Back to Journals » International Journal of Nanomedicine » Volume 11
Targeted modulation of cell differentiation in distinct regions of the gastrointestinal tract via oral administration of differently PEG-PEI functionalized mesoporous silica nanoparticles
Authors Desai D, Prabhakar N, Mamaeva V, Şen Karaman D ![]() , Lähdeniemi I, Sahlgren C, Rosenholm J
, Lähdeniemi I, Sahlgren C, Rosenholm J ![]() , Toivola D
, Toivola D
Received 11 August 2015
Accepted for publication 2 November 2015
Published 18 January 2016 Volume 2016:11 Pages 299—313
DOI https://doi.org/10.2147/IJN.S94013
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Diti Desai,1–4 Neeraj Prabhakar,2 Veronika Mamaeva,3,5 Didem Şen Karaman,2,4 Iris AK Lähdeniemi,1,6 Cecilia Sahlgren,3,7,* Jessica M Rosenholm,2,4,* Diana M Toivola1,6,*
1Cell Biology, Faculty of Science and Engineering, Åbo Akademi University, Turku, Finland; 2Pharmaceutical Sciences Laboratory, Faculty of Science and Engineering, Åbo Akademi University, Turku, Finland; 3Turku Centre for Biotechnology, University of Turku and Åbo Akademi University, Turku, Finland; 4Laboratory of Physical Chemistry, Faculty of Science and Engineering, Åbo Akademi University, Turku, Finland; 5Department of Clinical Science, University of Bergen, Bergen, Norway; 6Turku Center for Disease Modeling, Institute of Biomedicine, University of Turku, Turku, Finland; 7Department of Biomedical Engineering, Technical University of Eindhoven, Eindhoven, the Netherlands
*These authors contributed equally to this work
Abstract: Targeted delivery of drugs is required to efficiently treat intestinal diseases such as colon cancer and inflammation. Nanoparticles could overcome challenges in oral administration caused by drug degradation at low pH and poor permeability through mucus layers, and offer targeted delivery to diseased cells in order to avoid adverse effects. Here, we demonstrate that functionalization of mesoporous silica nanoparticles (MSNs) by polymeric surface grafts facilitates transport through the mucosal barrier and enhances cellular internalization. MSNs functionalized with poly(ethylene glycol) (PEG), poly(ethylene imine) (PEI), and the targeting ligand folic acid in different combinations are internalized by epithelial cells in vitro and in vivo after oral gavage. Functionalized MSNs loaded with γ-secretase inhibitors of the Notch pathway, a key regulator of intestinal progenitor cells, colon cancer, and inflammation, demonstrated enhanced intestinal goblet cell differentiation as compared to free drug. Drug-loaded MSNs thus remained intact in vivo, further confirmed by exposure to simulated gastric and intestinal fluids in vitro. Drug targeting and efficacy in different parts of the intestine could be tuned by MSN surface modifications, with PEI coating exhibiting higher affinity for the small intestine and PEI–PEG coating for the colon. The data highlight the potential of nanomedicines for targeted delivery to distinct regions of the tissue for strict therapeutic control.
Keywords: intestinal targeting, PEG-PEI copolymer, Notch inhibition
Introduction
Targeting specific biological pathways provides an opportunity to devise more specific and more effective approaches in cancer and stem cell therapy. In conjunction with improved molecular diagnostics, this is the foundation of personalized medicine.1,2 However, given the widespread and complex functions of biological pathways, increased specificity and tissue- and cell-targeted delivery of modulators are necessary. Nanotechnology-based drug delivery systems provide a solution to improve therapeutic efficacy with reduced side effects. Chemical design of nanoparticles offers control over bioavailability and biodistribution, but we still need to address the form and function relationships for any given administration.3,4
The Notch signaling pathway is the key regulator of stem cells in development and tissue homeostasis, and is deregulated in inflammatory intestinal disease and colon cancer.5,6 Clinical studies inhibiting Notch are focused on several types of cancers by mainly two approaches: use of antibodies against receptors and ligands, and γ-secretase inhibitors, which inhibit proteolytic processing and activation of the Notch receptor. However, Notch is essential for tissue homeostasis including that of the intestine and immune system. Further, while Notch is an oncogene in most biological systems, in the skin and vasculature, as well as in squamous epithelia, it acts as a tumor suppressor. Given these diverse actions, it is imperative that Notch modulation is spatially targeted.7 With novel drug delivery platforms, we are likely to see successful development toward the clinical use of Notch modulators.
Oral delivery is the most common method for drug administration because of its simplicity, noninvasive nature, and patient compliance. However, the major hurdles with oral delivery of many drugs are poor stability in the gastric environment, low aqueous solubility, as well as inadequate penetration through mucosal barriers resulting in poor oral bioavailability.8 Nanoparticulate delivery systems offer great promise and advantages in the administration of drugs also via the oral route because they are able to carry sufficient amounts of drugs that could be released at specific sites, at a specific pH value, be resistant toward digestive enzymes, and control the release of encapsulated or associated drug.9,10 Due to these advantages, nanoparticle formulation approaches have proven to be very useful for drug delivery applications, including oral formulations.11–13 Moreover, employing nanomedical concepts such as cellular targeting and intracellular drug release in oral delivery further offers the possibility of efficient and specific delivery to cells within the intestinal epithelium and thereby to specific regions of the gastrointestinal (GI) tract, offering more efficacious treatment possibilities for intestinal diseases such as colon cancer and inflammatory bowel diseases, not attainable via systemic drug delivery.9,10
In oral formulations, amorphous silicon dioxide (SiO2) or silica, has been used as a pharmaceutical excipient for >50 years and is classified by the US Food and Drug Administration as generally regarded as safe.14 Amorphous silica is well known to be stable at low pH and dissolves rapidly at higher pH.15 Among the silica materials explored for drug delivery purposes, mesoporous silica nanoparticles (MSNs) offer several attractive features for drug delivery. These include a uniform and tunable pore size, high loading capacity of guest molecules, easily modifiable surface properties, and ability of the nanosized porous channels to render the crystalline state of a drug amorphous, thereby increasing its apparent solubility.16 All these features allow for better control of drug loading and increased solubility, and should lead to efficient protection of drug molecules from the harsh conditions of the GI tract. This may lead to better oral bioavailability, a property that has also been exploited to some degree for bulk mesoporous silica.17,18 The nanoparticle containing the drug load could further be expected to penetrate the mucosal barrier, be taken up by the intestinal epithelial cells, and either release the drug intracellularly or further aid the drug permeability across the epithelial layers, thus offering some advantages not obtainable via traditional oral formulations.
In this study, we utilize the high positive charge density of poly(ethylene imine) (PEI) surface coating on MSNs for facilitating binding to the negatively charged cell surfaces, leading to cellular internalization. Pure PEI has been shown to result in toxic side effects directly related to the molecular weight of branched PEI, which would restrict its application for therapeutic drug delivery.19 However, derivatization or “charge capping” of PEI with other functional groups20,21 as well as when used as a constituent together with silica in the construction of hybrid materials22 has indicated that these drawbacks of pure PEI can be restrained. Poly(ethylene glycol) (PEG) is the most commonly used nonionic hydrophilic polymer in biomedical drug delivery design because it has been shown to reduce uptake by the reticuloendothelial system, provide good dispersibility in aqueous solvents, and diminish association with proteins.23,24 PEG has a low intrinsic toxicity that renders it very suitable for biomedical applications, and it is also considered to be a generally regarded as safe material (GRAS).25 PEG has been reported as mucoadhesive agent, providing a net neutral surface, which aids to avoid binding with intestinal mucin networks and enhances penetration of nanoparticles through the mucin barrier.26,27 Hence, PEGylated nanoparticles have shown a higher ability for the interaction with the intestinal mucosa than the stomach.28 Nevertheless, the type of surface conformation of PEG chains, molecular weight, grafting density, and even coupling chemistry may have a significant influence on the biological behavior of PEGylated nanoparticles. Thus, different conformations of the PEG chains could either facilitate the penetration of the PEGylated particles through the mucus layer or increase the residence time of the adhered fraction of particles in the mucosa.28 In our approach, PEGylation was achieved by attaching PEG either directly to the particle surface or via a grafted PEI layer.29 Further, folic acid (FA) was conjugated to the MSNs as a potential affinity ligand.
Our earlier reports have demonstrated targeted delivery of γ-secretase inhibitors, efficient Notch inhibitors, to breast tumors with consequent reduction in tumor growth, through intravenous administration of MSNs.30,31 We also evaluated the effect on small intestinal stem cell differentiation by oral delivery of γ-secretase inhibitor-loaded FA-PEI-MSNs and demonstrated enhanced cell fate switching as compared to free drug.30 However, the effect of different surface functionalizations, the interaction of the MSNs with the intestinal epithelium, and the effect in different GI regions were not evaluated. In this study, we took advantage of the same model system to evaluate the potential of a series of functionalized MSNs as drug carriers for cell-targeted drug delivery through the oral route, with focus on the effect of the different combinations of PEI, PEG, and FA surface modifications on intestinal targeting. We demonstrate enhanced intestinal cell targeting and therapeutic efficacy with the combination of PEG and PEI, and further show that different functionalizations target different regions of the intestinal tract. Our results should serve as a demonstration of the need for careful evaluation of the relationship between functionalization, site specificity/targetability, and therapeutic efficacy of nanomedicines through different administration routes.
Experimental
Synthesis, functionalization, and bioconjugation of MSNs
A detailed description of the synthesis, the surface functionalization, and conjugation procedures can be found in the “Supplementary materials” section. Briefly, MSNs were synthesized according to previously published protocols.22,32,33 Surfactant-extracted MSNs were functionalized with PEI by surface hyperbranching polymerization of PEI as previously reported to yield PEI-MSNs.34 Both plain MSNs and PEI-MSNs were further functionalized with PEG utilizing the primary amine groups of either the aminosilane (for plain MSNs) or terminal ends of PEI (for PEI-MSNs). Particles (50 mg) were dispersed in chloroform and were allowed to react with 12.5 mg hexamethylene diisocyanate (HMDI)-activated methoxy-PEG (mPEG)29,35 (Supplementary materials) in the presence of 5 μL N,N-diisopropylethylamine (DIPEA) overnight at 60°C under reflux. After the reaction, the particles were washed with ethanol and vacuum-dried to yield PEG-MSNs and PEG-PEI-MSNs. To obtain FA-conjugated MSNs, we followed the protocol previously described.36 To obtain FA-PEG-MSNs and FA-PEG-PEI-MSNs, maleimide (mal)-PEG-OH was reacted with HMDI to activate the −OH group of mal-PEG (similarly as mPEG activation) and further conjugated with folic acid-thiol.
Particle characterization methods
The particles were characterized according to previously reported methods.22,32,33 A detailed description can be found in the “Supplementary materials” section.
Cellular uptake studies by fluorescence-assisted cell sorting and confocal microscopy
Cellular uptake studies by fluorescence-assisted cell sorting and confocal microscopy were performed as previously described.32,33 A detailed description can be found in the “Supplementary materials” section.
Particle stability under simulated GI tract conditions
Stock suspensions of empty and DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester) loaded MSNs (PEG-PEI-MSNs and DAPT/PEG-PEI-MSNs) were prepared in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 7.2) at a concentration of 4 mg/mL. The untreated baseline samples (0 time point) were collected directly on transmission electron microscopy (TEM) grids and allowed to dry overnight. For the GI stability experiment, Spectra/Por® Float-A-Lyzer® G2 dialysis membrane of fixed 1 mL volume was used to transfer particles from HEPES to simulated gastric fluid (SGF), and further to simulated intestinal fluid (SIF). The dialysis membranes were washed and preconditioned according to the manufacturer’s guidelines. Additionally, the membranes were preincubated with SGF [2.0 g of sodium chloride and 3.2 g of purified pepsin (derived from porcine gastric mucosa; product number 77161; CAS Number 9001-75-6), Sigma; Sigma-Aldrich, St Louis, MO, USA] in 7.0 mL of hydrochloric acid and water up to 1,000 mL for an hour before use. The membranes were filled with 1 mL of PEG-PEI-MSN or DAPT/PEG-PEI-MSN suspension, and the outer chamber was filled with 9 mL of SGF. The buffer volume was selected according to the extracellular fluid volume in mice, which is approximately 5–8 mL (according to body weight).37 The samples were kept at 37°C with agitation for 2 hours in the SGF. After that, a small amount of samples were placed on a TEM grid and allowed to dry. The SGF in the outer chamber was collected for high-performance liquid chromatography (HPLC) analysis. Subsequently, the samples were moved to the chamber containing 9 mL of SIF (6.8 g of monobasic potassium phosphate in 250 mL of water and then 77 mL of 0.2 N sodium hydroxide and 500 mL of water; 10.0 g of pancreatin [from porcine pancreas, Sigma] is added, and the resulting solution is adjusted to a pH of 6.8±0.1 and finally diluted to 1,000 mL) and transferred back to 37°C with agitation for 6 hours. After the end of incubation, the samples were collected on a TEM grid for electron microscopy. The remaining sample in the membrane was collected for HPLC analysis of DAPT. The TEM analysis was done using JEM-1400 Plus Transmission Electron Microscope (JEOL Ltd., Tokyo, Japan), and the HPLC was done using an HPLC system (Agilent, Santa Clara, CA, USA).
In vivo oral delivery and sample collection
All animal experiments were approved and conducted in accordance with the institutional animal care policies of the University of Turku and Åbo Akademi University (ÅAU) (Turku, Finland) under the permits ESAVI/1197/04.10.07/2013 similarly as was reported in our previous study.30 For in vivo oral drug delivery studies, 18 FVB/N adult male mice were randomly allocated to six groups and subjected to gastric gavage once a day for 3 consecutive days, with group 1: dimethyl sulfoxide-vehicle solution, control MSN particles; group 2: FA-PEI-MSNs; group 3: FA-PEG-PEI-MSNs 100 mg/kg; group 4: free DAPT (10 mg/kg), and DAPT-loaded MSN particles; group 5: DAPT/FA-PEI-MSNs; and group 6: DAPT/FA-PEG-PEI-MSNs 100 mg/kg, 10 wt%. Total volume of administered substances was 0.5 mL per day. Stool samples (three mice per treatment) were collected directly from the animals, weighed, and dried overnight on the heating plate, and the water content (% hydration) was determined by the loss of weight upon drying. Goblet cell numbers were analyzed by regular periodic acid–Schiff staining and light microscopy by manual counting and by using ImageJ software, (US National Institutes of Health, Bethesda, MN, USA) as well as by measuring MUC2 mRNA content by reverse transcription polymerase chain reaction. Primers used are MUC2 forward primer 5′-CTT CTG TGC CAC CCT CGT-3′ and MUC2 reverse primer 5′-TTC GGG ATC TGG CTT CTT T-3′, and for normalization, β-actin forward primer 5′-TGG CTC CTA GCA CCA TGA AGA-3′ and β-actin reverse primer 5′-GTG GAC AGT GAG GCC AGG AT-3′.
For in vivo uptake studies with fluorescein isothiocyanate (FITC)-labeled MSNs, eight C57BL/6 adult male mice were separated randomly into four groups and subjected to gastric gavage with different nanoparticulate mixture and control HEPES solution. Group 1 received PEI-MSNs, group 2: PEG-PEI-MSNs, group 3: FA-PEG-MSNs, and group 4: HEPES. MSNs at 8 mg/mL were dispersed in sterile HEPES, and 0.5 mL of the solution was introduced to each animal by gastric gavage for 4 consecutive days. To increase the absorption rate in intestine, mice were fasted 10 hours before and 1 hour after gavage. At the end of all treatments, 1 hour after the last gavage, animals were killed by cervical dislocation and weighed. The whole GI tract was removed, divided into stomach, small intestine corresponding to duodenum, jejunum, and ileum, cecum, and proximal and distal colon (Figure S1). The sections were opened, and two-third of the section was collected in histology cassette and one-third of the section was snap frozen in liquid nitrogen. For the confocal microscopy, tissue sample were fixed in 4% paraformaldehyde (PFA) (in phosphate-buffered saline, pH 7.4) overnight and embedded in cryomold using optimum cutting temperature compound. The tissue samples were sectioned at 6 μm thickness by cryostat (CM3050 S, Leica, Nussloch, Germany), and the samples were imaged using Leica TCS SP5 confocal microscope (488 nm excitation). The frozen sections were analyzed by fluorescence spectroscopy and inductively coupled plasma mass spectrometry to determine the amount of internalized MSNs. All the experiments were done in low light to avoid possible loss for fluorescence.
Statistical analysis
The experiments were repeated a minimum of two to three times and represented as mean ± standard error of the mean. One-way analysis of variance or Student’s t-test (GraphPad Prism version 5.00 for Windows, GraphPad Software, La Jolla, CA, USA) was used to analyze the data: cell viability, stool hydration, goblet cell numbers, and MUC2 analysis experiments. The level of significance was set at probabilities of *P<0.05, **P<0.01, and ***P<0.001.
Results and discussion
Physicochemical characterization of functionalized MSNs
MSNs were synthesized as previously described adopting a co-condensation approach22,32,33 further modified by functionalization for cellular and oral delivery with PEI, PEG, and FA as described in the “Experimental” section, and summarized in Figure 1. The resulting particles and their physicochemical characteristics are summarized in Table 1. Dynamic light scattering demonstrated that particles modified with PEI were fully redispersible and colloidally stable in HEPES buffer (25 mM, pH 7.2) with a hydrodynamic size of 400–500 nm, whereas their counterparts without PEI either aggregated or sedimented, as indicated by their hydrodynamic size of >1,000 nm (Table 1) due to their near-neutral net charge under the studied conditions. To determine that these particles were not agglomerated, but could still be fully redispersed in aqueous solvent, the hydrodynamic diameter was also determined in pure water, revealing particle sizes in the same range as those modified with PEI in HEPES buffer (400–500 nm; Table 1 and Figure S2A). All particle suspension measurements had a low polydispersity index indicating that the particles were indeed present as single particles in suspension (Table 1).
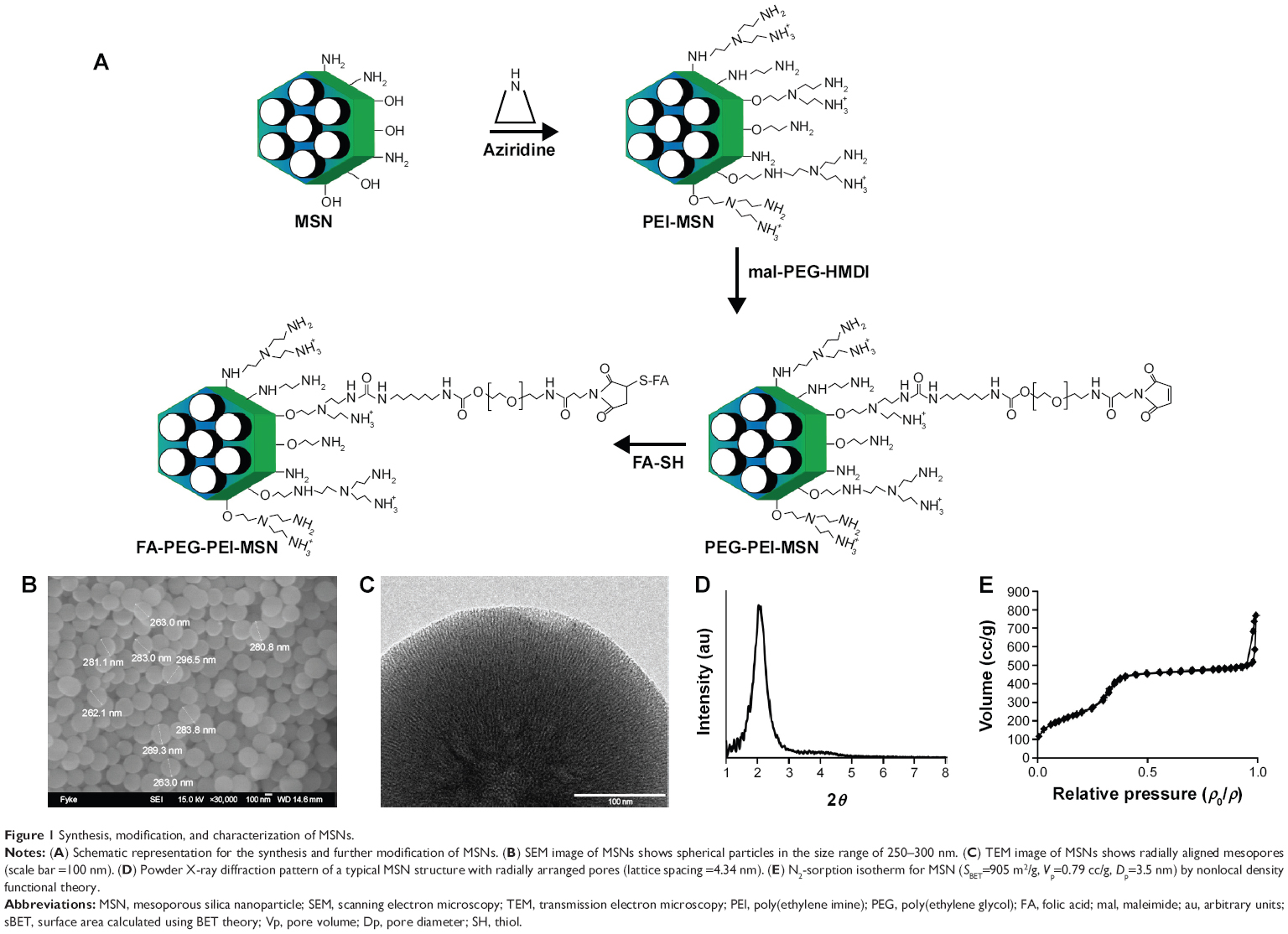 | Figure 1 Synthesis, modification, and characterization of MSNs. |
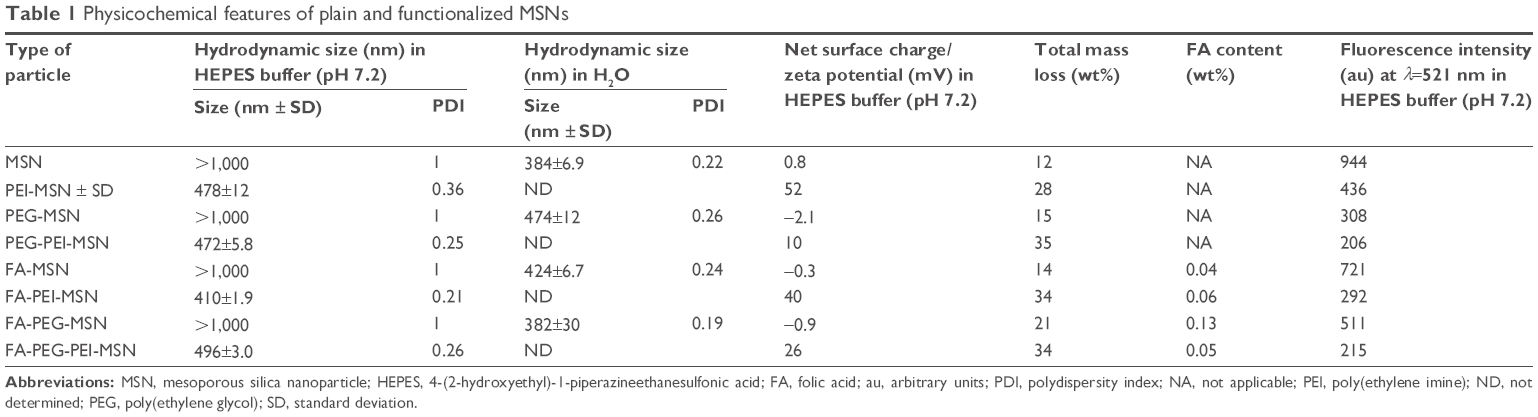 | Table 1 Physicochemical features of plain and functionalized MSNs |
The main difference in the characteristics of functionalized particles was observed in their zeta potential values, which indicates the net/effective surface charge of the particles at the studied conditions (Table 1). Non-coated MSN particles that displayed a net surface charge (zeta potential) of ~0 mV was shifted to ~50 mV upon PEI functionalization, indicative of successful grafting of PEI (Table 1 and Figure S2B). The total amount of organic functions for each particle type was estimated based on the mass loss up to 900°C from thermogravimetric analysis measurements (Table 1 and Figure S2C). Efficient removal of the surfactant template (CTAC) from the mesopores was confirmed by Fourier transform infrared spectroscopy (Figure S2D). FA content, determined with the aid of ultraviolet-visible spectrophotometry, showed that FA was successfully conjugated to the concerned particles (Table 1).
To be able to track the particle trafficking/distribution in vitro and in vivo, the starting particles (before functionalization) were fluorescently labeled in situ with fluorescein in the form of FITC. The fluorescence intensity of the particle suspensions in HEPES buffer at a particle concentration of 1 mg/mL is given in Table 1. The scanning and transmission electron microscopy images (Figure 1B and C) confirmed the mesoporous structure, spherical morphology, and the particle sizes, which were ~300 nm. Radially arranged mesopores can be discerned, with the radial arrangement of pores distinguishable in the center of the particle (Figure 1C; viewed from the top). The porous structure of the particle was further characterized by powder X-ray scattering and nitrogen sorption measurements (Figure 1D and E).
PEI-, PEG-, and FA-functionalized MSNs are nontoxic at therapeutic concentrations
The in vitro cytocompatibility of all produced MSNs was studied in colon cancer Caco-2 cells using the WST-1 assay, 48 hours after incubation with MSNs. All MSNs were non-toxic to Caco-2 cells up to 50 μg/mL concentration (Figure S3). At 100 μg/mL, most of the particles were still non-toxic, except for PEG-PEI-MSNs and FA-PEG-PEI-MSNs, which showed a 20%–30% decrease in cell viability compared to the vehicle dimethyl sulfoxide. This could be explained by a much higher uptake of these MSNs by the cells (Figure S3).The results show that all the produced MSNs are biocompatible at concentrations as high as 50 μg/mL. Further in vitro studies were performed at the concentration of 10 μg/mL.
MSNs can be readily internalized by colon cancer cells
The effect of surface functionalization on the internalization by two different human colon cancer cell lines (HT-29 and Caco-2 cells) was assessed by flow cytometry. As the fluorescence intensity of fluorescein varies depending on the surrounding pH38 as well as the particle functionalization regime22,32 (Table 1), we detected the percent of cells which have internalized particles as a measure of uptake efficiency (Figure 2A). In Caco-2 cells, the cellular uptake was determined as a function of cell confluency. This is important as the intestinal epithelial tissue comprises a sheet of interconnected cells. Particles were more efficiently internalized in non-confluent cells as compared to confluent cells (Figure 2A), as expected, since more surface area is available for uptake in non-confluent cells. Both HT-29 and Caco-2 are folate receptor-positive cell lines, but FA modification only enhanced the uptake of PEGylated particles (PEG-MSNs) and did not add further benefit to PEG-PEI-MSNs at the studied concentrations (Figure 2A and B).39 This might be because the inherent high uptake of PEI- and PEG-PEI-modified particles may have already reached the saturation levels,40 and consequently, further modification with FA has no advantage in these cases in terms of total uptake. A boosting effect of FA on cellular uptake was only, but clearly, detected in the case when FA was linked to PEGylated particles where PEG was conjugated to the MSNs directly, without an intermediate PEI layer; and for which the basal uptake was thus, was low. All particles functionalized with PEI were internalized more efficiently than their counter-particle without PEI, in accordance with our previous studies22,33 (Figure S4A and B), and we have previously shown that this type of particles were internalized by the clathrin-mediated endocytosis route.22 Particles without any polymeric coating or only modified with PEG showed low uptake probably associated with their almost neutral charge as well as tendency to aggregate at neutral conditions (Table 1).41 The combination of PEI and PEG showed the most efficient uptake (Figure 2A and B). The data was verified by normalizing the mean fluorescence intensity values against fluorescence intensity of these specific particles given in Table 1 (Figure 2B and S4B). Confocal microscopy further confirmed the cellular uptake (Figure 2C–L). Here, PEG-MSN particles appeared to be aggregated, in contrast to PEI-modified particles that exhibited a more dispersed intracellular pattern (Figure 2C–L). Taken together, these data showed superior performance of particles functionalized both with PEG and PEI in terms of maximizing the cellular internalization in the targeted cell lines.
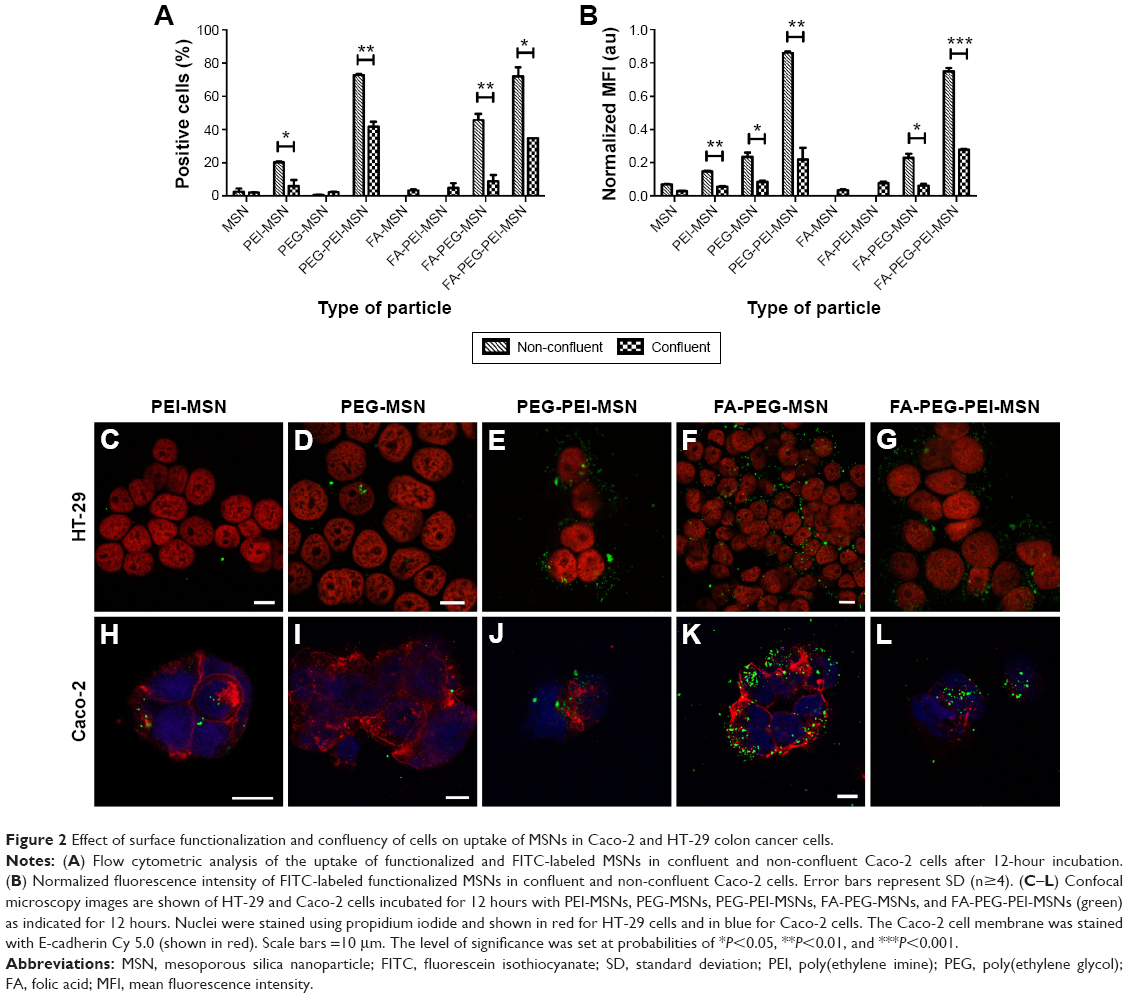 | Figure 2 Effect of surface functionalization and confluency of cells on uptake of MSNs in Caco-2 and HT-29 colon cancer cells. |
Enhancement of Notch inhibition and goblet cell differentiation in the colon is facilitated by PEG-functionalized and inhibitor-loaded MSNs in vivo
To determine the therapeutic potential of MSNs as drug delivery carriers in vivo for targeted oral drug delivery to the intestinal epithelium, we tested MSNs loaded with the γ-secretase inhibitor DAPT that inhibits the activity of Notch (Notch inhibitory particle).30 We have previously used DAPT-loaded FA-PEI-MSNs to successfully demonstrate their ability to control stem cell fate in the small intestine,30 where inhibiting Notch with DAPT increases the number of goblet cells resulting in increased stool hydration. To test if oral delivery and drug efficacy in vivo could be further improved by functionalization of the particles also with PEG, DAPT-loaded or control (unloaded) FA-PEI-MSNs, and FA-PEG-PEI-MSNs, DAPT alone or the vehicle control was fed to mice by oral gavage for 3 consecutive days. The uptake and effects were tested both in the small intestine, and colon, which is the target tissue in most intestinal diseases. The readout of Notch-mediated cell fate switches was monitored by the number of goblet cells using periodic acid–Schiff staining, MUC242 (the predominant intestinal mucin) mRNA analysis, and stool hydration. As expected, DAPT alone increased the number of goblet cells in colon (Figure 3A) and in the small intestine (Figure 4A and B). Importantly, the number of goblet cells in the colon of mice given DAPT/FA-PEG-PEI-MSNs were significantly higher than DAPT alone or control particles (FA-PEG-PEI-MSNs; Figure 3A and C). DAPT/FA-PEG-PEI-MSNs were also the only Notch inhibitory particles able to increase the expression of MUC2 in the colon (Figure 3B). DAPT/FA-PEI-MSNs were not able to improve the colonic Notch inhibition. Interestingly, a similar trend of improvement and goblet cell hyperplasia with PEG-PEI-coating was seen in the small intestine, whereas DAPT-loaded MSNs with FA and PEI (but not PEG) showed a significantly better inhibition of Notch compared to DAPT alone (Figure 4A and B). This effect was seen both in crypts and villi but more prominently in the villi, which have a bigger cell compartment and more goblet cells (Figure 4). These results are supported by an increased level of stool hydration only in mice fed with DAPT alone or DAPT/FA-PEG-PEI-MSNs (Figure S5), which reflects the Notch inhibition-induced cell fate switch from decreased numbers of water-absorbing colonocytes to mucus-producing goblet cells.43,44 The discrepancy between small intestine and colon could be related to the fact that the small intestine, being higher up in the GI tract, had been exposed to the particles for slightly longer time than the colon, and that DAPT/FA-PEI-MSNs were more efficiently internalized by the intestinal cells, subsequently leading to a more efficient drug delivery (as shown in increased uptake in cells over time; Figure S4). It is also likely that differences in colon and small intestine mucus layers influence particle penetration, for instance, the pore size and charge of mucin molecules may vary considerably along the GI tract.45 It has been shown that particles more easily penetrate the mucus in small intestine and hence there might be no beneficial effect of PEGylation, while this modification may be necessary in the colon which has much more goblet cells/area than the small intestine (Figures 3 and 4).46,47 Since the main intestinal disorders, colorectal cancer, and inflammatory bowel disease occur in the colon and not in the small intestine, the PEGylation of MSN should provide a useful tool for nanomedical drug development targeting the colon. In summary, MSNs in particular when functionalized with combination of PEG and PEI are efficient carriers for drug delivery and targeted Notch inhibition by oral administration, and significantly more efficient as compared to free drug administration, especially in the colon.
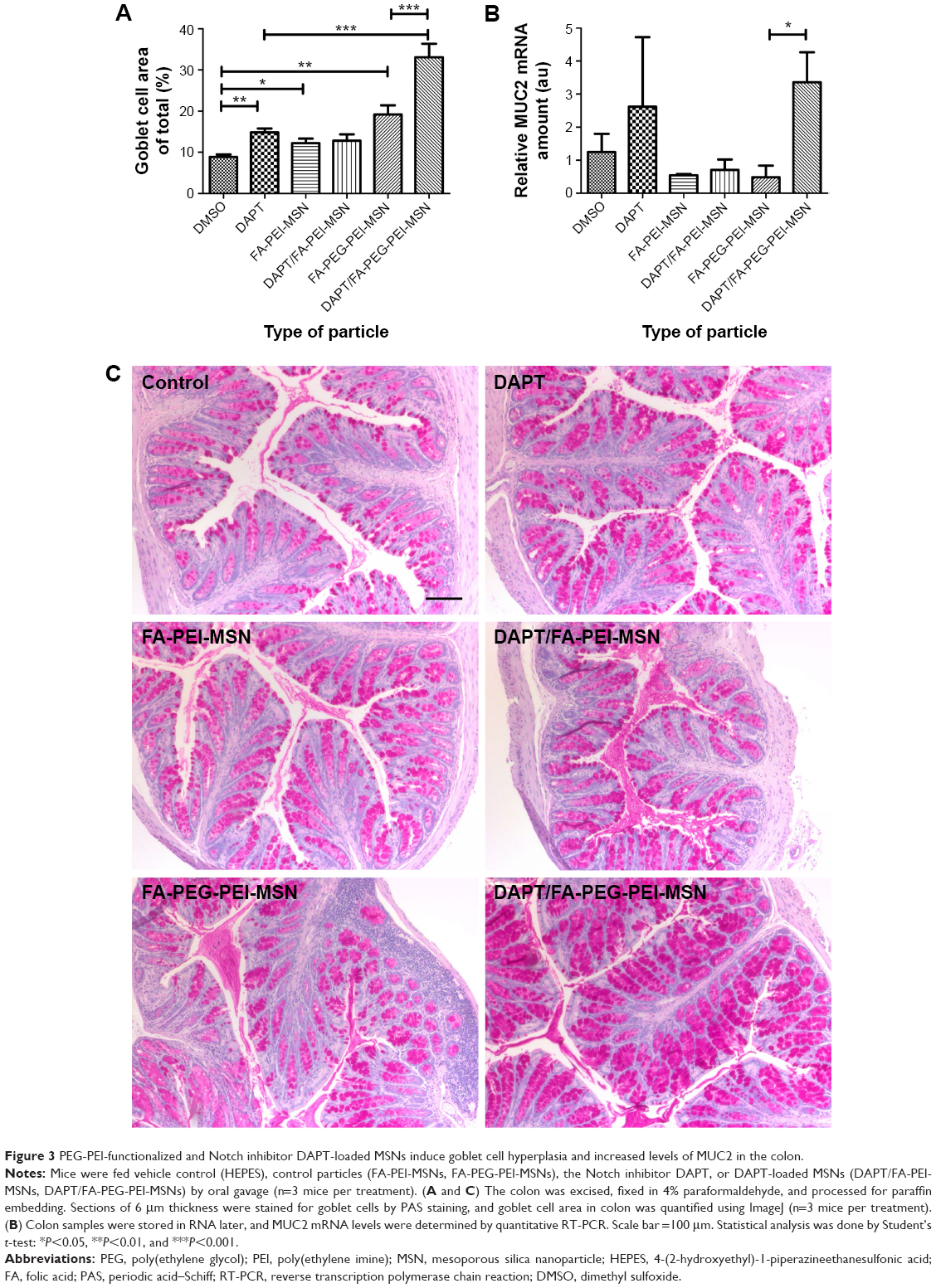 | Figure 3 PEG-PEI-functionalized and Notch inhibitor DAPT-loaded MSNs induce goblet cell hyperplasia and increased levels of MUC2 in the colon. |
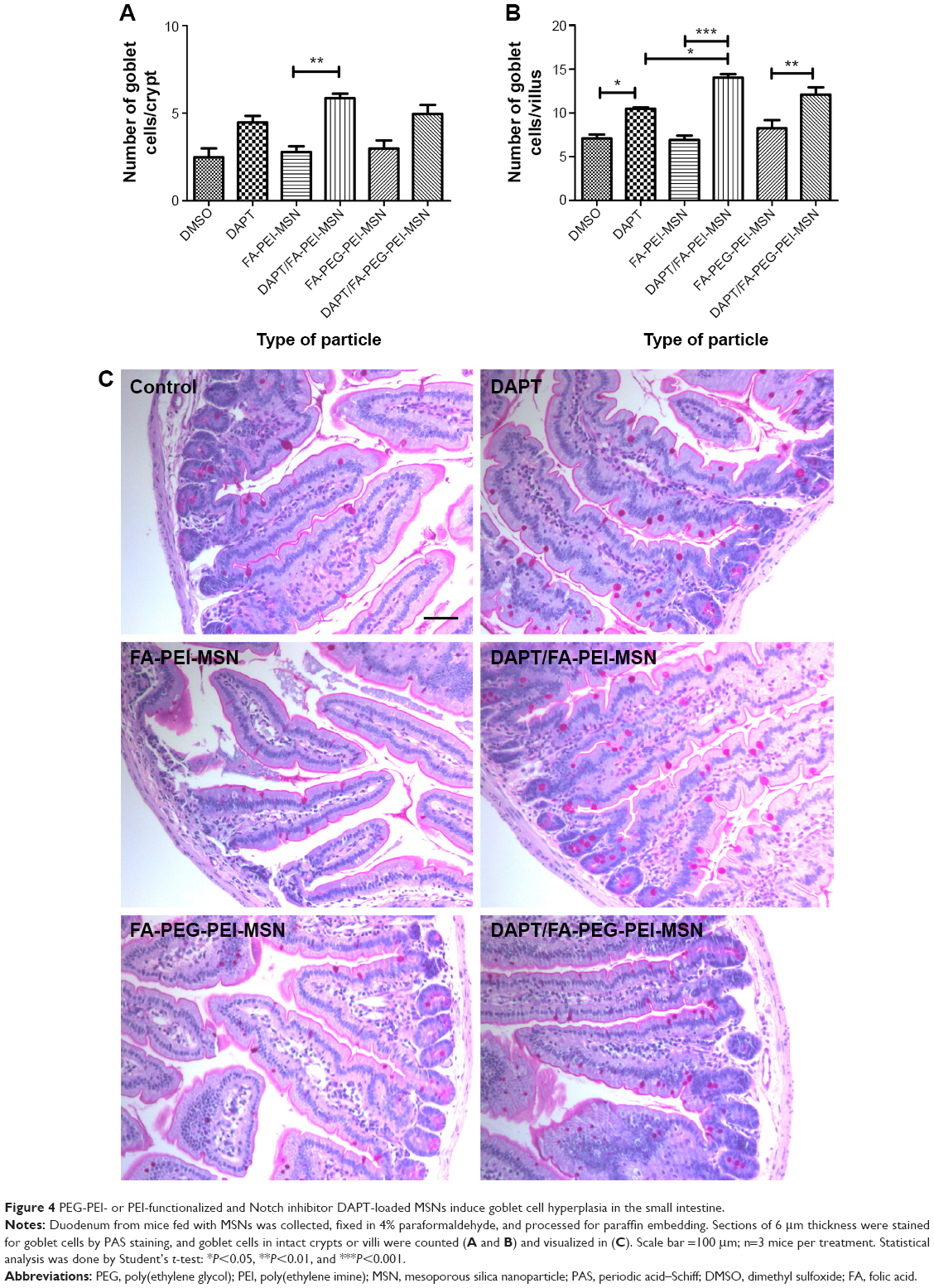 | Figure 4 PEG-PEI- or PEI-functionalized and Notch inhibitor DAPT-loaded MSNs induce goblet cell hyperplasia in the small intestine. |
Functionalized MSNs are stable in SGF and SIF
To evaluate whether PEG-PEI-functionalized MSNs are stable in the harsh conditions of GI tract and that they are able to protect the loaded drug/cargo, PEG-PEI-MSNs and DAPT/PEG-PEI-MSNs were exposed to SGF (containing the gastric enzyme pepsin and hydrochloric acid, pH 1.2) and SIF (containing, eg, the enzyme pancreatin, pH 6.8) whereafter the particle morphology was analyzed by TEM as described in the “Experimental” section. The TEM images show that the MSNs were intact and the structure was virtually unaltered both after the SGF (Figure 5D–F) and after the consequent SIF (Figure 5G–I) treatments compared to the untreated (starting) particles (Figure 5A–C). In order to analyze if MSNs can safely protect DAPT while passing the intestinal fluids, DAPT was eluted from MSNs and analyzed by chromatography before and after SGF and SIF incubation, which showed that DAPT is intact and has not been prematurely released nor degraded by exposure to SGF and SIF (Figure S6). Thus, functionalized MSNs can serve as an effective carrier to deliver cargo to the target site without losing its integrity even after passing through the GI environment with low pH and presence of digestive enzymes.
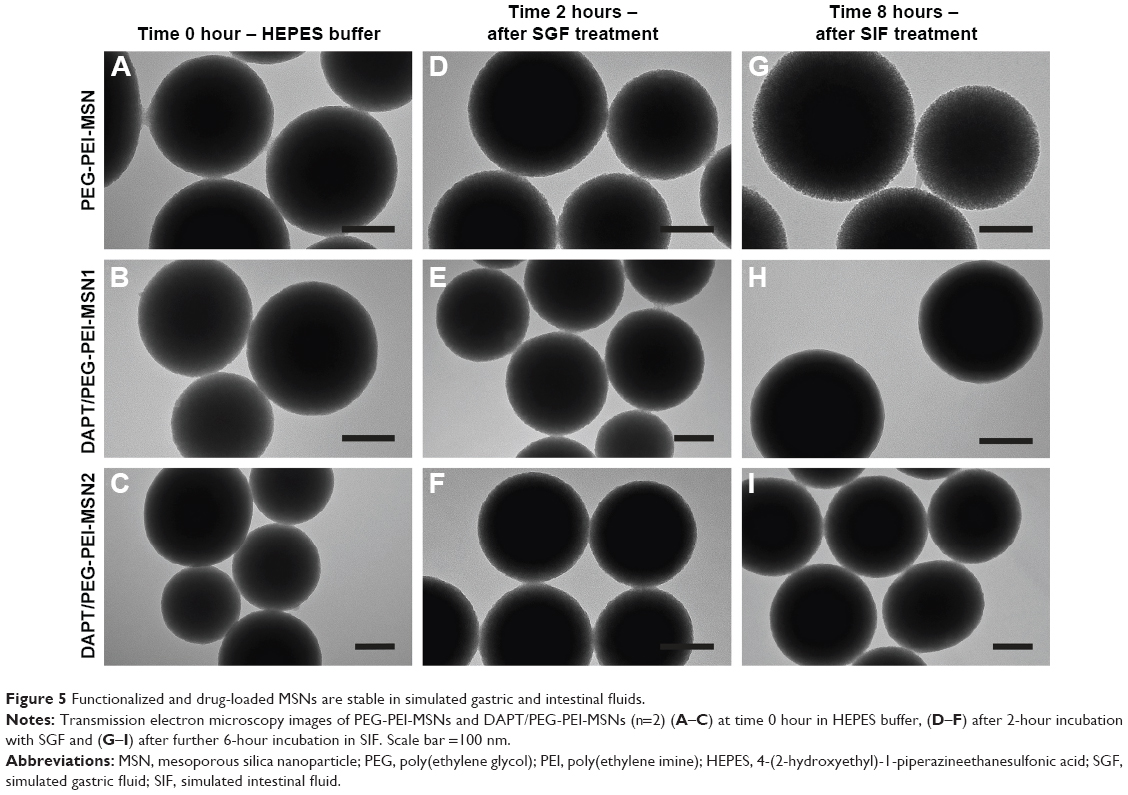 | Figure 5 Functionalized and drug-loaded MSNs are stable in simulated gastric and intestinal fluids. |
Functionalized MSNs can be internalized by intestinal epithelial in vivo
To determine whether orally administered MSNs with different types of polymeric coatings were taken up by the intestinal epithelial cells in vivo, we performed daily oral gavage for 4 days of PEI-MSNs, FA-PEG-MSNs, and PEG-PEI-MSNs. The mice were healthy during the treatment and were sacrificed at the end of the experiment. The biodistribution and in vivo uptake of MSNs in the GI tract (Figure S1) were assessed in tissue cryo-sections and by confocal microscopy. While tissue sections of the control mice gavaged with vehicle (HEPES buffer) alone show only autofluorescence from the tissue (Figure 6A–D), tissue sections from the mice fed with particles showed brighter green particulate fluorescence from the ~300 nm FITC-labeled MSNs either attached to the mucosal barrier or internalized by epithelial cells (Figure 6E–P; colon not shown). This was evident in all studied GI sections but easily discernible in the stomach and the small intestinal villi. Mice given PEG-PEI-MSNs appeared to have slightly more internalized particles in the epithelium, and this was further confirmed by staining for F-actin in tissue sections from mice given fluorescent MSNs to highlight epithelial apical microvilla toward the lumen and DRAQ5® to visualize nuclei (Figure 6Q–S). We further attempted to quantify the amount of fluorescent particles in dissected tissue sections; however, neither fluorescence spectroscopy of homogenized tissue samples (Figure S7) nor quantification of elemental silicon by inductively coupled plasma measurements (Figure S8) proved to be sensitive enough to detect changes in fluorescence or Si above the high baseline in control mice gavaged with vehicle (HEPES) alone due to autofluorescence and inherent Si content in the tissue. In summary, we have shown that PEI-, FA-PEG-, or PEG-PEI-functionalized MSNs can be taken up by intestinal epithelial cells in vivo after administration by oral gavage.
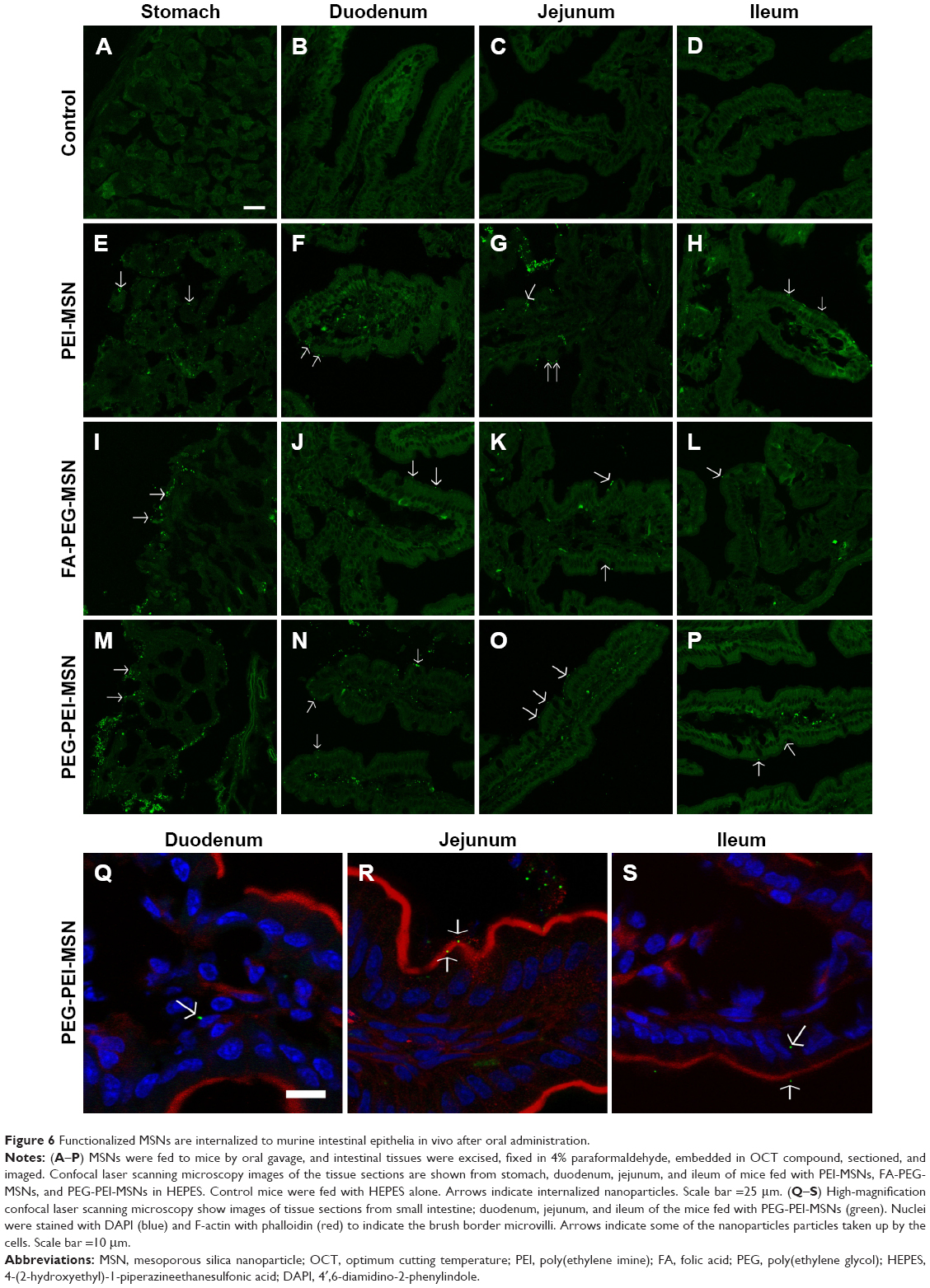 | Figure 6 Functionalized MSNs are internalized to murine intestinal epithelia in vivo after oral administration. |
There are a few studies demonstrating increased drug bioavailability and/or permeability for drug-loaded MSN formulations after oral administration48–51 relying on the enhanced solubility that can be attained for poorly soluble drugs upon loading into a porous matrix with subsequent release into the GI tract.52,53 To our knowledge, however, the present study is the first to demonstrate the potential of intestinal targeting of the MSN drug delivery carrier itself, with subsequent cellular uptake and intracellular release of a poorly soluble drug important for clinical use. Depending on the employed surface functionalization, the drug efficacy level varied in different parts of the GI tract, highlighting the potential of intestinal targeting imparted by nanomedicine.
Conclusion
We have developed a series of biocompatible MSNs with surface modifications suitable for oral drug delivery and intestinal targeting. The combination of PEI and PEG as particle surface modification is superior to enhance the internalization of MSNs to intestinal epithelial cells (Figure 7). Further, the carrier MSNs and loaded drug stay intact and functional after exposure to the harsh conditions of the stomach and the intestine. DAPT-loaded orally delivered FA-PEG-PEI-MSNs more efficiently decrease Notch signaling compared to free drug in the intestine, and the drug targeting can be further tuned by MSN surface modifications, since PEI coating exhibits higher affinity for the small intestine and PEG-PEI coating for the colon. These findings reveal significant potential for the use of MSNs as targeted oral drug delivery carriers through epithelial barriers, provided the right surface functionalization design is employed.
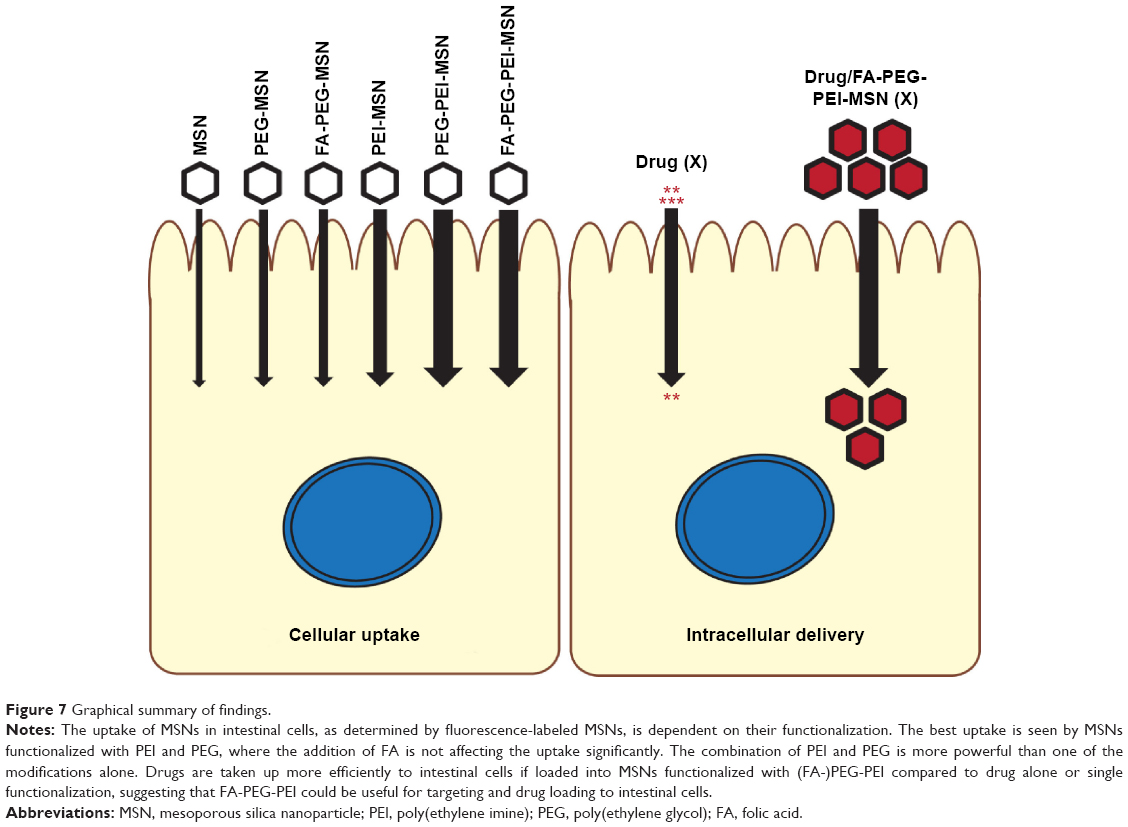 | Figure 7 Graphical summary of findings. |
Acknowledgments
The Cell Imaging Core at Turku Centre for Biotechnology, Helena Saarento, Catharina Alam, M Nadeem Asghar, Joel Nyström, and Julia Misiorek (Cell Biology/Biosciences, ÅAU) are thanked for technical support and advice. We are grateful to Dr Jussi Meriluoto, Biochemistry/Biosciences, ÅAU, for performing the HPLC analysis. Microscopy Service of Universidad Rey Juan Carlos (Spain) and Antonio Martín Rengel (Department of Chemical and Environmental Technology, Universidad Rey Juan Carlos, Madrid, Spain) are acknowledged for providing the TEM images. This work was financed by the Academy of Finland #140759/126161 (DT), #218062 (CS) and #137101/140193/260599/278812 (DD, DSK, NP, JMR), Sigrid Jusélius Foundation (DT, CS) and Liv och Hälsa foundation (DT), EU FP7 IRG (DT), Cancer Society of Finland and ÅAU Center of Excellence of Cell Stress and Molecular Aging (DT, CS). ERASMUS Experts III programme (DD), Centre of Excellence for Functional Materials (FunMat) (DSK), Doctoral Network in Materials Research at ÅAU (NP), and Turku Doctoral Programme in Molecular Biosciences at ÅAU and Swedish cultural foundation (IAKL) are greatly acknowledged for financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
Minko T, Rodriguez-Rodriguez L, Pozharov V. Nanotechnology approaches for personalized treatment of multidrug resistant cancers. Adv Drug Deliv Rev. 2013;65(13–14):1880–1895. doi:10.1016/j.addr.2013.09.017. | ||
Mura S, Couvreur P. Nanotheranostics for personalized medicine. Adv Drug Deliv Rev. 2012;64(13):1394–1416. doi:10.1016/j.addr.2012.06.006. | ||
Wong IY, Bhatia SN, Toner M. Nanotechnology: emerging tools for biology and medicine. Genes Dev. 2013;27(22):2397–2408. doi:10.1101/gad.226837.113. | ||
Rosenholm JM, Mamaeva V, Sahlgren C, Lindén M. Nanoparticles in targeted cancer therapy: mesoporous silica nanoparticles entering preclinical development stage. Nanomedicine. 2012;7(1):111–120. doi:10.2217/nnm.11.166. | ||
Laitman LE, Dahan S. Taking inflammatory bowel disease up a Notch. Immunol Res. 2012;54(1–3):69–74. doi:10.1007/s12026-012-8314-6. | ||
Suman S, Das TP, Ankem MK, Damodaran C. Targeting Notch signaling in colorectal cancer. Curr Colorectal Cancer Rep. 2014;10(4):411–416. doi:10.1007/s11888-014-0252-3. | ||
Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer Cell. 2014;25(3):318–334. doi:10.1016/j.ccr.2014.02.018. | ||
Lipinski CA. Poor aqueous solubility – an industry wide problem in drug discovery. Am Pharm Rev. 2002;5:82–85. | ||
Bawarski WE, Chidlowsky E, Bharali DJ, Mousa SA. Emerging nanopharmaceuticals. Nanomed Nanotechnol Biol Med. 2008;4(4):273–282. doi:10.1016/j.nano.2008.06.002. | ||
Laroui H, Wilson DS, Dalmasso G, et al. Nanomedicine in GI. Am J Physiol Gastrointest Liver Physiol. 2011;300(3):G371–G383. doi:10.1152/ajpgi.00466.2010. | ||
Shahbazi M-A, Santos HA. Improving oral absorption via drug-loaded nanocarriers: absorption mechanisms, intestinal models and rational fabrication. Curr Drug Metab. 2013;14(1):28–56. | ||
Plapied L, Duhem N, des Rieux A, Préat V. Fate of polymeric nanocarriers for oral drug delivery. Curr Opin Colloid Interface Sci. 2011;16(3):228–237. doi:10.1016/j.cocis.2010.12.005. | ||
Wilson DS, Dalmasso G, Wang L, Sitaraman SV, Merlin D, Murthy N. Orally delivered thioketal nanoparticles loaded with TNF-α-siRNA target inflammation and inhibit gene expression in the intestines. Nat Mater. 2010;9(11):923–928. doi:10.1038/nmat2859. | ||
Lewis & Harrison, LLC. Generally Recognized as Safe Determination for Silicon Dioxide When Added Directly and/or Indirectly to Human Food. January 2010. Available from: http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-foods-gen/documents/document/ucm269494.pdf. Accessed August 9, 2015. | ||
Iler RK. The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties, and Biochemistry. New York: John Wiley & Sons; 1979. | ||
Rigby SP, Fairhead M, van der Walle CF. Engineering silica particles as oral drug delivery vehicles. Curr Pharm Des. 2008;14(18):1821–1831. | ||
van Speybroeck M, Mellaerts R, Thi TD, et al. Preventing release in the acidic environment of the stomach via occlusion in ordered mesoporous silica enhances the absorption of poorly soluble weakly acidic drugs. J Pharm Sci. 2011;100(11):4864–4876. doi:10.1002/jps.22703. | ||
Mellaerts R, Mols R, Jammaer JAG, et al. Increasing the oral bioavailability of the poorly water soluble drug itraconazole with ordered mesoporous silica. Eur J Pharm Biopharm. 2008;69(1):223–230. doi:10.1016/j.ejpb.2007.11.006. | ||
Godbey WT, Wu KK, Mikos AG. Size matters: molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J Biomed Mater Res. 1999;45(3):268–275. | ||
Zintchenko A, Philipp A, Dehshahri A, Wagner E. Simple modifications of branched PEI lead to highly efficient siRNA carriers with low toxicity. Bioconjug Chem. 2008;19(7):1448–1455. doi:10.1021/bc800065f. | ||
Merdan T, Kunath K, Petersen H, et al. PEGylation of poly(ethylene imine) affects stability of complexes with plasmid DNA under in vivo conditions in a dose-dependent manner after intravenous injection into mice. Bioconjug Chem. 2005;16(4):785–792. doi:10.1021/bc049743q. | ||
Desai D, Karaman DS, Prabhakar N, et al. Design considerations for mesoporous silica nanoparticulate systems in facilitating biomedical applications. Mesoporous Biomater. 2014;1(1). | ||
Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed. 2010;49(36):6288–6308. doi:10.1002/anie.200902672. | ||
Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2(3):214–221. doi:10.1038/nrd1033. | ||
Lai SK, Wang Y-Y, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61(2):158–171. doi:10.1016/j.addr.2008.11.002. | ||
Huang Y, Leobandung W, Foss A, Peppas NA. Molecular aspects of muco- and bioadhesion: tethered structures and site-specific surfaces. J Control Release. 2000;65(1–2):63–71. | ||
Serra L, Doménech J, Peppas NA. Design of poly(ethylene glycol)-tethered copolymers as novel mucoadhesive drug delivery systems. Eur J Pharm Biopharm. 2006;63(1):11–18. doi:10.1016/j.ejpb.2005.10.011. | ||
Yoncheva K, Gómez S, Campanero MA, Gamazo C, Irache JM. Bioadhesive properties of pegylated nanoparticles. Expert Opin Drug Deliv. 2005;2(2):205–218. doi:10.1517/17425247.2.2.205. | ||
Şen Karaman D, Gulin-Sarfraz T, Hedström G, Duchanoy A, Eklund P, Rosenholm JM. Rational evaluation of the utilization of PEG-PEI copolymers for the facilitation of silica nanoparticulate systems in biomedical applications. J Colloid Interface Sci. 2014;418:300–310. doi:10.1016/j.jcis.2013.11.080. | ||
Mamaeva V, Rosenholm JM, Bate-Eya LT, et al. Mesoporous silica nanoparticles as drug delivery systems for targeted inhibition of Notch signaling in cancer. Mol Ther J Am Soc Gene Ther. 2011;19(8):1538–1546. doi:10.1038/mt.2011.105. | ||
Wittig R, Rosenholm JM, von Haartman E, et al. Active targeting of mesoporous silica drug carriers enhances γ-secretase inhibitor efficacy in an in vivo model for breast cancer. Nanomedicine. 2014;9(7):971–987. doi:10.2217/nnm.13.62. | ||
Rosenholm JM, Meinander A, Peuhu E, et al. Targeting of porous hybrid silica nanoparticles to cancer cells. ACS Nano. 2008;3(1):197–206. doi:10.1021/nn800781r. | ||
Karaman DS, Desai D, Senthilkumar R, et al. Shape engineering vs organic modification of inorganic nanoparticles as a tool for enhancing cellular internalization. Nanoscale Res Lett. 2012;7(1):1–14. doi:10.1186/1556-276X-7-358. | ||
Rosenholm JM, Duchanoy A, Lindén M. Hyperbranching surface polymerization as a tool for preferential functionalization of the outer surface of mesoporous silica†. Chem Mater. 2008;20(3):1126–1133. doi:10.1021/cm7021328. | ||
Petersen H, Fechner PM, Fischer D, Kissel T. Synthesis, characterization, and biocompatibility of polyethylenimine-graft-poly(ethylene glycol) block copolymers. Macromolecules. 2002;35(18):6867–6874. doi:10.1021/ma012060a. | ||
Nakajima N, Ikada Y. Mechanism of amide formation by carbodiimide for bioconjugation in aqueous media. Bioconjug Chem. 1995;6(1):123–130. doi:10.1021/bc00031a015. | ||
Chapman ME, Hu L, Plato CF, Kohan DE. Bioimpedance spectroscopy for the estimation of body fluid volumes in mice. Am J Physiol Renal Physiol. 2010;299(1):F280–F283. doi:10.1152/ajprenal.00113.2010. | ||
Sjöback R, Nygren J, Kubista M. Absorption and fluorescence properties of fluorescein. Spectrochim Acta A Mol Biomol Spectrosc. 1995;51(6):L7–L21. doi:10.1016/0584-8539(95)01421-P. | ||
Yang S-J, Lin F-H, Tsai K-C, et al. Folic acid-conjugated chitosan nanoparticles enhanced protoporphyrin IX accumulation in colorectal cancer cells. Bioconjug Chem. 2010;21(4):679–689. doi:10.1021/bc9004798. | ||
Davda J, Labhasetwar V. Characterization of nanoparticle uptake by endothelial cells. Int J Pharm. 2002;233(1–2):51–59. | ||
Hatakeyama H, Akita H, Harashima H. The polyethyleneglycol dilemma: advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol Pharm Bull. 2013;36(6):892–899. doi:10.1248/bpb.b13-00059. | ||
Tytgat KM, Büller HA, Opdam FJ, Kim YS, Einerhand AW, Dekker J. Biosynthesis of human colonic mucin: Muc2 is the prominent secretory mucin. Gastroenterology. 1994;107(5):1352–1363. | ||
Yeung TM, Chia LA, Kosinski CM, Kuo CJ. Regulation of self-renewal and differentiation by the intestinal stem cell niche. Cell Mol Life Sci. 2011;68(15):2513–2523. doi:10.1007/s00018-011-0687-5. | ||
van Es JH, van Gijn ME, Riccio O, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435(7044):959–963. doi:10.1038/nature03659. | ||
McConnell EL, Fadda HM, Basit AW. Gut instincts: explorations in intestinal physiology and drug delivery. Int J Pharm. 2008;364(2):213–226. doi:10.1016/j.ijpharm.2008.05.012. | ||
Ermund A, Schütte A, Johansson MEV, Gustafsson JK, Hansson GC. Studies of mucus in mouse stomach, small intestine, and colon. I. Gastrointestinal mucus layers have different properties depending on location as well as over the Peyer’s patches. Am J Physiol Gastrointest Liver Physiol. 2013;305(5):G341–G347. doi:10.1152/ajpgi.00046.2013. | ||
Johansson MEV, Sjövall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol. 2013;10(6):352–361. doi:10.1038/nrgastro.2013.35. | ||
Zhang Y, Wang J, Bai X, Jiang T, Zhang Q, Wang S. Mesoporous silica nanoparticles for increasing the oral bioavailability and permeation of poorly water soluble drugs. Mol Pharm. 2012;9(3):505–513. doi:10.1021/mp200287c. | ||
Popat A, Jambhrunkar S, Zhang J, et al. Programmable drug release using bioresponsive mesoporous silica nanoparticles for site-specific oral drug delivery. Chem Commun (Camb). 2014;50(42):5547–5550. doi:10.1039/c4cc00620h. | ||
Yoncheva K, Popova M, Szegedi A, et al. Functionalized mesoporous silica nanoparticles for oral delivery of budesonide. J Solid State Chem. 2014;211:154–161. doi:10.1016/j.jssc.2013.12.020. | ||
Kiekens F, Eelen S, Verheyden L, Daems T, Martens J, Van Den Mooter G. Use of ordered mesoporous silica to enhance the oral bioavailability of ezetimibe in dogs. J Pharm Sci. 2012;101(3):1136–1144. doi:10.1002/jps.23016. | ||
Doane T, Burda C. Nanoparticle mediated non-covalent drug delivery. Adv Drug Deliv Rev. 2013;65(5):607–621. doi:10.1016/j.addr.2012.05.012. | ||
Xu W, Riikonen J, Lehto V-P. Mesoporous systems for poorly soluble drugs. Int J Pharm. 2013;453(1):181–197. doi:10.1016/j.ijpharm. 2012.09.008. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.