")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Poly(styrene)-b-poly(DL-lactide) copolymer-based nanoparticles for anticancer drug delivery
Authors Lee J, Kim JS, Cho H, Kim D
Received 20 February 2014
Accepted for publication 9 April 2014
Published 3 June 2014 Volume 2014:9(1) Pages 2803—2813
DOI https://doi.org/10.2147/IJN.S62806
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Jae-Young Lee,1 Jung Sun Kim,2 Hyun-Jong Cho,3 Dae-Duk Kim1
1College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul, Republic of Korea; 2Division of Health Sciences, Dongseo University, Busan, Republic of Korea; 3College of Pharmacy, Kangwon National University, Chuncheon, Republic of Korea
Abstract: Poly(styrene)-b-poly(DL-lactide) (PS-PDLLA) copolymer-based nanoparticles (NPs) of a narrow size distribution, negative zeta potential, and spherical shape were fabricated for the delivery of docetaxel (DCT). The particle size was consistently maintained in serum for 24 hours and a sustained drug release pattern was observed for 10 days in the tested formulations. The cytotoxicity of the developed blank NPs was negligible in prostate cancer (PC-3) cells. Cellular uptake and distribution of the constructed NPs containing a hydrophobic fluorescent dye was monitored by confocal laser scanning microscopy (CLSM) for 24 hours. Anti-tumor efficacy of the PS-PDLLA/DCT NPs in PC-3 cells was significantly more potent than that of the group treated with commercially available DCT, Taxotere® (P<0.05). Blood biochemistry tests showed that no serious toxicity was observed with the blank NPs in the liver and kidney. In a pharmacokinetic study of DCT in rats, in vivo clearance of PS-PDLLA/DCT NPs decreased while the half-life in blood increased compared to the Taxotere-treated group (P<0.05). The PS-PDLLA NPs are expected to be a biocompatible and efficient nano-delivery system for anticancer drugs.
Keywords: docetaxel, prolonged blood circulation, prostate cancer
Introduction
Diverse approaches for cancer therapy and diagnosis continue to be pursued.1–3 Among the various formulation types, intravenous administration is the primary choice for cancer treatment, and several injectable formulations for anticancer drug delivery have recently been developed.4–7 In most of these cases, improving aqueous solubility and tumor targeting of anticancer agents have been the primary objectives in the formulation development. Nanovehicles, known to produce sufficient aqueous solubility, sustained release, and tumor targeting of anticancer drugs, have been developed based on diverse materials as one of these injectable formulations. Nevertheless, only a few among those developed have been approved for clinical applications due to toxicity of the materials and insufficient biodistribution of the nanovehicles to the tumors. Although the in vitro and in vivo toxicity of many organic and inorganic materials have been evaluated, a limited number of substances have been selected for clinical application so far.
In the current investigation, a poly(styrene)-b-poly(DL-lactide) (PS-PDLLA) copolymer (Figure 1A) was used for the preparation of nanoparticles (NPs) for docetaxel (DCT) delivery. Both PS and PDLLA have been used in various biomedical applications.8,9,15–17 PS is a synthetic aromatic polymer composed of the monomer styrene. Nano-sized vehicles based on PS have been widely investigated as a drug delivery system.8,9 Amorphous PDLLA, based on the polymerization of a racemic mixture of L- and D-lactides, has been used for drug delivery in itself or in its conjugate form.10 Taking the biomedical functionalities of each component (PS or PDLLA) into account, it seems that PS-PDLLA copolymer has merit to be used for the development of a drug delivery system. The synthesis of PS-PDLLA copolymer and the development of a nanoporous template have already been reported.11,12 In this study, the feasibility of PS-PDLLA for NP fabrication and its application to anticancer drug delivery are discussed for the first time, to the best of our knowledge.
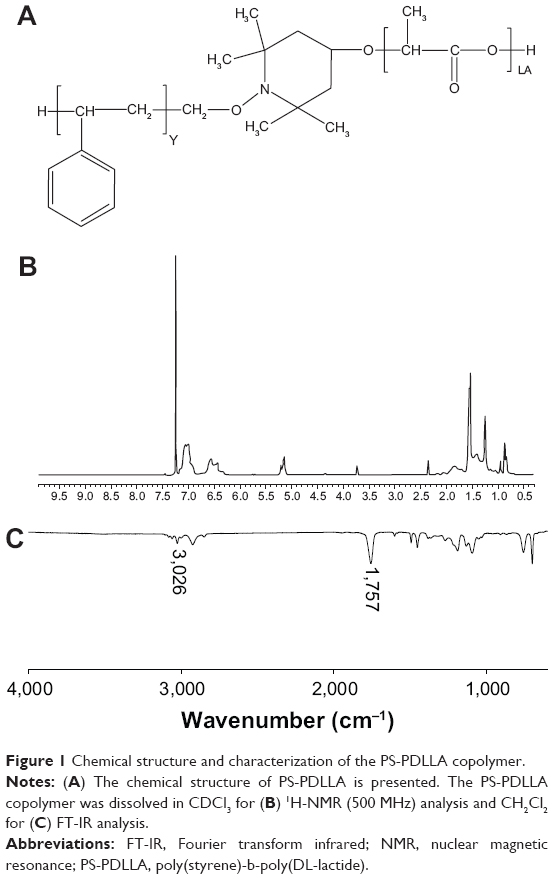 | Figure 1 Chemical structure and characterization of the PS-PDLLA copolymer. |
DCT, a second generation of taxoid, has anti-mitotic chemotherapeutic properties and has been used in breast, prostate, ovarian, and lung cancers. DCT is almost insoluble in water (4.93 μg/mL). Thus, drug solubilization techniques were used to prepare the commercial formulation Taxotere® (Sanofi S.A., Paris, France), which is marketed worldwide as an injectable formulation of DCT.13 However, although solubility of DCT has been improved for its clinical application, toxicity of the blank formulation and inaccurate tumor targeting still remain to be overcome. Various nanovehicles, such as liposomes, NPs, and micelles, have been developed and evaluated for DCT delivery to improve delivery efficiency and attenuate unwanted effects.14–16
Herein, we report on the PS-PDLLA copolymer-based NPs for the controlled delivery of water-insoluble anticancer drugs, using DCT as a model. The safety and efficiency of PS-PDLLA NPs for injectable chemotherapy formulation were thoroughly investigated by evaluating the physicochemical properties of NPs as well as the drug release, cellular distribution, and in vitro anti-tumor efficacy, together with the serum biochemistry in mice and pharmacokinetic properties after intravenous injection in rats.
Materials and methods
Materials
The PS-PDLLA was purchased from PolySciTech, a division of Akina, Inc. (West Lafayette, IN, USA). Taxotere was obtained from Sanofi S.A. Docetaxel (DCT) was acquired from LC Laboratories (Woburn, MA, USA). Paclitaxel (PTX) was purchased from Samyang Genex Corporation (Daejeon, Korea). Tween 80 was obtained from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Phosphate-buffered saline (PBS) was purchased from Biosesang (Seongnam, Korea). Dichloromethane (DCM) and dimethyl sulfoxide (DMSO) were purchased from Daejung Chemicals & Metals Co., Ltd. (Seoul, Korea). Poly(vinyl alcohol) (PVA), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) solution, and sodium bicarbonate (NaHCO3) solution were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). RPMI 1640 media, penicillin, streptomycin, and fetal bovine serum (FBS) were purchased from Gibco Life Technologies, Inc. (Thermo Fisher Scientific, Waltham, MA, USA). All other reagents were of analytical grade and were acquired from commercial sources.
Preparation and characterization of PS-PDLLA NPs
DCT-loaded PS-PDLLA NPs were prepared using an emulsification-solvent evaporation method with slight modification.17 Briefly, DCT and PS-PDLLA (4:20 and 4:40, mg, weight per weight [w/w]) were dissolved in 2 mL of DCM, and added to 20 mL of PVA solution (2%, weight per volume [w/v]). The emulsion was sonicated for 20 minutes using a probe sonicator (VC-750; Sonics & Materials, Inc., Newtown, CT, USA). The dispersion was stirred for 3 hours at room temperature and centrifuged at 13,200 rpm for 30 minutes. The NPs pellet was resuspended in 20 mL double-distilled water (DDW). The centrifuging and resuspending processes were repeated three times to remove PVA. The obtained DCT-loaded PS-PDLLA NPs were freeze-dried and stored at −70°C (DF8517; Ilshin Laboratory Co., Ltd., Seoul, Korea) for further experiments.
The encapsulation efficiency (EE) of DCT in PS-PDLLA NPs was measured by disrupting the NPs with DMSO (50× dilution). DCT concentration was determined using high-performance liquid chromatography (HPLC), equipped with a pump (Waters 1525; Waters Co., Milford, MA, USA), an automatic injector (Waters 717 plus), and dual λ absorbance detector (Waters 2487). A reverse phase C18 column (Gemini, 250×4.6 mm, 5 μm; Phenomenex, Torrance, CA, USA) was used and the mobile phase was composed of acetonitrile and DDW (65:35, volume per volume [v/v]). The detection wavelength, flow rate, and injection volume were 230 nm, 1.0 mL per minute, and 20 μL, respectively. The lower limit of quantitation (LLOQ) was 100 ng/mL and the inter- and intra-day variances of the HPLC method were within the acceptable range.
The morphology of DCT-loaded PS-PDLLA NPs was observed using a field emission-scanning electron microscope (FE-SEM; SUPRA 55VP; Carl Zeiss, Oberkochen, Germany). NPs were mounted on stubs and coated with Pt under vacuum. The particle size, polydispersity, and zeta potential of NPs were measured by electrophoretic light scattering method (ELS-Z; Otsuka Electronics, Tokyo, Japan). The change in the particle size and size distribution of developed nanoparticulate formulations in 50% (v/v) FBS solution was monitored for 24 hours.
In vitro release of DCT from NPs
Drug release from NPs was evaluated in vitro. DCT-loaded PS-PDLLA NPs dispersion (150 μL) was loaded into a mini GeBAflex-tube with a 12–14 kDa molecular weight cut-off (Gene Bio-Application Ltd., Kfar HaNagid, Israel). The dissolution medium (10 mL), PBS (pH 7.4) containing 0.5% (w/v) Tween 80, was agitated at 50 rpm using a shaking bath at 37°C. A 200 μL aliquot of the medium was collected at fixed times (3, 6, 9, 24, 48, 72, 96, 120, 144, 168, 192, 216, and 240 hours), and an equivalent volume of fresh media was replenished at each time point. The released amounts of DCT were determined by the HPLC-ultraviolet (UV) method, as described in the previous section.
In vitro cytotoxicity of blank PS-PDLLA NPs
PC-3 cells were obtained from the Korean cell line bank (Seoul, Korea) and cultured in RPMI 1640, containing 300 mg/L L-glutamine, 25 mM HEPES, and 25 mM NaHCO3, supplemented with 10% (v/v) heat-inactivated FBS, 1% (v/v) penicillin (100 U/mL), and streptomycin (0.1 mg/mL) in a 5% CO2 incubator with 95% relative humidity at 37°C.
The cytotoxicity of blank PS-PDLLA NPs in PC-3 cells was measured using an 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)-based assay. PC-3 cells (1.0×104 cells/well) were seeded in 96-well plates, and treated and incubated with freeze-dried blank PS-PDLLA NPs reconstituted with cell culture medium over a wide range of concentrations (2, 5, 10, 20, 50, 100, 200, 500, and 1,000 μg/mL) for 24, 48, and 72 hours at 37°C. After the incubation period, the medium was replaced with MTS-based CellTiter 96 Aqueous One Solution Cell Proliferation Assay Reagent (Promega Corp., Fitchburg, WI, USA) and incubated for 4 hours at 37°C according to the manufacturer’s protocol. The absorbance was measured at 490 nm using an EMax Precision Microplate Reader (Molecular Devices, Sunnyvale, CA, USA).
Cellular uptake study
DiI-loaded PS-PDLLA NPs were prepared using the same solvent evaporation method above (DiI:PS-PDLLA =1:40, w/w) to assess cellular uptake. The prepared DiI-loaded NPs were freeze-dried and stored at −70°C (DF8517; Ilshin Laboratory Co., Ltd.). The content of DiI in freeze-dried NPs was determined using an EMax Precision Microplate Reader at 560 nm by disrupting the NPs with DMSO.
The cellular uptake and distribution of NPs were observed by confocal laser scanning microscopy (CLSM). PC-3 cells were seeded in four-chamber culture slides (BD Biosciences, San Jose, CA, USA) at a density of 1.0×105 cells/well and incubated for 24 hours at 37°C. DiI (1 μg/mL)-loaded PS-PDLLA NPs were incubated for 2, 4, and 24 hours. The NPs suspensions were eliminated by washing with PBS (pH 7.4) three times. After washing, cells were fixed with 4% formaldehyde for 10 minutes. Fixed cells were treated with Vectashield® mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; H-1200; Vector Laboratories Inc., Burlingame, CA, USA) to prevent fading of fluorescence and to stain the nuclei blue. Cellular localization of DiI-loaded NPs was observed as a red color by CLSM (LSM 710; Carl Zeiss).
In vitro anti-tumor efficacy
Anti-tumor efficacy was tested in PC-3 cells using an MTS-based assay. PC-3 cells were seeded on 96-well plates at a density of 1.0×104/well and incubated overnight. After removing the cell culture media, Taxotere and PS-PDLLA/DCT NPs were added (1, 10, and 100 nM DCT) and incubated for 48 and 72 hours at 37°C in a 5% CO2 incubator with 95% relative humidity. Cells were treated with the MTS-based CellTiter 96® AQueous One Solution Cell Proliferation Assay Reagent and incubated at 37°C for 4 hours. The absorbance was measured at 490 nm with an EMax Precision Microplate Reader. Cell viability (%) was calculated from these absorbance values.
Serum biochemical analysis
Male BALB/c mice (10 weeks old; Charles River Laboratories, Wilmington, MA, USA) were used for serum biochemistry analysis of PS-PDLLA NPs. All mice were reared in a light-controlled room at 22°C±2°C and 55%±5% relative humidity (Animal Center for Pharmaceutical Research, College of Pharmacy, Seoul National University, Seoul, Korea). All animal experiments were approved by the Animal Care and Use Committee of the College of Pharmacy, Seoul National University. Blank (drug-unloaded) NPs were injected intravenously, at a dose of 25 mg/kg, every day for 1 week. Blood samples (0.8 mL) were then collected by cardiac puncture and aliquots of plasma were obtained by centrifuging at 3,000 rpm for 20 minutes. All serum biochemical parameters, such as total cholesterol (TCHO), alanine transaminase (ALT), aspartate aminotransferase (AST), total protein (TP), blood urea nitrogen (BUN), total bilirubin (TBIL), and inorganic phosphorus (IP), were obtained by using a Fuji Dri-Chem 3500s (Fujifilm Holdings Corp., Tokyo, Japan).
In vivo pharmacokinetic study in rats
Male Sprague-Dawley (SD) rats, weighing 250±5 g, were purchased from Orient Bio Inc. (Sungnam, Korea) to study in vivo pharmacokinetics. All rats were reared in a light-controlled room at 22°C±2°C and 55%±5% relative humidity (Animal Center for Pharmaceutical Research, College of Pharmacy, Seoul National University, Seoul, Korea). All animal experiments were approved by the Animal Care and Use Committee of the College of Pharmacy, Seoul National University. The femoral artery and vein were cannulated with Intramedic™ Polyethylene Tubing (PE-50; Becton, Dickinson & Co., Franklin Lakes, NJ, USA) under anesthesia with Zoletil® (Virbac S. A., Carros, France) at a dose of 50 mg/kg via intramuscular injection. Taxotere and DCT-loaded PS-PDLLA NPs at a dose of 1 mg/kg were administered via the femoral vein (intravenous bolus injection; 1 mL injection volume). Blood samples (~300 μL) were collected from the femoral artery at 1, 5, 15, 30, 45, 60, 90, 120, 180, 240, and 360 minutes after injection, followed by replenishment with a 0.9% sodium chloride injectable solution containing heparin (20 U/mL) at each time point to prevent blood clotting. After centrifugation of the blood samples (13,200 rpm for 3 minutes at 4°C), aliquots of plasma (~100 μL) were stored at -70°C until analysis. The concentration of DCT in rat plasma was measured with a liquid chromatography-tandem mass spectrometry (LC-MS/MS) system. Aliquots (50 μL) of the plasma samples were spiked with PTX as an internal standard in acetonitrile (10 μL volume, 1 μg/mL concentration), and mixed with acetonitrile (90 μL) for deproteinization. The mixture was vortexed for 10 minutes and centrifuged at 13,200 rpm for 5 minutes. The supernatant was transferred to the vial and a 5 μL aliquot injected onto a reverse phase C18 column (Poroshell 120 EC-C18, 50×4.6 mm, 2.7 μm; Agilent Technologies, Santa Clara, CA, USA). The mobile phase consisted of acetonitrile and DDW with 0.1% (v/v) formic acid (70:30, v/v) and the flow rate was 0.2 mL per minute. The column eluent was monitored using an Agilent LS-MS/MS system equipped with an Agilent Technologies 1260 Infinity HPLC system and Agilent Technologies 6430 Triple Quad LC/MS system. The optimized gas temperature, gas flow, nebulizer pressure, and capillary voltage were 350°C, 10 L per minute, 30 psi, and 6,000 V, respectively. The parameters manually optimized in the multiple reaction monitoring (MRM) mode with positive electrospray ionization (ESI) were m/z values of precursor to product ion, fragmentor voltage, and collision energy, which were 830.3 to 549.1, 168 V, and 17 eV for DCT and 876.2 to 591.0, 165 V, and 24 eV for PTX, respectively. The resolutions of the quadrupole Q1 and Q3 were set on wide/unit for DCT and unit/unit for PTX, respectively. The analytical data were processed using the MassHunter Workstation Software Quantitative Analysis (vB.05.00; Agilent Technologies). The retention times of DCT and PTX were 4.8 and 4.9 minutes, respectively. The lower limit of quantification (LLOQ) of DCT was 1 ng/mL, and precision and accuracy were within the acceptable range. The pharmacokinetic parameters of each formulation, area under the drug concentration in plasma–time curve from time zero to infinity (AUC), terminal half-life (t1/2), time-averaged total body clearance (CL), apparent volume of distribution at steady state (Vss), and mean residence time (MRT), were calculated using WinNonlin® (v3.1; Pharsight Corporation, Mountain View, CA, USA).
Statistical analysis
All experiments in this study were performed at least three times and the data presented as means ± standard deviation (SD). Two-tailed t-tests were performed for statistical analysis.
Results and discussion
Preparation and characterization of PS-PDLLA NPs
PS-PDLLA copolymer-based NPs were developed for the delivery of a model anticancer drug, DCT. Both PS and PDLLA, or their derivatives, have been widely used for biomedical applications, including drug delivery systems.8,18–20 In our previous reports, NPs based on biocompatible polymers were prepared and exhibited improved systemic exposure of the drug, in vivo anti-tumor efficacy, and cancer diagnosis.4,21–23 As a new polymer for drug delivery, PS-PDLLA was used for the fabrication of NPs to deliver DCT. 1H-nuclear magnetic resonance (NMR) and Fourier transform infrared (FT-IR) spectra revealed the characteristics of the PS-PDLLA copolymer when compared with reference (Figure 1).24–26 The 1H-NMR spectrum of the PS-PDLLA copolymer (Figure 1B) showed the peaks for phenyl protons of styrene and protons of lactide (-CO-CH[CH3]-O-) at 7.3 ppm and 5.2 ppm, respectively. FT-IR spectrum of PS-PDLLA (Figure 1C) showed that the C-H stretching band of the aromatic ring of PS and the C=O stretching band of the lactide were 3,026 cm−1 and 1,757 cm−1, respectively. It was expected that the ester linkage in the PDLLA backbone could be degraded via esterase-catalyzed hydrolysis in biological fluids.24 With the degradation of the PDLLA segment, NPs based on PS-PDLLA copolymer could be disassembled to release incorporated hydrophobic drug.
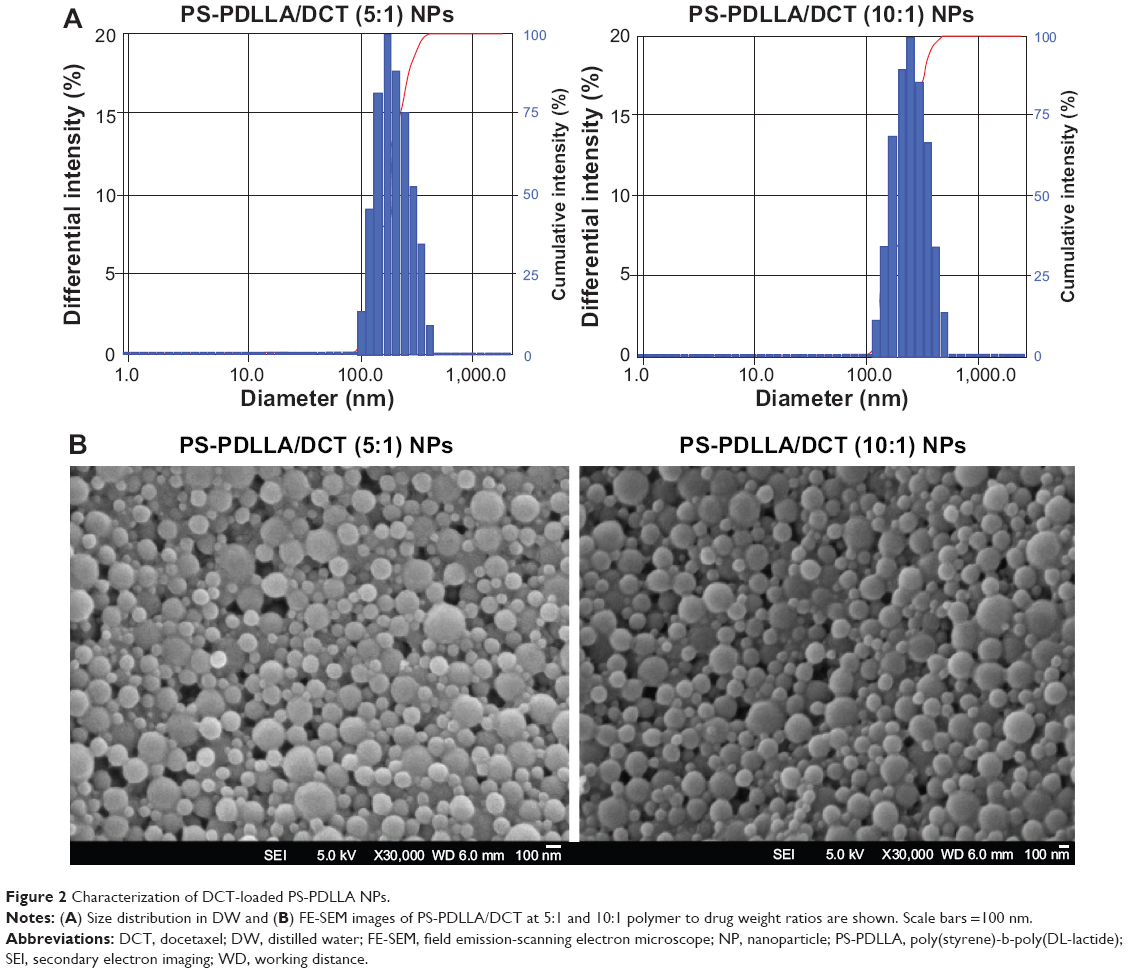
As shown in Table 1 and Figure 2, we prepared DCT-loaded NPs with a 220-240 nm mean diameter, narrow size distribution, negative zeta potential value, and >80% drug EE. The uniform size of the developed NPs could improve passive tumor targeting via the enhanced permeability and retention (EPR) effect. The weight ratio between polymer and drug was set at 5:1 and 10:1, and slightly different physicochemical properties of DCT-loaded NPs, including particle size and zeta potential, were observed. Spherical shape and similar particle size, compared to the hydrodynamic size (Table 1), of NPs are shown in FE-SEM images (Figure 2B). Moreover, the mean diameter and size distribution of the developed NPs containing DCT were consistently maintained for 24 hours in 50% (volume per volume) FBS (Figure 3). Proteins can bind to the surface of polymeric NPs after intravenous injection of NPs, forming a protein corona.28 Diverse proteins in biological fluids can participate in the formation of protein corona, acting as opsonins related to cellular uptake by a mononuclear phagocyte system (MPS).29 Thus, the interaction between proteins and a polymeric NP surface can modulate the pharmacokinetic properties of the drug and the fate of NPs.30 Stability testing in serum (Figure 3) indicated that DCT-loaded NPs could reach the tumor region without aggregation or precipitation in the blood stream and rapid clearance by the reticuloendothelial system.
 | Table 1 Characterization of DCT-loaded PS-PDLLA copolymer-based NPs |
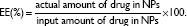
 | Figure 2 Characterization of DCT-loaded PS-PDLLA NPs. |
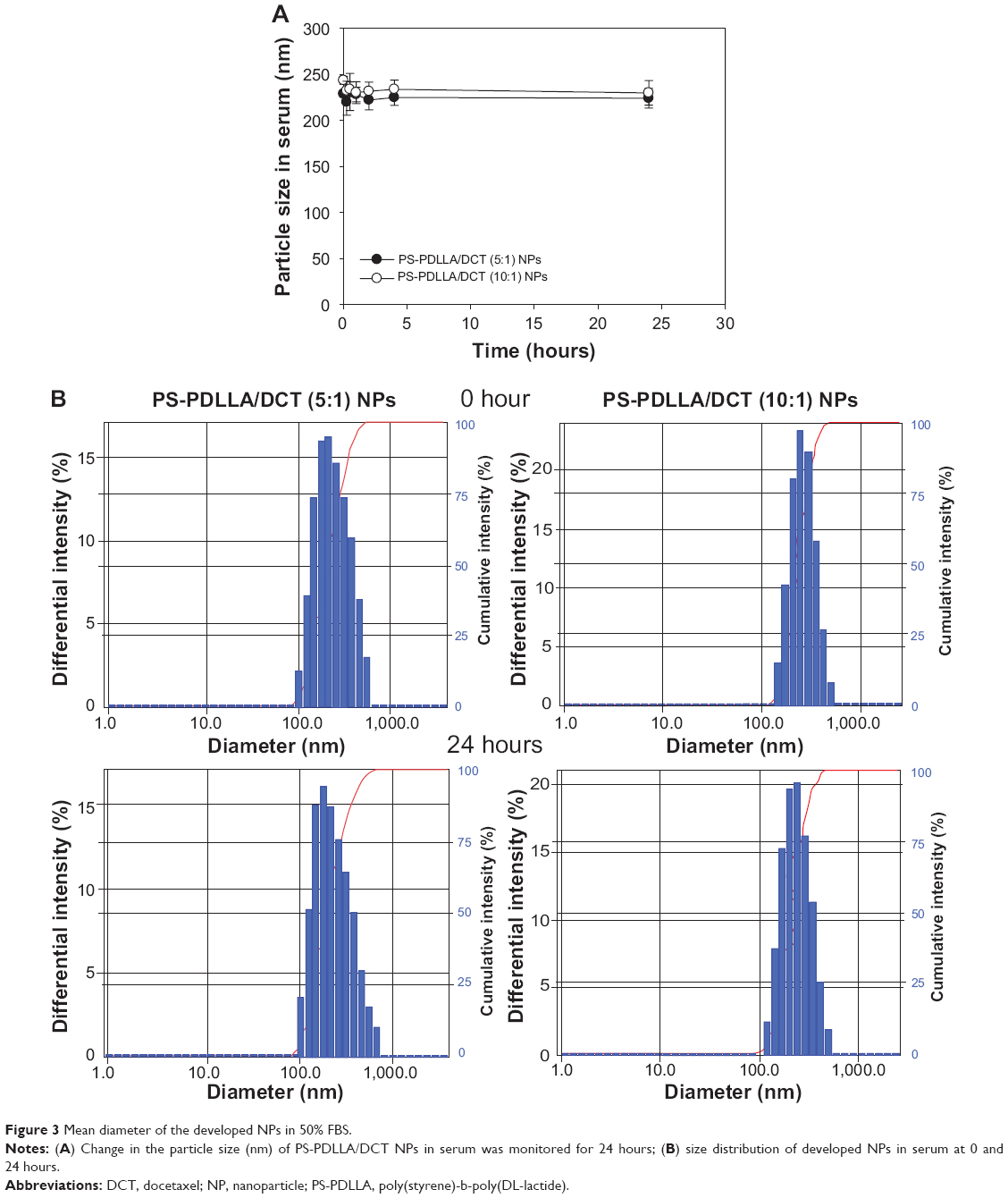 | Figure 3 Mean diameter of the developed NPs in 50% FBS. |
In vitro drug release
DCT release from NPs was assessed in vitro. A sustained release pattern is critical in anticancer drug delivery to decrease in vivo clearance and improve retention time in the bloodstream. It may contribute to reduced intravenous injection frequency, enhanced pharmacological efficacy, and improved patient compliance. As shown in Figure 4, DCT release from NPs at pH 7.4 was continued for 10 days. In DCT-loaded NPs with a 5:1 polymer:drug weight ratio, 77.06%±9.76% of DCT was released from NPs after 10 days. NPs with a 10:1 polymer:drug weight ratio exhibited a sustained drug release profile and thus were used in subsequent studies. A sustained drug release pattern can improve systemic exposure of the drug after intravenous injection. Degradation of ester linkages in the PDLLA segment of the copolymer could be one of the mechanisms of drug release from NPs in biological fluids.27 It is assumed that the existence of esterases in biological fluids can facilitate disruption of NPs, thereby eliminating the copolymers from the body, and releasing the drug.
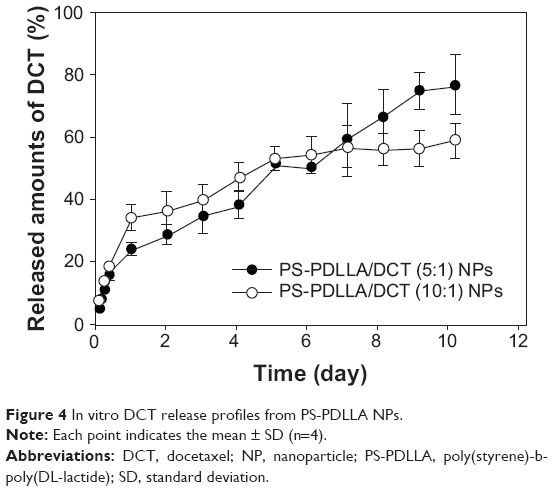 | Figure 4 In vitro DCT release profiles from PS-PDLLA NPs. |
In vitro cytotoxicity of blank NPs
Cytotoxicity of blank (without drug loading) NPs was evaluated in PC-3 cells. PC-3 cell viability (%) was assessed after 24, 48, and 72 hours of incubation at 0–1,000 μg/mL of the PS-PDLLA copolymer (Figure 5). The developed blank NPs exhibited negligible cytotoxicity in PC-3 cells under these conditions. Biocompatibility of PS-PDLLA can be estimated from the biomedical application of each polymer component as reported.27,31 The result of the cytotoxicity test supports the feasibility of PS-PDLLA NPs as a safe drug delivery vehicle.
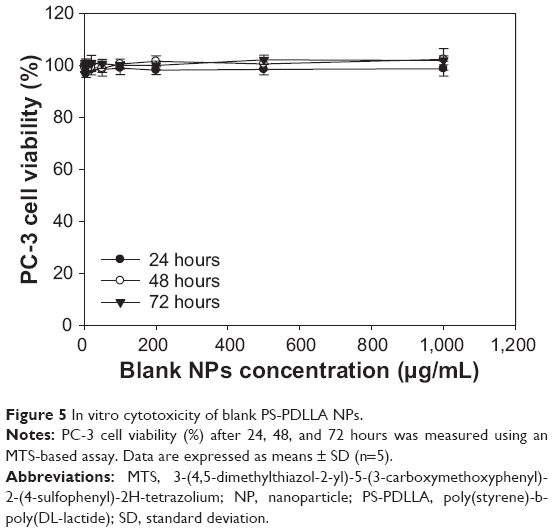 | Figure 5 In vitro cytotoxicity of blank PS-PDLLA NPs. |
Cellular uptake
Cellular uptake and the distribution pattern of the developed NPs were observed by CLSM (Figure 6). DiI, as a hydrophobic fluorescent dye, was incorporated into the nanovesicles instead of DCT for monitoring the intracellular movement of NPs for 24 hours in PC-3 cells. The fluorescence intensity was stronger at 24 hours compared to the 2- and 4-hour incubated groups. As incubation time increased, cellular uptake of NPs increased. Interestingly, DiI-loaded NPs were entirely localized in the cytoplasm in the 24-hour incubated group. Although the PS-PDLLA copolymer does not include a tumor-targeting moiety, NPs based on the PS-PDLLA copolymer were expected to be efficiently taken up into the cells via endocytosis. Cellular uptake efficiency of the developed NPs can improve cancer therapy after the arrival of NPs in the tumor region via passive tumor targeting, based on the EPR effect.
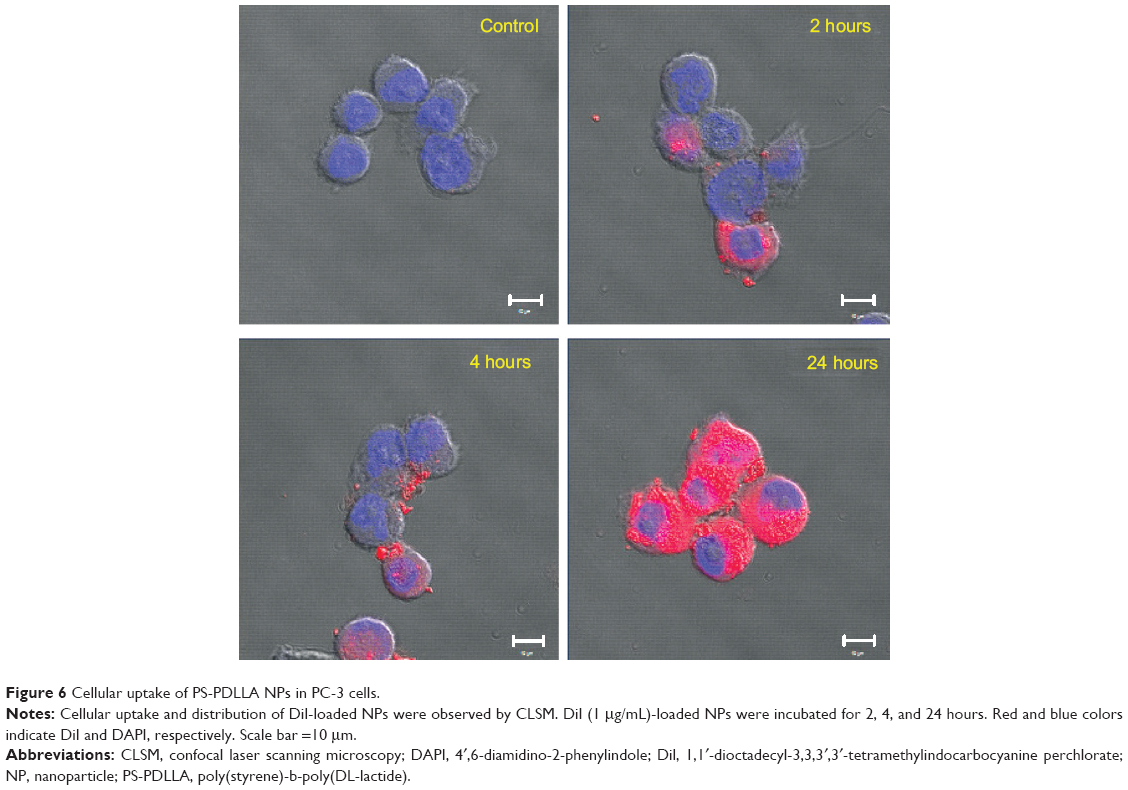 | Figure 6 Cellular uptake of PS-PDLLA NPs in PC-3 cells. |
In vitro anti-tumor efficacy
Anti-tumor efficacy of DCT-loaded PS-PDLLA NPs was evaluated in PC-3 cells (Figure 7). Various approaches have been used for DCT delivery for prostate cancer therapy.32,33 DCT concentration (1-100 nM) in this study was set up considering its reported IC50 values in PC-3 cells.34–36 Cell viability was measured using an MTS-based assay after 48 and 72 hours of incubation of Taxotere and PS-PDLLA/DCT NPs. At all DCT concentration (1, 10 and 100 nM) and incubation time (48 and 72 hours) groups, the cell viability (%) of the PS-PDLLA/DCT NP-treated group was lower than that of the Taxotere group (P<0.05). Cytotoxicity testing of blank NPs suggested that they did not induce any serious cytotoxicity in PC-3 cells (Figure 5). Therefore, the observed cytotoxicity in Figure 7 seems to be based on the performance of DCT-incorporated PS-PDLLA NPs. In particular, in vitro anti-tumor efficacy of the developed NPs was higher than that of the commercial formulation, Taxotere. The lower cell viability of the developed DCT-loaded NPs also indicates their improved cellular uptake efficiency. Furthermore, improved inhibition of cancer cell growth could lead to enhanced in vivo anti-tumor efficacy, based on the stability of the developed NPs in serum (Figure 3).
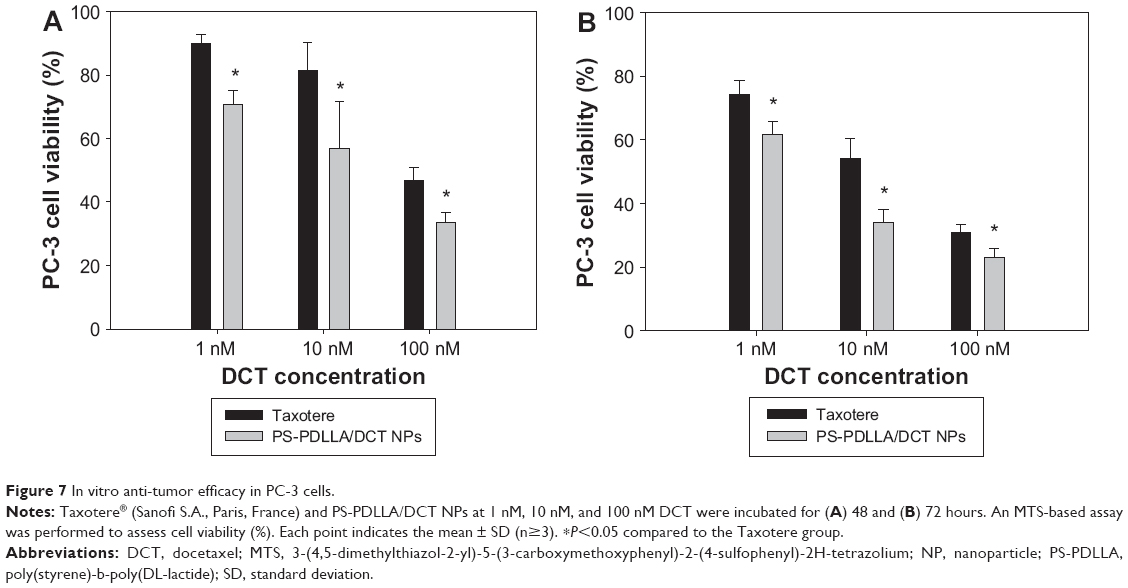 | Figure 7 In vitro anti-tumor efficacy in PC-3 cells. |
Serum biochemistry analysis
In vivo toxicity of blank PS-PDLLA NPs was assessed in a mouse model, based principally on serum biochemical analysis. Numerous NPs based on each copolymer component (PS or PDLLA) were fabricated and their characteristics, including toxicity, were investigated.27,31 It was reported that the cytotoxicity of PS NPs was influenced by the functional group attached on the surface of the NPs, the particle size, and the presence of enzymes or serum.37 PDLLA has been used as a biocompatible polymer, alone or in a conjugated form, for drug delivery and tissue engineering.38,39 Nonetheless, the in vivo toxicity of PS-PDLLA NPs has not been previously investigated. Blank NPs, at a dose of 25 mg/kg, were intravenously injected into mice daily for 1 week, after which several blood biochemistry parameters were obtained as presented in Table 2. These parameters are for assessing toxicity or abnormality in the hepatobiliary system (TCHO, ALT, AST, TP, albumin, and TBIL), the kidney (BUN, creatinine), and others (Ca2+, IP). There was no significant difference between the control and the NP-treated group, indicating that intravenous injection of the developed PS-PDLLA NPs at the times and doses used resulted in no serious toxicities. Together with the cytotoxicity results (Figure 5), this demonstrates the biocompatibility of the developed NPs and supports their use as an injection formulation for anticancer drug delivery.
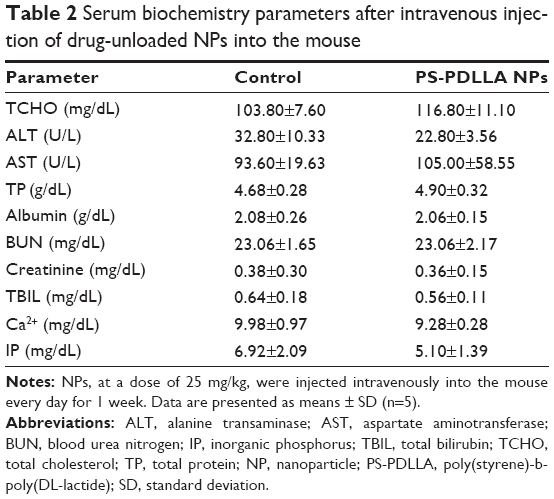 | Table 2 Serum biochemistry parameters after intravenous injection of drug-unloaded NPs into the mouse |
In vivo pharmacokinetics
Pharmacokinetic properties of PS-PDLLA/DCT NPs were compared with those of Taxotere in rats after intravenous administration. Though serum biochemical analysis was performed in mice, pharmacokinetic study was done in rats considering their special characteristics, such as enough blood volume for multiple sampling compared to mice. The DCT concentration in plasma over time is shown in Figure 8, and several pharmacokinetic parameters were obtained (Table 3). In several reports,40–42 polymeric NPs containing DCT were fabricated; improved in vivo pharmacokinetic properties were observed compared to DCT solution or commercial formulations. However, toxicity and other factors have hindered their clinical application, despite their considerable in vivo performance in animal models. NPs based on the PS-PDLLA copolymer were prepared and the pharmacokinetics of DCT investigated. As shown in Table 3, clearance (CL) of the Taxotere-treated group was lower than that of the PS-PDLLA/DCT (10:1) group (P<0.05). AUC, t1/2, and MRT values of the PS-PDLLA/DCT group were 2.31-, 2.33-, and 4.16-fold higher than those of the Taxotere group, respectively (P<0.05). Decreased in vivo clearance of DCT appeared to be the result of sustained drug release and in vitro stability in serum. As reported in other studies regarding intravenous DCT delivery, in vivo clearance of DCT from developed NPs was decreased compared to Taxotere in this investigation.40,42 Prolonged circulation time of drug may contribute to the decreased dosing frequency and improved patient compliance. Moreover, it is expected that prolonged drug circulation could improve in vivo anti-tumor efficacy via passive tumor targeting and elevated systemic exposure.
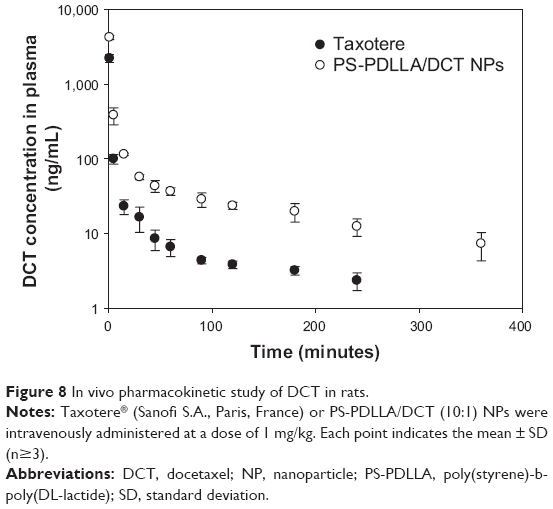 | Figure 8 In vivo pharmacokinetic study of DCT in rats. |
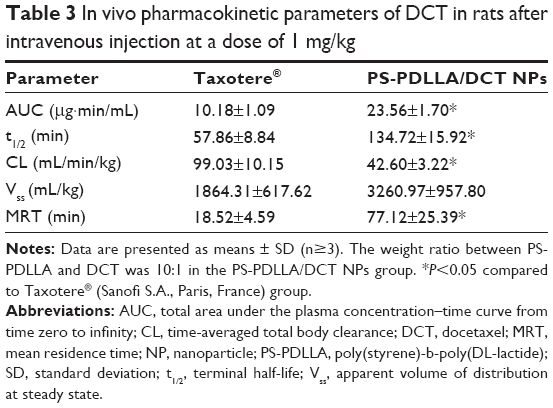 | Table 3 In vivo pharmacokinetic parameters of DCT in rats after intravenous injection at a dose of 1 mg/kg |
Conclusion
We developed NPs based on a PS-PDLLA copolymer for delivery of DCT via intravenous injection. We fabricated NPs with a narrow size distribution, negative zeta potential value, spherical shape, and high drug EE. The particle size of the NPs was adequately maintained in serum and a sustained drug release pattern was observed. Blank NPs did not induce any serious cytotoxicity in PC-3 cells. Cellular uptake and localization of fluorescence dye-loaded NPs were confirmed by CLSM. Improved inhibition of cancer cell growth was shown in the PS-PDLLA/DCT NP-treated group compared to the Taxotere-treated group. Decreased in vivo drug clearance, elevated systemic exposure, and prolonged circulation of the drug were verified in a pharmacokinetic study in rats. All of these results support the use of the PS-PDLLA copolymer for fabrication of NPs in anticancer drug delivery.
Acknowledgment
This research was supported by the National Research Foundation of Korea funded by the Korean government (MSIP) (2009-0083533 and NRF-2012R1A1A1038944).
Disclosure
The authors report no conflicts of interest in this work.
References
Arvizo RR, Saha S, Wang E, Robertson JD, Bhattacharya R, Mukherjee P. Inhibition of tumor growth and metastasis by a self-therapeutic nanoparticle. Proc Natl Acad Sci U S A. 2013;110(17):6700–6705. | ||
Muthu MS, Feng SS. Theranostic liposomes for cancer diagnosis and treatment: current development and pre-clinical success. Expert Opin Drug Deliv. 2013;10(2):151–155. | ||
Yoon G, Park JW, Yoon IS. Solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs): recent advances in drug delivery. J Pharm Invest. 2013;43(5):353–362. | ||
Cho HJ, Yoon HY, Koo H, et al. Self-assembled nanoparticles based on hyaluronic acid-ceramide (HA-CE) and Pluronic® for tumor-targeted delivery of docetaxel. Biomaterials. 2011;32(29):7181–7190. | ||
Park J, Wrzesinski SH, Stern E, et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater. 2012;11(10):895–905. | ||
Termsarasab U, Cho HJ, Kim DH, et al. Chitosan oligosaccharide-arachidic acid-based nanoparticles for anti-cancer drug delivery. Int J Pharm. 2013;441(1–2):373–380. | ||
Whitehead KA, Matthews J, Chang PH, et al. In vitro-in vivo translation of lipid nanoparticles for hepatocellular siRNA delivery. ACS Nano. 2012;6(8):6922–6929. | ||
Kulkarni SA, Feng SS. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm Res. 2013;30(10):2512–2522. | ||
Mohammad AK, Amayreh LK, Mazzara JM, Reineke JJ. Rapid lymph accumulation of polystyrene nanoparticles following pulmonary administration. Pharm Res. 2013;30(2):424–434. | ||
Lee SY, Kim S, Tyler JY, Park K, Cheng JX. Blood-stable, tumor-adaptable disulfide bonded mPEG-(Cys)4-PDLLA micelles for chemotherapy. Biomaterials. 2013;34(2):552–561. | ||
Keen I, Yu A, Cheng HH, et al. Control of the orientation of symmetric poly(styrene)-block-poly(D,L-lactide) block copolymers using statistical copolymers of dissimilar composition. Langmuir. 2012;28(45): 15876–15888. | ||
Lo KH, Chen MC, Ho RM, Sung HW. Pore-filling nanoporous templates from degradable block copolymers for nanoscale drug delivery. ACS Nano. 2009;3(9):2660–2666. | ||
Yin YM, Cui FD, Mu CF, et al. Docetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluation. J Control Release. 2009;140(2):86–94. | ||
Muthu MS, Kulkarni SA, Raju A, Feng SS. Theranostic liposomes of TPGS coating for targeted co-delivery of docetaxel and quantum dots. Biomaterials. 2012;33(12):3494–3501. | ||
Tong R, Chiang HH, Kohane DS. Photoswitchable nanoparticles for in vivo cancer chemotherapy. Proc Natl Acad Sci U S A. 2013;110(47): 19048–19053. | ||
Zheng C, Zheng M, Gong P, et al. Polypeptide cationic micelles mediated co-delivery of docetaxel and siRNA for synergistic tumor therapy. Biomaterials. 2013;34(13):3431–3438. | ||
Yang R, Yang SG, Shim WS, et al. Lung-specific delivery of paclitaxel by chitosan-modified PLGA nanoparticles via transient formation of microaggregates. J Pharm Sci. 2008;98(3):970–984. | ||
Hu Q, Gao X, Gu G, et al. Glioma therapy using tumor homing and penetrating peptide-functionalized PEG-PLA nanoparticles loaded with paclitaxel. Biomaterials. 2013;34(22):5640–5650. | ||
Larson N, Greish K, Bauer H, Maeda H, Ghandehari H. Synthesis and evaluation of poly(styrene-co-maleic acid) micellar nanocarriers for the delivery of tanespimycin. Int J Pharm. 2011;420(1):111–117. | ||
Thomas C, Rawat A, Hope-Weeks L, Ahsan F. Aerosolized PLA and PLGA nanoparticles enhance humoral, mucosal and cytokine responses to hepatitis B vaccine. Mol Pharm. 2011;8(2):405–415. | ||
Cho HJ, Yoon HY, Koo H, et al. Hyaluronic acid-ceramide-based optical/MR dual imaging nanoprobe for cancer diagnosis. J Control Release. 2012;162(1):111–118. | ||
Cho HJ, Yoon IS, Yoon HY, et al. Polyethylene glycol-conjugated hyaluronic acid-ceramide self-assembled nanoparticles for targeted delivery of doxorubicin. Biomaterials. 2012;33(4):1190–1200. | ||
Jin YJ, Termsarasab U, Ko SH, et al. Hyaluronic acid derivative-based self-assembled nanoparticles for the treatment of melanoma. Pharm Res. 2012;29(12):3443–3454. | ||
Guo Q, Li X, Ding Q, et al. Preparation and characterization of poly(pluronic-co-L-lactide) nanofibers for tissue engineering. Int J Biol Macromol. 2013;58:79–86. | ||
Wang W, Zhang H, Geng W, et al. Synthesis of poly(methyl methacrylate)-b-polystyrene with high molecular weight by DPE seeded emulsion polymerization and its application in proton exchange membrane. J Colloid Interface Sci. 2013;406:154–164. | ||
Zhao ZX, Gao SY, Wang JC, et al. Self-assembly nanomicelles based on cationic mPEG-PLA-b-Polyarginine(R15) triblock copolymer for siRNA delivery. Biomaterials. 2012;33(28):6793–6807. | ||
Samarajeewa S, Ibricevic A, Gunsten SP, et al. Degradable cationic shell cross-linked knedel-like nanoparticles: synthesis, degradation, nucleic acid binding, and in vitro evaluation. Biomacromolecules. 2013;14(4): 1018–1027. | ||
Cedervall T, Lynch I, Lindman S, et al. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci U S A. 2007;104(7): 2050–2055. | ||
Owens DE 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. | ||
Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134(4):2139–2147. | ||
Ekkapongpisit M, Giovia A, Follo C, Caputo G, Isidoro C. Biocompatibility, endocytosis, and intracellular trafficking of mesoporous silica and polystyrene nanoparticles in ovarian cancer cells: effects of size and surface charge groups. Int J Nanomedicine. 2012;7: 4147–4158. | ||
Kobayashi D, Kawai N, Sato S, et al. Thermotherapy using magnetic cationic liposomes powerfully suppresses prostate cancer bone metastasis in a novel rat model. Prostate. 2013;73(9):913–922. | ||
Xiao Z, Levy-Nissenbaum E, Alexis F, et al. Engineering of targeted nanoparticles for cancer therapy using internalizing aptamers isolated by cell-uptake selection. ACS Nano. 2012;6(1):696–704. | ||
Ting HJ, Hsu J, Bao BY, Lee YF. Docetaxel-induced growth inhibition and apoptosis in androgen independent prostate cancer cells are enhanced by 1alpha,25-dihydroxyvitamin D3. Cancer Let. 2007;247(1):122–129. | ||
Williams JF, Muenchen HJ, Kamradt JM, Korenchuk S, Pienta KJ. Treatment of androgen-independent prostate cancer using antimicrotubule agents docetaxel and estramustine in combination: an experimental study. Prostate. 2000;44(4):275–278. | ||
O’Neill AJ, Prencipe M, Dowling C, et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol Cancer. 2011;10:126. | ||
Clift MJ, Bhattacharjee S, Brown DM, Stone V. The effects of serum on the toxicity of manufactured nanoparticles. Toxicol Lett. 2010;198(3): 358–365. | ||
Lee JS, Feijen J. Biodegradable polymersomes as carriers and release systems for paclitaxel using Oregon Green® 488 labeled paclitaxel as a model compound. J Control Release. 2012;158(2):312–318. | ||
Ren J, Zhao P, Ren T, Gu S, Pan K. Poly(D,L-lactide)/nano-hydroxyapatite composite scaffolds for bone tissue engineering and biocompatibility evaluation. J Mater Sci Mater Med. 2008;19(3):1075–1082. | ||
Ernsting MJ, Tang WL, MacCallum NW, Li SD. Preclinical pharmacokinetic, biodistribution, and anti-cancer efficacy studies of a docetaxel-carboxymethylcellulose nanoparticle in mouse models. Biomaterials. 2012;33(5):1445–1554. | ||
Ho KS, Aman AM, Al-awar RS, Shoichet MS. Amphiphilic micelles of poly(2-methyl-2-carboxytrimethylene carbonate-co-D,L-lactide)-graft-poly(ethylene glycol) for anti-cancer drug delivery to solid tumours. Biomaterials. 2012;33(7):2223–2229. | ||
Mu CF, Balakrishnan P, Cui FD, et al. The effects of mixed MPEG-PLA/Pluronic copolymer micelles on the bioavailability and multidrug resistance of docetaxel. Biomaterials. 2010;31(8):2371–2379. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.