Back to Journals » Journal of Inflammation Research » Volume 18
Emerging Role and Function of Th9 Cells in Allergic Inflammation
Authors Kaminuma O ![]() , Kitamura N, Gotoh M
, Kitamura N, Gotoh M
Received 1 October 2025
Accepted for publication 8 November 2025
Published 22 November 2025 Volume 2025:18 Pages 16385—16397
DOI https://doi.org/10.2147/JIR.S546234
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Osamu Kaminuma,1,* Noriko Kitamura,2,* Minoru Gotoh3
1Department of Disease Models, Research Institute for Radiation Biology and Medicine, Hiroshima University, Hiroshima, Japan; 2Department of Pollinosis Science, Nippon Medical School, Tokyo, Japan; 3Department of Otorhinolaryngology, Nippon Medical School, Tokyo, Japan
*These authors contributed equally to this work
Correspondence: Osamu Kaminuma, Department of Disease Models, Research Institute for Radiation Biology and Medicine, Hiroshima University, Hiroshima, Japan, Tel +81-82-257-5819, Email [email protected]
Abstract: Th9 cells have emerged as pivotal orchestrators of allergic inflammation across the airway, skin, and nasal mucosa, constituting a mechanistically distinct axis beyond canonical Th2 immunity. This review specifically highlights: (i) the Th9 axis as a unifying driver in asthma, atopic dermatitis, and allergic rhinitis; (ii) key mechanistic programs, including signal transducer and activator of transcription (STAT) 5/STAT6 licensing of the IL9 locus, peroxisome proliferator-activated receptor (PPAR) γ-mammalian target of rapamycin complex (mTORC) 1 metabolic wiring, and the IL-9-monocarboxylate transporter (MCT) 1 feedback loop; (iii) organ-level phenotypes such as eosinophil-independent bronchial hyperresponsiveness (BHR) and variable steroid responsiveness; and (iv) therapeutic implications, including biomarker-guided endotyping, Janus kinase (JAK) inhibition, TNF-like ligand (TL) 1A/death receptor (DR) 3 blockade, and metabolic or airway smooth muscle (ASM) tone modulation. Differentiating under the combined influence of interleukin (IL)‑4 and tumor growth factor (TGF)-β, Th9 cells secrete IL‑9, a pleiotropic cytokine that drives mast‑cell proliferation, goblet cell metaplasia, and airway remodeling. Their transcriptional program is epigenetically licensed by STAT5/STAT6, which opens chromatin at the IL9 locus and is metabolically sustained by a PPARγ-mechanistic/mTORC1-dependent glycolytic state. This bioenergetic wiring establishes an IL‑9-MCT1 feedback loop that reinforces effector function and durability. Clinically, Th9 signatures align with BHR, which can be eosinophil-independent and variably responsive to inhaled corticosteroids; experimental models further demonstrate that Th9‑mediated BHR persists in eosinophil-deficient contexts and displays relative glucocorticoid resistance. Within the broader landscape of bronchial asthma, a chronic inflammatory disease marked by reversible airway obstruction, mucus hypersecretion, and BHR, these insights help explain the non‑Th2 endotypes that respond poorly to standard anti-inflammatory therapies. Although anti-IL‑9 monoclonal antibodies have not improved lung function in unselected asthma cohorts, aggregate evidence argues for biomarker‑guided endotyping and upstream pathway intervention, including tumor necrosis factor-like cytokine TL1A/DR3 blockade and metabolic modulation, as more rational strategies to disrupt Th9 pathogenic circuits. Importantly, the Th9 axis also represents one of the non-IgE‑mediated hypersensitivity mechanisms pertinent to allergic conditions, a perspective that enhances clinical discoverability and bench‑to‑bedside translation. This review integrates foundational mechanistic and pharmacologic knowledge with recent advances, positioning Th9 cells as a unifying driver across asthma, atopic dermatitis, and allergic rhinitis, and delineates therapeutic avenues that target epigenetic, metabolic, and cytokine networks sustaining Th9‑dependent diseases.
Keywords: allergic rhinitis, asthma, atopic dermatitis, bronchial hyperresponsiveness, non-IgE-mediated hypersensitivity
Introduction
The immunopathology of allergic diseases has evolved beyond the classical Th2 framework, revealing multiple T cell subsets that shape distinct inflammatory circuits. Among these, Th9 cells represent a unique lineage with specialized effector functions.1,2 Induced by interleukin (IL)‑4 in combination with tumor growth factor (TGF)‑β, Th9 cells acquire a transcriptional program centered on IL‑9 secretion, a cytokine with broad tissue effects, including mast‑cell expansion, goblet‑cell metaplasia, epithelial remodeling, and modulation of airway smooth‑muscle tone.1,3,4 Unlike Th2 cells, which primarily drive eosinophilic inflammation, Th9 cells contribute to disease phenotypes that are often severe, persistent, and variably responsive to corticosteroids.5 This paradigm is particularly relevant to bronchial asthma, a chronic inflammatory disorder affecting over 330 million individuals worldwide and characterized by recurrent wheeze, dyspnea, chest tightness, and physiologic hallmarks of reversible airway obstruction, mucus hypersecretion, and bronchial hyperresponsiveness (BHR).6 While asthma has long been conceptualized as a Th2‑dominant disease, clinical and molecular studies over the past two decades have revealed striking heterogeneity and multiple mechanistic endotypes that better explain variable natural history and treatment response.7,8 Experimental evidence further demonstrates that Th2‑independent pathways, including Th9, can sustain airway inflammation and BHR, even in eosinophil‑deficient contexts, underscoring the need for a broader immunologic map to guide therapy.9 Recent advances have clarified how Th9 cells maintain pathogenicity across tissues. Epigenetically, signal transducer and activator of transcription (STAT) 5 and STAT6 remodel chromatin at the IL9 locus, enabling bystander IL‑9 production in response to paracrine IL‑2/IL‑4 without T-cell receptor (TCR) restimulation.10,11 Metabolically, a peroxisome proliferator-activated receptor (PPAR) γ-mechanistic/mammalian target of rapamycin complex (mTORC) 1-driven glycolytic circuit establishes an IL‑9-monocarboxylate transporter (MCT) 1 feedback loop that stabilizes effector function.12,13 These intrinsic programs intersect with extrinsic cues, such as tumor necrosis factor-like cytokine (TL) 1A/death receptor (DR) 3 signaling, which promotes epithelial injury and barrier loss, and Piezo1‑mediated mechanotransduction, linking matrix stiffness to Th9 differentiation and airway responsiveness.14,15
Therapeutically, the failure of anti-IL‑9 monoclonal antibody therapy (MEDI‑528) to improve lung function or exacerbation rates in uncontrolled asthma highlights the limitations of cytokine‑centric strategies and reinforces the need for biomarker‑guided endotyping.16 Candidate biomarkers include STAT5/6 transcriptional signatures, IL‑9⁺CD4⁺ frequencies, and PPARγ/MCT1 metabolic modules.11,12,17 Upstream or parallel interventions, such as Janus kinase (JAK) inhibition, TL1A/DR3 blockade, and airway‑smooth‑muscle-targeted approaches, offer promising avenues to blunt BHR irrespective of granulocyte burden.18–20
In this review, we have integrated foundational and emerging insights into Th9 biology in asthma, atopic dermatitis, and allergic rhinitis through the comprehensive review of recent publications. Inclusion criteria comprised peer‑reviewed mechanistic, translational, or clinical studies in English and major guidelines/position statements; we excluded non‑peer‑reviewed commentaries, single‑patient case reports unless mechanistically informative, and duplicate cohorts. When findings conflicted, we prioritized higher‑quality evidence and biological plausibility and noted uncertainty explicitly.
We begin by delineating the transcriptional, epigenetic, and metabolic networks governing Th9 differentiation, then examine the organ‑level pathobiology and mechanisms of hyperresponsiveness, including the RhoA/Rho-associated coiled-coil containing protein kinase (ROCK)-myosin light chain phosphatase (MLCP) axis, and conclude with a therapeutic and endotyping framework tailored to Th9‑high allergic diseases.1,21 In alignment with the modern nomenclature proposed by the European Academy of Allergy and Clinical Immunology (EAACI) position paper,22 hypersensitivity mechanisms extend beyond classical IgE‑mediated pathways and encompass T‑cell-driven and tissue‑ or metabolism‑linked endotypes. Situating Th9‑driven inflammation within these non‑IgE or mixed mechanisms provides a clearer clinical bridge from bench to bedside and a rationale for biomarker‑guided endotyping.
Differentiation and Regulatory Networks of Th9
IL‑9 is a pleiotropic cytokine that drives mast‑cell proliferation, goblet‑cell hyperplasia, induction of IL‑13, influx and local maturation of eosinophils, and the emergence of BHR.5,23,24 Historically, IL‑9 has been regarded as a Th2 cytokine and implicated in allergic asthma as well as parasitic infections.25,26 In 2008, a CD4⁺ T‑helper subset that preferentially produces IL‑9 was identified and designated Th9.3 Th9 cells arise from naïve T cells under the concerted influence of IL‑4 and TGF-β.3,27 Although their full transcriptional circuitry remains incompletely resolved, several signature factors, including STAT6, GATA binding protein (GATA) 3, purine-rich box (PU).1, and interferon regulatory factor (IRF) 4, have been shown to orchestrate Th9 polarization.1,2,28 IL‑4 activates the STAT6 pathway, which in turn induces GATA3, a master regulator of Th2 identity, together with IRF4. In multiple, though not all, studies, GATA3 augments IL‑9 production in Th9 cells.27,29 GATA3 may also antagonize the forkhead transcription factor Foxp3, the lineage determinant of regulatory T (Treg) cells.30,31 Notably, both Treg and Th9 development require TGF‑β; this signaling induces PU.1, which enhances IL‑9 during Th9 differentiation and counterbalances GATA3 function.28,32 TGF‑β further induces IRF4, and STAT6, together with IRF4, has been shown to bind directly to the IL9 promoter.4,28,33 Although IRF4, STAT6, and GATA3 are indispensable across several Th lineages, the specific combinations and sequence of cytokine cues that engage this network are crucial for committing cells to a Th9 fate.2,28,33 The differentiation and regulatory networks of Th9 cells have also been comprehensively depicted in several recent reviews and original articles.28,33,34
Th9 cells have been linked to a spectrum of conditions, including autoimmunity and pathogen‑mediated immunomodulatory disorders, while multiple studies have suggested that Th9 cells play a prominent role in anti-tumor immunity.35,36 Purwar et al. demonstrated that Th9 cells elicit stronger anti‑tumor activity than Th1 or Th17 cells in adoptive‑transfer mouse models.37 Th9 cells potentiate adaptive anti‑tumor responses via IL‑9, which activates mast cells endowed with tumor growth-suppressive functions.37,38 Lu et al. reported that Th9 cells promote robust host CD8⁺ cytotoxic T‑lymphocyte (CTL) responses by recruiting dendritic cells (DCs) to tumor sites through C-C motif chemokine ligand (CCL) 20/C-C motif chemokine receptor (CCR) 6‑dependent pathways.38,39
Although the capacity of Th9 cells to kill cancer cells directly remains debated, Purwar et al. showed that OT‑II-derived Th9 cells killed ovalbumin (OVA)‑expressing tumor cells and expressed high levels of granzyme B; down‑regulating granzyme B diminished their anti‑melanoma efficacy.37 Beyond STAT6, IRF4, and PU.1, cooperative Ets factors (E26 transformation-specific (ETS) transcription factors-related gene (ERG)/ETS variant 5 (ETV5)) together with the Dual-specificity phosphatase (DUSP) 8-Pur‑α axis further refine IL‑9 transcriptional control; critically, STAT5 is required for enhancer accessibility and for Basic leucine zipper transcription factor, ATF-like (BATF)‑mediated activation.28,40
Organ-Level Pathobiology: Asthma, Dermatitis, and Rhinitis
Bronchial Asthma
Bronchial asthma is a chronic inflammatory airway disease characterized by reversible obstruction, mucus hypersecretion, and BHR.6 Traditionally, type 2 cytokines, IL-4, IL-5, and IL-13, have been considered central to its pathogenesis, driving IgE class switching, eosinophil recruitment, and airway remodeling (Table 1).5,53,54 IL-4 promotes IgE production and mast cell activation,53,55 while IL-13 induces goblet cell hyperplasia and airway smooth muscle changes.54,56 IL-5 is essential for eosinophil maturation and survival, and eosinophil-derived mediators contribute to tissue damage and BHR.57–60 Eosinophil levels often correlate with disease severity and exacerbation frequency.61 However, studies using eosinophil-deficient mice have shown that BHR can occur independently of eosinophils, indicating heterogeneity in asthma mechanisms.5,62,63 In addition to Th2 pathways, Th1 and Th17 cells can also induce BHR, often associated with neutrophilic inflammation and steroid resistance.9,64–66 These findings support the concept of asthma as a spectrum of endotypes, with variable contributions from Th2, Th1, and Th17 cells, and highlight the need for tailored therapeutic approaches.7,8,67 Beyond these traditional and recently proposed pathophysiological mechanisms, the pivotal role of the Th9-IL-9 axis in asthma pathogenesis, particularly in the development of BHR, is discussed in the following section.
 |
Table 1 Th9 Axis Across Allergic Diseases: Pathobiology, Biomarkers, and Therapeutic Implications |
Atopic Dermatitis
Atopic dermatitis (AD) represents a chronic, relapsing inflammatory skin disorder characterized by epidermal barrier dysfunction, pruritus, and heightened susceptibility to microbial colonization.68,69 While traditionally associated with type 2 cytokines such as IL‑4 and IL‑13, emerging evidence implicates Th9 cells as critical amplifiers of cutaneous inflammation (Table 1).46,47 Th9 cells, through robust IL‑9 secretion, exert pleiotropic effects on mast cells, keratinocytes, and resident immune cells, fostering a microenvironment conducive to chronic allergic inflammation.42,43 IL‑9 promotes mast-cell proliferation and activation, enhancing histamine release and protease secretion, which aggravate pruritus and tissue remodeling.70–72 Furthermore, IL‑9 synergizes with IL‑4 and IL‑13 to reinforce epithelial barrier disruption and mucus-like glycoprotein deposition, features increasingly recognized in severe AD phenotypes.44,73
Recent mechanistic studies revealed that the Th9 effector function in the skin is metabolically gated by the PPARγ-mTORC1 axis, which drives glycolytic reprogramming and selectively augments IL‑9 expression without proportionally increasing IL‑13.12,13 This metabolic dependency is further stabilized by an IL‑9-MCT1 feedback loop, enabling sustained lactate flux and proliferation under nutrient-variable conditions typical of inflamed dermis.13,74 Clinically, elevated Th9 signatures and serum IL‑9 correlate with disease severity, and microbial cues, such as Staphylococcus aureus superantigens, can potentiate Th9 polarization, linking dysbiosis to immune amplification.46,47,75
These insights suggest that metabolic and cytokine-targeted interventions, including PPARγ modulators and IL‑9 pathway inhibitors, are promising adjuncts to conventional barrier-restorative and biological therapies in AD.45,76
Allergic Rhinitis
Allergic rhinitis (AR) is a prevalent upper-airway disorder marked by nasal congestion, rhinorrhea, sneezing, and mucosal hyperreactivity.77,78 Historically attributed to IgE-mediated mast-cell activation and type 2 cytokines, AR pathogenesis is now understood to involve a broader T-helper repertoire, including Th9 cells.77,79 Experimental transfer models demonstrate that Th9 cells can induce nasal hyperresponsiveness (NHR) independent of eosinophilic infiltration, underscoring their capacity to modulate airway tone through noncanonical pathways.80,81 IL‑9 produced by Th9 cells amplifies mast-cell density within nasal mucosa, augments local histamine release, and promotes goblet-cell hyperplasia, collectively intensifying secretory responses and mucosal edema (Table 1).48,49,82 Mechanistically, IL‑9 acts in concert with epithelial-derived alarmins and type 2 cytokines to sustain chronic inflammation, while epigenetic and metabolic programs, STAT5/STAT6-mediated chromatin licensing, and PPARγ-driven glycolysis, mirror those observed in lower-airway disease.11–13,82 Notably, NHR exhibits tissue-specific pharmacodynamics: whereas bronchial Th9-driven BHR often resists corticosteroid suppression, nasal models reveal partial dexamethasone responsiveness, suggesting differential glucocorticoid sensitivity across airway compartments.41,80,81 This heterogeneity likely reflects variations in local cytokine gradients, vascular permeability, and stromal cell composition.83
Clinically, elevated IL‑9 levels in nasal secretions and increased Th9 frequency in peripheral blood have been documented in patients with AR, correlating with symptom burden.49–51 These findings advocate for endotype-driven strategies that integrate IL‑9 blockade or upstream modulators (eg, TL1A/DR3 antagonists) with established intranasal corticosteroids and allergen immunotherapy to achieve durable disease control.14,52
Mechanisms of Hyperresponsiveness and Tissue Mechanics
Th9‑linked BHR: Clinical and Experimental Lines of Evidence
In addition to their anti-tumor activity, Th9 cells also play a substantial role in allergic diseases. Like Th2 cells, Th9 cells can provoke airway eosinophilic inflammation accompanied by BHR, yet the routes by which Th9 cells drive BHR are multifactorial (Table 2).5,87 In an original mouse transfer model, allergen challenge of Th9‑recipient mice induced both BHR and eosinophil infiltration, mirroring the phenotype seen after transfer of in vitro-differentiated allergen‑specific Th2 cells.9,41 Upstream drivers are diverse: tumor necrosis factor receptor superfamily, member 4 (TNFRSF4/OX40) signaling in T cells promotes Th9 differentiation and airway inflammation,88 and chronic exposure to Aspergillus fumigatus augments Th9 development in murine lungs.89 Clinically, peripheral blood from patients with allergic asthma contains higher Th9 frequencies and elevated IL‑9 relative to healthy subjects.90,91 Against this background, a randomized, placebo‑controlled, double‑blind, multicenter trial of an anti‑IL‑9 monoclonal antibody in uncontrolled moderate‑to‑severe asthma (2013) failed to improve predicted forced expiratory volume 1 (FEV1), underscoring that IL‑9 neutralization alone is insufficient in an unselected cohort.16 Organ‑level pathways linking Th9 programs to bronchial and nasal hyperresponsiveness (including chemokine‑guided trafficking and airway‑smooth‑muscle tone interfaces) are summarized in previously published figures and reviews.92,93
 |
Table 2 Th-Subset–Driven Hyperresponsiveness Across Airway Compartments: Dependency, Steroid Response, Bronchoactive/ASM Pathways |
Consistent with these findings, our mouse study showed Th9‑mediated BHR was not abrogated by IL‑9 blockade; IL‑10 was dispensable, as IL‑10-deficient Th9 cells still induced BHR; and, notably, Th9‑dependent BHR was markedly increased in eosinophil‑deficient mice, contrasting with the eosinophil requirement observed in Th2‑mediated BHR.9 Although previous studies by Kung et al and Cheng et al demonstrated that anti-IL-9 antibody treatment reduced airway inflammation and BHR in allergic mouse models, differences in experimental design, such as mouse strain and allergen challenge duration, may account for the heightened IL-9 dependency observed in those settings.94,95 In parallel, steroid responses diverge by lineage: despite similar glucocorticoid‑receptor expression and dexamethasone‑sensitive cytokine output in vitro, dexamethasone suppressed allergen‑induced eosinophilia and BHR in Th2‑transfer mice but not in Th9‑transfer mice.41 Tracking allergen‑specific cells revealed dexamethasone curtailed Th2, but not Th9, migration into the lung, suggesting steroid efficacy in Th2‑driven BHR may stem from reductions in Th2 cell trafficking rather than direct eosinophil depletion.96
Together, these data suggest that Th9 cells can sustain BHR through pathways that do not strictly rely on IL‑9 or eosinophils and respond variably to steroids, reinforcing the need for endotype-aware interventions.5,9,41
Eosinophil-T‑cell Crosstalk, Bronchoactive Mediators, and the Airway Smooth Muscle (ASM) Contractile Set‑point
T cells and their cytokines, especially IL‑5, are necessary for eosinophil accumulation in allergic lungs, yet eosinophils themselves shape T‑cell dynamics.97,98 Beyond canonical CCR3⁺/Siglec‑F⁺ profiles, functionally distinct eosinophil subsets have been identified; Siglec‑F⁺Gr1hi cells accumulate after allergen challenge and maintain T‑cell-active cytokines.99 In eosinophil‑deficient mice, adoptive transfer of eosinophils plus CCL11 instillation rescued BHR and T‑cell infiltration, and independent work showed that accumulated T cells contribute to eosinophil‑dependent BHR.5,100 Differences in BHR steroid‑responsiveness between Th2 and Th9 contexts may therefore reflect subset‑specific chemotaxis and cellular choreography within inflamed tissue.5 Importantly, BHR can also arise without eosinophils: transfer models using Th1, Th9, or Th17 cells each produced eosinophil‑independent BHR, implying the presence of shared bronchoactive factors released by T cells (Table 2).9,64,66
T cells can synthesize acetylcholine, and we identified a high‑molecular‑weight T‑cell-derived activity that contracts bronchial smooth muscle.101,102 Moreover, allergen‑induced late‑phase airway obstruction occurred after transfer of OVA‑reactive T‑cell clones.103 In parallel, mechanotransduction via Piezo1 links matrix stiffness to Th9 output and airway responsiveness, providing a physical cue that complements RhoA/ROCK-MLCP‑mediated Ca2⁺ sensitization.15,20 These convergent routes place the T cell, not solely the eosinophil, at the center of dynamic tone control in the allergic airway.5,15,64,66
Tissue Specificity and Nasal Hyperresponsiveness: Physiology, Pharmacology, and Mechanics
With respect to hyperresponsiveness and commonality across Th subsets, allergen‑induced NHR, quantified by increased sneezing responses to non‑specific stimuli, was elicited not only by Th2 transfer but also by Th1 and Th17 transfer in mice (Table 2).84 In striking contrast to the eosinophil requirement typical of Th2‑mediated BHR, eosinophils were dispensable for NHR even in Th2‑transfer models, indicating that upper‑airway hyperreactivity can be sustained by T‑cell programs independent of granulocyte burden.84 In immunized and Th2‑ or Th17-transferred mice, allergen‑induced NHR was suppressed by dexamethasone, though the impact on Th1‑driven NHR remains to be determined.81,84,86 These observations argue for a tissue‑specific lens: bronchial smooth‑muscle contraction and the sneeze reflex are distinct physiological outputs with overlapping but non‑identical upstream controllers.83,104
Mapping these outputs onto local mechanics clarifies the picture: Piezo1‑based sensing of matrix stiffness can tune Th9 effector output and airway responsiveness, while the RhoA/ROCK-MLCP axis modulates Ca2⁺ sensitivity at the myocyte level, together defining a dual control system in which immune programming and tissue biomechanics co‑produce the hyperresponsive state.15,20 This framework helps explain differential steroid responsiveness between bronchi and nose and supports endotype‑ and site‑specific strategies for intervention.5,80,84
Therapeutics and Endotyping
Guideline Backbone and Modulators of the Th2 Cascade
The foundation of asthma management remains the Global Initiative for Asthma (GINA) stepwise/track-based approach, centered on inhaled corticosteroids (ICS) and, as needed, add-on therapies such as long-acting beta-agonists (LABA), leukotriene receptor antagonists (LTRA), and theophylline. Recent updates emphasize ICS-containing reliever regimens to minimize short-acting beta-agonist (SABA)-only exposure.6 From a systems perspective, cysteinyl leukotrienes (CysLTs) are potent endogenous bronchoconstrictors, and CysLT1 antagonists (eg, montelukast) have demonstrated efficacy in randomized trials, serving as adjuncts in mild-to-moderate or non-type 2 asthma.85,105 Building on this standard, endotype-driven biologic therapies have become established. The IL-5/IL-5 receptor (IL-5R) axis is particularly effective in type 2-high phenotypes, marked by elevated blood/sputum eosinophils or oral corticosteroid (OCS) dependence. Mepolizumab and benralizumab show robust OCS-sparing effects and sustained efficacy in real-world and extension studies.106–108 However, IL-5-independent endotypes also exist, necessitating careful stratification.106 Targeting IL-4/IL-13 has proven more nuanced.109 While single-cytokine blockade yielded inconsistent results, dual inhibition via IL-4 receptor (IL-4R) α (dupilumab) suppresses both IL-4 and IL-13 signaling, delivering reproducible reductions in exacerbations and sustained lung function improvements, especially in type 2-high patients identified by fractional exhaled nitric oxide (FeNO) and blood eosinophils. These biomarkers have proven useful for predicting and stratifying dupilumab responses in both clinical trials and real-world practice.110–112
Beyond Th2: Th9‑axis Strategies and an Endotype‑guided Roadmap
Anti-IL‑9 monoclonal antibody therapy did not improve outcomes in uncontrolled asthma, indicating that downstream neutralization may be insufficient without endotype enrichment (Table 3).16 In contrast, the STAT5/STAT6‑licensed bystander IL‑9 program in Th9 cells, together with patient data, positions JAK inhibition as a rational option for Th9‑high endotypes.11 Upstream interference with the TL1A/DR3 pathway and metabolic modulation of Th9 biology (PPARγ/mTORC1; acetyl-CoA carboxylase (ACC) 1) are similarly compelling avenues,12–14,113 while ROCK2 inhibition or MLCP activation offers endotype‑agnostic control of airway tone by attenuating Ca2⁺ sensitization in airway smooth muscle.20,21
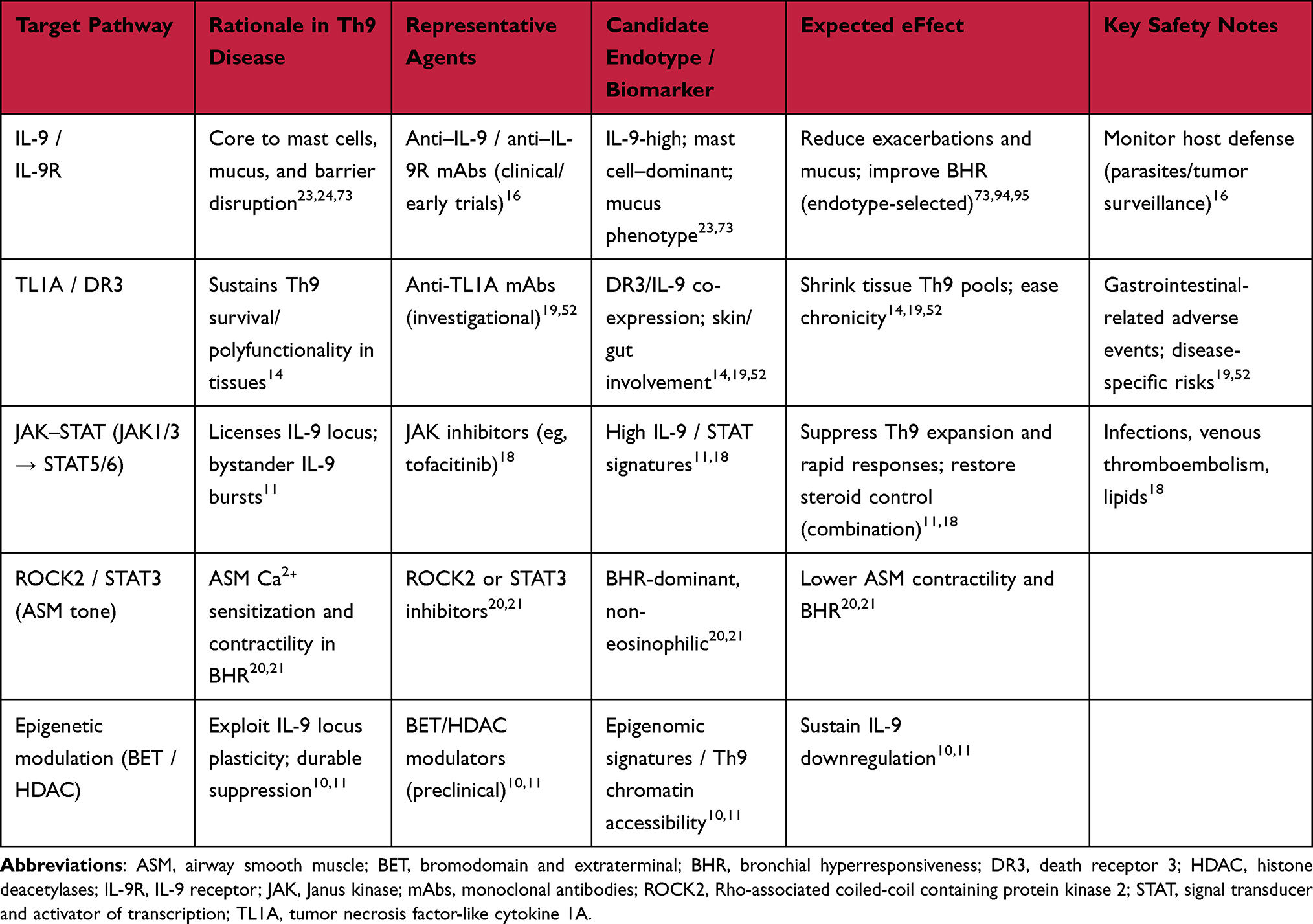 |
Table 3 Therapeutic Targets Related to Th9 Biology (Candidate Agents, Endotypes, and Safety) |
Practically, an endotyping framework would integrate (i) clinical markers (exacerbation pattern, steroid responsiveness), (ii) cellular/molecular readouts (blood and airway Th2/Th9 signatures; IL‑5/IL‑13/IL‑9 axes; STAT5/STAT6 gene sets), and (iii) physiologic surrogates (FeNO, sputum eosinophils, bronchial hyperresponsiveness) to match patients to IL‑5/IL‑5R, IL‑4Rα, or JAK/TL1A‑targeted approaches, adding ASM‑directed therapy when tone dysregulation predominates.8,18,114,115 Notably, a subset of patients with asthma, rhinitis, or atopic dermatitis exhibit “intrinsic” (non-IgE-mediated) allergic phenotypes, which may be overlooked by conventional IgE-based diagnostics. The application of the leukocyte adherence inhibition test and related assays has enabled the identification of non-IgE-mediated immunoreactivity in such patients, reinforcing the need for endotype-driven approaches in allergy diagnosis and management.116 This layered strategy leverages type 2 successes, acknowledges IL‑5-independent diseases, and incorporates Th9‑specific circuitry to personalize control across the asthma spectrum.8,114
Safety and Tolerability
From a safety perspective, downstream IL‑9 neutralization has shown neutral efficacy signals without new major safety concerns in unselected asthma cohorts;16 in contrast, JAK inhibition warrants infection, lipid, and thromboembolism monitoring,18 and investigational TL1A/DR3 blockade has reported gastrointestinal‑related adverse events in disease‑specific contexts.19,52 Epigenetic modulators (BET/HDAC) and ASM‑tone targeted approaches (eg, ROCK2/STAT3 axis) carry class‑typical risks and remain under active evaluation. These safety considerations are summarized alongside putative endotypes in Table 3.
Clinical Translation and Decision-Making
A pragmatic pathway links (i) suspected endotype features (exacerbation pattern, eosinophil-independence, variable steroid responsiveness; bronchial vs nasal compartment) with (ii) molecular readouts (blood/airway Th9 signatures; IL-9⁺CD4⁺ frequencies; STAT5/STAT6 gene sets; metabolic modules) and (iii) therapeutic choices (IL-5/IL-5R or IL-4Rα for type 2-high; JAK or TL1A/DR3 targeting for Th9-high; ASM-tone modulation when Ca2⁺ sensitization predominates). This bench-to-bedside mapping is intended to guide trial enrichment and individualized care.
Limitations
The evidence base for Th9-axis interventions remains heterogeneous across tissues (bronchus versus nose) and disease endotypes; downstream IL-9 neutralization has yielded inconsistent benefits in unselected cohorts, underscoring the need for biomarker-enriched trial designs. Mechanistic inferences drawn from preclinical models may not fully capture human tissue pharmacodynamics, and standardized readouts for Th9 signatures require further validation across platforms.
Conclusions and Future Directions
In summary, while type 2‑eosinophilic inflammation remains a central mechanism of asthma, non-type 2 endotypes are common and clinically consequential. Therefore, we posit novel, eosinophil‑independent BHR mechanisms in which various Th subsets, particularly Th9, drive hyperresponsiveness through immune‑metabolic‑mechanical crosstalk. Aligning endotyping, upstream targeting (STAT/JAK, TL1A/DR3), metabolic control (PPARγ/mTORC1, ACC1), and ASM-directed modulation (ROCK2/MLCP) offers a coherent, precision-ready strategy to expand benefits across the full spectrum of allergic airway diseases.
Future studies should integrate single-cell epigenomics under steroid and JAK inhibitor exposure, map nutrient and stiffness gradients that set IL‑9 thresholds (PPARγ/MCT1; Piezo1), and prospectively test biomarker‑enriched cohorts for JAK or TL1A/DR3 blockade combined with ASM-directed therapy.
Abbreviations
ACC1, acetyl-CoA carboxylase 1; ASM, airway smooth muscle; BATF, basic leucine zipper transcription factor, ATF-like; BHR, bronchial hyperresponsiveness; DR3, death receptor 3; EAACI, European Academy of Allergy and Clinical Immunology; ECAR, extracellular acidification rate; FeNO, fractional exhaled nitric oxide; GINA, Global Initiative for Asthma; HDAC, histone deacetylase; IL, interleukin; IL-9R, interleukin-9 receptor; IRF4, interferon regulatory factor 4; JAK, Janus kinase; mAb, monoclonal antibody; MLCP, myosin light chain phosphatase; MCT1, monocarboxylate transporter 1; mTORC1, mechanistic/mammalian target of rapamycin complex 1; NHR, nasal hyperresponsiveness; OCS, oral corticosteroid; TNFRSF4/OX40, tumor necrosis factor receptor superfamily, member 4 ; PPARγ, peroxisome proliferator-activated receptor gamma; ROCK2, Rho-associated coiled-coil containing protein kinase 2; STAT, signal transducer and activator of transcription; TL1A, TNF-like ligand 1A; Treg, regulatory T cell.
Data Sharing Statement
Data availability is not applicable as no new data was generated for this paper.
Author Contributions
Osamu Kaminuma: Conceptualization, Writing – original draft, Funding acquisition, Investigation, Project administration, Visualization. Noriko Kitamura: Writing – review and editing, Funding acquisition, Validation. Minoru Gotoh: Conceptualization, Writing – review and editing, Supervision.
All authors gave final approval of the version to be published; approved on the journal to which this article was submitted; and agree to be accountable to the content of this article.
Funding
This work was supported by a Grant-in-Aid for JSPS KAKENHI (No. 22H00398 to O.K.), Triangle Project Grant (O.K.) and a Joint Research Grant (N.K.) from the Research Center for Radiation Disaster Medical Science, and the Japan Foundation for Applied Enzymology (O.K.), Institute for Fermentation, Osaka (O.K.), KOSÈ Cosmetology Research Foundation (O.K.).
Disclosure
All authors declare that the research was conducted in the absence of any commercial or financial relationships that could be considered potential conflicts of interest.
References
1. Goswami R. Th9 Cells: new member of T helper cell family. Methods Mol Biol. 2017;1585:1–19. doi:10.1007/978-1-4939-6877-0_1
2. Jia L, Wu C. Differentiation, regulation and function of Th9 cells. Adv Exp Med Biol. 2014;841:181–207. doi:10.1007/978-94-017-9487-9_7
3. Veldhoen M, Uyttenhove C, van Snick J, et al. Transforming growth factor-β ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9(12):1341–1346. doi:10.1038/ni.1659
4. Goswami R, Jabeen R, Yagi R, et al. STAT6-dependent regulation of Th9 development. J Immunol. 2012;188(3):968–975. doi:10.4049/jimmunol.1102840
5. Saeki M, Nishimura T, Kitamura N, et al. Potential mechanisms of T cell-mediated and eosinophil-independent bronchial hyperresponsiveness. Int J Mol Sci. 2019;20(12):2980. doi:10.3390/ijms20122980
6. Global Initiative for Asthma (GINA). 2024 GINA Main Report. Available from: https://ginasthma.org/2024-report/.
7. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18(5):716–725. doi:10.1038/nm.2678
8. Pelaia C, Crimi C, Vatrella A, et al. Molecular targets for biological therapies of severe asthma. Front Immunol. 2020;11:603312. doi:10.3389/fimmu.2020.603312
9. Saeki M, Kaminuma O, Nishimura T, et al. Th9 cells elicit eosinophil-independent bronchial hyperresponsiveness in mice. Allergol Int. 2016;65:S24–29. doi:10.1016/j.alit.2016.05.003
10. Fu Y, Wang J, Panangipalli G, et al. STAT5 promotes accessibility and is required for BATF-mediated plasticity at the Il9 locus. Nat Commun. 2020;11(1):4882. doi:10.1038/s41467-020-18648-6
11. Son A, Meylan F, Gomez-Rodriguez J, et al. Dynamic chromatin accessibility licenses STAT5- and STAT6-dependent innate-like function of Th9 cells to promote allergic inflammation. Nat Immunol. 2023;24(6):1036–1048. doi:10.1038/s41590-023-01501-5
12. Bertschi NL, Steck O, Luther F, et al. PPARγ regulates the effector function of human T helper 9 cells by promoting glycolysis. Nat Commun. 2023;14(1):2471. doi:10.1038/s41467-023-38233-x
13. Peesari S, McAleer JP. Regulation of human Th9 cell differentiation by lipid modulators targeting PPARγ and acetyl-CoA-carboxylase 1. Front Immunol. 2024;15:1509408. doi:10.3389/fimmu.2024.1509408
14. Richard AC, Tan C, Hawley ET, et al. The TNF-family ligand TL1A and its receptor DR3 promote T cell-mediated allergic immunopathology by enhancing differentiation and pathogenicity of IL-9-producing T cells. J Immunol. 2015;194(8):3567–3582. doi:10.4049/jimmunol.1401220
15. Yang Q, Cao Y, Wang L, et al. Mechanical force receptor Piezo1 regulates Th9 cell differentiation. Cell Rep. 2025;44(1):115136. doi:10.1016/j.celrep.2024.115136
16. Oh CK, Leigh R, McLaurin KK, et al. A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respir Res. 2013;14(1):93. doi:10.1186/1465-9921-14-93
17. Li J, Chen S, Xiao X, et al. IL-9 and Th9 cells in health and diseases-from tolerance to immunopathology. Cytokine Growth Factor Rev. 2017(37):47–55. doi:10.1016/j.cytogfr.2017.07.004
18. Georas SN, Donohue P, Connolly M, et al. JAK inhibitors for asthma. J Allergy Clin Immunol. 2021;148(4):953–963. doi:10.1016/j.jaci.2021.08.013
19. Steele H, Sachen K, McKnight AJ, et al. Targeting TL1A/DR3 signaling offers a therapeutic advantage to neutralizing IL13/IL4Rα in muco-secretory fibrotic disorders. Front Immunol. 2021;12:692127. doi:10.3389/fimmu.2021.692127
20. Yasuda Y, Wang L, Chitano P, et al. Rho-kinase inhibition of active force and passive tension in airway smooth muscle: a strategy for treating airway hyperresponsiveness in asthma. Biology. 2024;13(2):115. doi:10.3390/biology13020115
21. Álvarez-Santos MD, Álvarez-González M, Estrada-Soto S, et al. Regulation of myosin light-chain phosphatase activity to generate airway smooth muscle hypercontractility. Front Physiol. 2020;11:701. doi:10.3389/fphys.2020.00701
22. Jutel M, Agache I, Zemelka-Wiacek M, et al. Nomenclature of allergic diseases and hypersensitivity reactions: adapted to modern needs: an EAACI position paper. Allergy. 2023;78(11):2851–2874. doi:10.1111/all.15889
23. Townsend JM, Fallon GP, Matthews JD, et al. IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity. 2000;13(4):573–583. doi:10.1016/s1074-7613(00)00056-x
24. Temann UA, Laouar Y, Eynon EE, et al. IL9 leads to airway inflammation by inducing IL13 expression in airway epithelial cells. Int Immunol. 2007;19(1):1–10. doi:10.1093/intimm/dxl117
25. Zhou Y, McLane M, Levitt RC. Th2 cytokines and asthma. Interleukin-9 as a therapeutic target for asthma. Respir Res. 2001;2(2):80–84. doi:10.1186/rr42
26. Chakraborty S, Kubatzky KF, Mitra DK. An update on Interleukin-9: from its cellular source and signal transduction to its role in immunopathogenesis. Int J Mol Sci. 2019;20(9):2113. doi:10.3390/ijms20092113
27. Dardalhon V, Awasthi A, Kwon H, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. 2008;9(12):1347–1355. doi:10.1038/ni.1677
28. Kaplan MH. The transcription factor network in Th9 cells. Semin Immunopathol. 2017;39(1):11–20. doi:10.1007/s00281-016-0600-2
29. Koch S, Sopel N, Finotto S. Th9 and other IL-9-producing cells in allergic asthma. Semin Immunopathol. 2017;39(1):55–68. doi:10.1007/s00281-016-0601-1
30. Wohlfert EA, Grainger JR, Bouladoux N, et al. GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J Clin Invest. 2011;121(11):4503–4515. doi:10.1172/JCI57456
31. Mantel PY, Kuipers H, Boyman O, et al. GATA3-driven Th2 responses inhibit TGF-β1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol. 2007;5(12):e329. doi:10.1371/journal.pbio.0050329
32. Chang HC, Sehra S, Goswami R, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol. 2010;11(6):527–534. doi:10.1038/ni.1867
33. Yazdani R, Shapoori S, Rezaeepoor M, et al. Features and roles of T helper 9 cells and interleukin 9 in immunological diseases. Allergol Immunopathol. 2019;47(1):90–104. doi:10.1016/j.aller.2018.02.003
34. Huang M, j D. Critical roles of balanced T helper 9 cells and regulatory T cells in allergic airway inflammation and tumor immunity. J Immunol Res. 2021;2021:8816055. doi:10.1155/2021/8816055
35. Chen J, Guan L, Tang L, et al. T helper 9 cells: a new player in immune-related diseases. DNA Cell Biol. 2019;38(10):1040–1047. doi:10.1089/dna.2019.4729
36. Xu WD, Chen YY, Li YW, et al. Targeting Th9 cells in autoimmune diseases: a narrative review. Front Immunol. 2025;16:1615611. doi:10.3389/fimmu.2025.1615611
37. Purwar R, Schlapbach C, Xiao S, et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat Med. 2012;18(8):1248–1253. doi:10.1038/nm.2856
38. Lu Y, Hong S, Li H, et al. Th9 cells promote antitumor immune responses in vivo. J Clin Invest. 2012;122(11):4160–4171. doi:10.1172/JCI65459
39. Lu Y, Yi Q. Utilizing Th9 cells as a novel therapeutic strategy for malignancies. Oncoimmunology. 2013;2:e23084. doi:10.4161/onci.23084
40. Jabeen R, Goswami R, Awe O, et al. Th9 cell development requires a BATF-regulated transcriptional network. J Clin Invest. 2013;123(11):4641–4653. doi:10.1172/JCI69489
41. Saeki M, Kaminuma O, Nishimura T, et al. Th9 cells induce steroid-resistant bronchial hyperresponsiveness in mice. Allergol Int. 2017;66S:S35–S40. doi:10.1016/j.alit.2017.07.001
42. Niehues H, Smits JPH, Rodijk-Olthuis D, et al. Keratinocyte proliferation and differentiation on IL-9 stimulation: an explorative in vitro study. Acta Derm Venereol. 2017;97(6):741–742. doi:10.2340/00015555-2643
43. Clark RA, Schlapbach C. TH9 cells in skin disorders. Semin Immunopathol. 2017;39(1):47–54. doi:10.1007/s00281-016-0607-8
44. D’Avino P, Kiykim A, Sokolowska M, et al. The epithelial barrier theory and its associated diseases. Allergy. 2024;79(12):3192–3237. doi:10.1111/all.16318
45. Briganti S, Mosca S, Nardo AD, et al. New insights into the role of PPARγ in skin physiopathology. Biomolecules. 2024;14(6):728. doi:10.3390/biom14060728
46. Shafi T, Rasool R, Bhat R, et al. Exploring the role of the Th9/IL-9 axis in atopic dermatitis: analysis of Th9 cells, PU.1 expression and serum IL-9 levels. Immunol Res. 2025;73(1):114. doi:10.1007/s12026-025-09671-0
47. Ma L, Xue HB, Guan XH, et al. Possible pathogenic role of T helper type 9 cells and interleukin (IL)-9 in atopic dermatitis. Clin Exp Immunol. 2014;175(1):25–31. doi:10.1111/cei.12198
48. Hauber HP, Bergeron C, Toda M, et al. Up-regulation of Interleukin-9 and the Interleukin-9-associated calcium-activated chloride channel hCLCA1 in nasal mucosa following In vivo allergen challenge. Allergy Asthma Clin Immunol. 2007;3(1):19–23. doi:10.1186/1710-1492-3-1-19
49. Nouri-Aria KT, Pilette C, Jacobson MR, et al. IL-9 and c-Kit+ mast cells in allergic rhinitis during seasonal allergen exposure: effect of immunotherapy. J Allergy Clin Immunol. 2005;116(1):73–79. doi:10.1016/j.jaci.2005.03.011
50. Liu F, Wang B, Mao C. Changes in peripheral blood IL-9, Th9, and BAFF levels in patients with allergic rhinitis and their clinical implications. Technol Health Care. 2024;32(6):4571–4580. doi:10.3233/THC-240756
51. Wang XQ, Hu GH, Kang HY, et al. High frequency of T helper type 9 cells in Chinese patients with allergic rhinitis. Asian Pac J Allergy Immunol. 2015;33(4):301–307. doi:10.12932/AP0609.33.4.2015
52. Niese ML, Pajulas AL, Rostron CR, et al. TL1A priming induces a multi-cytokine Th9 cell phenotype that promotes robust allergic inflammation in murine models of asthma. Mucosal Immunol. 2024;17(4):537–553. doi:10.1016/j.mucimm.2024.03.006
53. Poulsen LK, Hummelshoj L. Triggers of IgE class switching and allergy development. Ann Med. 2007;39(6):440–456. doi:10.1080/07853890701449354
54. Pelaia C, Heffler E, Crimi C, et al. Interleukins 4 and 13 in asthma: key pathophysiologic cytokines and druggable molecular targets. Front Pharmacol. 2022;13:851940. doi:10.3389/fphar.2022.851940
55. Kubo M. Mast cells and basophils in allergic inflammation. Curr Opin Immunol. 2018;54:74–79. doi:10.1016/j.coi.2018.06.006
56. Zhu Z, Homer RJ, Wang Z, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103(6):779–788. doi:10.1172/JCI5909
57. Takatsu K. Interleukin-5 and IL-5 receptor in health and diseases. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87(8):463–485. doi:10.2183/pjab.87.463
58. Corren J. Anti-interleukin-5 antibody therapy in asthma and allergies. Curr Opin Allergy Clin Immunol. 2011;11(6):565–570. doi:10.1097/ACI.0b013e32834c3d30
59. Buchheit KM, Shaw D, Chupp G, et al. Interleukin-5 as a pleiotropic cytokine orchestrating airway type 2 inflammation: effects on and beyond eosinophils. Allergy. 2024;79(10):2662–2679. doi:10.1111/all.16303
60. Travers J, Rothenberg ME. Eosinophils in mucosal immune responses. Mucosal Immunol. 2015;8(3):464–475. doi:10.1038/mi.2015.2
61. Bousquet J, Chanez P, Lacoste JY, et al. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323(15):1033–1039. doi:10.1056/NEJM199010113231505
62. Fattouh R, Al-Garawi A, Fattouh M, et al. Eosinophils are dispensable for allergic remodeling and immunity in a model of house dust mite-induced airway disease. Am J Respir Crit Care Med. 2011;183(2):179–188. doi:10.1164/rccm.200905-0736OC
63. Yang M, Hogan SP, Henry PJ, et al. Interleukin-13 mediates airways hyperreactivity through the IL-4 receptor-alpha chain and STAT-6 independently of IL-5 and eotaxin. Am J Respir Cell Mol Biol. 2001;25(4):522–530. doi:10.1165/ajrcmb.25.4.4620
64. Hansen G, Berry G, DeKruyff RH, et al. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J Clin Invest. 1999;103(2):175–183. doi:10.1172/JCI5155
65. Ashino S, Wakita D, Shiohama Y, et al. A T(h)17-polarized cell population that has infiltrated the lung requires cells that convert to IFN-{gamma} production in order to induce airway hyperresponsiveness. Int Immunol. 2010;22(6):503–513. doi:10.1093/intimm/dxq034
66. McKinley L, Alcorn JF, Peterson A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181(6):4089–4097. doi:10.4049/jimmunol.181.6.4089
67. Newcomb DC, Peebles RS Jr. Th17-mediated inflammation in asthma. Curr Opin Immunol. 2013;25(6):755–760. doi:10.1016/j.coi.2013.08.002
68. Guttman-Yassky E, Renert-Yuval Y, Brunner PM. Atopic dermatitis. Lancet. 2025;405(10478):583–596. doi:10.1016/S0140-6736(24)02519-4
69. Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242(1):233–246. doi:10.1111/j.1600-065X.2011.01027.x
70. Roy DN, Goswami R. IL-9 signaling pathway: an update. Methods Mol Biol. 2017;1585:37–50. doi:10.1007/978-1-4939-6877-0_3
71. Sismanopoulos N, Delivanis DA, Alysandratos KD, et al. IL-9 induces VEGF secretion from human mast cells and IL-9/IL-9 receptor genes are overexpressed in atopic dermatitis. PLoS One. 2012;7(3):e33271. doi:10.1371/journal.pone.0033271
72. Yosipovitch G, Rosen JD, Hashimoto T. Itch: from mechanism to (novel) therapeutic approaches. J Allergy Clin Immunol. 2018;142(5):1375–1390. doi:10.1016/j.jaci.2018.09.005
73. Vermeer PD, Harson R, Einwalter LA, et al. Interleukin-9 induces goblet cell hyperplasia during repair of human airway epithelia. Am J Respir Cell Mol Biol. 2003;28(3):286–295. doi:10.1165/rcmb.4887
74. Nakajima T, Kanno T, Ueda Y, et al. Fatty acid metabolism constrains Th9 cell differentiation and antitumor immunity via the modulation of retinoic acid receptor signaling. Cell Mol Immunol. 2024;21(11):1266–1281. doi:10.1038/s41423-024-01209-y
75. Badolati I, van der Heiden M, Brodin D, et al. Staphylococcus aureus-derived factors promote human Th9 cell polarization and enhance a transcriptional program associated with allergic inflammation. Eur J Immunol. 2023;53(3):e2250083. doi:10.1002/eji.202250083
76. Schärli S, Luther F, Domizio JD, et al. IL-9 sensitizes human TH2 cells to proinflammatory IL-18 signals in atopic dermatitis. J Allergy Clin Immunol. 2025;155(2):491–504. doi:10.1016/j.jaci.2024.10.027
77. Guo LP, Yan M, Niu RB, et al. Role of Th2, Th17 and Treg cells and relevant cytokines in pathogenesis of allergic rhinitis. Allergy Asthma Clin Immunol. 2024;20(1):40. doi:10.1186/s13223-024-00905-8
78. Baraniuk JN. Pathogenesis of allergic rhinitis. J Allergy Clin Immunol. 1997;99(2):S763–72. doi:10.1016/s0091-6749(97)70125-8
79. He Y, Chen Y, Xu S, et al. Pathogenesis and key cells in allergic rhinitis. Int Arch Allergy Immunol. 2025;186(5):418–429. doi:10.1159/000541666
80. Nishimura T, Kaminuma O, Saeki M, et al. Effects of anti-allergic drugs on T cell-mediated nasal hyperresponsiveness in a murine model of allergic rhinitis. Allergol Int. 2018;67S:S25–31. doi:10.1016/j.alit.2018.05.002
81. Koyama T, Miura K, Yamasaki N, et al. Suppressive effect of dexamethasone on murine Th9 cell-mediated nasal eosinophilic inflammation. Asia Pac Allergy. 2021;11(3):e25. doi:10.5415/apallergy.2021.11.e25
82. Gao YD, Wang ZJ, Ogulur I, et al. The evolution, immunopathogenesis and biomarkers of type 2 inflammation in common allergic disorders. Allergy. 2025;80(7):1848–1877. doi:10.1111/all.16620
83. Cho HJ, Ha JG, Lee SN, et al. Differences and similarities between the upper and lower airway: focusing on innate immunity. Rhinology. 2021;59(5):441–450. doi:10.4193/Rhin21.046
84. Nishimura T, Kaminuma O, Saeki M, et al. Essential contribution of CD4+ T cells to antigen-induced nasal hyperresponsiveness in experimental allergic rhinitis. PLoS One. 2016;11(1):e0146686. doi:10.1371/journal.pone.0146686
85. Barnes NC, Piper PJ, Costello JF. Comparative effects of inhaled leukotriene C4, leukotriene D4, and histamine in normal human subjects. Thorax. 1984;39(7):500–504. doi:10.1136/thx.39.7.500
86. Ueda S, Miura K, Kawasaki H, et al. Th17-dependent nasal hyperresponsiveness is mitigated by steroid treatment. Biomolecules. 2022;12(5):674. doi:10.3390/biom12050674
87. Temann UA, Ray P, Flavell RA. Pulmonary overexpression of IL-9 induces Th2 cytokine expression, leading to immune pathology. J Clin Invest. 2002;109(1):29–39. doi:10.1172/JCI13696
88. Xiao X, Balasubramanian S, Liu W, et al. OX40 signaling favors the induction of TH9 cells and airway inflammation. Nat Immunol. 2012;13(10):981–990. doi:10.1038/ni.2390
89. Kerzerho J, Maazi H, Speak AO, et al. Programmed cell death ligand 2 regulates TH9 differentiation and induction of chronic airway hyperreactivity. J Allergy Clin Immunol. 2013;131(4):1048–1057. doi:10.1016/j.jaci.2012.09.027
90. Jones CP, Gregory LG, Causton B, et al. Activin A and TGF-β promote T(H)9 cell-mediated pulmonary allergic pathology. J Allergy Clin Immunol. 2012;129(4):1000–1010. doi:10.1016/j.jaci.2011.12.965
91. Hoppenot D, Malakauskas K, Lavinskienė S, et al. Peripheral blood Th9 cells and eosinophil apoptosis in asthma patients. Medicina. 2015;51(1):10–17. doi:10.1016/j.medici.2015.01.001
92. Soroosh P, Doherty TA. Th9 and allergic disease. Immunology. 2009;127(4):450–458. doi:10.1111/j.1365-2567.2009.03114.x
93. Bick F, Blanchetot C, Lambrecht BN, et al. A reappraisal of IL-9 in inflammation and cancer. Mucosal Immunol. 2025;18(1):1–15. doi:10.1016/j.mucimm.2024.10.003
94. Kung TT, Luo B, Crawley Y, et al. Effect of anti-mIL-9 antibody on the development of pulmonary inflammation and airway hyperresponsiveness in allergic mice. Am J Respir Cell Mol Biol. 2001;25(5):600–605. doi:10.1165/ajrcmb.25.5.4533
95. Cheng G, Arima M, Honda K, et al. Anti-interleukin-9 antibody treatment inhibits airway inflammation and hyperreactivity in mouse asthma model. Am J Respir Crit Care Med. 2002;166(3):409–416. doi:10.1164/rccm.2105079
96. Kara EE, Comerford I, Bastow CR, et al. Distinct chemokine receptor axes regulate Th9 cell trafficking to allergic and autoimmune inflammatory sites. J Immunol. 2013;191(3):1110–1117. doi:10.4049/jimmunol.1203089
97. Rosenberg HF, Phipps S, Foster PS. Eosinophil trafficking in allergy and asthma. J Allergy Clin Immunol. 2007;119(6):1303–1310. doi:10.1016/j.jaci.2007.03.048
98. Jacobsen EA, Zellner KR, Colbert D, et al. Eosinophils regulate dendritic cells and Th2 pulmonary immune responses following allergen provocation. J Immunol. 2011;187(11):6059–6068. doi:10.4049/jimmunol.1102299
99. Percopo CM, Brenner TA, Ma M, et al. SiglecF+Gr1hi eosinophils are a distinct subpopulation within the lungs of allergen-challenged mice. J Leukoc Biol. 2017;101(1):321–328. doi:10.1189/jlb.3A0416-166R
100. Walsh ER, Sahu N, Kearley J, et al. Strain-specific requirement for eosinophils in the recruitment of T cells to the lung during the development of allergic asthma. J Exp Med. 2008;205(6):1285–1292. doi:10.1084/jem.20071836
101. Mashimo M, Iwasaki Y, Inoue S, et al. Acetylcholine released from T cells regulates intracellular Ca2+, IL-2 secretion and T cell proliferation through nicotinic acetylcholine receptor. Life Sci. 2017;172:13–18. doi:10.1016/j.lfs.2016.12.015
102. Mori A, Kouyama S, Abe A, et al. T cell induced-bronchoconstriction in vitro and in vivo. In: Allergies: Current Challenges and Solutions, Proceedings of the 30th Symposium of the Collegium Internationale Allergologicum. Pisa, Italy: Pancini Editore S.r.l; 2016:169–171.
103. Ohtomo T, Kaminuma O, Kitamura N, et al. Murine Th clones confer late asthmatic response upon antigen challenge. Int Arch Allergy Immunol. 2009;149 Suppl 1:2–6. doi:10.1159/000210646
104. Rui Y, Xin T, Chen Y, et al. The sneeze reflex in physiological and pathological states: a mini review. Front Neurosci. 2025;19:1598027. doi:10.3389/fnins.2025.1598027
105. Reiss TF, Chervinsky P, Dockhorn RJ, et al. Montelukast, a once-daily leukotriene receptor antagonist, in the treatment of chronic asthma: a multicenter, randomized, double-blind trial. montelukast clinical research study group. Arch Intern Med. 1998;158(11):1213–1220. doi:10.1001/archinte.158.11.1213
106. Nair P, Pizzichini MM, Kjarsgaard M, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360(10):985–993. doi:10.1056/NEJMoa0805435
107. Nair P, Wenzel S, Rabe KF, et al. Oral glucocorticoid-sparing effect of benralizumab in severe asthma. N Engl J Med. 2017;376(25):2448–2458. doi:10.1056/NEJMoa1703501
108. Menzies-Gow A, Gurnell M, Heaney LG, et al. Oral corticosteroid elimination via a personalised reduction algorithm in adults with severe, eosinophilic asthma treated with benralizumab (PONENTE): a multicentre, open-label, single-arm study. Lancet Respir Med. 2022;10(1):47–58. doi:10.1016/S2213-2600(21)00352-0
109. Sahnoon L, Bajbouj K, Mahboub B, et al. Targeting IL-13 and IL-4 in asthma: therapeutic implications on airway remodeling in severe asthma. Clin Rev Allergy Immunol. 2025;68(1):44. doi:10.1007/s12016-025-09045-2
110. Le Floc’h A, Allinne J, Nagashima K, et al. Dual blockade of IL-4 and IL-13 with dupilumab, an IL-4Rα antibody, is required to broadly inhibit type 2 inflammation. Allergy. 2020;75(5):1188–1204. doi:10.1111/all.14151
111. Blaiss M, Bleecker ER, Jacob-Nara J, et al. Real-world effectiveness of dupilumab in patients with asthma: findings from the US ADVANTAGE study. Ann Allergy Asthma Immunol. 2024;132(4):463–468. doi:10.1016/j.anai.2023.11.006
112. Pavord ID, Deniz Y, Corren J, et al. Baseline FeNO independently predicts the dupilumab response in patients with moderate-to-severe asthma. J Allergy Clin Immunol Pract. 2023;11(4):1213–1220. doi:10.1016/j.jaip.2022.11.043
113. Meylan F, Gomez-Rodriguez J. T cell receptor and co-stimulatory signals for Th9 generation. Methods Mol Biol. 2019;2017(1585):59–71. doi:10.1007/978-1-4939-6877-0_5
114. Hamilton D, Lehman H. Asthma phenotypes as a guide for current and future biologic therapies. Clin Rev Allergy Immunol. 2020;59(2):160–174. doi:10.1007/s12016-019-08760-x
115. Gao J, Wu F. Association between fractional exhaled nitric oxide, sputum induction and peripheral blood eosinophil in uncontrolled asthma. Allergy Asthma Clin Immunol. 2018;14:21. doi:10.1186/s13223-018-0248-7
116. Olivier CE, Pinto DG, Teixeira APM, et al. Immunoreactivity against dermatophagoides pteronyssinus assessed by the leukocyte adherence inhibition test in patients with intrinsic atopic dermatitis and correlated “intrinsic” non-IgE-mediated allergic conditions. Eur J Clin Med. 2021;2(6):45–50. doi:10.24018/ejclinicmed.2021.2.6.153
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.