Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
A Dose-Ranging Study of the Novel Inhaled Dual PDE 3 and 4 Inhibitor Ensifentrine in Patients with COPD Receiving Maintenance Tiotropium Therapy
Authors Ferguson GT ![]() , Kerwin EM
, Kerwin EM ![]() , Rheault T, Bengtsson T, Rickard K
, Rheault T, Bengtsson T, Rickard K ![]()
Received 18 February 2021
Accepted for publication 8 April 2021
Published 22 April 2021 Volume 2021:16 Pages 1137—1148
DOI https://doi.org/10.2147/COPD.S307160
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Gary T Ferguson,1 Edward M Kerwin,2 Tara Rheault,3 Thomas Bengtsson,4 Kathleen Rickard3
1Pulmonary Research Institute of Southeast Michigan, Farmington Hills, MI, USA; 2Altitude Clinical Consulting, Medford, OR, USA; 3Verona Pharma, Plc, Raleigh, NC, USA; 4Stat Mind AB, Lund, Sweden
Correspondence: Tara Rheault
Verona Pharma, 8045 Arco Corporate Drive, Suite 130, Raleigh, NC, 27617, USA
Tel +1 646-530-2126
Email [email protected]
Purpose: Ensifentrine is an inhaled dual inhibitor of phosphodiesterase (PDE) 3 and 4 that has shown bronchodilatory effects and symptom improvement in clinical studies in patients with chronic obstructive pulmonary disease (COPD), and anti-inflammatory effects in healthy volunteers in a model of COPD-like inflammation. This manuscript reports on the results of the clinical study examining if ensifentrine provides meaningful improvements in lung function when added on to tiotropium over 4 weeks in patients with COPD who have impaired lung function and symptoms despite treatment with tiotropium.
Patients and Methods: This randomized, double-blind, placebo-controlled, parallel-group, dose-ranging study recruited patients with moderate-to-severe COPD. Patients were randomized to open-label tiotropium once daily (QD) plus (+) blinded escalating doses of ensifentrine or placebo twice daily (BID). Effects on lung function, symptoms and quality of life (QoL) were assessed over 4 weeks.
Results: A total of 416 COPD patients were randomized and 413 received at least one dose of blinded study medication + tiotropium. All ensifentrine doses produced a significant and dose-dependent increase in peak forced expiratory volume in 1 second (FEV1) from baseline to Week 4, with placebo-corrected differences of 77.5 mL when added to tiotropium (0.375 mg; 95% CI: 4.8, 150.1 mL; p=0.037) to 124.2 mL (3 mg; 95% CI: 51.0, 196.8 mL; p< 0.001). A significant increase in average FEV1 (0– 12h) was shown at Week 4 with the 3 mg dose (87.3 mL; 95% CI: 20.0, 154.5 mL; p=0.011). Clinically meaningful and statistically significant improvements in the St. George’s Respiratory Questionnaire – COPD (SGRQ-C) additive to tiotropium were observed at Week 4, exceeding the minimally clinically important difference of 4 units with the 1.5 and 3 mg doses. Adverse events were similar in frequency between the ensifentrine and placebo arms.
Conclusion: This clinical study demonstrated that nebulized ensifentrine added on to tiotropium produced clinically important improvements in lung function and QoL over 4 weeks in COPD patients receiving tiotropium who demonstrated symptoms and lung function impairment, with a safety profile similar to placebo.
Keywords: dual PDE3 and 4 inhibitor, phosphodiesterase, COPD, tiotropium, bronchodilation
Plain Language Summary
This study evaluated if nebulized ensifentrine provides additional improvements in lung function when added to tiotropium in COPD patients who have impaired lung function and symptoms despite treatment with tiotropium. Study results showed that, when added to tiotropium, ensifentrine produced clinically meaningful and statistically significant bronchodilation with a dose-dependent increase from baseline to peak FEV1 after 4 weeks. Interpretation: When nebulized ensifentrine was added on to tiotropium maintenance therapy, it produced dose-dependent, clinically important improvements in lung function over 4 weeks in COPD patients receiving tiotropium who demonstrated symptoms and lung function impairment.
Introduction
The World Health Organization listed chronic obstructive pulmonary disease (COPD) as the third leading cause of death worldwide in 2019.1 COPD is characterized by chronic, irreversible airflow obstruction and persistent airway inflammation leading to symptoms such as dyspnea, cough and sputum production and resulting in a reduced quality of life (QoL).2 Current standard of care, including treatment with inhaled long-acting bronchodilators and corticosteroids, has been shown to improve lung function, COPD symptoms and health-related QoL and to reduce exacerbation frequency. However, even with maximal inhaled therapies, many patients remain symptomatic and functionally impaired.2–4 Thus, new treatment options are urgently needed for patients with COPD which can provide additional bronchodilation and anti-inflammatory effects targeting the chronic inflammatory pathology associated with COPD.
Phosphodiesterases (PDEs) are enzymes that impact a range of cellular functions by modulating intracellular levels of cyclic nucleotide signaling molecules. PDE3 regulates cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) concentrations in airway smooth muscle, such that inhibition results in airway smooth muscle relaxation.5–7 PDE4 regulates cAMP concentrations and is involved in inflammatory cell activation; consequently, inhibition has anti-inflammatory effects.8–10 There is evidence to suggest that combined inhibition of PDE3 and PDE4 may have additive or synergistic effects with respect to both anti-inflammatory and bronchodilator activity given the expression of both PDE isoforms on inflammatory cells and airway smooth muscles, respectively.11
Ensifentrine is a first-in-class inhaled dual inhibitor of PDE3 and 4. In a 4-week study, nebulized ensifentrine alone demonstrated significant bronchodilatory effects and symptom improvement and was well tolerated, with an adverse event profile similar to placebo, in COPD patients at doses from 0.75 to 6 mg twice daily.12 When added on to standard classes of bronchodilators in short-term studies in patients with COPD, ensifentrine showed rapid and meaningful improvement in lung function and a reduction in lung volumes, which may indicate an effect on small airways and a physiological mechanism for symptom relief.13 Anti-inflammatory effects including a significant reduction in inflammatory cell types (ie, macrophages, neutrophils, lymphocytes and eosinophils) were demonstrated in the sputum of healthy volunteers challenged with the antigen lipopolysaccharide (a model of COPD-like inflammation) after 6-days of dosing with ensifentrine.14
This Phase IIb study examined the efficacy and safety of nebulized ensifentrine in doses ranging from 0.375 to 3 mg twice daily (BID) added to a once-daily (QD) long-acting muscarinic antagonist (LAMA; tiotropium) over 4 weeks and in patients with COPD who continued to have impaired lung function and significant dyspnea following a 2-week run-in on tiotropium.
Patients and Methods
Tiotropium (delivered via Spiriva® Respimat®, two puffs 2.5 µg QD) was selected as a commonly prescribed LAMA bronchodilator to provide a uniform background maintenance treatment for all patients across the run-in and treatment periods. LAMAs are a recommended treatment for symptomatic COPD patients.2 Symptomatic patients with a pre-dose FEV1 of 30–70% and a modified Medical Research Council (mMRC) dyspnea scale score ≥2 after 2 weeks of daily treatment with tiotropium were selected to examine the benefits of added nebulized ensifentrine in this population. The study was approved by independent ethics committees at each institution and was performed in accordance with the Declaration of Helsinki and Good Clinical Practice (ICH/CPMP/135/95). Written informed consent was obtained from all patients. The study is registered at ClinicalTrials.gov (NCT03937479). All participating sites used Copernicus Group Independent Review Board (CGIRB) of Cary, NC, USA.
Design
This was a randomized, double-blind, placebo-controlled, 5-arm parallel group study examining the dose-dependent effects of nebulized ensifentrine BID on lung function in patients with moderate-to-severe COPD. Eligible patients at screening entered a 2-week run-in period with open-label tiotropium. All patients received short-acting bronchodilators (albuterol metered dose inhaler) for rescue use that was withheld at least 6 hours prior to spirometry.
Following the run-in period, patients demonstrated pre-dose forced expiratory volume in 1 second (FEV1) of 30–70% predicted and ≥2 on the mMRC dyspnea scale for randomization eligibility. Patients were stratified at entry based on V1 reversibility status following treatment with albuterol (“reversible”: ≥12% and ≥200 mL increase in FEV1; or “non-reversible”: <12% or <200 mL increase in FEV1; each stratum capped at 50%) and randomized equally to receive double-blinded nebulized study medication BID added to open-label tiotropium QD for 4 weeks.
Patients were 40–80 years old, and had baseline post-bronchodilator (four puffs of albuterol) FEV1/FVC ratio of ≤0.70 and FEV1 ≥30% and ≤70% of predicted normal (NHANES III) after meeting a 48-hour washout of any background maintenance bronchodilators, a score of ≥2 on the mMRC dyspnea scale, and current or former smoking status with smoking history of ≥10 pack years. Patients with a history of asthma or other pulmonary disease; oral COPD therapy within 3 months (eg, oral steroids, theophylline, and roflumilast), inhaled corticosteroids (ICS) therapy within 4 weeks or a history of any COPD exacerbation requiring treatment with systemic corticosteroids or antibiotics within 3 months, or a severe COPD exacerbation within 6 months of screening were excluded. The Supplementary Materials display the complete study design (Supplement Figure 1), eligibility and randomization criteria (Supplement Tables 1–3), and study procedures (Supplement Table 4).
At randomization, baseline (pre-study medication dose) data were collected for spirometry (FEV1), St. George’s Respiratory Questionnaire – COPD (SGRQ-C), Baseline Dyspnea Index (BDI), electrocardiogram (ECG) and vital signs (eg, blood pressure, pulse rate). All baseline measurements were assessed with patients on steady state tiotropium at the morning trough (pre-dose), including spirometry. Patients were administered tiotropium in the clinic followed by double-blind nebulized study medication after completion of pre-dose assessments. Spirometry was assessed pre-dose at −30 minutes (min) and up to 3 hours (h) post-dose (+30min, 1, 2, and 3h) on Weeks 1, 2 and 3, and up to 12h post-dose (+30min, 1, 2, 3, 4, 6, 8 and 12h) on Day 1 and Week 4. Transition Dyspnea Index (TDI) and SGRQ-C, were assessed pre-dose at Weeks 2 and 4. Daily throughout the study (including run-in) patients used an e-diary to record rescue medication use and COPD symptoms (Evaluating Respiratory Symptoms [E-RS™: COPD] questionnaire). Permissions were obtained for the use of the instruments. Vital signs and 12-lead ECGs were assessed pre- and post-dose on all visits. Adverse events (AEs) were captured over the study duration.
Outcomes
Efficacy
The primary endpoint was the change from baseline to Week 4 in peak FEV1 (maximum value during 3h post-dose, 0–3h), placebo and tiotropium adjusted.
Secondary endpoints included the placebo and tiotropium adjusted change from baseline in: average area under the curve (AUC) 0–3h FEV1, average AUC0-12h FEV1 at Day 1 and Week 4, peak FEV1 at Day 1 and Weeks 1–3; morning trough FEV1 at Weeks 1–4. QoL and symptom improvement was assessed with the placebo and tiotropium adjusted change from baseline in: the SGRQ-C at Weeks 2 and 4; mean weekly values over Weeks 1 to 4 in COPD symptoms, as measured by E-RS™: COPD; TDI at Weeks 2 and 4; and mean weekly values over Weeks 1 to 4 in rescue medication use. Compliance with blinded study medication was assessed via returned vials and captured in the eDiary. Compliance with tiotropium dosing was captured in the eDiary. The time of dosing was not collected.
Safety
Ensifentrine safety was assessed by evaluation of incidence of treatment-emergent adverse events (TEAEs), laboratory safety tests, pre- and post-dose 12-lead ECG data (including mean heart rate and QTcF interval), and vital signs.
Statistical Analysis
The standard deviation for the change in peak FEV1 was estimated to be 200 mL. With a 2-sided test at a 5% significance level and 73 evaluable patients per group, there was 80% power to detect a true difference of 93 mL between any two treatments. This detectable limit was considered sufficient to identify an effective dose of ensifentrine added on to tiotropium. Assuming a 10% early withdrawal rate, 80 patients per group were randomized.
Efficacy data analysis was performed on the full analysis set, which comprised all randomized and treated patients with sufficient data collected after intake of study medication to compute the pharmacodynamic parameters based on FEV1 on at least one occasion. The primary endpoint (Week 4 peak FEV1) was analyzed using a restricted maximum likelihood-based mixed model for repeated measured (MMRM), including fixed effects for treatment, visit and treatment by visit interaction, patient as random effect, baseline value as covariate and covariance structure by visit. Ensifentrine–placebo differences with 95% confidence intervals and corresponding two-sided p-values were calculated. To control for the familywise error rate, a fixed-sequence testing strategy was employed, with the highest ensifentrine dose (3 mg) tested vs placebo. If a statistically significant difference was found at the two-sided α level of 5%, the testing proceeded with the next lower dose. If a test was non-significant, testing stopped and the remaining null hypotheses accepted. The average FEV1 endpoints were calculated using the linear trapezoidal method as the area under the curve divided by the length of the time interval of interest.
A similar MMRM method was used to analyze most of the secondary efficacy endpoints, with the same hierarchical testing within endpoint, although endpoints were tested independently. In the analysis of TDI, BDI was used as baseline in the model and the dependent variable was the TDI total score. Weekly mean values were used for the MMRM analysis of E-RS™: COPD and the use of rescue medication and baseline for both measures was computed over the last 7 days of the run-in period. Values at Weeks 2 and 4 were used for the MMRM analysis of TDI, as well as the change from baseline in SGRQ-C.
Results
The study was conducted between May and November 2019 at 46 study centers across the United States (USA), although 49 centers consented at least one patient.
Disposition
Overall, 416 patients were randomized. Of those, 413 (99.3%) received at least one dose of blinded study medication and were included in the full analysis set as well as the safety analysis set. A total of 373 (89.7%) patients completed the 4-week study. Figure 1 illustrates patient disposition for the study.
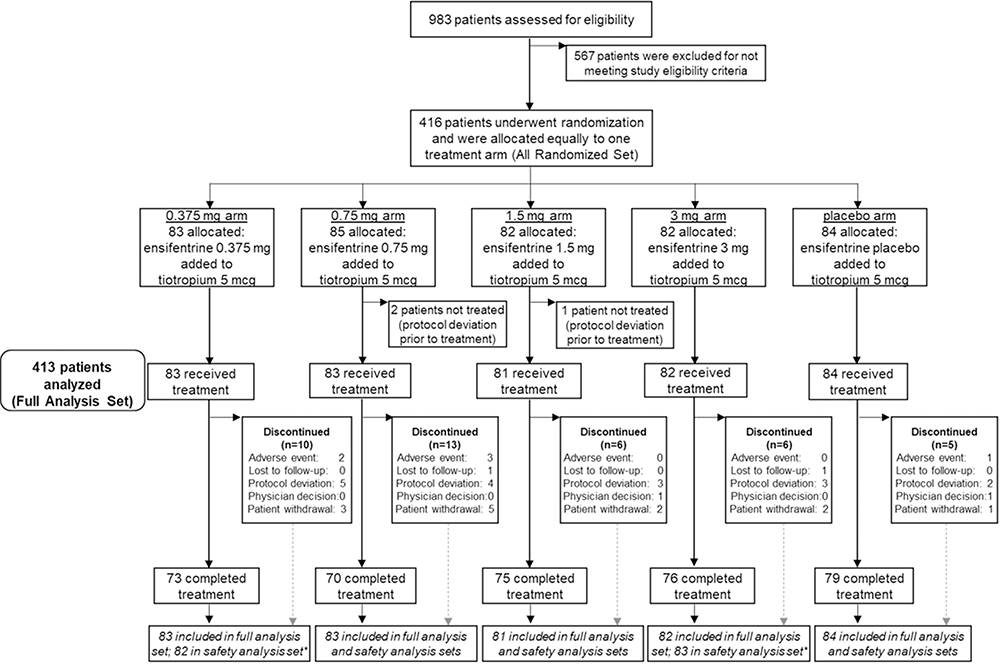 |
Figure 1 Patient flow through the study. Abbreviations: FEV1, forced expiratory volume in 1second; n, number of patients. Notes: Tiotropium: open-label tiotropium (two puffs 2.5 µg; once daily). Ensifentrine: double-blind study medication (via jet nebulizer with compressor; 0.375 mg, 0.75 mg, 1.5 mg, 3 mg, or placebo; twice daily). Full analysis set: all randomized patients with sufficient data collected after intake of blinded study medication to compute the pharmacodynamic parameters based on FEV1 on at least one occasion (n=413). Safety analysis set: all patients that received at least one dose of study medication (n=413). *One patient was dispensed 3mg drug dosing rather than 0.375 mg. |
The most common reasons for early study discontinuation were protocol deviations (n =20, 46.5%), largely driven by patients not meeting randomization criteria (eg, recent COPD exacerbation, asthma or other respiratory disorder, or intolerance to tiotropium), and withdrawal by patient (n =13, 30.2%). A higher proportion of patients in the 0.75 mg arm discontinued the study early compared to other treatments arms (n =15, 17.6% vs 6.0–12.0%). There were no deaths during the study.
Compliance
Based on the number of vials dispensed and returned, mean treatment compliance was high (≥97.7%) and similar across arms. Median duration of exposure was 29.0 days across all groups.
Characteristics
Baseline population characteristics for the full analysis set (n =413) are listed in Table 1. The majority of patients were White (90.1%), with a mean age of 64.3 years (range: 45 to 80 years). Over half (57.5%) were female and 52.2% were less than 65 years of age. There were 214 non-reversible (33.2% male) and 199 reversible (52.8% male) patients.
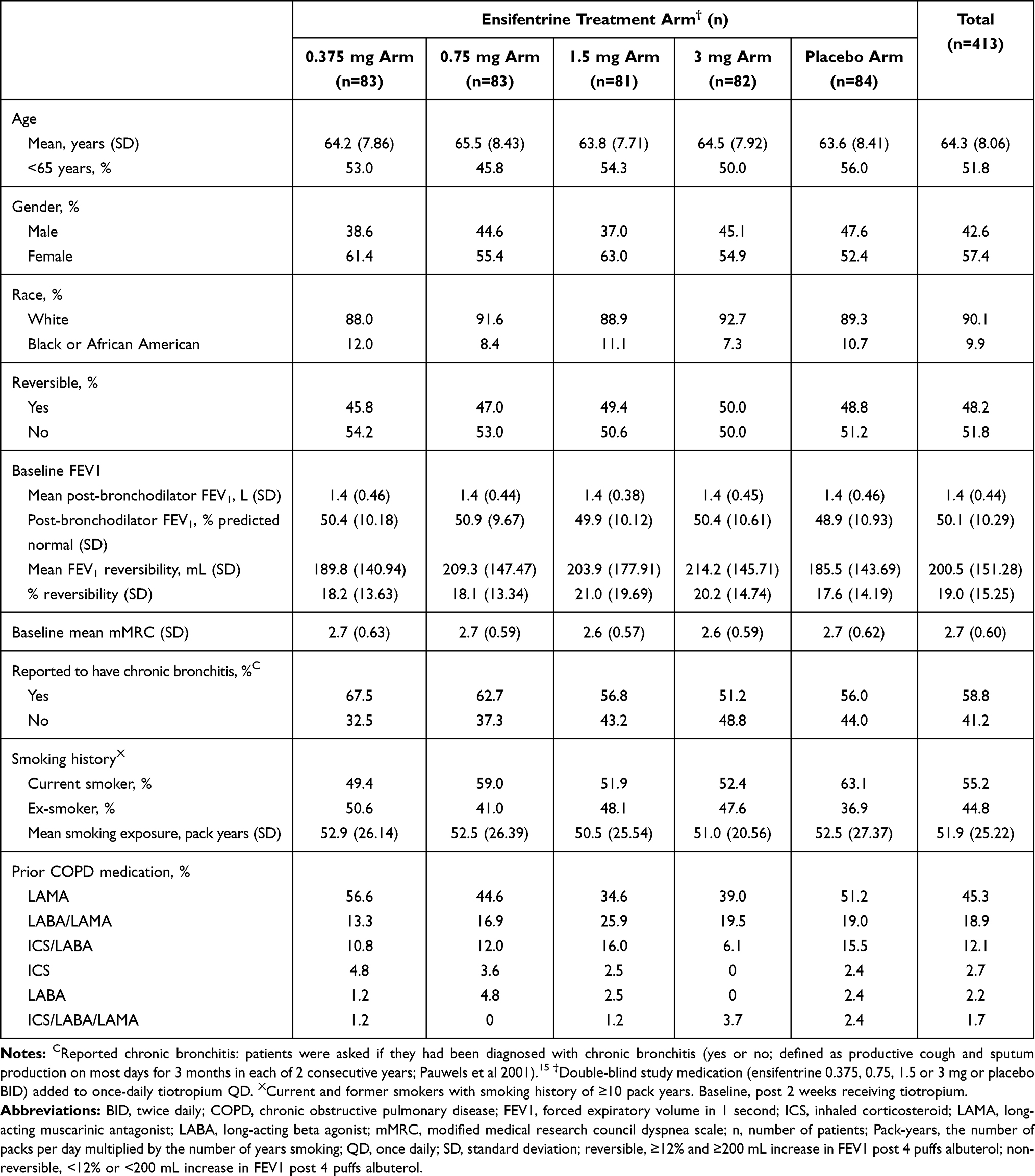 |
Table 1 Baseline Demographics and Disease Characteristics in the Full Analysis Set |
Baseline disease and clinical characteristics are also displayed in Table 1. Mean pre-bronchodilator FEV1 of 1.20 L, mean post-bronchodilator FEV1 of 1.40 L (50.2% of predicted normal FEV1) and an FEV1/FVC ratio of 0.52. Mean baseline reversibility was 87.0 and 321.4 mL for non-reversible and reversible strata, respectively. The 0.375 mg arm had the highest percentage of patients that self-reported a diagnosis of chronic bronchitis (67.5% vs 51.2–62.7%) as well as previous LAMA users (56.6% vs 34.6–51.2%). The placebo arm had the highest percentages of males (47.6% vs 37.0–45.1%) and current smokers (63.1% vs 49.4–59.0%), in addition to highest baseline mean PRO scores (SGRQ-C and E-RS™: COPD total scores) and rescue medication use (Table 2).
 |
Table 2 Baseline Patient-Reported Outcome (PRO) Scores and Rescue Medication Use for Patients with Baseline and Week 4 Values Reported in the Full Analysis Set |
Primary Endpoint
Ensifentrine resulted in a dose-dependent, statistically significant improvement in peak FEV1, compared to the placebo arm when added to tiotropium. Least-squares mean (LSM) placebo-corrected differences for the 0.375, 0.75, 1.5 and 3 mg arms were as follows: 77.5 (p=0.037; 95% CI: 4.8, 150.1); 91.2 (p=0.015; 95% CI: 18.0, 164.3); 107.2 (p=0.004; 95% CI: 34.4, 180.0); and 124.2 mL (p<0.001; 95% CI: 51.7, 196.8) respectively (Table 3, Figure 2).
 |
Table 3 Change from Baseline to Week 4 in Peak FEV1 (Over 3 Hours), Average FEV1 (0–12 Hours) and Morning Trough FEV1 in the Full Analysis Set |
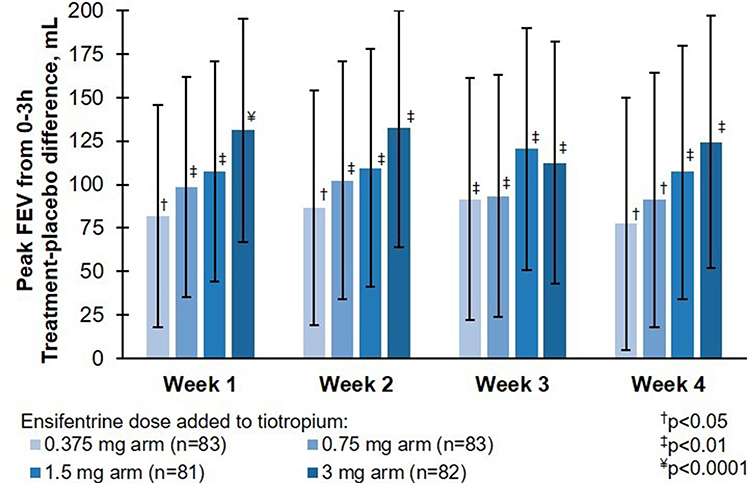 |
Figure 2 Peak FEV1 between 0 and 3 h post-dose in the full analysis set. Abbreviations: CI, confidence interval; FEV1, forced expiratory volume in 1second; p, p-value vs placebo; placebo, placebo ensifentrine added to tiotropium. Notes: Mean change from baseline FEV1 and standard error of mean are shown. Data are least squares means treatment – placebo differences and 95% CI of the least squares mean difference. †P<0.05 vs placebo; ‡P<0.01 vs placebo; ¥P<0.0001 vs placebo. Least squares mean changes from baseline in the placebo ensifentrine added to tiotropium arm in the full analysis set (placebo, n=84) were 104, 110, 107, and 119 mL at Weeks 1, 2, 3 and 4, respectively. Baseline, post 2 weeks receiving tiotropium. |
Other Endpoints
A statistically significant improvement was observed in peak FEV1 at Day 1 and Weeks 1–3 in all ensifentrine arms compared to the placebo arm (LSM placebo-corrected difference, all p<0.02, Figure 2, Supplement Table 5).
A dose-ordered response in the change from baseline in average FEV1 over 12h at Week 4 was also observed with a statistically significant improvement for the 3 mg arm (LSM placebo-corrected difference; 87.3 mL; 95% CI: 20.0, 154.5; p=0.011; Table 2). Separation from placebo was maintained over 12h with the 3 mg arm as well, as demonstrated in the 12h spirometry profile at Week 4 (Supplement Figure 2).
A nominal improvement from baseline in morning trough FEV1 at Week 4 was observed with the 3 mg arm compared to the placebo arm (27.2 mL; Table 3). However, there were no statistically significant improvements from baseline in trough FEV1 for any of the treatment arms (Table 3).
Post-hoc analysis of average FEV1 6–12h from baseline to Week 4 with blinded study drug added to tiotropium revealed nominal improvements. LSM placebo-corrected differences were 5 (0.375 mg arm; p=0.884, 95% CI: −59, 69); 33 (0.75 mg arm; p=0.314, 95% CI: −31, 97); 27 (1.5 mg arm; p=0.407, 95% CI: −37, 92); and 58 mL (3 mg arm; p=0.078, 95% CI: −6, 122).
In COPD health-related QoL and symptoms, ensifentrine showed statistically significant improvements in SGRQ-C (LSM change from baseline) compared to placebo, exceeding the minimal clinically important difference (MCID) of 4 units at Week 4 with the 1.5 and 3 mg arms (LSM placebo-corrected differences; both p<0.05, Table 4). Compared to the placebo arm, ensifentrine generally provided numerically greater improvements at Week 4 in the E-RS™: COPD total score and TDI score, but did not reach statistical significance (Table 4). There were minimal changes from baseline in rescue medication use compared to the placebo arm (Supplement Table 6).
 |
Table 4 Change from Baseline to Week 4 in SGRQ-C Total Score, E-RS™: COPD Total Score and TDI Score in the Full Analysis Set |
Safety
Overall, the AE profile of all ensifentrine treatment arms were similar to the placebo arm. Ensifentrine was well tolerated at all dose levels and there were no patterns or relationship to ensifentrine dose observed. Gastrointestinal and cardiac disorders were rare overall.
Table 5 provides the overall summary of TEAEs as well as those most commonly reported in more than one patient overall by preferred term. The most commonly reported TEAE in patients that received ensifentrine was headache (1.8% [6/329] vs 1.2% [1/84] that received placebo). In patients that received ensifentrine, 0.91% (3/329) reported serious TEAE; however, there were no reports of serious drug-related TEAE. There were no meaningful changes in vital signs, blood pressure or pulse-rate and no dose-related or meaningful changes in heart rate nor to QTcF interval, pre-dose or 2h post-dose. Baseline, Week 4 and change from baseline to Week 4 values of ECG mean heart rate and ECG QTcF interval are shown in Supplement Tables 7 and 8.
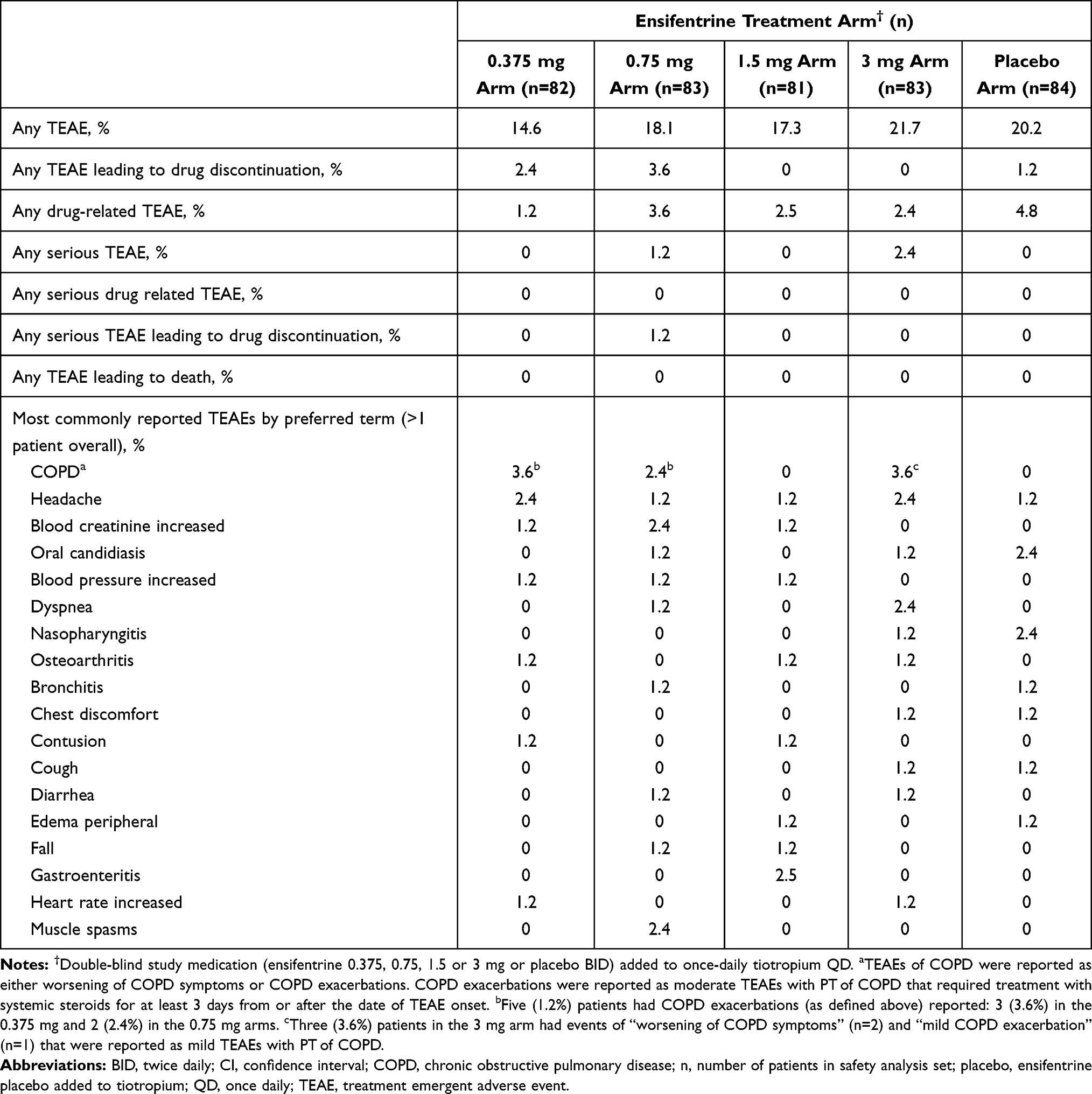 |
Table 5 Proportion of Treatment Emergent Adverse Events Summary Listed by Prevalence in the Safety Analysis Set |
Discussion
In this double-blind, placebo-controlled, randomized, dose-ranging study, twice-daily, nebulized ensifentrine (0.375–3 mg) improved peak FEV1 (0–3h) in a dose-ordered, statistically significant manner with all ensifentrine doses added to tiotropium QD compared to placebo added to tiotropium QD after 4 weeks (0.375 mg: 78 mL, p=0.037; 0.75 mg: 91 mL, p=0.015; 1.5 mg: 107 mL, p=0.004; 3 mg: 124 mL, p<0.001). This outcome is similar to the previously reported dose-ranging study of ensifentrine monotherapy (0.75 to 6 mg or placebo) in COPD patients, where ensifentrine significantly improved peak FEV1 (0–3h) compared to placebo for all doses after 4 weeks of treatment and showed a similar dose response up to the 3 mg arm.12
There were dose-dependent improvements in lung function observed for average AUC0-12h FEV1, including a statistically significant improvement of 87 mL with the 3 mg arm, which appeared to be the most efficacious dose for airflow improvements over the 12h serial spirometry. Nominal, but consistent improvements in trough FEV1 with the 3 mg arm were also observed, which were smaller in magnitude in this study compared to prior Phase 2b study CO-203 due to the background tiotropium use in the current study. These results are also consistent with those observed in the earlier ensifentrine monotherapy study, where statistically significant improvements of 119 mL versus placebo (average FEV1 AUC 0–12h) and 68 mL (morning trough FEV1) were shown with the 3 mg arm.12
Ensifentrine added on to tiotropium produced a robust, clinically meaningful and statistically significant improvement in QoL (measured with SGRQ-C) after 4 weeks that were supported by numerical improvements in COPD symptoms (measured by E-RSTM: COPD and TDI) compared to placebo added on to tiotropium. This represents an unprecedented improvement in quality of life over only 4 weeks of treatment in patients with COPD who continued to have impaired lung function and remain highly symptomatic despite maintenance use of tiotropium. The magnitude of improvement observed in quality of life added on to tiotropium, compared to placebo added on to tiotropium appears to be greater than the incremental improvements in lung function would predict and included improvements in all SGRQ-C sub-scales, with the largest improvements demonstrated in the Activity and Impacts domains. This suggests that the dual PDE3/4 mechanism by which ensifentrine acts is providing benefits beyond bronchodilation, and possibly related to anti-inflammatory effects. The data on lung function, symptoms and quality of life improvement described herein, support the twice-daily dosing of ensifentrine in patients with moderate-to-severe COPD as a potential add-on to maintenance bronchodilators for symptomatic COPD patients who may benefit from escalation of therapy.
As observed with the earlier ensifentrine monotherapy study, delivery of ensifentrine directly to the airways through nebulization in addition to tiotropium was well tolerated at all doses in this study with an adverse event profile similar to the placebo arm, including the cardiovascular and gastrointestinal effects.
This study was not enriched with exacerbation-prone patients, and the 4-week treatment duration was too brief to assess ensifentrine doses for potential prevention of COPD exacerbations. However, another approved oral PDE4 antagonist, roflumilast 500mg daily, has reported decreases in COPD exacerbation rates in at least some subgroups of COPD patients,16 suggesting ensifentrine might also favorably modify COPD exacerbations in a long-term COPD-exacerbation prone population. In delivering ensifentrine directly to the airways through nebulization, the medication appeared well tolerated. There were very few reports of treatment-related nausea and headache or other adverse events often reported for roflumilast or other systemically delivered phosphodiesterase therapies.
Limitations
The run-in period with tiotropium prior to randomization was 2 weeks in order to achieve steady state on bronchodilation, and it is possible that this was not long enough to establish a robust baseline for patient-reported outcome measures and rescue medication use. Furthermore, this study enrolled 50% of patients who showed evidence of reversibility to albuterol at screening in order to better characterize responsiveness to ensifentrine in patients with COPD. The impact of this design feature combined with the relatively short run-in on tiotropium on study outcomes will be described in a subsequent publication. Trough FEV1 was assessed on a background of tiotropium, thus does not represent a true trough effect of ensifentrine. Additionally, as assessed in this study, the pre-dose morning trough assessment cannot be confirmed as 12-hours post evening dose as the timing of the evening dose prior to spirometry was not collected.
This dose-ranging study was powered for improvements in lung function, not symptom or QoL improvement. With approximately 80 patients randomized per arm, differences in baseline values for E-RS™: COPD and SGRQ-C across treatment arms were observed, which may limit the interpretation of dose-responsiveness in these scales known to be impacted by baseline severity. Additionally, this was a short-term study over a 4-week treatment period; thus, longer-term efficacy and safety needs to be established. In addition, potential benefits for nebulized PDE3/4 inhibitors such as ensifentrine for prevention of COPD exacerbations could not be evaluated in this short trial, and would require longer trials in COPD patients at risk for COPD exacerbations.
Strengths included the measurement of multiple endpoints to monitor airflow, health status and symptoms as well as safety, and the normalization of background maintenance therapy with sponsor provided tiotropium across the run-in and treatment periods.
Conclusion
Ensifentrine provided a dose-dependent, statistically significant and clinically meaningful bronchodilation (peak FEV1) of 78 mL (0.375 mg), 91 mL (0.75 mg), 107 mL (1.5 mg) and 124 mL (3 mg) when administered BID in addition to tiotropium (all p<0.05 compared to placebo+tiotropium), in COPD patients who remained symptomatic while taking tiotropium. Additionally, this novel, inhaled inhibitor of PDE3 and PDE4, provided significant improvement in QoL and has a safety profile similar to placebo.
Data Sharing Statement
Individual participant data will not be shared. The study protocol and statistical analysis plan are shared at ClinicalTrials.gov (NCT03937479).
Ethics Approval
This study was approved by an institutional review board (Copernicus Group IRB of Cary, NC, USA).
Acknowledgments
This research was funded by Verona Pharma plc. The sponsor developed the study and funded the writing of the manuscript. Verona Pharma is the guarantor of the content of the manuscript, including the data and analysis.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
Drs Ferguson and Kerwin served as an investigator for the study, received study grant from Verona Pharma, has received consulting fees from Verona Pharma. Dr Ferguson reports grants, personal fees, and/or non-financial support from Boehringer Ingelheim, GlaxoSmithKline, Novartis, Sunovion, Theravance/Mylan, AstraZeneca, Innoviva, Circassia, Sanofi, Altvant, Knopp, Teva, Orpheris, DevPro, and Galderma, outside the submitted work. Dr Kerwin reports personal fees and non-financial support for consulting, advisory board membership, and travel reimbursement from Amphastar, AstraZeneca, Boehringer Ingelheim, Chiesi, Connect Biopharma, GlaxoSmithKline, Mylan, Novartis, Sunovion, and Theravance, outside the submitted work. Drs Rheault and Rickard are employees and shareholders for Verona Pharma. Mr Bengtsson serves as the biostatistical contractor for Verona Pharma. The authors report no other conflicts of interest in this work.
References
1. World Health Organization. Available from: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
2. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease; 2020. Available from: https://goldcopd.org/gold-reports/.
3. Vestbo J, Papi A, Corradi M, et al. Single inhaler extrafine triple therapy versus long-acting muscarinic antagonist therapy for chronic obstructive pulmonary disease (TRINITY): a double-blind, parallel group, randomised controlled trial. Lancet. 2017;389(10082):1919–1929. doi:10.1016/S0140-6736(17)30188-5
4. Mahler D, Decramer M, D’Urzo A, et al. Dual bronchodilation with QVA149 reduces patient-reported dyspnoea in COPD: the BLAZE study. Eur Respir J. 2014;43(6):1599–1609. doi:10.1183/09031936.00124013
5. Banner KH, Press NJ. Dual PDE3/4 inhibitors as therapeutic agents for chronic obstructive pulmonary disease. Br J Pharmacol. 2009;157(6):892–906. doi:10.1111/j.1476-5381.2009.00170.x
6. De Boer J, Philpott AJ, Van Amsterdam RG, Shahid M, Zaagsma J, Nicholson CD. Human bronchial cyclic nucleotide phosphodiesterase isoenzymes: biochemical and pharmacological analysis using selective inhibitors. Br J Pharmacol. 1992;106(4):1028–1034. doi:10.1111/j.1476-5381.1992.tb14451.x
7. Page CP, Spina D. Phosphodiesterase inhibitors in the treatment of inflammatory diseases. In: Francis S, Conti M, Houslay M, editors. Phosphodiesterases as Drug Targets. Handbook of Experimental Pharmacology. Berlin: Springer; 2011:391–414.
8. Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ. M2-124 and M2-125 Study Groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009;374(9691):685–694. doi:10.1016/S0140-6736(09)61255-1
9. Singh D, Nandeuil MA, Pigeon-Francisco C, et al. Efficacy and safety of CHF6001, a novel inhaled PDE4 inhibitor in COPD: the Pioneer Dose Finding Study. Am J Respir Crit Care Med. 2019;199:A4529.
10. DALIRESP® (roflumilast) [prescribing information]. Wilmington. DE: AstraZeneca Pharmaceuticals LP; January, 2018.
11. Zuo H, Cattani-Cavalieri I, Musheshe N, Nikolaev V, Schmidt M. Phosphodiesterases as therapeutic targets for respiratory diseases. Pharmacol Ther. 2019;197:225–242.
12. Singh D, Martinez FJ, Watz H, Bengtsson T, Maurer BT. A dose-ranging study of the inhaled dual phosphodiesterase 3 and 4 inhibitor ensifentrine in COPD. Respir Res. 2020;21(1):47. doi:10.1186/s12931-020-1307-4
13. Singh D, Abbott-Banner K, Bengtsson T, Newman K. The short-term bronchodilator effects of the dual PDE3 and PDE4 inhibitor RPL554 in COPD. Eur Respir J. 2018;52(5):1801074. doi:10.1183/13993003.01074-2018
14. Franciosi LG, Diamant Z, Banner KH, et al. Efficacy and safety of RPL554, a dual PDE3 and PDE4 inhibitor, in healthy volunteers and in patients with asthma or chronic obstructive pulmonary disease: findings from four clinical trials. Lancet Respir Med. 2013;1(9):714–727. doi:10.1016/S2213-2600(13)70187-5
15. Pauwels RA, Buist AS, Calverley PM, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) workshop summary. Am J Respir Crit Care Med. 2001;163(5):1256–1276. doi:10.1164/ajrccm.163.5.2101039
16. Rennard SI, Calverley PM, Goehring UM, Bredenbröker D, Martinez FJ. Reduction of exacerbations by the PDE4 inhibitor roflumilast–the importance of defining different subsets of patients with COPD. Respir Res. 2011;12(1):18. doi:10.1186/1465-9921-12-18
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.