Back to Journals » Journal of Inflammation Research » Volume 13
Therapeutic Effect of C-C Chemokine Receptor Type 1 (CCR1) Antagonist BX471 on Allergic Rhinitis
Authors Feng S ![]() , Ju L
, Ju L ![]() , Shao Z, Grzanna M, Jia L, Liu M
, Shao Z, Grzanna M, Jia L, Liu M
Received 25 March 2020
Accepted for publication 26 June 2020
Published 21 July 2020 Volume 2020:13 Pages 343—356
DOI https://doi.org/10.2147/JIR.S254717
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Video abstract presented by Suoyi Feng.
Views: 378
Suoyi Feng,1,2 Longzhu Ju,1 Ziqi Shao,1,3 Mark Grzanna,2 Lu Jia,4 Ming Liu5
1College of Veterinary Medicine, Northeast Agricultural University, Harbin, Heilongjiang Province 150030, People’s Republic of China; 2Science Department, The John Carroll School, Bel Air, Maryland, USA; 3College of Veterinary Medicine, Huazhong Agricultural University, Wuhan, Hubei Province 430070, People’s Republic of China; 4School of Basic Medical Science, Shanxi University of Traditional Chinese Medicine, Jinzhong, Shanxi Province 030619, People’s Republic of China; 5State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute of Chinese Academy of Agricultural Sciences, Harbin, Heilongjiang Province 150069, People’s Republic of China
Correspondence: Suoyi Feng
Science Department, The John Carroll School, 703 E Churchville Road, Bel Air, Maryland 21014, USA
Email [email protected]
Ming Liu
State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute of Chinese Academy of Agricultural Sciences, 678 Haping Road, Harbin, Heilongjiang Province 150069, People’s Republic of China
Email [email protected]
Objective and Design: Allergic rhinitis (AR) is an immunoglobulin E (IgE)-mediated inflammatory respiratory hypersensitivity characterized by elevated Th2 cytokines and infiltration of inflammatory cells to nasal tissues. BX471 is a small-molecule C-C chemokine receptor type 1 (CCR1) antagonist involved in suppression of inflammation via blocking of primary ligands. In this study, we examined the anti-inflammatory effect of BX471 on ovalbumin (OVA)-induced AR mice model.
Materials and Methods: Levels of OVA-specific IgE and Th1 cytokines were determined by enzyme-linked immunosorbent assay (ELISA). Nasal expression of proinflammatory mediators was assessed by real-time polymerase chain reaction (RT-qPCR). Nasal-cavity sections were stained with hematoxylin and eosin (HE) and periodic acid-Schiff (PAS) to study eosinophil infiltration and goblet cell metaplasia. Relative protein levels of Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-kB), Toll-like Receptor 4 (TLR4) and Toll-like-receptor 2 (TLR2) were assessed by Western Blot. Percentage of CD4+CD25+Foxp3+ T regulatory cells (Treg) was measured by flow cytometry.
Results: Mice treated with BX471 showed significantly relieved sneezing and nasal-rubbing behaviors. The expression of nasal proinflammatory factors was significantly downregulated by BX471, and protein levels of tumor necrosis factor alpha (TNF- α) and NF-kB were suppressed. Blockade of CCR1 ligands inhibited eosinophil recruitment in nasal cavity. In addition, Treg cells population were upregulated in BX471-treated mice.
Conclusion: BX471 exerts anti-inflammatory effects in a mouse model of AR by inhibiting CCR1-mediated TNF-α production, which subsequently suppresses NF-kB activation in inflammatory cells, leading to a decrease in Th2 cytokines, IL-1β, VCAM-1, GM-CSF, RANTES, and MIP-1α expression levels, thus inhibiting eosinophil recruitment to nasal mucosa. In addition, BX-471 exhibits anti-allergic effect by increasing Treg cell population. Overall, BX471 represents a promising therapeutic strategy against AR.
Keywords: allergic rhinitis (AR), inflammation, C-C chemokine receptor type 1 antagonist, cytokines, chemokines
Introduction
Allergic rhinitis (AR) is a type of immunoglobulin E (IgE)-mediated chronic inflammatory respiratory disease characterized by production of Th2 cytokines, elevated IgE levels, histamine production, recruitment of inflammatory cells (such as eosinophils, basophils, mast cells, and neutrophils). Symptoms of AR include nasal congestion, sneezing, watery discharge, and nasal itching. AR is also defined as inflammation of the lining of nasal passages and sensory-nerve hypersensitivity triggered by inhaled antigens.1–3 AR affects 10–30% of adult population and over 40% of children.4 According to the World Health Organization, approximately 400 million people globally suffer from AR.4 In addition to symptoms involving the nasal cavity, AR is associated with sleep disorder, emotional problems, social isolation, poor concentration, and diminished quality of life (QOL).5,6 Current medications, such as glucocorticosteroids and antihistamines, provide only temporary relief. Therefore, new therapeutic approaches that addressing the root of AR pathogenesis are in great demand.
AR pathogenesis includes sensitization and generation of nasal symptom upon re-exposure to the specific allergens.2 During sensitization, dendritic cells uptake and present allergens to MHC class II peptide T-cell receptors. Additionally, allergens activate type 2 innate lymphoid cells and basophils, which release Th2-driving cytokines such as interleukin (IL)-13 and IL-4. Activated Th2 cells produce IL-4 and IL-13, stimulating B cells to undergo an immunoglobulin isotype switch. The immunoglobulin isotype switch results in the production of IgE, which binds to mast cells and basophils.2,7 Then, allergens binds to IgE and cross-link IgE receptors (FCeRI complex) on mast cells and basophils.2 When allergens crosslink a critical number of IgE receptors, mast cells and basophils release inflammatory cytokines IL-4, IL-5, IL-6, IL-8, IL-10, IL-13, and tumor necrosis factor alpha (TNF-α), as well as chemokines and histamine.8 The inflammatory cytokines induce Th2-mediated inflammation, ultimately resulting in the symptoms of AR.8
C-C chemokine receptor 1 (CCR1) is expressed on eosinophils, basophils, dendritic cells, monocytes, and natural killer T (NKT) cells. CCR1 has ligands MIP-1α, RANTES, MCP-3, C-C motif chemokine ligand (CCL)13, CCL16, and CCL23, which are upregulated in patients with AR.9–11 MIP-1α and RANTES serve as major eosinophil chemotactic proteins, leading to activation and accumulation of eosinophils at sites of allergic airway inflammation.12,13 MIP-1α and RANTES are also directly involved in stimulating antigen-specific IgE production and antigen-specific Immunoglobulin G4 (IgG4) production by B cells in vitro.14 Additionally, in AR, RANTES regulates mast-cell migration into the nasal epithelia, stimulates histamine release in basophils,15,16 and mediates basophil and neutrophil infiltration into the nasal mucosa.17 Furthermore, MCP-3 is highly expressed in AR and may serve as a chemotactic protein.18 CCR1 deficient mouse model of chronic fungal allergic asthma indicates decreased levels of IL-4, IL-13, decreased level of Th2-induced chemokines, and increased levels of Interferon gamma (IFN-γ) in the lung.19 Ligands of CCR1 play important roles in pathogenesis of AR, indicating that CCR1 may be a promising pharmaceutical target for therapies against AR.
BX471 is a small non-peptide small antagonist of CCR1 that can displace MIP-1α, RANTES, and MCP-3 by directly binding to CCR1, thereby inhibiting Ca2+ mobilization, extracellular acidification, CD11 expression, and leukocyte migration.20 The structure of BX471 was first identified by Liang et al.20 BX471 showed anti-inflammatory effects in mouse models of renal fibrosis by reducing the levels of CCR1 and CCR5 mRNA at a dose of 20 mg/kg in cyclodextrin.21 Carpenter et al22 demonstrated that BX471 downregulated TNF-α, CCL22, toll-like receptor (TLR) 2, and TLR6 expression in vitro (in macrophage culture). Carpenter et al22 further suggested that BX471 attenuated airway inflammation, hyper‐responsiveness, and remodeling in murine model of asthma. To better understand the underlying mechanism of BX471 in treating allergic airway inflammation in vivo, we assessed the effects of CCR1 antagonist BX471 in a murine model of AR.
Materials and Methods
Animals
Female BALB/C mice, approximately 8-weeks-old, were purchased from the Animal Center of Heilongjiang University of Chinese Medicine (Harbin, China). The mice were housed in a pathogen-free facility of the Northeast Agricultural University (Harbin, China) at 22–26°C and 50%±5% humidity with 12-hour light-dark cycle. Sterile food and water were provided ad libitum. All experimental procedures were reviewed and approved by Northeast Agricultural University (Harbin, China), and followed the Guide for the Care and Use of Laboratory Animals of the Northeast Agricultural University (Harbin, China).
Preparation of the Injectable BX471 Solution and the Vehicle Solution
The injectable BX471 solution was prepared according to a previously used protocol by Anders et al.21 A 40% cyclodextrin solution was prepared using unbuffered saline and dissolved overnight. The 40% cyclodextrin solution was filtered through a 0.45-µm filter, and the pH was adjusted to 3.30 by using hydrochloric acid (HCl). The BX471 (MedChem Express, USA) solution was prepared at a concentration of 20 mg/mL of 40% cyclodextrin solution. The injectable BX471 solution was prepared by adjusting pH to 4.5 using 1N KOH. The injectable BX471 solution had a concentration of 46 millimolar. The vehicle solution was identical to injectable BX471 solution except for adding BX471.
Ovalbumin-Induced AR Mouse Model and Administration of BX471 Injectable Solution
Our experiment utilized 30 female BALB/c mice, divided equally into three treatment groups: the normal group, the vehicle control group, and the BX471-treated group. The normal group consisted of normal, healthy mice treated with saline alone. In the vehicle control group, an AR mouse model was generated by ovalbumin (OVA)-induction and was treated with the vehicle solution. In the BX471-treated group, OVA-induced mice were treated with injectable BX471 solution. The OVA-induced AR mouse model by sensitization and nasal challenge was performed following the work of Ikeda et al.23 OVA solution for sensitization was prepared by adding 0.5 mg OVA (Sigma, USA) and 20 mg aluminum hydroxide to every milliliter of saline. The vehicle control group and the BX471-treated group of mice were sensitized by intraperitoneally (IP) injecting 200 µL OVA solution on days 0, 7, and 14 (Figure 1). The normal group of mice were IP injected with saline on days 0, 7, and 14 following the same procedures (Figure 1). OVA solution for challenge was prepared at a concentration of 40 mg/mL of saline. The vehicle control group and the BX471-treated group of mice were challenged by nasal dripping 20 µL of concentrated OVA solution once a day from day 21 to 28 (Figure 1). The normal mice were challenged by nasal dripping 20 µL saline once a day from day 21 to 28 following same procedure (Figure 1). The BX471-treated group of mice were injected with injectable BX471 solution at a dose of 1 mL per kilogram of body weight twice a day on day 21 to 28 (Figure 1). An injection was administrated 30 min before the nasal challenge at 12 hours intervals. The vehicle control group and the normal group of mice were injected with vehicle solution or saline solution respectively at same volume using the same injection schedule and the same procedure (Figure 1). Nasal symptoms were assessed by counting the frequency of sneezes and nasal-rubbing behaviors during the 15-min period 10 min after administration of the last nasal challenge on day 28. The mice were humanely sacrificed 24 hours after administration of the last nasal challenge. Their brachial lymph nodes and spleens were then removed and processed for flow cytometry and Western blotting, respectively. Nasal tissues were isolated for immunological and histological studies.
 |
Figure 1 OVA-induced murine model of AR: sensitization, challenge, and administration of injectable BX471 solution or vehicle solution or saline. The vehicle control group and the BX471-treated group of mice were sensitized with OVA solution on days 0, 7, and 14. On days 21–28, these mice were challenged using concentrated OVA solution. Mice designated as the normal group were sensitized and challenged with saline using the same volume as those used for the vehicle control and the BX471-treated groups. BX471 was administrated on days 21–28 twice a day; vehicle control group was administered using the same schedule and volume of vehicle solution as those used for BX471-treated mice. Mice administered as the normal group were only injected with saline using the same injection schedule and volume as that used for BX471-treated and vehicle control mice. |
ELISA Immunoassay
Serum was collected from mice immediately after sacrifice, and levels of OVA-specific IgE were measured using mouse OVA-specific IgE enzyme-linked immunosorbent assay (ELISA) kit (AE98207mu, Sangon Biotech, China). IFN-γ levels were measured using IFN-γ high sensitivity ELISA kit (EK280HS, MultiSciences Biotech, China). TNF-α levels were measured using TNF-α high sensitivity ELISA kit (EK282HS, MultiSciences Biotech, China). All kits were used in accordance with instructions of respective manufacturers. The plates were read using BioTek elx800 microplate reader (BioTek, USA).
Western Blot Analysis
Spleens were collected immediately after sacrificing the mice. Samples were stored at −80°C until analyzed. Samples were processed with 100 mg/mL RIPA lysis buffer (Beyotime Biotech, China) and 1000 mg/mL PMSF (Beyotime Biotech, China). Supernatant was collected after centrifugation at 4°C and was analyzed by Western blot. Protein samples were electrophoresed through 10% of SDS-PAGE gels (Sangon, China) by electrophoresis apparatus (Bio-Rad, USA) under condition 80 volts for 15 min and 140 volts for 50 min. Loading buffer (Beyotime Biotech, China) was purchased and the running buffer (Tris 3.03g, Glycine 18.8g, SDS 1g, and deionized water up to 1 liter) was made. The proteins on the SDS-PAGE were transferred to 0.45µm Immobilon PVDF membrane (Sigma, USA). Membranes were blocked with 5% nonfat dried milk for 1 hour, followed by washing with TBST buffer. Samples were subsequently treated with anti β-actin antibody (BM3873, Boster Biological Technology, China), NF-kB p65 monoclonal antibody (A10609, Abclonal, USA.), TLR4 antibody (BA1717, Boster Biological Technology, China), or TLR2 antibody (BM4001, Boster Biological Technology, China), and were incubated overnight at 4°C. Following incubation, secondary antibodies were added to each membrane. NF-kB p65 membranes were probed with HRP-conjugated goat anti-mouse IgG secondary antibody (BA1050, Boster Biological Technology, China), while β-actin membranes, TLR4 membranes, and TLR2 membranes were probed with HRP-conjugated goat anti-rabbit IgG secondary antibody (BA1054, Boster Biological Technology, China). Finally, blots were visualized with the Azure Biosystem C300 (Azure, USA). The relative expression was calculated by dividing target gene by house-keeping gene (β-actin).
Real-Time PCR
Nasal tissues were removed from the nasal cavities immediately after sacrificing the mice, placed in centrifuge tubes, and stored in liquid nitrogen until analyzed. Total RNA was extracted and purified with TaKaRa MiniBEST Universal RNA Extraction Kit (No. 9767, TaKaRa, Japan). For every sample, 3 µg total RNA was reverse-transcribed using TaKaRa PrimeScript RT reagent Kit Perfect Real Time (RR037B, TaKaRa, Japan) according to the manufacturer’s instructions. Relative expression of MIP-1α, RANTES, thymus and activation regulated chemokine (TARC), CCL22, IL-10, vascular cell adhesion molecule 1 (VCAM-1), Granulocyte-macrophage colony-stimulating factor (GM-CSF), TNF-α, IL-1β, IL-4, IL-5, IL-13, and β-actin were assessed using primers listed in Table 1. Primer sequences of MIP-1α, RANTES, TARC, GM-CSF, TNF-α, IL-1β, IL-4, IL-5, IL-13, and β-actin were obtained from previous articles.24–30 Primer sequences of CCL22, IL-10, and VCAM-1 were designed by using NCBI primer blast. Real-time polymerase chain reaction (RT-qPCR) was performed using TB Green Fast qPCR Mix (RR430B, TaKaRa, Japan) in a Real-time PCR System (Tianlong, China). Conditions for primer pairs with Tm between 56°C and 60°C were as follows: pre-denaturation at 95°C for 30 s; three-step amplification via 95°C for 5 s, 57°C for 30 s, and 72°C for 10 s, for 40 cycles; followed by melt–curve analysis. Same conditions were used for primer pairs with Tm below 56°C except for the second step of amplification, which was conducted at 55°C for 30 s. For primer pairs with Tm above 60°C, two-step amplification was used instead as follows: 95°C for 5 s, and 62°C for 10 s. The amplification curve was used to evaluate specificity. Relative expression of target genes was normalized to that of housekeeping gene β-actin. Relative expression was calculated via comparative cycle threshold (2−ΔΔCt) method.
 |
Table 1 Sequence of Primer Pairs Used in RT-qPCR |
Histological Analysis
After the sacrifice, the heads of the mice were removed and fixed in 4% paraformaldehyde for 48 hours. Specimens were decalcified with paraformaldehyde-EDTA decalcifying solution (G2520, Solarbio, China) for 5 days. Following fixation, heads were dehydrated, embedded in paraffin, and sectioned at 4 µm. Nasal-cavity sections were stained with hematoxylin and eosin (HE) (G1120, Solarbio, China) to obtain an eosinophil count in the lamina propria along the nasal septum under respiratory epithelium and measurement of epithelium thickness at same place of epithelium affiliated to nasal septum. Additionally, this section of epithelium in the lamina propria was measured for thickness in all treatment groups. All the stained tissues were scanned using 3DHistech P250 High Capacity Slide Scanner (3DHISTECH Ltd., Hungary). CaseViewer V2.3 (3DHISTECH Ltd., Hungary) was utilized to view the scanned slides. Frame selection function in CaseViewer V2.3 was used to randomly select 1 square millimeter rectangle area of lamina propria along the nasal septum under respiratory epithelium for each sample. Two researchers blinded of the group and sample information counted the number of eosinophils in areas described above in each sample. To measure the nasal thickness, we used distance measurement function in CaseViewer V2.3 to measure the distance between the epithelium outer surface and epithelium inner surface above nasal septum in scanned slide of each sample. Nasal cavity sections also were stained for glycogen using periodic acid-Schiff (PAS) stain (G1218, Solarbio, China) to identify goblet cells.
Flow Cytometry
Brachial lymph nodes were removed from mice immediately after sacrifice and were placed into a tissue culture dish containing 2 mL PBS. A plunger of a 5-mL syringe was then used to grind the tissues into a homogeneous suspension at 4°C; the suspension was filtered through a 70-µm cell filter (Solarbio, China) and centrifuged at 400 g for 7 min at 4°C. The supernatant was discarded, and 1 × 107 cells were placed into a flow cytometry tube and stained using mouse regulatory T cell staining kit (KTR201, MultiSciences Biotech, China) according to the manufacturer’s instructions to identify CD4+CD25+Foxp3+ T regulatory cells (Treg). The stained cell suspension was evaluated using BD Accuri C6 Plus flow cytometer (BD Bioscience, USA), and data were analyzed using FlowJo V10 software.
Statistical Analysis
All data were analyzed using Microsoft Excel and GraphPad Prism V8.2.1. One-way ANOVA and Tukey’s Test post hoc analysis was performed on GraphPad Prism V8.2.1 (GraphPad Software, La Jolla California, USA). The statistical comparisons were the normal group versus the vehicle control group and the BX471-treated group versus the vehicle control group. If statistical significance exists between the normal group and the vehicle control group, a symbol indicating statistical significance was added on the top of the normal group. If statistical significance exists between the BX471-treated group and the vehicle control group, a symbol indicating statistical significance was added on the top of the BX471-treated group. p<0.05 was considered statistically significant. All data are presented as means ± SEM.
Results
Treatment with BX471 Attenuated Nasal Symptoms
The number of sneezes and the number of nasal-rubbing behaviors were different significantly between the normal group, the vehicle control group, and the BX471-treated group. As shown in Figure 2A, the number of sneezes significantly increased in the vehicle control group (20.9±1.5) compared with that of the normal group (1.1±0.7) during the 15 min period after the challenge (p<0.001). As shown in Figure 2B, the number of nasal-rubbing behaviors also significantly increased in the vehicle control group (149.3±9.5) compared with that of the normal group (73.6±9.9) during the 15 min period after the challenge (p<0.001). The number of sneezes in the BX471-treated group during the 15 min period after the challenge was 9.5±1.8, which was significantly attenuated compared with that of the vehicle control group (p<0.001) (Figure 2A); the number of nasal-rubbing behaviors of the BX471-treated group was 107.6±14.0, which was significantly lower than that of the vehicle control group (p<0.05) (Figure 2B). The symptom scores indicate that BX471 treatment alleviated the symptoms of AR in mice.
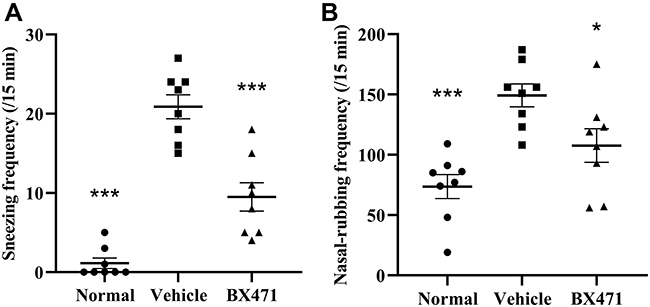 |
Figure 2 Effect of BX471 administration on symptoms of AR. (A) Number of sneezes during a 15-min interval was counted starting at 10 min after administration of the last nasal challenge on day 28 (n=8 per group). (B) the number of nasal-rubbing behaviors was counted during the 15-min interval, starting at 10 min after administration of the last nasal challenge on day 28 (n=8 per group). All values are represented as mean ± SEM. * p<0.05 and *** p<0.001 versus the vehicle control group. |
BX471 Suppressed TNF-α Levels
The BX471-treated group showed significantly lower levels of TNF-α in serum than the vehicle control group. No statistical significance of OVA-specific IgE levels and IFN-γ levels was found between BX471-treated group and the vehicle control group. As shown in Figure 3A, the vehicle control group showed significantly higher levels of OVA-specific IgE than those of the normal group (p<0.001). No statistically significant difference of IgE levels was detected between the vehicle control group and the BX471-treated group (p>0.05) (Figure 3A). Serum TNF-α levels were significantly (p<0.01) reduced in BX471 group compared with the vehicle control group (Figure 3B). The serum concentration of TNF-α in the vehicle control group was 27.54±4.39 pg/mL, whereas in the BX471-treated group that was 12.08±1.11 pg/mL (Figure 3B). No statistically significant difference in serum IFN-γ levels was observed among the normal group, the vehicle control group, and the BX471-treated group (Figure 3C). The ELISA results indicated that the BX471-treated group had lower serum TNF-α levels than that of vehicle control group, but BX471 treatment did not influence serum levels of OVA-specific IgE and IFN-γ.
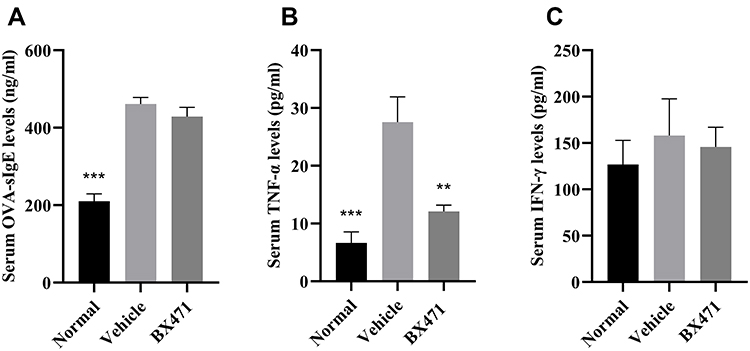 |
Figure 3 Effect of BX471 on peripheral OVA-specific IgE and Th1 cytokine levels. Mice were sacrificed 24 hours after administration of the last nasal challenge, and sera were collected. (A) Serum levels of OVA-specific IgE were measured using ELISA for each treatment group (n=6 per group). (B) Serum levels of TNF-α were measured using ELISA for each treatment group (n=6 per group). (C) Serum levels of IFN-γ were measured using ELISA for each treatment group (n=6 per group). Values are represented as mean ± SEM. ** p<0.01 and *** p<0.001 versus the vehicle control group. |
BX471 Suppresses the Relative Expression of NF-kB P65
Protein levels of Nuclear Factor kappa-light-chain-enhancer of activated B cells p65 (NF-kB p65) were significantly different between the BX471-treated group and the vehicle control group, but TLR4 and TLR2 levels did not differ significantly between the BX471-treated group and the vehicle control group (Figure 4A–D). Relative expression level of NF-kB p65 is 0.31±0.02 in the normal group, 0.44±0.06 in the vehicle control group, and 0.27±0.02 in the BX471-treated group (Figure 4B). The relative expression level is statistically different (p < 0.05) between the vehicle control and the BX471-treated group (Figure 4B). Relative expression levels of TLR4 and TLR2 show no statistically significant difference between the vehicle control group and the BX471-treated group (Figure 4C and D). These results indicate BX471 treatment reduces relative expression level of NF-kB p65 but not that of TLR4 and TLR2.
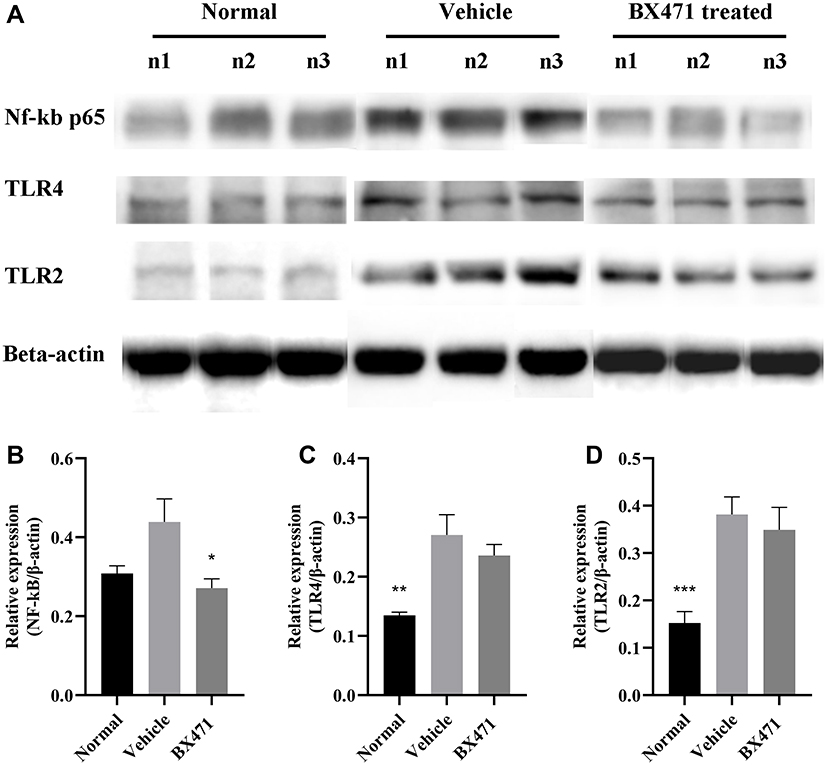 |
Figure 4 Relative quantification of NF-kB p65, TLR4, and TLR2 protein expression levels. (A) Western blot analysis for NF-kB p65, TLR4, and TLR2 relative expression from groups indicated. (B) summary graph for relative expression of NF-kB (n=8 per group). (C) summary graph for relative expression of TLR4 (n=8 per group). (D) summary graph for relative expression of TLR2 (n=8 per group). All values are represented as mean ± SEM. * p<0.05; ** p<0.01; and *** p<0.001 versus the vehicle control group. |
Effect of BX471 on Levels of Chemokines and Cytokines in the Nasal Tissues
Generally, cytokine profiles indicated significant differences between the BX471-treated group and the vehicle control group. TNF-α levels were lower in the BX471-treated group, 0.5±0.1 fold, than in the vehicle control group, 8.8±2.4 fold (p<0.01) (Figure 5). Additionally, nasal expression levels of IL-4, IL-5, IL-13 were also significantly lower in BX471-treated group compared with the vehicle control group (Figure 5). Fold expression of IL-4 in the vehicle control group was 452.9±85.3 fold, whereas in the BX471-treated group it was 38.5±15.7 fold (p<0.001). Fold expression of IL-5 in the vehicle control group was 10.3±1.6 fold, whereas in the BX471-treated group it was 1.1±0.2 fold (p<0.001) (Figure 5). Fold expression of IL-13 was 116.3±38.5 fold in the vehicle control group and 5.5±1.1 fold in the BX471-treated group (p<0.05) (Figure 5).
 |
Figure 5 Relative expression of proinflammatory chemokines and cytokines in the nasal cavity. Total RNA, obtained from the different groups of mice, was used to measure the expression levels of TNF-α, IL-4, IL-5, IL-13, MIP-1α, RANTES, VCAM-1, GM-CSF, IL-1β, IL-10, TARC, CCL22 via RT-qPCR (n=5 per group). All values are represented as mean ± SEM. * p<0.05; ** p<0.01; and *** p<0.001 versus the vehicle control group. |
MIP-1α and RANTES, which play important roles in regulation of Th2 and other inflammatory cytokines, are primary targets of BX471. As shown in Figure 5, mRNA expression of MIP-1α in the BX471-treated group was 1.2±0.3 fold, significantly lower than that of the vehicle control group which was 61.6±20.2 fold (p<0.01). The mRNA expression of RANTES in the BX471-treated group was downregulated by 0.9±0.03 fold compared with that of the vehicle control group, 6.6±1.8 fold (p<0.01) (Figure 5).
Other proinflammatory factors involved in the inflammatory pathway of AR were also assessed. Results indicated significantly lower expression levels of VCAM-1 (p<0.01), GM-CSF (p<0.05), IL-1β (p<0.01), IL-10 (P<0.01), TARC (P<0.05), and CCL22 (p<0.05) in the BX471-treated group compared with the vehicle group (Figure 5). The RT-PCR results confirm the ameliorated AR in BX471 treated group of mice compared with vehicle control group.
Histological Changes in the Nasal Cavity Caused by Administration of BX471
Numbers of eosinophils per square millimeter in the lamina propria were counted (Figure 6A, B, C, and G). The number of eosinophils per every square millimeter of nasal tissue was 2.5±1.0 for the normal group, 54.0±6.5 for the vehicle control group, and 26.8±8.7 for the BX471-treated group. Statistically significant differences were found between the normal group and the vehicle control group (p<0.01) and between the vehicle control group and the BX471-treated group (p<0.05) (Figure 6G). Goblet cell density was significantly higher in the vehicle control group, 46.8±19.7, than the normal group, 1.0±0.7 (p<0.05) (Figure 6D, E, F, and H). No statistical significance of the number of goblet cells in respiratory epithelium was found between vehicle control group and BX471-treated group (Figure 6D, E, F, and H). Epithelial thickness was significantly higher in the vehicle control group, 67.9±3.9 µm, than in the normal group, 50.5±2.9 µm (p<0.05) (Figure 6I). No statistically significant difference in the thickness of nasal mucosa was observed between the vehicle control group and BX471-treated group (Figure 6I). Results indicated that BX471 treatment prevent the eosinophil recruitment in AR but has no influence on the epithelium thickness and the goblet cell metaplasia.
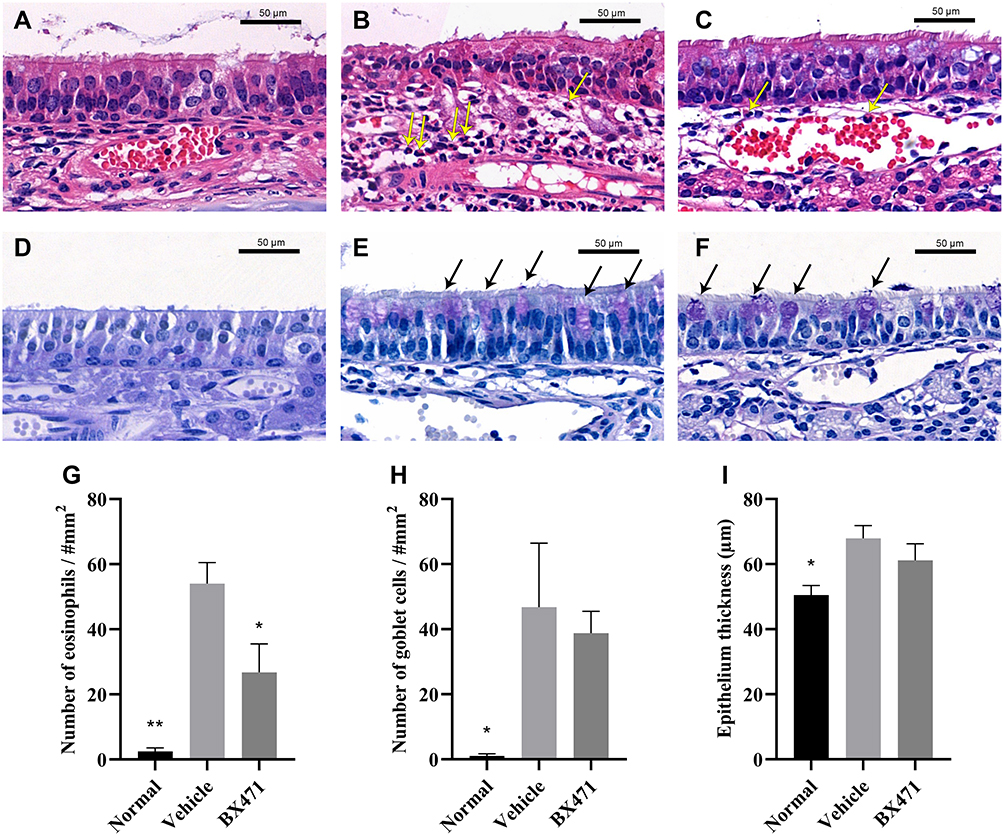 |
Figure 6 Histological examination of nasal mucosa. Hematoxylin and eosin stains (magnification, ×650) of nasal mucosa from the normal group (A), vehicle control group (B), and BX471-treated group (C). The yellow arrows indicate the identified eosinophils in lamina propria. PAS stains (magnification, ×650) of nasal mucosa from the normal group (D), vehicle control group (E), and BX471-treated group (F). We counted the number of eosinophils in the lamina propria (G) and that of goblet cells in the respiratory epithelium (H), (n=4 per group); we also measured the thickness of respiratory epithelium (I), (n=4 per group). All values are represented as mean ± SEM. * p<0.05 and ** p<0.01 versus the vehicle control group. Bar indicates 50µm. |
BX471 Upregulates the Population of Treg Cells
Treg cells were analyzed using flow cytometry for each of the treatment groups (Figure 7A, B, C, and E), and unstained control was shown as well (Figure 7D). The percentage of CD25+ FOXP3+ cells in CD4+ population was 11.00±0.69% in the normal group, 4.83±1.06% in the vehicle control group, and 10.57±0.97% in the BX471-treated group (Figure 7E). The vehicle control group showed a significantly lower percentage of Treg cells than did the normal group (p<0.01) (Figure 7E). The percentage of Treg cells in the BX471 group was significantly higher than that of the vehicle control group (p<0.01) (Figure 7E). These results clearly indicate that BX471 treatment increases the population of CD25+ FOXP3+ cells in CD4+ cells population.
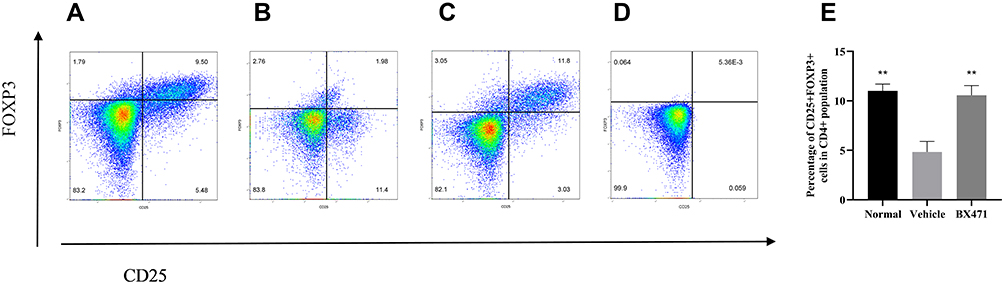 |
Figure 7 Flow cytometry analysis of Treg cells. Lymphocytes were gated first for CD4+ T cells and then for CD25+ FOXP3+ cells in (A) the normal group, (B) the vehicle-control group, and (C) the BX471-treated group. (D) Unstained control. (E) The percentage of CD25+ FOXP3+ cells in CD4+ cell population was assessed in all treatment groups (n=5 per group). All values are represented as mean ± SEM. ** p<0.01 versus the vehicle control group. |
Discussion
Previous studies on therapeutic effects of BX471 in allergic asthma showed the potential of targeting CCR1 in allergic-airway disease.22 In this study, by using OVA-induced murine model (Figure 1), the effect of BX471 on AR symptoms was examined and underlying mechanisms were explored. Administration of BX471 demonstrated reduced inflammatory chemokine and cytokine expression, suppressed eosinophil recruitment, relieved nasal symptoms, and an increased Treg cell population. These results suggest that BX471 reduced NF-kB level by regulating CCR1 mediated TNF-α secretion. The suppressed NF-kB levels may contribute to downregulation of Th2 cytokines, VCAM-1, GM-CSF, IL-1β, RANTES, and MIP-1α. Furthermore, the downregulation of Th2 cytokines and RANTES levels may explain reduced eosinophil infiltration into the nasal mucosa.
Counting the number of sneezes and nasal-rubbing behaviors during a time interval is an effective way to evaluate AR symptoms, and is used in various AR studies.3,31–33 In the current study, the vehicle control group showed increased sneezing and nasal-rubbing behaviors in the time interval following OVA sensitization and challenge (Figure 2A and B). Treatment with BX471 lowered the symptom score for sneezing and nasal-rubbing behaviors, indicating that symptoms of AR were attenuated (Figure 2A and B).
Many studies have demonstrated that the NF-kB pathway plays key roles in regulating expression of proinflammatory factors and allergic airway inflammation.34–37 Present study investigated the protein levels of NF-kB, TLR2, and TLR4 by Western blot to better understand effect of BX471 treatment on the NF-kB activation pathway (Figure 4A–D). Nigo et al38 and Carta et al39 suggested that TLR2 and TLR4 activated NF-kB to produce cytokines in airway inflammation. Interestingly, NF-kB p65, which is a major heterodimer of NF-kB p50, was significantly downregulated by BX471 (Figure 4B), while TLR2 and TLR4 protein expression levels were not affected (Figure 4C and D). Decreased levels of NF-kB p65 but not TLR2 and TLR4 protein in BX471-treated group indicate that the BX471 may not be acting on the toll-like receptor activated NF-kB pathway. This intriguing result may be explained by the decreased levels of TNF-α after BX471 treatment (Figure 3B), which fails to activate NF-kB pathway. Previous studies suggest that CCR1 mediates production of TNF-α both in vitro via mast cells and in vivo, and increased levels of TNF-α contribute to airway inflammation and Th2 cytokines production.40–42 In the present study, the knowledge of relationship of CCR1 and the TNF-α/NF-kB pathway has been expanded in vivo in a mouse model of AR. The findings of this study indicate that BX471 treatment reduced TNF-α protein level and subsequently reduced NF-kB production. These findings suggest that BX471 treatment induces an anti-inflammatory effect through the TNF-α activated NF-kB pathway. While previous ideas of targeting AR have focused on downstream factors such as IgE and histamine, the present study provides new insights of attenuating AR via upstream pathway: the TNF-α activated NF-kB pathway.
Additionally, RT-qPCR results showed decreased expression levels of IL-4, IL-5, IL-13 m-RNA in the nasal cavity in the BX471-treated group compared to the vehicle control group (Figure 5), which indicates reduced Th2-mediated inflammation. A previous investigation demonstrated that mice with a NF-kB p50 knockout gene failed to develop allergic airway inflammation by inhibiting IL-4, IL-5, IL-13, MIP-1α, and RANTES expression.36,37 Since NF-kB serves as an upstream factor for Th2 cytokines, it is possible that the lower levels of Th2 cytokines expression in the BX471-treated group could be attributed to the inactivation of NF-kB. These findings further suggest that TNF-α could play a role in regulating NF-kB activation in Th2 cytokines production in AR. Furthermore, decreased levels of Th2 cytokines in the BX471-treated group suggest that BX471 inhibits AR via the TNF-α/NF-kB pathway. In accordance with previous studies, the present study demonstrates that CCR1-/- mice with chronic allergic airway disease exhibited significantly lower levels of IL-4 and IL-13 compared with those of wild-type controls.19 It has been reported that BX471 may serves as a competitive inhibitor, and previous studies report that BX471 treatment could modulate RANTES and MIP-1α at the site of inflammation.20,22 The current results show downregulation of RANTES and MIP-1α expression in the nasal cavity (Figure 5). Lower levels of RANTES and MIP-1α expression are consistent with a previous finding that NF-kB regulates RANTES and MIP-1α expression. As NF-kB fails to activate upon BX471 treatment, it could result in reduced RANTES and MIP-1α expression levels, which could be a reason why BX471 treated mice produce less RANTES and MIP-1α. In AR, Th2 cytokines, RANTES, and MIP-1α have been reported to contribute to eosinophil recruitment. This study shows that BX471 treatment inhibited eosinophil recruitment in the BX471-treated group (Figure 6G), which supports the proposed mechanism of action through the TNF-α activated NF-kB pathway. Reduced eosinophil recruitment correlates with results shown by previous studies on BX471 administration in asthma.22
In AR, IgE production is typically triggered by IL-4- and IL-13-mediated isotype switching.34,35 IFN-γ can prevent this isotype switch, thereby preventing antigen-specific IgE production, while IL-10 inhibits the activity of IFN-γ and restores the isotype-switching process.36 In present study, the group of mice treated with BX471 showed reduced levels of IL-4, IL-13, and IL-10 (Figure 5); however, we did not detect any changes in IgE levels in the BX471-treated group (Figure 3A), and no changes in IFN-γ protein levels were observed (Figure 3C). A previous study showed that IgE production is not dependent on CCR1.19 The reason for this remains unclear, and why CCR1 affects the levels of Th2 cytokines, but not those of IgE, should be investigated in future studies.
VCAM‐1 and GM-CSF serve as important players in eosinophilic recruitment in allergic inflammation.8,43 It was indicated previously that the TNF-α activated NF-kB protein pathway triggers expression of VCAM‐1 in inflammatory response, and NF-kB regulates expression of GM-CSF and IL-1β.44,45 The findings of present study corroborates with results from a previous study showing a downregulation of VCAM‐1, GM-CSF, and IL-1β expression in BX471-treated group (Figure 5). These results indicate that BX471 suppressed VCAM‐1, GM-CSF, and IL-1β via TNF-α/NF-kB pathway. CCL22 and TARC, which are the two principle ligands of CCR4, contribute to infiltration of Th2 cells.46,47 The reduced level of CCL22 and TARC may be due to reduced eosinophil infiltration, which could potentially decrease production of CCL22 and TARC.48 Administration of BX471 in allergic asthma results in reduced levels of CCR4 in murine asthma.22 Although no direct relationship between CCR1 and ligands of CCR4 has been found, the decreased levels of these two factors shown by the BX471-treated group in our study, may be attributed to the reduced Th2 inflammation and eosinophil recruitment. The relationship between CCR1 and ligands of CCR4 should be further investigated in future studies.
Recent evidence shows that Treg cells in allergic-airway disease modulate the immune response, and that FOXP+ expressing cells show antigen tolerance.49–52 In present study, the vehicle control group showed a decreased percentage of CD25+ FOXP3+ expression cells in the CD4+ cell population by compared to the normal group upon sensitization and challenge (Figure 7E); the BX471-treated group showed an increased percentage of CD25+ FOXP3+ expression cells in the CD4+ cell population (Figure 7E), indicating that BX471 may inhibit AR by increasing the numbers of Treg cells. Our results suggest that CCR1 blockade may boost the population of Treg cells. This indicates that controlling the levels of Treg cells may be a promising therapeutic strategy for treating AR.
Conclusion
BX471 exerts anti-inflammatory effects in a mouse model of AR. This study expands the knowledge of therapeutic effect of CCR1 antagonist and possible underlying mechanisms. The possible mechanism of action of CCR1 antagonist BX471 attenuating OVA-induced AR is that BX471 inhibits expression of proinflammatory mediators and eosinophil recruitment by inhibiting the TNF-α activated NF-kB pathway. In addition, BX471 also functions to attenuate AR by increasing the natural-occurring T cell population. Overall, present study demonstrates that BX471 represents a promising therapeutic strategy against AR.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article.
Acknowledgments
We would like to thank Dr. Andrew Ketchum and Dr. Mingchun Gao for proofreading the manuscript and providing valuable advice. Additionally, we appreciate Dr. Ming Liu from the Morgan State University for providing supports.
Author Contributions
All authors made substantial contributions to conception and design, execution, acquisition of data, analysis and interpretation of data, or more than one of these areas; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Disclosure
All authors have no competing interests to declare.
References
1. Seidman MD, Gurgel RK, Lin SY, et al. Clinical practice guideline: allergic rhinitis. Otolaryngol Head Neck Surg. 2015;152(1 Suppl):
2. Wheatley LM, Togias A. Clinical practice. Allergic rhinitis. N Engl J Med. 2015;372(5):456–463. doi:10.1056/NEJMcp1412282
3. Hiromura Y, Kishida T, Nakano H, et al. IL-21 administration into the nostril alleviates murine allergic rhinitis. J Immunol. 2007;179(10):
4. Pawankar R, Canonica GW, Holgate ST, Lockey RF, Blaiss M. The WAO White Book on Allergy; 2013. Available from: https://www.worldallergy.org/UserFiles/file/WhiteBook2-2013-v8.pdf.
5. Loekmanwidjaja J, Carneiro ACF, Nishinaka MLT, et al. Sleep disorders in children with moderate to severe persistent allergic rhinitis. Braz J Otorhinolaryngol. 2018;84(2):
6. Meltzer EO. Quality of life in adults and children with allergic rhinitis. J Allergy Clin Immunol. 2001;108(1 Suppl):
7. Lebman DA, Coffman RL. Interleukin 4 causes isotype switching to IgE in T cell-stimulated clonal B cell cultures. J Exp Med. 1988;168(3):
8. Pawankar R, Mori S, Ozu C, Kimura S. Overview on the pathomechanisms of allergic rhinitis. Asia Pac Allergy. 2011;1(3):
9. Pease JE, Williams TJ. Chemokines and their receptors in allergic disease. J Allergy Clin Immunol. 2006;118(2):
10. Zhang RX, Yu SQ, Jiang JZ, Liu GJ. Complementary DNA microarray analysis of chemokines and their receptors in allergic rhinitis. J Investig Allergol Clin Immunol. 2007;17(5):
11. Bonecchi R, Galliera E, Borroni EM, Corsi MM, Locati M, Mantovani A. Chemokines and chemokine receptors: an overview. Front Biosci. 2009;14:
12. Lukacs NW, Standiford TJ, Chensue SW, Kunkel RG, Strieter RM, Kunkel SL. C-C chemokine-induced eosinophil chemotaxis during allergic airway inflammation. J Leukoc Biol. 1996;60(5):
13. Alam R, Stafford S, Forsythe P, et al. RANTES is a chemotactic and activating factor for human eosinophils. J Immunol. 1993;150(8Pt 1):
14. Kimata H, Yoshida A, Ishioka C, Fujimoto M, Lindley I, Furusho K. RANTES and macrophage inflammatory protein 1 alpha selectively enhance immunoglobulin (IgE) and IgG4 production by human B cells. J Exp Med. 1996;183(5):
15. Ozu C, Pawankar R, Takizawa R, Yamagishi S, Yagi T. Regulation of mast cell migration into the allergic nasal epithelium by RANTES and not SCF [abstract]. J Allergy Clin Immunol. 2004;113(2):S28. doi:10.1016/j.jaci.2003.12.057
16. Kuna P, Reddigari SR, Schall TJ, Rucinski D, Viksman MY, Kaplan AP. RANTES, a monocyte and T lymphocyte chemotactic cytokine releases histamine from human basophils. J Immunol. 1992;149(2):
17. Kuna P, Alam R, Ruta U, Gorski P. RANTES induces nasal mucosal inflammation rich in eosinophils, basophils, and lymphocytes in vivo. Am J Respir Crit Care Med. 1998;157(3 Pt 1):
18. Christodoulopoulos P, Wright E, Frenkiel S, Luster A, Hamid Q. Monocyte chemotactic proteins in allergen-induced inflammation in the nasal mucosa: effect of topical corticosteroids. J Allergy Clin Immunol. 1999;103(6):
19. Blease K, Mehrad B, Standiford TJ, et al. Airway remodeling is absent in CCR1-/- mice during chronic fungal allergic airway disease. J Immunol. 2000;165(3):
20. Liang M, Mallari C, Rosser M, et al. Identification and characterization of a potent, selective, and orally active antagonist of the CC chemokine receptor-1. J Biol Chem. 2000;275(25):
21. Anders HJ, Vielhauer V, Frink M, et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J Clin Invest. 2002;109(2):
22. Carpenter KJ, Ewing JL, Schuh JM, et al. Therapeutic targeting of CCR1 attenuates established chronic fungal asthma in mice. Br J Pharmacol. 2005;145(8):
23. Ikeda Y, Kaneko A, Yamamoto M, Ishige A, Sasaki H. Possible involvement of suppression of Th2 differentiation in the anti-allergic effect of Sho-seiryu-to in mice. Jpn J Pharmacol. 2002;90(4):
24. Rojas A, Gueorguieva P, Lelutiu N, Quan Y, Shaw R, Dingledine R. The prostaglandin EP1 receptor potentiates kainate receptor activation via a protein kinase C pathway and exacerbates status epilepticus. Neurobiol Dis. 2014;70:
25. Mgrditchian T, Arakelian T, Paggetti J, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A. 2017;114(44):
26. Inoue J, Aramaki Y. Suppression of skin lesions by transdermal application of CpG-oligodeoxynucleotides in NC/Nga mice, a model of human atopic dermatitis. J Immunol. 2007;178(1):
27. Ishida H, Imai T, Suzue K, et al. IL-23 protection against Plasmodium berghei infection in mice is partially dependent on IL-17 from macrophages. Eur J Immunol. 2013;43(10):
28. Park JG, Kim SC, Kim YH, et al. Anti-Inflammatory and Antinociceptive Activities of Anthraquinone-2-Carboxylic Acid. Mediators Inflamm. 2016;2016:1903849. doi:10.1155/2016/1903849
29. Liu Y, Zhang Y, Zheng X, et al. Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J Neuroinflammation. 2018;15(1):112. doi:10.1186/s12974-018-1141-5
30. Xu J, Han R, Kim DW, et al. Role of Interleukin-10 on nasal polypogenesis in patients with chronic rhinosinusitis with nasal polyps. PLoS One. 2016;11(9):e0161013. doi:10.1371/journal.pone.0161013
31. Shin SH, Kim YH, Kim JK, Park KK. Anti-allergic effect of bee venom in an allergic rhinitis mouse model. Biol Pharm Bull. 2014;37(8):
32. Takamura K, Fukuyama S, Nagatake T, et al. Regulatory role of lymphoid chemokine CCL19 and CCL21 in the control of allergic rhinitis. J Immunol. 2007;179(9):
33. Li J, Wang B, Luo Y, Bian Y, Wang R. Effect of artemisinin and neurectomy of pterygoid canal in ovalbumin-induced allergic rhinitis mouse model. Allergy Asthma Clin Immunol. 2018;14(1):22. doi:10.1186/s13223-018-0249-6
34. Poynter ME, Irvin CG, Janssen-Heininger YM. Rapid activation of nuclear factor-kappaB in airway epithelium in a murine model of allergic airway inflammation. Am J Pathol. 2002;160(4):
35. Lilly CM, Tateno H, Oguma T, Israel E, Sonna LA. Effects of allergen challenge on airway epithelial cell gene expression. Am J Respir Crit Care Med. 2005;171(6):
36. Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A critical role for NF-kappa B in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol. 2001;2(1):
37. Yang L, Cohn L, Zhang DH, Homer R, Ray A, Ray P. Essential role of nuclear factor kappaB in the induction of eosinophilia in allergic airway inflammation. J Exp Med. 1998;188(9):
38. Nigo YI, Yamashita M, Hirahara K, et al. Regulation of allergic airway inflammation through Toll-like receptor 4-mediated modification of mast cell function. Proc Natl Acad Sci U S A. 2006;103(7):
39. Carta S, Silvestri M, Rossi GA. Modulation of airway epithelial cell functions by Pidotimod: NF-kB cytoplasmatic expression and its nuclear translocation are associated with an increased TLR-2 expression. Ital J Pediatr. 2013;39:29. doi:10.1186/1824-7288-39-29
40. Fifadara NH, Aye CC, Raghuwanshi SK, Richardson RM, Ono SJ. CCR1 expression and signal transduction by murine BMMC results in secretion of TNF-alpha, TGFbeta-1 and IL-6. Int Immunol. 2009;21(8):
41. Amat M, Benjamim CF, Williams LM, et al. Pharmacological blockade of CCR1 ameliorates murine arthritis and alters cytokine networks in vivo. Br J Pharmacol. 2006;149(6):
42. Nakae S, Ho LH, Yu M, et al. Mast cell-derived TNF contributes to airway hyperreactivity, inflammation, and TH2 cytokine production in an asthma model in mice. J Allergy Clin Immunol. 2007;120(1):
43. Peric A, Spadijer-Mirkovic C, Matkovic-Jozin S, Jovancevic L, Vojvodic D. Granulocyte-Macrophage Colony-Stimulating Factor Production and Tissue Eosinophilia in Chronic Rhinitis. Int Arch Otorhinolaryngol. 2016;20(4):
44. Lee CW, Lin WN, Lin CC, et al. Transcriptional regulation of VCAM-1 expression by tumor necrosis factor-alpha in human tracheal smooth muscle cells: involvement of MAPKs, NF-kappaB, p300, and histone acetylation. J Cell Physiol. 2006;207(1):
45. Schuliga M. NF-kappaB Signaling in Chronic Inflammatory Airway Disease. Biomolecules. 2015;5(3):
46. Sekiya T, Miyamasu M, Imanishi M, et al. Inducible expression of a Th2-type CC chemokine thymus- and activation-regulated chemokine by human bronchial epithelial cells. J Immunol. 2000;165(4):
47. Yanai M, Sato K, Aoki N, et al. The role of CCL22/macrophage-derived chemokine in allergic rhinitis. Clin Immunol. 2007;125(3):
48. Jacobsen EA, Ochkur SI, Pero RS, et al. Allergic pulmonary inflammation in mice is dependent on eosinophil-induced recruitment of effector T cells. J Exp Med. 2008;205(3):
49. Ling EM, Smith T, Nguyen XD, et al. Relation of CD4+CD25+ regulatory T-cell suppression of allergen-driven T-cell activation to atopic status and expression of allergic disease. Lancet. 2004;363(9409):
50. Bellinghausen I, Klostermann B, Knop J, Saloga J. Human CD4+CD25+ T cells derived from the majority of atopic donors are able to suppress TH1 and TH2 cytokine production. J Allergy Clin Immunol. 2003;111(4):
51. Suto A, Nakajima H, Kagami SI, Suzuki K, Saito Y, Iwamoto I. Role of CD4(+) CD25(+) regulatory T cells in T helper 2 cell-mediated allergic inflammation in the airways. Am J Respir Crit Care Med. 2001;164(4):
52. Ostroukhova M, Seguin-Devaux C, Oriss TB, et al. Tolerance induced by inhaled antigen involves CD4(+) T cells expressing membrane-bound TGF-beta and FOXP3. J Clin Invest. 2004;114(1):
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.