included in several projects aimed at the creation of platforms that are interoperable with HIS, that integrate app stores based on IQVIA recommendations, and that provide decision support tools for the prescription of mHealth applications.9,23,24
This background knowledge about mHealth usage indicates that the prescription of mHealth applications depends on the interaction between the regulatory, legal, and medical domains. Accordingly, our study correlated data from the scientific literature with data on regulatory developments in the US, the EU, and France with the aim of identifying the conditions for the prescription of mHealth applications.
In this paper, we begin by providing an overview of the international organizations responsible for the regulation and standardization of MDs and, in particular, of mHealth applications. Second, we describe the common regulatory framework developed by different international organizations and national bodies to ensure the global circulation of MDs. Third, we trace the implementation of international regulations in the US, the EU, and France. Fourth and last, we discuss the improvements that are still needed to reach the objective of widespread mHealth adoption taking into consideration the integration of artificial intelligence and machine learning in mHealth applications.
Materials and Methods
The present study on mHealth prescribing is based on the collection of data from several disciplines: medicine, public health policy, and regulatory and normative policy. The consultation of different multidisciplinary databases required knowledge of their organization and a command of the specific mHealth vocabulary used in each.
As the growing number of empirical studies increases knowledge in the area of mHealth, systematic reviews are needed to evaluate their findings and to provide a balanced and objective summary of the evidence they provide. To achieve this purpose, we adopted the systematic literature review process proposed by Brereton et al,25 which is commonly used in software engineering. This method was the most appropriate for our purposes as it can be applied to a wide range of domains, including clinical research.
Following Brereton et al, all empirical evidence from a specific field of study must be gathered and then evaluated to answer one or several predefined research questions. The systematic literature review process consists of three major steps that can be adapted to achieve an evidence-based paradigm/model. In our study, these three steps were as follows: article search, article selection, and data correlation. The article search consisted in translating our research problem into a series of answerable questions and in searching for the best evidence with which to answer these questions. The article selection process consisted in choosing the documents most likely to answer all or part of the questions. The data correlation process consisted in analyzing data published by international and national regulatory bodies in light of scientific data obtained from the WoS, PubMed, and ScienceDirect databases.
Article Search
We translated the need for information into the following four questions and searched for the best evidence with which to answer them.
Question 1: What is an mHealth application?
One of the first publications to use the term “mHealth application” dates from 2004,26 which is also the year when the word “mHealth” first appeared in the literature.27 Numerous other descriptors were employed to describe these new MDs until the term “mHealth application” was integrated in the MeSHas unique ID : D063731 in 2014. These different descriptors were included in our query.
Question 2: What regulations have international organizations established regarding mHealth?
We collected information on the membership and missions of the World Health Organization (WHO) and the International Medical Device Regulators Forum (IMDRF). The organizational chart and mode of functioning of these organizations were examined to determine which documents contained the information we needed. Policy papers, reports on the definition or clinical evaluation of mHealth, and procedures for marketing mHealth applications were included in the analysis. Session proceedings were excluded.
Question 3: How are the regulations of international organizations implemented by their Member States, more specifically the US, the EU, and France?
We identified the health agencies responsible for implementing international regulations in the US, the EU, and France. These were, respectively, the FDA, the European Medicines Agency (EMA), and the French National Authority for Health (HAS, Haute Autorité de Santé). We collected the following data from these agencies’ websites: reports, recommendations, and guidelines for filing marketing authorization applications. Conventions signed between the US, the EU, and France were also collected.
Question 4: How do scientific advances contribute to the evolution of regulations and how are scientific developments impacted by regulations? What synergy exists between the regulatory and scientific domains?
Two multidisciplinary databases, WoS and PubMed, were searched to gather scientific evidence using the same query. One advantage of the WoS database is that it covers not only the field of medicine, but also those of manufacturing, computer technologies, and security and technical standards. This allowed us to explore a wide range of literature addressing technical and normative issues beyond the medical domain. Another advantage is that Wos publications can be filtered according to their content by using the “ANALYZE” command on the bibliographic record’s heading. We used the “medical informatics” heading in our analysis.
Article Selection
The WoS and PubMed databases were queried for the period between 1 January 1975 and 30 November 2020. Two concepts were searched in MeSH descriptors and in article titles, abstracts, and keywords. The first concept, « mHealth,” was searched with the following terms: Mhealth OR m-health OR mobile health OR health devices OR health device OR digital health OR mobile Apps OR mobile app OR medical app OR medical Apps OR smart device OR smart devices OR mobile phone OR mobile phones OR mobile technology OR mobile technologies OR mobile device OR mobile devices OR connected health OR mobile application OR phone app OR phone Apps OR health application OR health applications OR mobile healthcare OR smartphone app OR smartphone Apps OR mobile applications OR smartphone Apps OR telecare technolog*. The second concept, “prescription” or “recommendation,” was searched with the following terms: prescription OR prescrib* OR recommandation.
A total of 3872 and 1913 references, respectively, were retrieved from WoS and PubMed and stored in a database created for this purpose. The titles and abstracts of the 4544 references were automatically analyzed using an SQL procedure: Only the references containing the concepts “mHealth” and “prescription” or “recommendation” in the same sentence were retained, for a total of 1143 references. After reading the abstracts and keywords of these 1143 references, we excluded those focusing on applications dedicated to wellbeing, administrative functions (eg appointment scheduling) or general health information, as well as technical articles that described mHealth without studying its impact on diseases. A total of 552 references were retained. After reading the full text of these 552 references, we selected 15 articles that dealt explicitly with the prescription of mHealth and 150 articles that dealt indirectly with prescribability in the context of clinical practice or research (eg RCTs evaluating mHealth applications, studies describing mHealth quality criteria such as MARS or UMARS or addressing usability, ethical issues, security, privacy, user perceived value). The article selection process is shown in Figure 1.
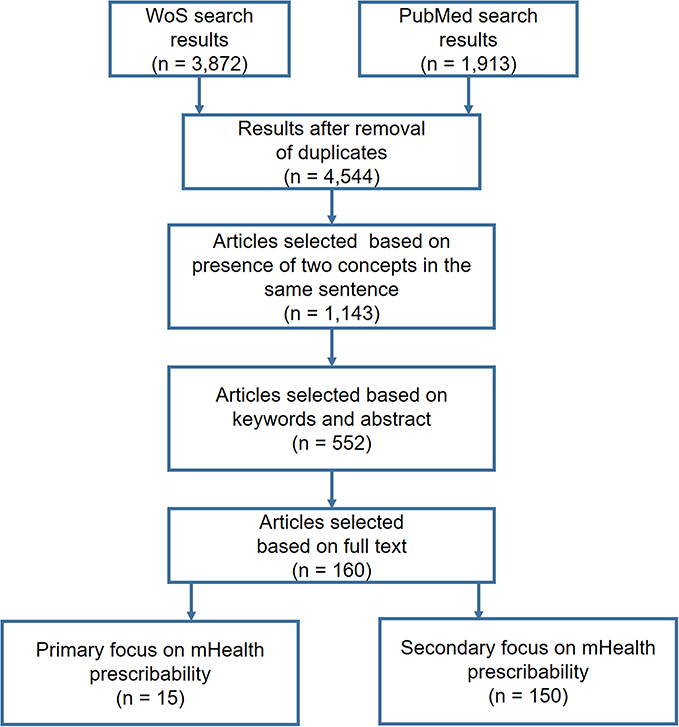 |
Figure 1 Article selection flowchart. |
The 15 articles dealing explicitly with the prescription of mHealth applications are listed in Table 1.
 |
Table 1 Articles Dealing Explicitly with the Prescription of Mobile Health Applications |
For the literature on mHealth regulations, we used a narrative, descriptive approach to examine thematic and geographical aspects of the prescription of mHealth applications.
Data Correlation
From the list of selected articles, we identified those that focused on regulation and those that dealt with prescription. We performed a systematic reading of the references cited in the most relevant articles obtained with the “mHealth prescription” query to identify additional references on mHealth regulation. Conversely, we scanned the bibliography of the articles obtained with the “mHealth regulation” query to find additional references on mHealth prescription. The continuous back and forth between these two sets of data allowed us to refine our research questions and, consequently, to avoid collecting unnecessary material.
Lastly, the ScienceDirect database was consulted to complement the collected data when needed. The PubMed database was consulted for the same purpose as late as May 2021, ie a few months after the end of the search period.
The Regulatory Environment
Regulations and Standards
It is essential to clearly define the notions of regulation and standardization, both of which are widely used in the field of MDs.
Regulation is the set of rules established by national or international organizations that defines the participation of people or companies in a market or sector of activity. In the case of MDs, these rules concern circulation and control in the areas of health safety, contribution to care, and health care reimbursement. National and international organizations can agree to grant the right to market products, which implies the creation of labels or markings (eg CE marking, etc.), nomenclatures, and common clinical research programs in the case of MDs. Regulations provide actors in the field (manufacturers, researchers, healthcare personnel, patients, etc.) with a specific framework in line with public policy and public health objectives. Unlike standards, regulations are mandatory.
Standardization is the process of implementing technical standards in manufacturing for the purpose of limiting risks and ensuring safety of use (eg AFNOR in France and ISO at the global level). For some MDs and most mHealth applications, standards also concern the security of patient data. The key standarts are: EN ISO 13485:2016, medical devices -quality management systems - Requirements for regulatory purposes. EN ISO 14971:2012, Application of risk management. IEC/TR 80002-1:2009 medical device software. Since standards are voluntary, manufacturers are free to choose which ones to use. The two most important standards for MD manufacturers are ISO 9001 (quality management systems) and ISO 13485 (quality management systems for organizations involved in one or more stages of the MD life cycle). Both of these standards are regularly updated.
International Organizations
Two international organizations, the WHO and the IMDRF.28 have been developing regulations for MDs in collaboration with a group of Member States.
World Health Organization
The WHO considers that health technologies, which include MDs, are essential to the proper functioning of health systems. In particular, mHealth applications are seen as playing a crucial role in the prevention, diagnosis, and treatment of diseases and in the rehabilitation of patients.
In 2003, the WHO published a document to provide guidance to Member States wishing to create or modify their own MD regulatory systems, while acknowledging that a single model would be difficult to achieve.29
Recognizing the important role of health technologies, the World Health Assembly adopted resolution WHA60.29 in May 200730 to address the problems arising from the inappropriate deployment and use of health technologies. The resolution also highlighted the need to establish priorities for the selection, regulation, and evaluation of health technologies.
Thus, in 2012, the WHO published the National eHealth Strategy Toolkit in collaboration with the International Telecommunication Union (ITU).31 Over the years, it published several other documents to strengthen research and implementation in the field of health technologies.32
The 2018 World Health Assembly unanimously adopted a resolution calling on WHO leadership to develop a global strategy on mHealth to support national efforts toward universal health coverage. This strategy is to be implemented between 2020 and 2025.33
Lastly, Dr. Tedros announced the creation of the Department of Digital Health on 6 March 2019. The aim was to give the WHO a greater role in assessing digital technologies and helping Member States prioritize, integrate, and regulate these technologies.34
International Medical Device Regulators Forum
The IMDRF is a forum of volunteer countries that regulates existing and future MDs and builds on the work of the Global Harmonization Task Force (GHTF) on Medical Devices. The IMDRF was established in October 2011 in Ottawa by representatives of MD regulations and standards authorities from Australia, Brazil, Canada, China, Japan, the US, and the EU, in collaboration with the WHO. Other international organizations involving other jurisdictions are regularly invited, including the Asia-Pacific Economic Cooperation (APEC) Life Sciences Innovation Forum (LSIF).35
The IMDRF proposes strategies, policies, and orientations for the deployment of MDs which draw on the expertise of different working groups that are stakeholders in the field (industry, academia, health professionals, consumer and patient representatives).
Some of the work items that have been completed by the IMDRF – quality of international standards, unique device identification (UDI) application guide, integration of MDs in patient registries, definition of SaMDs – are common to all MDs, whether or not these are based on information technology (IT).36,37
The Development of a Common Regulatory Framework
In an international context marked by the barrier of multilingualism, a common regulatory framework has been developed to ensure the global circulation of MDs. In addition to common standards, this framework includes a common classification system, a common identification system, and a common nomenclature.
Classification
The WHO has established an international classification of MDs related to patient safety. This classification is based on the following criteria:
- Duration of use.
- Degree of invasiveness.
- Possibility of reuse.
- Therapeutic or diagnostic purpose.
- Dependence on an energy source.
- Part of the body in contact with the MD.
Medical devices are classified according to four levels of risk, from the product presenting the lowest risk to patients to the intrusive product presenting the highest risk. Each class of risk determines the applicable regulations for marketing in all countries.38 These classes are as follows:
- Class A (lowest risk) – eg, corrective glasses, vehicles for disabled people, crutches, etc.
- Class B (moderate/medium risk) – eg, contact lenses, ultrasound devices, dental crowns, etc.
- Class C (high/important risk) – eg, condoms, lens disinfectants, etc.
- Class D (highest risk) – eg, breast implants, stents, hip replacements, etc.
The four classes of risk defined by the WHO have been adopted in France by the National Agency for the Safety of Medicines and Health Products (ANSM, Agence Nationale de la Sécurité du Médicament et des Produits de Santé)39 and in the EU by the Directorate-General for Health.40 They have been renamed classes I, IIa, IIb, and III in both jurisdictions (Table 2).
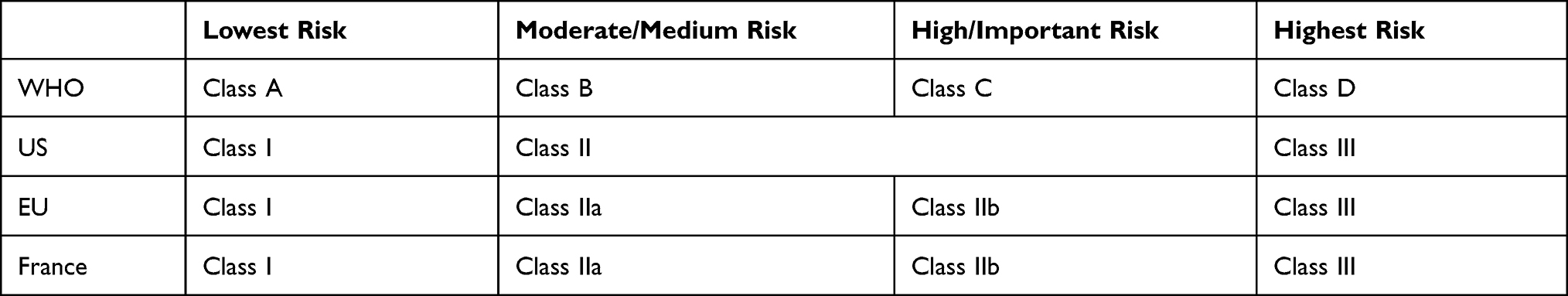 |
Table 2 Classification of MDs According to Geographic Area |
In the US, the FDA41 uses three classes of risk named I, II, and III. Class II corresponds to the moderate/medium and high/important risk levels defined by the WHO (Classes B and C), and Class III corresponds to the highest risk level (Class D). The Center for Devices and Radiological Health (CDRH) is responsible for MD approval under the authority of the FDA.42
The class of a new MD is initially proposed by its manufacturer based on the classification rules established by the relevant jurisdiction in accordance with the product’s claimed medical purpose.
In all countries, classification is the first step in filing for MD approval: It determines the pathway to follow and the documents to provide.
Identification
A UDI system common to all countries has also been developed to facilitate the traceability and marketing of MDs.
In 2012, the FDA created the UDI for US manufacturers to use on all MDs after receiving approval.43 The FDA then proposed to lead an international working group under the auspices of the IMDRF with a view to expanding, harmonizing, and streamlining the UDI system at the global level. In 2017, the EU included the UDI in the MDR (EU) 2017/745 directive44 aimed at establishing the European Database on Medical Devices (EUDAMED).
In 2014, the FDA published a guide entitled Global Unique Device Identification Database (GUDID): Guidance for Industry.43 This guide specifies which information about MDs must be entered into the new GUDID database: description, identification, manufacturer, user instructions, and malfunctions (as reported by the national authorities responsible for monitoring these devices).
The EUDAMED database was created to compile information on MDs similar to the GUDID database. Yet, despite existing agreements, differences remained between the two databases that prompted the US and the EU to sign a new agreement in July 2018 for the purpose of aligning their content. In addition, four international bodies were designated by the European Commission and the FDA to supervise the production and allocation of UDI codes.45
The GUDID and EUDAMED databases allow the traceability of MDs and ensure security through incident reporting in all participating countries.
Nomenclature
In 2001, the Global Medical Device Nomenclature (GMDN) agency46 was created as a collaboration between the EU, Canada, and the US to develop and regularly update internationally agreed descriptors for MDs. Medical device experts from all over the world (manufacturers, health authorities, and regulators) have since compiled the GMDN nomenclature in accordance with the requirements of ISO 15225. This nomenclature is recommended by the IMDRF and is now used in more than 70 countries, including the US and EU countries.
The GMDN nomenclature allows to find “predicates” or equivalents of a new MD, simplifying applications for approval. It also ensures traceability and safety, both of which are essential to achieve prescribability.
Implementation of Regulations on Medical Devices and Mobile Health Applications in the United States, The European Union, and France
The intertwining of medical, technical, economic, and legislative dimensions leads to complexity in the implementation of MD regulations. First, despite international harmonization efforts, different countries have their own legislative frameworks that must be taken into consideration in the implementation process. Second, there is heterogeneity in the MD sector in terms of technologies (eg bandages, medical imaging devices, mHealth applications), medical purposes (eg diagnosis, care, prevention), and associated risks (eg biocompatibility of materials, data security issues). Third, economic imperatives require the rapid introduction of digital health and its constant innovations into the healthcare system.47
In this context, the MD sector is undergoing regulatory renewal not only at the international level but also in the US, the EU, and France. We shall now review the guidelines for obtaining MD marketing approval, the initiatives for the promotion of mobile health, and the data protection and privacy rules that have been implemented in each of these geographical areas.
Marketing of Medical Devices in the United States
In the US, the FDA is responsible for regulating the marketing of products intended for human use. Manufacturers who wish to have a product recognized as an MD must select one of the following four procedures depending on the classification they deem appropriate.48
Filing an approval application or Premarket Notification (510(k))49 for Class I or II MDs with one or more equivalents on the market. A 510(k) is a premarket submission made to the FDA to demonstrate that the MD is at least as safe and effective as an already legally marketed device. A “Substantially Equivalent” (SE) letter is issued when the FDA determines that the MD is substantially equivalent to the already legally marketed device. For Class I MDs (lowest risk), there is no need to perform effectiveness studies. For Class II MDs (moderate/medium risk), effectiveness studies must be carried out after receiving approval from a committee in charge of verifying compliance with good clinical practices including ethical rules.
Filing a Premarket Approval (PMA) application50 for Class III MDs with one or more equivalents (ie SE devices) on the market. The PMA is the FDA’s scientific and regulatory review process to evaluate the security and effectiveness of Class III MDs. These MDs are those that support or sustain human life, play an important role in the prevention of harm to human health, or create an unreasonable risk of disease or injury. Clinical studies of MDs presenting a high/important risk must be approved by the FDA and an ethics committee before they begin.
Submitting a De Novo Classification Request for Class I, II, or III MDs that have no equivalent on the market. A “Not Substantially Equivalent” (NSE) letter is issued when the FDA determines that the MD is not substantially equivalent to the already legally marketed device.51 In practice, De Novo MDs are classified as Class III by default, although they may be reclassified as Class I or II when sufficient controls based on several criteria are applied.52
Requesting a Humanitarian Device Exemption (HDE)48 for MDs intended to benefit patients with a disease that affects less than 8000 people per year in the US. This approval process allows applicants to market MDs without providing evidence of clinical effectiveness. The applicant must demonstrate that there is no unreasonable or substantial risk of disease or injury, that the likely health benefits outweigh the risks, and that there are no comparable devices on the market.
Initiatives for the Promotion of Mobile Health in the United States
In 2017, the FDA released an action plan aimed at fostering innovation in digital health.53 This action plan was to amend legislative provisions regarding medical software through issuing a new policy on clinical decision support software and proposing a Digital Health Software Precertification (Pre-Cert) Program.54
The Pre-Cert program, currently in a pilot phase, is an FDA experiment intended to speed up the process of SaMD marketing. Currently, the FDA approval process for an MD takes approximately 90 to 180 days, and this timeframe can increase up to two-fold depending on delays or changes to the application file. A new certification process is therefore needed that can keep up with the speed of software innovation.
As its name indicates, the Pre-Cert program allows for the precertification of a new SaMD based on the certification of the company and the processes it currently uses to manufacture SaMDs. Five “excellence principles” must be met for certification to be awarded: patient safety, product quality, clinical responsibility, cybersecurity responsibility, and proactive culture. Once a company is certified, it can manufacture new “precertified” SaMDs without having to go through the entire certification process again. It can undergo “simplified reviews” or, in some cases, be exempted from the entire review process if the product is low risk.
In 2007, the FDA and Duke University entered into a public-private partnership called the Clinical Trial Transformation Initiative (CTTI) to promote the use of mHealth applications in clinical research. The CTTI has since noted that few interventional studies - particularly RCTs – are using mobile devices, which is especially striking given the speed of technological advances in the field.55,56 The CTTI has recently created a database of 275 publications evaluating MDs or mHealth applications for data capture. This database compiles information on medical context and choice of mobile technology, details about sensors, algorithms, and study samples, as well as results obtained by digital measurement.
Through developing recommendations and resources, the CTTI provides practical, user-friendly solutions that promote the conduct of RCTs using digital health and mHealth applications.
Data Protection and Privacy in the United States
In the US, the Health Insurance Portability and Accountability (HIPAA) Act of 199657 governs privacy protection through ensuring secure access to health care and patient information. The Act requires that patients be given clear written information on how their health information may be used, stored, or disclosed.
The legislation on the security of health information was strengthened in 2009 with the adoption of the Health Information Technology for Economic and Clinical Health (HITECH) Act.58,59 The HITECH Act, which is aimed at promoting the adoption and meaningful use of health IT, includes provisions concerning privacy and security that reinforce the HIPAA Act in the area of electronic transmission of health data. It also provides a system for alerting patients, healthcare professionals, and manufacturers about hardware or software malfunctions.
Marketing of Medical Devices in the European Union
Created in 1995, the EMA60 aims to protect public and animal health in EU Member States by ensuring that all health products placed on the EU market are safe, effective, and of high quality.
In the EU, MDs are currently governed by three directives: the Active Implantable Medical Devices Directive 90/385/EEC (1990), the Medical Devices Directive 93/42/EEC (1993) and the In Vitro Diagnostic Medical Devices Directive 98/79/EC (1998). While these three directives set objectives, the form and means of implementation remain under national authority. In 2017, two new regulations, 2017/745/EU on MDs61 and 2017/746/EU on in vitro diagnostic MDs,62 were adopted to ensure better protection of public health and patient safety. These regulations came into force on 25 May 2017 and are intended to replace the existing directives following a transition period lasting until 2022.
These last two legislative texts recommend maintaining the four-level risk classification proposed by the WHO, to adopt the UDI system for the traceability and circulation of MDs, and to use the common GMDN nomenclature. Lastly, these regulations are accompanied by a set of guidelines intended for all actors in the MD sector and for experts who accompany them in their application for MD approval.63
The single application procedure covers all four classes of risk, integrates quality system management (QSM) practices, and requires proof of compliance with ethical rules (respect of patient privacy) as well as evidence of effectiveness. For Classes I and IIa, the requested evidence is mainly documentary. For Classes IIb and III, all data collected during tests and trials must be provided, especially those concerning patient safety. All controls are carried out by Notified Bodies (NBs) which are proposed by EU Member States via the EMA and appointed by the Medical Device Coordination Group (MDCG) (see below).
For Classes IIb and III, the EU recommends that clinical studies on MDs be conducted in collaboration between different European research teams. A manufacturer may cite clinical studies on a product similar to his or her own as evidence of effectiveness. The manufacturer must monitor the MD so as to report any adverse event and eventually withdraw it from the market. Lastly, he or she must report to the MDCG any changes in the manufacturing process.
The MD is marketed after issuance of the CE marking, which includes the UDI, the name of the manufacturer, and the country of manufacture.
A regulatory system has been set up that involves several organizations and a website for the sharing of information, the monitoring of certification procedures, and the gathering of data on MDs. It is specifically composed of:
Notified Bodies, proposed by EU Member States via the EMA and appointed by the MDCG (see below). In addition to assessing compliance with regulations,40 these bodies are responsible for market surveillance and for the monitoring of incidents caused by certified MDs.
The MDCG,64 composed of experts from all disciplines appointed by the EU. The MDCG appoints the NBs, settles disputes related to classification, evaluates preventive health protection measures, verifies compliance with standards, and oversees the harmonization and implementation of regulations.
The EUDAMED website,44 currently under construction, whose mission is to list MDs placed on the market. This tool, which will be shared between Member States, will serve to reference clinical studies on MDs. In its upcoming version, the website will facilitate the exchange of information between European and American actors. The long-term objective is to create a coherent authorization pathway that will allow better circulation of MDs between the EU and the US.65
Initiatives for the Promotion of Mobile Health in the European Union
The EU’s position on MDs and mHealth applications is the result of two action plans – eHealth 2004–201166 and eHealth 2012–202067 — that cover broad and distinct areas of digital health.
In 2016, the EU published a document on SaMDs based on a literature search and a study conducted in several EU countries; it also organized a seminar entitled “Safety of non-embedded software, including on safety of health, lifestyle, and wellbeing apps”.68 Both the study and the seminar highlighted the need to improve transparency regarding the security of health and wellbeing applications and to create a common EU framework for SaMDs (as several EU countries were developing their own frameworks). No general framework is yet in place, but many guidelines have been published concerning a similar set of activities: medical content, security and privacy, ease of use, and effectiveness.
In 2017, the EU funded the WHO-ITU-mHealth project,69 whose objectives are to develop mHealth interventions in selected EU Member States and to create and maintain a “Knowledge and Innovation Hub for mHealth.” The results of the project are presented on the European mHealth Hub website.70 Among other things, the European mHealth Hub will produce a set of knowledge tools providing advice and guidance on the large-scale implementation of mHealth services and interventions (a first toolkit has been published that focuses on the evaluation frameworks for mHealth applications adopted in 12 EU countries). This project follows the Working Group on mHealth assessment guidelines, which defined six criteria: privacy, transparency, reliability, validity, interoperability, safety in 2016 and then 7 more criteria: technical stability, effectiness, accessibility, usabiity, scalability, user experience, security in 2018.
In 2020, the EU recommended using SaMDs as a means to empower patients to take care of their health, to encourage prevention, and to enable feedback and interaction between care users and providers.71
Data Protection and Privacy in the European Union
The General Data Protection Regulation (GDPR),72 applicable since 25 May 2018, replaces Directive 95/46/EC. The GDPR governs the processing of data (including health data) from all EU citizens. The European Data Protection Board (EDPB) ensures the consistent implementation of the GDPR and ePrivacy73 directives.
In order to comply with the GDPR, mHealth applications74 must integrate the concepts of privacy by design and privacy by default right from their conception. Protection measures must be taken systematically, and the level of security of personal data must be preconfigured in order to minimize risks. Note, however, that the GDPR only applies when data are collected and stored on an information system (ie it does not apply in the absence of data transmission.75
The European Network and Information Security Agency (ENISA) is responsible for network and information security (NIS) in EU Member States and other European countries.76 The ENISA works to improve capabilities in cybersecurity and in critical information infrastructure management in the health sector.
Marketing of Medical Devices in France
France has introduced EU regulations concerning MDs into its health law. Thus, any MD, whether it is aimed solely at the French market or more widely at the European market, must obtain a CE marking and a UDI and must refer to the common EU nomenclature in order to be placed on the market.
In France, the organizations responsible for implementing MD regulations and marketing MDs are the ANSM and the HAS.77
As in other EU countries, manufacturers must implement the following actions for their MDs to be placed on the market:
Demonstrate compliance with technological standards by producing detailed documentation.
Implement a quality approach for manufacturing and suitability for use.
Comply with the GDPR78 and the French Data Protection Act (LIL, Loi Informatique et Libertés).79
Conduct a risk analysis and develop an incident management system for the duration of the MD’s life cycle.
Report product changes and provide instructions for use in several languages.
Conduct clinical studies if the selected level of risk (Classes I, IIa, IIb, or III) requires it.
For MDs to be reimbursed by the French health insurance system, they must be registered on the List of Reimbursable Products and Services (LPPR, Liste des produits et prestations remboursables). This requires submitting a registration application to the Medical Device and Health Technology Evaluation Committee (CNEDIMTS, Commission Nationale d’Evaluation des Dispositifs Médicaux et des Technologies de Santé),80 which is the French NB appointed by the MDCG. The HAS has recently published a guide to help manufacturers submit registration applications for different types of MDs.81
The CNEDIMTS evaluates MDs based on the improvement they provide compared to a designated equivalent product admitted or not to reimbursement and considered as a reference in the scientific literature.82 The level of improvement provided is determined by the ratio between expected service and delivered service.83
For innovative MDs (ie those that provide significant clinical benefit or a reduction in costs), the HAS may grant exceptional and temporary funding (Innovation Pass).
Exceptional and temporary funding is conditional on the applicant conducting a clinical study for the purpose of confirming the significant benefit of this new technology for health.84
As an EU Member-State, France (via the ANSM) is expected to integrate its data on MDs into the EUDAMED database by 2022. Currently, the ANSM distributes this information via several documents and tables available on its website.85
The HAS has recently proposed a classification comprising 11 types of digital health applications. These are classified into four levels – A, B, C, and D – according to purpose of use, capacity to offer a personalized response, and level of autonomy in decision-making (ie “capacity to operate with or without human intervention”). The classification is aimed at promoting the introduction of digital applications into the healthcare system.86
Initiatives for the Promotion of Mobile Health in France
The ANSM distributes a range of information to help economic actors determine whether an application should be considered a wellbeing device or an MD.87 Software and mobile applications related to health are presented in the form of a flowchart, taking into consideration regulatory provisions40 and the decisions of the European Court of Justice (ECJ) concerning decision support software.
As new regulations are introduced in the EU, the HAS publishes documents to support manufacturers and other economic actors. For example, in 2016, the HAS issued a guide on “Good practice guidelines on health apps and smart devices (mobile health or mHealth)” for manufacturers as well as a list of good practices for healthcare actors.88 One of its latest publications (from 2019) describes the evaluation process that needs to be followed for an MD to receive CE marking and to become eligible for reimbursement by the French health insurance system.81
Data Protection and Privacy in France
The GDPR legislation harmonizes EU regulations by providing a common legal framework for professionals working with data and aims to foster the development of digital activities within the EU based on user trust. In France, the GDPR legislation prompted the adaptation of the pre-existing French Data Protection Act of 6 January 1978. One of the main changes to the Act was the extension of the powers of control and sanction of the French National Commission on Information Technology and Liberties (CNIL, Commission Nationale de l’Informatique et des Libertés). Accordingly, the CNIL now cooperates closely with the EDPB.
In the area of cybersecurity, the French National Agency for the Security of Information Systems (ANSSI, Agence Nationale de la Sécurité des Systèmes d’Information), which reports to the General Secretariat for Defense and National Security (SGDSN, Secrétaire Général de la Défense et de la Sécurité Nationale), is responsible for proposing rules for the protection of state IT systems and for verifying the implementation of adopted measures. The protection of French healthcare IT systems therefore depends in part on the decisions and future prospects of the ANSSI. Initially intended for IT security professionals, the guides and recommendations issued by the ANSSI constitute useful methodological bases for healthcare structures and actors89 Table 3.
 |
Table 3 Overview of the Regulatory Framework for Medical Devices in the United States, The European Union, and France |
Discussion
This study correlated data from the scientific literature with data on regulatory developments in the US, the EU, and France with the aim of identifying the conditions for the prescription of mHealth applications. Our main findings are as follows. The IMDRF has ensured the international structuring of the regulatory field in collaboration with participating countries. The creation and updating of databases have allowed the tracking of MD versions/upgrades and incidents. The regulatory organizations of the US, the EU, and France are currently consulting healthcare personnel, manufacturers, and patients to establish evaluation criteria for usability and quality of instructions for use that take into consideration patients’ level of literacy. These organizations are also providing support to manufacturers who wish to file marketing applications. Marketing, privacy, and cybersecurity measures are evolving with developments in technology and state cooperation policies. The prescription of mHealth applications will gain social acceptance only if consistency and coordination are ensured at all stages of the process: from pre-design, through verification of medical effectiveness, to ethical consideration during data collection and use, and on to marketing. Achieving this will require increased collaboration among physicians, MD manufacturers, and “IT stakeholders”.90–94
Two important medical aspects of mHealth prescribability were not addressed in the 165 included studies: the economic costs and benefits of mHealth innovations and the barriers to the prescription of mHealth applications. We shall now briefly discuss how these aspects have been approached in other studies.
In view of the steady increase in health care expenditures in all countries, health economics researchers are paying more and more attention to the economic impact of mHealth use. In their 2007 literature review of economic evaluations of mHealth, Iribarren et al95 identified 39 studies conducted in 19 countries (most of them high-income countries or upper middle-income countries): In 29 of these studies (74.3%), mHealth interventions were found to be cost-effective, economically beneficial, and/or cost-saving.
Allenby and al shed light on biopharmaceutical companies’ interest in acquiring mHealth start-ups. Indeed, biopharmaceutical companies are looking for opportunities to take advantage of new ways of organizing care based on digital technologies. They are also trying to position themselves as third parties in the care relationship by providing, in addition to classic medication, applications that facilitate patient-doctor interaction and applications that allow for care monitoring either by the patient him/herself or by a healthcare professional.96
Several barriers to the prescription of mHealth applications have been identified in the rest of the literature. One of the most common is that general practitioners have insufficient time to explain to their patients how to use mHealth applications.97,98 To overcome this problem, Rijcken C. recommends that instructions on use be given by pharmacists instead of physicians (as is already the case for drugs).99
Despite all the developments that have occurred since the introduction of connected devices, the prescription and reimbursement of mHealth applications remain rare.
What improvements are still needed?
Most importantly, there is a need for rigorous evaluation of mHealth applications. The lack of transparency, the instability of emerging technologies, and the gap between the long timeframe of clinical studies and the short timeframe of technological development should be addressed by adopting the Consolidated Standards of Reporting Trials of Electronic and Mobile HEalth Applications and onLine TeleHealth (CONSORT-EHEALTH).100 For applications incorporating artificial intelligence (AI), the recent Consolidated Standards of Reporting Trials-Artificial Intelligence (CONSORT-AI) protocols should be preferred.101 Application of these standards would allow for comparisons between trials.
The evaluation of mHealth applications based exclusively on traditional RCTs is a hindrance to the penetration of innovations into the world of healthcare. For this reason, researchers102 have proposed a hybrid trial methodology that combines simulation studies using data from patient registries (which provide information on patient characteristics and help to identify simulation parameters) with RCTs conducted on a limited number of patients. This approach has proven successful for regulatory decision-making linked to drug effectiveness in some contexts.103
In addition to evaluations based on clinical data, the EU104 recommends the inclusion of patient input in regulatory decisions on the use of health technologies. In this regard, EU regulatory bodies have developed several initiatives that range from direct patient involvement in the regulatory decision-making process to the use of patient information as evidence in empirical studies.
In the US and the EU, a regulatory framework is now being built for digital technologies, with a particular focus on the introduction of artificial intelligence/machine learning (AI/ML) in MDs. Muehlematter et al observe that there is currently no clear approval process and no specific regulatory pathway for AI/ML-based MDs in these jurisdictions.105 Accordingly, they recommend greater transparency regarding approval and regulation procedures with a view to improving public trust as well as the effectiveness, security, and quality of AI/ML-based MDs.
Should mHealth applications not be evaluated differently from classic medication?
In practice, mHealth applications are evaluated in a manner similar to classic medication. Thus, in a study from 2016, Van Norman106 found similarities between the processes of approval for drugs and MDs. While this study examined MDs in general, it was especially relevant with regards to mHealth devices.
One proposal to achieve mHealth prescribability is to create a specific framework for mHealth applications while still considering them as part of MDs, which would entail defining these applications with greater precision than is currently done with MDs. Other proposals are to build databases of applications that are accessible to all (from citizens to health personnel), to adopt a homogeneous UDI system, to provide a more clinically oriented description of applications based on specific medical terminology, to consider evaluations by external organizations, and to ensure quality use as requested by many researchers.107,108 Lastly, patient registries such as electronic health records (EHRs) and personal health records (PHRs) should be made interoperable with decision support modules to help promote the recommendation and prescription of mHealth devices.
The main limitation of our study is that our search focus on prescribability caused us to miss references that address the economic costs and barriers to using mHealth. This focus, however, was justified by the subject matter. Moreover, these issues were explored in the Discussion along with avenues for improving mHealth perscribability.
Conclusion
This review of the literature has identified the following conditions for mHealth prescribability: the adaptation of international regulation by the different states, the state provision of marketing support, and the evaluation of mHealth applications. It has also shown that states are increasingly collaborating to ensure the global circulation of MDs. For mHealth to gain social acceptance, increased collaboration among physicians, MD manufacturers, and “IT stakeholders” is needed. Once this is achieved, MHealth can become the cornerstone of successful health care reform.
Abbreviations and Acronyms
mHealth, Mobile health; SaMD, Software as a medical device; SiMD, Software in a medical device; MD, Medical device; IMS, Institute for Healthcare Informatics; HIS, Health information system; FDA, Food and Drug Administration; RCT, Randomized clinical trial; IQVIA, IQVIA doesn’t have a full form as such. It is pronounced as I-Q-VIA, where I is taken from IMS Health or can be interpreted as Intelligence, Q comes from Quintiles or can be interpreted as Quotient and VIA is basically the path of transformation or a helping hand to achieve something. https://www.quora.com/What-is-the-full-form-of-IQVIA; WHO, World Health Organization; IMDRF, International Medical Device Regulators Forum; EMA, European Medicines Agency; HAS, French National Authority for Health; ITU, International Telecommunication Union; GHTF, Global Harmonization Task Force; APEC, Asia-Pacific Economic Cooperation; LSIF, Life Sciences Innovation Forum; IT, Information technology; UDI, Unique device identification; ANSM, French National Agency for the Safety of Medicines and Health Products; CDRH, Center for Devices and Radiological Health; EUDAMED, European Database on Medical Devices; GUDID, Global Unique Device Identification Database; GMDN, Global Medical Device Nomenclature; SE, Substantially equivalent; PMA, Premarket Approval; NSE, Not substantially equivalent; HDE, Humanitarian Device Exemption; HUD, Humanitarian use device; Pre-Cert, Precertification; CTTI, Clinical Trial Transformation Initiative; HIPAA Act, Health Insurance Portability and Accountability Act; HITECH Act, Health Information Technology for Economic and Clinical Health Act; QSM, Quality system management; NB, Notified Body; MDCG, Medical Device Coordination Group; GPDR, General Data Protection Regulation; EDPB, European Data Protection Board; ENISA, European Network and Information Security Agency; NIS, Network and information security; LPPR, French List of Reimbursable Products and Services; CNEDIMTS, French Medical Device and Health Technology Evaluation Committee; ECJ, European Court of Justice; CNIL, French National Commission on Information Technology and Liberties; ANSSI, French National Agency for the Security of Information Systems; SGDSN, French General Secretariat for Defense and National Security; CONSORT-EHEALTH, Consolidated Standards of Reporting Trials of Electronic and Mobile HEalth Applications and onLine TeleHealth; AI, Artificial intelligence; CONSORT-AI, Consolidated Standards of Reporting Trials-Artificial Intelligence; AI/ML, Artificial intelligence/machine learning; EHR, Electronic health record; PHR, Personal health record.
Acknowledgments
This work was supported by the French National Research Agency (reference #ANR-17-CE19-002).
Disclosure
Dr Jean Charles Dufour reports grants from French National Research Agency (reference #ANR-17-CE19-002) during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. WHO Global Observatory for eHealth, World Health Organization. MHealth: new horizons for health through mobile technologies. World Health Organization; 2011. Available from: http://www.who.int/goe/publications/goe_mhealth_web.pdf.
2. European Commission. Green paper on mobile health (“mHealth”). Shaping Europe’s digital future; 2014. Available from: https://ec.europa.eu/digital-single-market/en/news/green-paper-mobile-health-mhealth.
3. IMDRF SaMD Working Group. Software as a Medical Device (SaMD): key definitions; 2013:9. Available from: http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-131209-samd-key-definitions-140901.pdf.
4. Payne HE, Lister C, West JH, Bernhardt JM. Behavioral functionality of mobile apps in health interventions: a systematic review of the literature. JMIR MHealth UHealth. 2015;3(1):e20. doi:10.2196/mhealth.3335
5. Gagnon M-P, Ngangue P, Payne-Gagnon J, Desmartis M. m-Health adoption by healthcare professionals: a systematic review. J Am Med Inform Assoc. 2016;23(1):212–220. doi:10.1093/jamia/ocv052
6. Cajita MI, Hodgson NA, Lam KW, Yoo S, Han H-R. Facilitators of and barriers to mhealth adoption in older adults with heart failure. Comput Inform Nurs. 2018;36(8):376–382. doi:10.1097/CIN.0000000000000442
7. Rowland SP, Fitzgerald JE, Holme T, Powell J, McGregor A. What is the clinical value of mHealth for patients? NPJ Digit Med. 2020;3:4. doi:10.1038/s41746-019-0206-x
8. IMS Institute for health care informatics. patient-adoption-of-mhealth.pdf; 2015. Available from: https://www.iqvia.com/-/media/iqvia/pdfs/institute-reports/patient-adoption-of-mhealth.pdf.
9. Lopez Segui FF, Pratdepadua Bufill C, Abdon Gimenez N, Martinez Roldan J, Garcia Cuyas F. The prescription of mobile apps by primary care teams: a pilot project in Catalonia. JMIR MHealth UHealth. 2018;6(6):e10701. doi:10.2196/10701
10. Ferguson C, Jackson D. Selecting, appraising, recommending and using mobile applications (apps) in nursing. J Clin Nurs. 2017;26(21–22):3253–3255. doi:10.1111/jocn.13834
11. Perry B, Herrington W, Goldsack JC, et al. Use of mobile devices to measure outcomes in clinical research, 2010–2016: a systematic literature review. Digit Biomark. 2018;2(1):11–30. doi:10.1159/000486347
12. Forsström J, Perry B, Herrington W, Goldsack J. Why certification of medical software would be useful? Int J Med Inf. 1997;47(3):143–152. doi:10.1016/s1386-5056(97)00098-1
13. Onodera RR. Shintaro. Innovation process of mHealth: an overview of FDA-approved mobile medical applications. Int J Med Inf. 2018;118:65–71. doi:10.1016/j.ijmedinf.2018.07.004
14. Austin RR, Hull S. The power of mobile health technologies and prescribing apps. Comput Inform Nurs. 2014;32(11):513–515. doi:10.1097/CIN.0000000000000120
15. Aungst TD, Clauson KA, Misra S, Lewis TL, Husain I. How to identify, assess and utilise mobile medical applications in clinical practice. Int J Clin Pract. 2014;68(2):155–162. doi:10.1111/ijcp.12375
16. Terry K. Prescribing mobile apps: what to consider. Med Econ. 2015;92(12):35–38, 40.
17. Berkowitz CM, Zullig LL, Koontz BF, Smith SK. Prescribing an app? Oncology providers’ views on mobile health apps for cancer care. Jco Clin Cancer Inform. 2017;1. doi:10.1200/CCI.17.00107.
18. Fox BI, Umphress DA, Hollingsworth JC. Development and delivery of an interdisciplinary course in mobile health (mHealth). Curr Pharm Teach Learn. 2017;9(4):585–594. doi:10.1016/j.cptl.2017.03.005
19. Daifi C, Bahrami S, Kaakeh R, Kaakeh Y. Evolving frontier: a review of the role of mobile medical application prescribing. J Pharm Technol. 2016;32(3):91–97. doi:10.1177/8755122516633873
20. Byambasuren O, Beller E, Hoffmann T, Glasziou P. Barriers to and facilitators of the prescription of mhealth apps in Australian general practice: qualitative Study. JMIR MHealth UHealth. 2020;8(7). doi:10.2196/17447
21. Byambasuren O, Sanders S, Beller E, Glasziou P. Prescribable mHealth apps identified from an overview of systematic reviews. NPJ Digit Med. 2018;1(1):1–12. doi:10.1038/s41746-018-0021-9
22. The IQVIA Institute. The growing value of digital health; 2021. Available from: https://www.iqvia.com/insights/the-iqvia-institute/reports/the-growing-value-of-digital-health.
23. López Seguí FF, Pratdepàdua Bufill C, Rius Soler A, et al. Prescription and integration of accredited mobile apps in Catalan health and social care: protocol for the appsalut site design. JMIR Res Protoc. 2018;7(12):e11414. doi:10.2196/11414
24. Dufour J-C, Grosjean J, Darmoni S, et al. ApiAppS: a project to study and help practitioners in recommending mhealth apps and devices to their patients. Stud Health Technol Inform. 2019;264:1919–1920. doi:10.3233/SHTI190713
25. Brereton P, Kitchenham BA, Budgen D, Turner M, Khalil M. Lessons from applying the systematic literature review process within the software engineering domain. J Syst Softw. 2007;80(4):571–583. doi:10.1016/j.jss.2006.07.009
26. Yu P, Yu H Lessons learned from the practice of mobile health application development.
27. Zhang Y, Koch S. Mobile health apps in Sweden: what do physicians recommend? Stud Health Technol Inform. 2015;210:793–797.
28. IMDRF. About IMDRF. IMDRF; 2020. Available from: http://www.imdrf.org/about/about.asp.
29. WHO. Medical device regulations: global overview and guiding principles. World Health Organization; 2003. Available from: https://www.who.int/medical_devices/publications/fr/MD_Regulations.pdf.
30. World Health Organization. Health technologies1. WHO Health System; 2007. Available from: https://www.who.int/healthsystems/WHA60_29.pdf?ua=1.
31. World Health Organization. OMS | L’UIT et l’OMS lancent une initiative en faveur de la santé sur mobile pour lutter contre les maladies non transmissibles. World Health Organization; 2020. Available from: https://www.who.int/mediacentre/news/releases/2012/mHealth_20121017/fr/.
32. World Health Organization 2021. Global strategy on digital health 2020–2025; 2015. Available from: https://cdn.who.int/media/docs/default-source/documents/gs4dhdaa2a9f352b0445bafbc79ca799dce4d.pdf?sfvrsn=f112ede5_75.
33. WHO OMS. 71e Assemblée mondiale de la Santé – principaux débats du 25 mai; 2018. Available from: https://www.who.int/fr/news/item/25-05-2018-seventy-first-world-health-assembly-update-25-may.
34. WHO OMS. L’OMS publie les premières lignes directrices sur les interventions de santé numérique; 2020. Available from: https://www.who.int/fr/news-room/detail/17-04-2019-who-releases-first-guideline-on-digital-health-interventions.
35. APEC, Asia-Pacific Economic Cooperation. Life sciences innovation forum. APEC; 2020. Available from: https://www.apec.org/Groups/Committee-on-Trade-and-Investment/Life-Sciences-Innovation-Forum.
36. IMDRF. Work items. IMDRF; 2020. Available from: http://www.imdrf.org/workitems/work.asp.
37. IMDRF. Title: software as a Medical Device (SaMD): clinical Evaluation Authoring Group: software as a Medical Device Working Group Date: 21 September 2017; 2017. Available from: http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-170921-samd-n41-clinical-evaluation_1.pdf.
38. WHO Regional Publications, Eastern Mediterranean Series. Regulation of medical devices A step-by-step guide. World Health Organization; 2016. Available from: https://applications.emro.who.int/dsaf/emropub_2016_EN_18962.pdf?ua=1&ua=1&ua=1.
39. Nos missions - Les dispositifs médicaux et les dispositifs médicaux de diagnostic in vitro [Our missions - Medical devices and in vitro diagnostic medical devices]. ANSM. Available from: https://ansm.sante.fr/qui-sommes-nous/notre-perimetre/les-dispositifs-medicaux-et-les-dispositifs-medicaux-de-diagnostic-in-vitro/p/les-dispositifs-medicaux-et-les-dispositifs-medicaux-de-diagnostic-in-vitro-dmdiv. Accessed November 16, 2021.
40. Public Health- European Commission. Available from: https://ec.europa.eu/health/home_en.
41. Learn if a medical device has been cleared by FDA for marketing. FDA; 2018. Available from: https://www.fda.gov/medical-devices/consumers-medical-devices/learn-if-medical-device-has-been-cleared-fda-marketing.
42. CDRH Health. Center for devices and radiological health. FDA; 2020. Available from: https://www.fda.gov/about-fda/fda-organization/center-devices-and-radiological-health.
43. CDRH Health. UDI rule and guidances, training, resources, and dockets. FDA; 2020. Available from: https://www.fda.gov/medical-devices/unique-device-identification-system-udi-system/udi-rule-and-guidances-training-resources-and-dockets.
44. Cardoen G. Medical devices - EUDAMED. European Commission-public health; 2020. Available from: https://ec.europa.eu/health/md_eudamed/overview_en.
45. Commission Implementing Decision (EU) 2019/939 of 6 June 2019. Designating the Allocation Entities Responsible for Implementing a Unique Device Identifier (UDI) Allocation System in the Field of Medical Devices ; 2020. Available from: https://eur-lex.europa.eu/legal-content/FR/TXT/HTML/?uri=CELEX:32019D0939&from=EN.
46. Global Medical Device Nomenclature Agency. Base de données GMDN - GMDN Agency. GMDN; 2020. Available from: https://www.gmdnagency.org/services/gmdn.
47. Jiang X, Ming W-K, You JH. The cost-effectiveness of digital health interventions on the management of cardiovascular diseases: systematic review. J Med Internet Res. 2019;21(6):e13166. doi:10.2196/13166
48. HDE Approvals. FDA; 2018. Available from: https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/hde-approvals.
49. CDRH Health. 510(k) Clearances. FDA; 2019. Available from: https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/510k-clearances.
50. CDRH Health. PMA approvals. FDA; 2019. Available from: https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/pma-approvals.
51. CDRH Health. De novo classification request. FDA; 2019. Available from: https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request.
52. CDRH Health. De novo classification process (Evaluation of Automatic Class III Designation). FDA; 2019. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/de-novo-classification-process-evaluation-automatic-class-iii-designation.
53. CDRH Health. digital health innovation action plan. FDA; 2017. Available from: https://www.fda.gov/media/106331/download.
54. Alon N, Stern AD, Torous J. Assessing food and drug administration’s risk-based framework for software precertification with top U.S. Health apps: quality improvement study. JMIR MHealth UHealth. 2020;8:e20482. doi:10.2196/20482
55. Bakker JP, Goldsack JC, Clarke M, et al. A systematic review of feasibility studies promoting the use of mobile technologies in clinical research. NPJ Digit Med. 2019;2:47. doi:10.1038/s41746-019-0125-x
56. CTTI. CTTI unveils new database of feasibility studies on mobile technologies in clinical research; 2019. Available from: https://www.ctti-clinicaltrials.org/news/ctti-unveils-new-database-feasibility-studies-mobile-technologies-clinical-research.
57. U.S. Department of Health & Human Servic. HIPAA for Professionals. HHS.gov; 2015. Available from: https://www.hhs.gov/hipaa/for-professionals/index.html.
58. ONC. ONC HITECH Programs | healthIT.gov. ONC; 2020. Available from: https://www.healthit.gov/topic/onc-hitech-programs.
59. FDA Office of the. FDA informs patients, providers and manufacturers about potential cybersecurity vulnerabilities for connected medical devices and health care networks that use certain communication software. FDA; 2020. Available from: https://www.fda.gov/news-events/press-announcements/fda-informs-patients-providers-and-manufacturers-about-potential-cybersecurity-vulnerabilities.
60. EMA. European Medicines Agency; 2020. Available from: https://www.ema.europa.eu/en.
61. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on Medical Devices, Amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and Repealing Council Directives 90/385/EEC and 93/42/EEC (Text with EEA Relevance). Vol 117.; 2017. Available from http://data.europa.eu/eli/reg/2017/745/oj/eng. Accessed November 14, 2021.
62. Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in Vitro Diagnostic Medical Devices and Repealing Directive 98/79/EC and Commission Decision 2010/227/EU (Text with EEA Relevance). Vol 117.; 2017. Available from http://data.europa.eu/eli/reg/2017/746/oj/eng. Accessed November 14, 2021.
63. IMDRF documents [Internet]. Available from: http://www.imdrf.org/documents/documents.asp.
64. European Commission. Register of commission expert groups and other similar entities; 2020. Available from: https://ec.europa.eu/transparency/regexpert/index.cfm?do=groupDetail.groupDetail&groupID=35.
65. Bianchini E, Francesconi M, Testa M, Tanase M, Gemignani V. Unique device identification and traceability for medical software: a major challenge for manufacturers in an ever-evolving marketplace. J Biomed Inform. 2019;93:103150. doi:10.1016/j.jbi.2019.103150
66. European commission. e-Health - making healthcare better for European citizens: an action plan for a European e-Health Area {SEC}; 2004. Available from: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=COM:2004:0356:FIN:EN:PDF.
67. European Commission. Communication from the Commission to The European Parliament, the Council, The European Economic and Social Committee and The Committee of the regions ehealth action plan 2012–2020 - innovative healthcare for the 21st century; 2012. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:52012DC0736&from=FR.
68. European Commission. Safety of non-embedded software, including on safety of health, lifestyle and wellbeing apps. Shaping Europe’s digital future - European Commission; 2019. Available from: https://ec.europa.eu/digital-single-market/en/news/safety-non-embedded-software-including-safety-health-lifestyle-and-wellbeing-apps.
69. European commission. WHO - ITU mHealth Hub in EU | WHO-ITU-mHealth Project | H2020 | CORDIS | European Commission. CORDIS; 2020. Available from: https://cordis.europa.eu/project/id/737427.
70. European Commission. European mhealth hub | mHealth Hub work areas. Available from: https://mhealth-hub.org/work-areas.
71. European Commission. Health, Demographic Change and Wellbeing. Horizon 2020 - European Commission; 2013. Available from: https://ec.europa.eu/programmes/horizon2020/en/h2020-section/health-demographic-change-and-wellbeing.
72. EUR-Lex. L_2016119EN.01000101.xml. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:32016R0679&rid=3.
73. EUR-Lex. EUR-Lex - 32002L0058 - EN - EUR-Lex. Available from: https://eur-lex.europa.eu/legal-content/FR/ALL/?uri=celex%3A32002L0058.
74. European Commission. Report of the Working Group on mHealth assessment guidelines | shaping Europe’s digital future. Available from: https://digital-strategy.ec.europa.eu/en/library/report-working-group-mhealth-assessment-guidelines.
75. Anonymous. Code of Conduct on privacy for mHealth apps has been finalised. Shaping Europe’s digital future - European Commission; 2016. Available from: https://ec.europa.eu/digital-single-market/en/news/code-conduct-privacy-mhealth-apps-has-been-finalised.
76. Cyber security agency ENISA maps good practice in Europe. ENISA; 2011. Available from: https://www.enisa.europa.eu/news/enisa-news/cyber-security-agency-enisa-maps-good-practice-in-europe. Accessed November 16, 2021.
77. HAS.Référentiel de bonnes pratiques sur les applications et les objets connectés en santé (mobile Health ou mHealth) [Repository of good practices on applications and connected objects in health (mobile Health or mHealth).]; 2020. Available from: https://www.has-sante.fr/jcms/c_2681915/fr/referentiel-de-bonnes-pratiques-sur-les-applications-et-les-objets-connectes-en-sante-mobile-health-ou-mhealth.
78. CNIL. Donnée personnelle | CNIL. CNIL; 2020. Available from: https://www.cnil.fr/fr/definition/donnee-personnelle.
79. CNIL. Donnée sensible | CNIL. Available from: https://www.cnil.fr/fr/definition/donnee-sensible.
80. HAS.Commission nationale d’évaluation des dispositifs médicaux et des technologies de santé [National Commission for the Evaluation of Medical Devices and Health Technologies. High Authority of Health] Available from: https://www.has-sante.fr/jcms/c_419486/fr/commission-nationale-d-evaluation-des-dispositifs-medicaux-et-des-technologies-de-sante.
81. HAS CNEDiMTS. guide_sur_les_specificites_devaluation_clini- que_dun_dmc_en_vue_de_son_acces_au_remboursement.pdf. [Evaluation of medical devices by CNEDiMTS; Guide to the specificities of the clinical evaluation of a connected medical device medical device (CMD) with a view to its access to reimbursement .pdf]. 2019. Available from: https://www.has-sante.fr/upload/docs/application/pdf/2019-02/guide_sur_les_specificites_devaluation_clinique_dun_dmc_en_vue_de_son_acces_au_remboursement.pdf.
82. Gautier A. Dispositifs médicaux: quelques repères pour mieux comprendre ce secteur devenu incontournable.[Medical devices: some points of reference to better understand this sector which has become essential].] J Imag Diagn Interv. 2019;2(1):46–51. doi:10.1016/j.jidi.2018.11.003
83. Legifrance. Section 2: procédures Relatives à l’établissement de La Liste Prévue à l’article L. 165–1 et à La Fixation Des Tarifs et Des Prix (Articles R165-7 à R165-16) - Légifrance [Procedures Relating to the Establishment of the List Provided for in Article L. 165-1 and the Fixing of Tariffs and Prices (Articles R165-7 to R165-16)] - Légifrance 2020. Available from: https://www.legifrance.gouv.fr/codes/id/LEGIARTI000042270843/2020-08-28/.
84. HAS. Forfait innovation [Innovation package] ; 2020. Available from: https://www.has-sante.fr/jcms/c_2035788/fr/forfait-innovation.
85. ANSM.Listes et répertoires - Dispositifs médicaux [Lists and directories - Medical devices] . ANSM; 2020. Available from https://www.ansm.sante.fr/Mediatheque/Publications/Listes-et-repertoires-Dispositifs-medicaux.
86. HAS. HAS propose la 1ère classification des solutions numériques utilisées en santé [The HAS proposes the 1st classification of digital solutions used in health];2021. Available from: https://www.has-sante.fr/jcms/p_3238368/fr/la-has-propose-la-1ere-classification-des-solutions-numeriques-utilisees-en-sante.
87. ANSM.Logiciels et applications mobiles en santé. [Software and mobile application in health]; 2020 .Available from: https://www.ansm.sante.fr/Activites/Mise-sur-le-marche-des-dispositifs-medicaux-et-dispositifs-medicaux-de-diagnostic-in-vitro-DM-DMIA-DMDIV/Logiciels-et-applications-mobiles-en-sante/(offset)/3#paragraph_78807.
88. HAS. Good practice guidelines on health apps and smart devices (mobile health or mhealth). Haute Autorité de Santé. Available from: https://www.has-sante.fr/jcms/c_2681915/en/good-practice-guidelines-on-health-apps-and-smart-devices-mobile-health-or-mhealth.
89. ANSSI Cyber veille-santé.l'ANSSI publie son rapport annuel de2018 | accompagnement Cybersécurité des Structures de Santé.[ANSSI publishes its 2018 annual report | Cybersecurity Support for Health Structures [ANSSI cyber health watch. ANSSI publishes its 2018 annual report | Cybersecurity support for Health Structures]. ANSSI; 2019. Available from: https://www.cyberveille-sante.gouv.fr/cyberveille/1258-lanssi-publie-son-rapport-annuel-de-2018-2019-04–18.
90. Shuren J, Patel B, Gottlieb S. FDA regulation of mobile medical apps. JAMA. 2018;320(4):337–338. doi:10.1001/jama.2018.8832
91. Moshi M, Tooher MT, Merlin T. Development of a health technology assessment module for evaluating mobile medical applications. Int J Technol Assess Health Care. 2020;36(3):252–261. doi:10.1017/S0266462320000288
92. Fraser AG, Butchart EG, Szymański P. The need for transparency of clinical evidence for medical devices in Europe. Lancet. 2018;392(10146):521–530. doi:10.1016/S0140-6736(18)31270-4
93. Amrani LE, Engberink AO, Ninot G, Hayot M, Carbonnel F. Connected health devices for health care in french general medicine practice: cross-Sectional Study. JMIR MHealth UHealth. 2017;5(12):e193. doi:10.2196/mhealth.7427
94. Vogt F, Seidl F, Santarpino G, et al. Healthcare IT utilization and penetration among physicians: novel it solutions in healthcare - use and acceptance in hospitals. Eur Surg Res Eur Chir Forsch Rech Chir Eur. 2018;59(1–2):100–113. doi:10.1159/000490241
95. Iribarren S, Cato K, Falzon L, Stone PW. What is the economic evidence for mHealth? A systematic review of economic evaluations of mHealth solutions. PLoS One. 2017;12(2):e0170581. doi:10.1371/journal.pone.0170581
96. Allenby CE, Babiash ES, Blank PN, et al. Analysis of the market, regulatory landscape, and current state of clinical trials pertaining to digital health. Technol Transf Entrep. 2018;5(1):21–34. doi:10.2174/2213809905666180816115016
97. Bates DW, Landman A, Levine DM. Health apps and health 1490 policy: what is needed? JAMA. 2018;320(19):1975–1976. doi:10.1001/jama.2018.14378
98. Gordon WJ, Dw B. Beyond validation: getting health apps into clinical practice. NPJ Digit Med. 2020;3. doi:10.1038/s41746-019-0212-z
99. Rijcken C. Pharmaceutical Care in Digital Revolution: Insights Towards Circular Innovation. Academic Press; 2019.
100. Eysenbach G; CONSORT-EHEALTH Group. CONSORT-EHEALTH: improving and standardizing evaluation reports of Web-based and mobile health interventions. J Med Internet Res. 2011;13(4):e126. doi:10.2196/jmir.1923
101. Liu XX, Rivera SC, Moher D, Calvert MJ, Denniston AK. Reporting guidelines for clinical trial reports for interventions involving artificial intelligence: the CONSORT-AI extension. Nat Med. 2020;26(9):1364–1374. doi:10.1038/s41591-020-1034-x
102. Guo C, Ashrafian H, Ghafur S, Fontana G, Gardner C, Prime M. Challenges for the evaluation of digital health solutions—A call for innovative evidence generation approaches. NPJ Digit Med. 2020;3(1):110. doi:10.1038/s41746-020-00314-2
103. Baumfeld AE, Reynolds R, Caubel P, Azoulay L, Dreyer NA. Trial designs using real-world data: the changing landscape of the regulatory approval process. Pharmacoepidemiol Drug Saf. 2019. doi:10.1002/pds.4932
104. Mühlbacher AC, Juhnke C, Beyer AR, Garner S. Patient-focused benefit-risk analysis to inform regulatory decisions: the European Union perspective. Value Health J Int Soc Pharmacoecon Outcomes Res. 2016;19(6):734–740. doi:10.1016/j.jval.2016.04.006
105. Muehlematter UJ, Daniore P, Vokinger KN. Approval of artificial intelligence and machine learning-based medical devices in the USA and Europe (2015–20): a comparative analysis. Lancet Digit Health. 2021;3(3):e195–e203. doi:10.1016/S2589-7500(20)30292-2
106. Van Norman GA. Drugs, devices, and the FDA: part 2: an overview of approval processes: FDA approval of medical devices. J Am Coll Cardiol. 2016;1(4):277–287. doi:10.1016/j.jacbts.2016.03.009
107. Larson RS. A path to better-quality mHealth apps. JMIR MHealth UHealth. 2018;6(7):e10414. doi:10.2196/10414
108. Stoyanov SR, Hides L, Kavanagh DJ, Zelenko O, Tjondronegoro D, Mani M. Mobile app rating scale: a new tool for assessing the quality of health mobile apps. JMIR MHealth UHealth. 2015;3(1):e27. doi:10.2196/mhealth.3422
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.