Back to Journals » Journal of Inflammation Research » Volume 18
Perampanel Regulates Neuroinflammation and Ferroptosis via Activating FSP1 Following Brain Ischemia
Authors Lv JM, Pan YJ, Wang X, Zhang MM, Li W, Liu J, Wang T
Received 4 June 2025
Accepted for publication 3 October 2025
Published 17 October 2025 Volume 2025:18 Pages 14423—14437
DOI https://doi.org/10.2147/JIR.S544785
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Dharmappa Krishnappa
Jian-Meng Lv, Ya-Juan Pan, Xuan Wang, Mei-Mei Zhang, Wei Li, Juan Liu, Tao Wang
Department of Neurology, Shaanxi Provincial People’s Hospital, Xi’an, Shaanxi, 710068, People’s Republic of China
Correspondence: Tao Wang, Department of Neurology, Shaanxi Provincial People’s Hospital, 256 Youyi West Road, Xi’an, Shaanxi, 710068, People’s Republic of China, Email [email protected]
Purpose: Ischemic stroke remains a leading cause of global disability and mortality, with neuroinflammation and ferroptosis emerging as critical contributors to secondary neuronal damage. Perampanel, a non-competitive α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist, exhibits neuroprotective properties in neurological disorders, yet its mechanisms in ischemic stroke remain incompletely understood. This study investigated the therapeutic potential of post-injury perampanel administration in both in vivo and in vitro models, focusing on neuroinflammation, ferroptosis, and the role of ferroptosis suppressor protein 1 (FSP1).
Methods: Rats received intraperitoneal perampanel (1.5 mg/kg) 10– 15 minutes post-reperfusion for 3 days and exposed to middle cerebral artery occlusion (MCAO) for 60 minutes. Neurological function, neuronal survival, and markers of neuroinflammation and ferroptosis were assessed via immunostaining, Western blot, and behavioral tests. The in vitro ischemia model was mimicked by oxygen glucose deprivation (OGD) in primary cultured cortical neurons.
Results: Perampanel significantly attenuated MCAO-induced neuronal loss (NeuN+ cells) and improved motor coordination in rotating pole tests. It suppressed microglial (Iba-1+) and astrocytic (GFAP+) activation, indicating its anti-inflammatory effects. Mechanistically, perampanel reversed the MCAO-driven downregulation of ferroptosis markers FTH-1 and GPX-4, while enhancing neuronal FSP1 expression. Crucially, the FSP1 inhibitor icFSP1 abolished perampanel-mediated neuroprotection, neuronal preservation, and ferroptosis suppression, supporting the FSP1-dependent mechanisms. In in vitro conditions, perampanel exerted protective effects in a dose- and time-dependent manner. The results of immunostaining and Western blot showed that perampanel attenuated neuronal ferroptosis via activation of FSP1.
Conclusion: These findings demonstrate that perampanel mitigates post-ischemic brain injury by inhibiting neuroinflammation and neuronal ferroptosis via FSP1 activation. This study highlights FSP1 as a novel therapeutic target and positions perampanel as a promising candidate for ischemic stroke treatment, leveraging its established safety profile and clinical availability.
Keywords: stroke, neuroinflammation, ferroptosis, FSP1
Introduction
Ischemic stroke, also known as brain ischemia, is one of the leading causes of disability and mortality all over the world.1 It causes neurological dysfunction due to the ischemic and hypoxic damage induced by the interruption of blood flow to a part of the brain. Recent years have witnessed significant advancements in our understanding of the pathophysiological mechanisms underlying ischemic stroke, but the exact molecular mechanisms remain undetermined.2,3 More and more drugs with neuroprotective potential, which have been proved in experimental models, need to be further investigated in clinical trials.
Glutamate is the major excitatory neurotransmitter in the brain, and its excessive release, followed by the over activation of glutamate receptors, occurs after ischemic stroke. AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors, a subtype of ionotropic glutamate receptors, play a crucial role in the pathophysiology of ischemic stroke.4 Overactivation of AMPA receptor (AMPAR) leads to a massive influx of sodium and calcium ions, resulting in neuronal swelling and death, and many AMPAR antagonists, including NBQX, DNQX, and CNQX, have been demonstrated to be neuroprotective in experimental ischemic stroke models.5,6 The highly selective and non-competitive AMPAR antagonist perampanel is a third-generation antiepileptic drug that is effective in treating various types of epilepsy.7 Several previous studies have shown that perampanel can alleviate brain damage in other neurological disorders, such as traumatic brain injury (TBI) and ischemic stroke.8–10 Pretreatment with perampanel at 10 mg/kg was shown to attenuate brain edema, neuronal apoptosis, and inflammatory cytokines release following focal cerebral ischemia in rats.11 In addition, intraperitoneal administration with perampanel at a dose of 1.5 mg/kg decreased infarct volumes and preserved motor function in transient middle cerebral artery occlusion (MCAO) injured rats.12 Our previous in vitro study also demonstrated that perampanel preserved the blood-brain barrier (BBB) permeability in murine brain endothelial cells (mBECs) after oxygen glucose deprivation (OGD).13 However, the molecular mechanisms underlying the neuroprotective effects induced by perampanel against ischemic stroke have not been fully determined.
In the present study, we investigated the protective effects of post-injury administration of perampanel in an ischemic stroke model. We also investigated the potential molecular mechanism with focus on neuroinflammation and neuronal ferroptosis. Our data demonstrated that perampanel dually regulated neuroinflammation and ferroptosis after ischemic stroke and identified FSP1 as a key mediator of perampanel’s neuroprotective effects. This study validated the therapeutic efficacy of perampanel in both in vivo and in vitro ischemia models with clinically relevant timing.
Materials and Methods
Animals and Chemicals
Male Sprague-Dawley (SD) rats (weight: 300–350 g) were housed in a regular 12 h light/dark cycle with free access to food and water at a relatively constant temperature (approximately 22 °C). The study was performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, approved by the Shaanxi Provincial People’s Hospital Animal Care and Use Committee (Xi’an, China, No. 2022–054). Perampanel was purchased from Santa Cruz (sc-477647, CA, USA), and icFSP1 was obtained from DeZai Biotechnology Company (L8920224, Wuxi, China).
Middle Cerebral Artery Occlusion (MCAO)
Brain ischemia was induced in rats using the intraluminal filament technique as previously described with modifications. Briefly, animals were anesthetized with 5% isoflurane in oxygen and maintained at 2–2.5% during surgery. Following a midline neck incision, the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were carefully exposed. A 4–0 silicone-coated nylon monofilament was introduced into the ECA lumen and advanced 18–20 mm distal to the carotid bifurcation to occlude the middle cerebral artery origin. A >70% reduction from baseline cerebral blood flow was confirmed using laser Doppler flowmetry. After 60 min of occlusion, reperfusion was initiated by filament withdrawal under anesthesia. Sham-operated controls received identical procedures without filament insertion.
Experimental Design
In vivo experiment 1. The animals were divided into three groups: sham, MCAO and MCAO + perampanel. The sham group rats were subjected to surgical procedures without artery occlusion. The MCAO group rats were subjected to MCAO. The rats in MCAO + perampanel group were subjected to MCAO and treated with 1.5 mg/kg perampanel 10–15 min post reperfusion for 3 days.
In vivo experiment 2. The animals were divided into four groups: sham, MCAO, MCAO + perampanel and MCAO + perampanel + icFSP1. The sham group rats were subjected to surgical procedures without artery occlusion. The MCAO group rats were subjected to MCAO. The rats in MCAO + perampanel group were subjected to MCAO and treated with 1.5 mg/kg perampanel 10–15 min post reperfusion for 3 days. The rats in MCAO + perampanel group were subjected to MCAO and treated with 1.5 mg/kg perampanel and 50 mg/kg icFSP1.
In vitro experiment 1. The cultured neurons were divided into five groups: control, OGD, OGD + 1 μM perampanel, OGD + 3 μM perampanel and OGD + 10 μM perampanel. The control neurons were cultured without injury, and the neurons in OGD group were subjected to OGD. The neurons in the last three groups were treated with 1, 3 or 10 μM perampanel after OGD.
In vitro experiment 2. The cultured neurons were divided into five groups: control, OGD, OGD + perampanel 0 h, OGD + perampanel 3 h and OGD + perampanel 6 h. The control neurons were cultured without injury, and the neurons in OGD group were subjected to OGD. The neurons in the last three groups were treated with 10 μM perampanel at 0, 3 or 6 h after OGD.
In vitro experiment 3. The cultured neurons were divided into four groups: control, OGD, OGD + perampanel and OGD + perampanel + Erastin. The control neurons were cultured without injury, and the neurons in OGD group were subjected to OGD. The neurons in OGD + perampanel group were subjected to OGD and treated with 10 μM perampanel. The neurons in OGD + perampanel + Erastin group were subjected to OGD and treated with 10 μM perampanel and 10 μM Erastin.
In vitro experiment 4. The cultured neurons were divided into four groups: control, OGD, OGD + perampanel and OGD + perampanel + icFSP1. The control neurons were cultured without injury, and the neurons in OGD group were subjected to OGD. The neurons in OGD + perampanel group were subjected to OGD and treated with 10 μM perampanel. The neurons in OGD + perampanel + icFSP1 group were subjected to OGD and treated with 10 μM perampanel and 2.5 μM icFSP1.
Immunohistochemistry
Coronal sections (20 μm) from paraformaldehyde-perfused brains were collected using a cryostat (Leica CM1950) at −20°C. After antigen retrieval in citrate buffer (pH 6.0, 95°C, 20 min), sections were blocked with 10% normal goat serum in 0.3% Triton X-100/PBS for 1 h at room temperature (RT). Primary antibody incubation with anti-NeuN (1:200, ab177487), anti-Iba-1 (1:100, ab153696), anti-GFAP (1:100, ab7260), or anti-FSP1 (1:50, sc376987) was performed overnight at 4°C in a humidified chamber. Following three PBS washes (5 min each), sections were incubated with Alexa Fluor 594- or 488-conjugated IgG (1:1000, Invitrogen, A-11005) for 2 h at RT. Nuclei were counterstained with DAPI (5 μg/mL, Sigma, 5 min), and slides were mounted with anti-fade medium. Fluorescence images were captured using a confocal microscope (Zeiss LSM 900) with identical exposure settings. ImageJ software was used to perform automated quantification, ensuring consistency and accuracy.
Rotating Pole Test
Sensorimotor coordination was evaluated using a rotating pole apparatus according to established protocols with modifications.14 Animals were required to traverse a 150-cm-long horizontal metallic rod (diameter 3 cm) rotating at predetermined angular velocities (3 or 6 revolutions per minute). Following three consecutive training trials (1 rpm, non-recorded), each subject underwent two timed test trials at increasing speeds with 15-min inter-trial intervals.
Primary Culture of Cortical Neurons
Primary cortical neurons were cultured from pregnant female Sprague-Dawley rats at embryonic day 18 (E18). Pregnant dams were euthanized via CO2 asphyxiation, and embryos were extracted under sterile conditions. Cerebral cortices were dissected in ice-cold Hank’s Balanced Salt Solution (HBSS, Thermo Fisher), minced, and digested with 0.25% trypsin-EDTA (Sigma-Aldrich) for 15 min at 37°C. Enzymatic activity was neutralized using Dulbecco’s Modified Eagle Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, HyClone). Tissues were triturated gently with a fire-polished Pasteur pipette to achieve single-cell suspension. Cells were centrifuged, resuspended in Neurobasal medium (Gibco) containing 2% B-27 supplement, 0.5 mM L-glutamine, and 1% penicillin/streptomycin, and plated onto poly-D-lysine (PLL, 50 µg/mL, Sigma)-coated plates at a density of 1 × 106 cells/mL. Cultures were maintained at 37°C in a 5% CO2 humidified incubator, with half-medium changes every 3 days. Cytarabine (5 µM, Sigma) was added at day in vitro 3 to inhibit glial proliferation.
In vitro Ischemia Model
In vitro ischemia model was established using the oxygen glucose deprivation (OGD) method. For OGD induction, the medium was replaced with Neurobasal Medium-no D-glucose (Thermo Fisher Scientific), and cultures were transferred to a hypoxic chamber flushed with 5% CO2 and 95% N2 for 1 h at 37°C. Reoxygenation was initiated by replacing the medium with standard glucose-containing Neurobasal medium and returning cultures to normoxic conditions. The control group was cultured according to the routine procedures during this period.
LDH Release Measurement
Neuronal injury in vitro was detected by measuring LDH release into the culture medium using a commercial kit according to the manufacturer’s protocol (Jian-Cheng Biotec, Nanjing, China).
Calcein Signal Measurement
Neuronal survival of cortical neurons after OGD was determined by measuring Calcein AM signal using a kit according to the manufacturer’s protocol (Enzo Life Sciences, Farmingdale, NY, USA). Briefly, neurons were maintained under standard conditions and subjected to the indicated treatments. At specific time points, the medium was replaced with Calcein AM solution (2 μM, serum-free) and cells were incubated at 37 °C for 30 min in the dark. During this period, intracellular esterases converted the non-fluorescent probe into green-emitting Calcein. After incubation, cells were rinsed with PBS to eliminate residual dye. Fluorescence was then recorded using a microplate reader at 485/530 nm. Cell viability was calculated as the relative fluorescence percentage compared with untreated controls, enabling reliable quantitative assessment.
Measurement of Intracellular Iron Content
Intracellular iron levels were quantified using a commercial Iron Colorimetric Assay Kit (E1042-100, Applygen, Beijing, China). Following various treatments, cortical neurons were washed twice with ice-cold PBS and lysed in RIPA buffer containing protease inhibitors. Lysates were centrifuged at 12,000 × g for 15 min at 4°C to remove debris. For iron detection, 100 μL of each sample was aliquoted into a 96-well plate alongside the Blank control (100 μL assay buffer), Standard curve (100 μL iron standards), and Test samples (100 μL normalized lysates). Reagent A (100 μL; iron-chelating agent) was added to all wells, followed by incubation at 60°C for 60 min. After cooling to room temperature, 30 μL of chromogenic working solution (Reagent B) was added to each well. Plates were incubated for 30 min at 25°C in the dark. Absorbance was measured at 550 nm using a microplate reader. Iron content was calculated from the standard curve and normalized to total protein.
Measurement of Intracellular ROS
Intracellular ROS levels were assessed using the cell-permeable fluorogenic probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; D6883, Sigma-Aldrich) according to the manufacturer’s protocol. Cortical neurons were gently washed with pre-warmed (37°C) HBSS and incubated with 10 μM DCFH-DA in Neurobasal medium (without phenol red or antioxidants) for 30 min at 37°C in the dark. Cells were rinsed twice with HBSS to remove excess probe and returned to conditioned medium for 15 min to permit complete de-esterification. Fluorescence images were acquired using a confocal microscope with 488 nm excitation/525 nm emission filters.
Immunocytochemistry
For immunostaining in vitro, cortical neurons were cultured in PLL-coated coverslips and exposed to various treatments as mentioned above. After being fixed with 4% paraformaldehyde for 30 min, neurons were washed with PBS three times and blocked with 5% BSA for 30 min. Incubation with the MAP-2 (1:80, ab5392) and FSP1 (1:50, sc376987) primary antibodies was performed at 4 °C overnight. After being washed by PBS three times, the samples were incubated with the secondary antibody for 1 h at 37 °C. Then, incubation with DAPI was performed to stain the nuclei, and the images were obtained using a fluorescence microscope. ImageJ software was used to perform automated quantification, ensuring consistency and accuracy.
Western Blot
Equal amounts of protein (30 μg/lane) were separated on 10% SDS-PAGE gels and transferred to 0.45 μm PVDF membranes at 300 mA for 90 min. Membranes were blocked with 5% non-fat milk in TBST (Tris-buffered saline with 0.1% Tween-20) for 1 h at RT, then incubated overnight at 4°C with the following primary antibodies: anti-FTH-1 (1:1000, CST#3998), anti-GPX-4 (1:1000, CST#59735) and anti-Tubulin (1:5000, Abcam, ab7291). After three TBST washes (10 min each), membranes were probed with HRP-conjugated secondary antibodies for 1 h at RT. Protein bands were visualized using a chemiluminescent detection system.
Statistical Analysis
All animals and cell samples were randomly assigned to experimental groups using a computer-generated randomization sequence. During behavioral testing, immunostaining, and subsequent data acquisition, investigators were blinded to group allocation to minimize bias. Data analysis was performed by independent researchers who were unaware of the treatment conditions. Statistical analysis was performed using SPSS 16.0. Statistical evaluation of the data was performed by one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparisons.
Results
Effects of Perampanel on Brain Damage After Brain Ischemia
To investigate the potential protective effects of perampanel in vivo, rats were intraperitoneally treated with 1.5 mg/kg 10–15 min post reperfusion for 3 days. Immunostaining was performed using NeuN antibody to detect neurons in the brain cortex following brain ischemia (Figure 1A). The result showed that MCAO significantly decreased the number of NeuN-positive cells, which was partially prevented by perampanel (Figure 1B). In addition, rotating pole test was used to evaluate the effects of perampanel on functional neurological recovery following brain ischemia. MCAO induced an increased time needed for traversing the rotating pole in both 3 rpm and 6 rpm tests. These motor-coordination deficits were attenuated by perampanel at 3 d in 3 rpm test (Figure 1C) or at 7 d in 6 rpm test (Figure 1D).
 |
Figure 1 Effects of perampanel on brain damage after brain ischemia. (A and B) Immunostaining (A) and quantitative analysis (B) showed that perampanel increased the number of NeuN positive cells after brain ischemia. (C and D) Perampanel promoted post-stroke motor-coordination recovery in the rotating pole test at 3 rpm (C) and 6 rpm (D). Scale bar = 50 μm. n=5 (A and B) n=6 (C and D) in each group. Data are shown as mean ± SD. #p < 0.05 vs Sham group. *p < 0.05 vs MCAO group. |
Effects of Perampanel on Neuroinflammation After Brain Ischemia
To investigate the effect of perampanel on microglial activation after brain ischemia, we performed immunostaining using Iba-1 antibody (Figure 2A). The results showed that the increased number of Iba-1 positive cells induced by MCAO was alleviated by perampanel (Figure 2B). We also performed immunostaining with GFAP antibody to detect activation of astrocytes (Figure 2C), and the number of GFAP positive cells in MCAO + perampanel group was lower than that in MCAO group (Figure 2D).
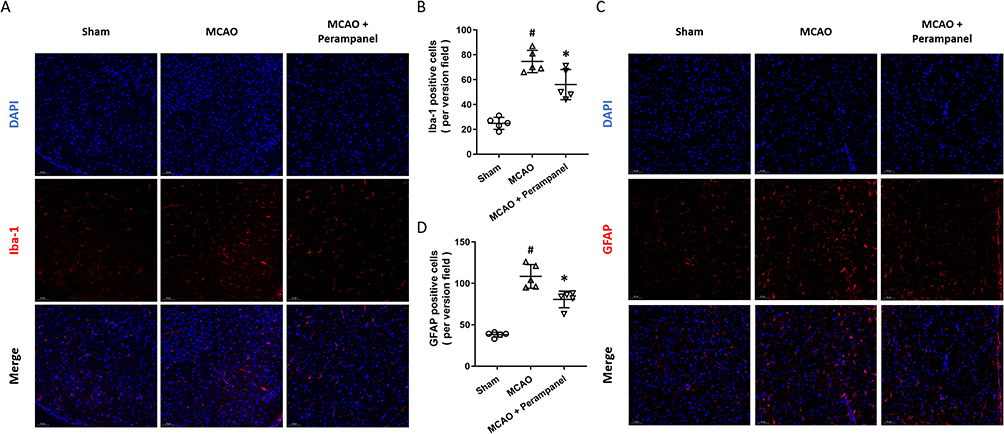 |
Figure 2 Effects of perampanel on neuroinflammation after brain ischemia. (A and B) Immunostaining (A) and quantitative analysis (B) showed that perampanel decreased the number of Iba-1 positive cells after brain ischemia. (C and D) Immunostaining (C) and quantitative analysis (D) showed that perampanel decreased the number of GFAP positive cells after brain ischemia. Scale bar = 50 μm. n=5 in each group. Data are shown as mean ± SD. #p < 0.05 vs Sham group. *p < 0.05 vs MCAO group. |
Effects of Perampanel on Neuronal Ferroptosis After Brain Ischemia
To investigate the effect of perampanel on neuronal ferroptosis, we performed Western blot to detect the expression of FTH-1 and GPX-4, two molecular markers of ferroptosis (Figure 3A). The results showed that MCAO significantly decreased the expression of FTH-1, which was partially prevented by perampanel (Figure 3B). As shown in Figure 3C, a similar result on GPX-4 expression was also observed.
 |
Figure 3 Effects of perampanel on neuronal ferroptosis after brain ischemia. (A–C) Western blot (A) and quantitative analysis (B and C) showed that perampanel increased the expression of FTH-1 (B) and GPX-4 (C) after brain ischemia. n=5 in each group. Data are shown as mean ± SD. #p < 0.05 vs Sham group. *p < 0.05 vs MCAO group. |
Effects of Perampanel on FSP1 Expression After Brain Ischemia
Then, we performed immunostaining using FSP1 and NeuN antibodies to detect the expression of FSP1 in cortical neurons (Figure 4). The results showed that FSP1 is predominantly expressed in the cytoplasm and processes of cortical neurons, and its expression increased significantly after MCAO. Perampanel treatment further promoted FSP1 expression in neurons following brain ischemia.
 |
Figure 4 Effects of perampanel on FSP1 expression after brain ischemia. Immunostaining with FSP1 and NeuN antibodies showed the expression of FSP1 protein in neurons after brain ischemia and perampanel treatment. Scale bar = 50 μm. |
Effects of the FSP1 Inhibitor icFSP1 on Perampanel-Induced Protection
To further confirm the involvement of FSP1 in our findings, we repeated above experiments after pretreatment with the FSP1 inhibitor icFSP1. The cortical neuronal loss was measured by immunostaining with NeuN (Figure 5A), and the preserved neurons induced by perampanel were abolished by icFSP1 (Figure 5B). As shown in Figure 5C, the perampanel-induced increases in the expression of FTH-1 (Figure 5D) and GPX-4 (Figure 5E) were both attenuated by icFSP1.
 |
Figure 5 Effects of the FSP1 inhibitor icFSP1 on perampanel-induced protection. (A and B) Immunostaining (A) and quantitative analysis (B) showed that the increased number of NeuN positive cells induced by perampanel after MCAO was prevented by icFSP1. (C–E) Western blot (C) and quantitative analysis (D and E) showed that the increased expression of FTH-1 (D) and GPX-4 (E) induced by perampanel after MCAO were prevented by icFSP1. Scale bar = 50 μm. n=5 in each group. Data are shown as mean ± SD. *p < 0.05. |
Perampanel Attenuates Neuronal Injury Following OGD in vitro
To mimic neuronal injury in brain ischemia in vitro, cultured cortical neurons were treated with OGD. The results of immunostaining showed that the OGD-induced neuronal loss, as evidenced by reduced number of MAP-2-positive cells, was attenuated by perampanel (Figure 6A). To evaluate dose-dependent protection, perampanel was tested at multiple concentrations. The results showed that 10 μM perampanel, but not 1 and 3 μM, significantly reduced LDH release after OGD (Figure 6B). Consistently, the decreased calcein signal induced by OGD was increased by 10 μM perampanel (Figure 6C). Furthermore, we repeated the above experiments using 10 μM perampanel at 0, 3 or 6 h after OGD to determine the therapeutic time window. The results showed that the perampanel-induced decrease in LDH release could be observed when it was added within 3 h (Figure 6D). As shown in Figure 6E, treatment with perampanel at 3 h after OGD seemingly preserved calcein signal, but it was not statistically significant. Then, we used the ferroptosis inducer Erastin to investigate the potential involvement of ferroptosis in our data. The results showed that the decreased LDH release (Figure 6F) and increased calcein signal (Figure 6G) induced by perampanel were both canceled by Erastin.
 |
Figure 6 Perampanel attenuated neuronal injury following OGD in vitro. (A) Perampanel preserved neuronal survival after OGD in cortical neurons. Scale bar = 50 μm. (B) Perampanel at 10 μM decreased LDH release after OGD. (C) Perampanel at 10 μM increased Calcein signal after OGD. (D) Treatment with 10 μM perampanel at 0 and 3 h after OGD decreased LDH release. (E) Treatment with 10 μM perampanel at 0 h after OGD increased Calcein signal. (F) The decreased LDH release induced by perampanel after OGD was increased by Erastin. (G) The increased Calcein signal induced by perampanel after OGD was decreased by Erastin. n=5 in each group. Data are shown as mean ± SD. #p < 0.05 vs Control group. *p < 0.05 vs OGD group. |
Perampanel Inhibits Neuronal Ferroptosis After OGD
We measured intracellular iron levels to detect ferroptosis in cortical neurons, and the results showed that the increased iron levels induced by OGD was attenuated by perampanel (Figure 7A). Then the immunostaining with DCFH-DA was performed to detect intracellular ROS generation (Figure 7B). The ROS fluorescence intensity was increased by OGD, which was partially reversed by perampanel treatment (Figure 7C). Then we performed Western blot to detect the expression of FTH-1 and GPX-4 in cortical neurons (Figure 7D). The results showed that the decreased expression of FTH-1 (Figure 7E) and GPX-4 (Figure 7F) following OGD were both increased by perampanel. In addition, the OGD-induced increase in 4-HNE levels was reduced by perampanel (Figure 7G).
 |
Figure 7 Perampanel inhibited neuronal ferroptosis after OGD. (A) Perampanel decreased the intracellular iron levels after OGD in cortical neurons. (B and C) Immunostaining (B) and quantitative analysis (C) showed that perampanel reduced the fluorescence intensity of ROS in cortical neurons after OGD. (D–F) Western blot (D) and quantitative analysis (E and F) showed that perampanel increased the expression of FTH-1 (E) and GPX-4 (F) after OGD. (G) Perampanel decreased the 4-HNE levels after OGD in cortical neurons. Scale bar = 50 μm. n=5 in each group. Data are shown as mean ± SD. #p < 0.05 vs Control group. *p < 0.05 vs OGD group. |
Perampanel Regulates Neuronal Ferroptosis via FSP1 in vitro
To investigate the potential involvement of FSP1 in vitro, we performed immunostaining using FSP1 antibody in neuronal slides (Figure 8A). The results showed that the FSP1 expression was increased by OGD, and further enhanced by perampanel. We also repeated the LDH release (Figure 8B) and calcein (Figure 8C) assays after inhibiting FSP1 signaling with icFSP1. The perampanel-induced protection in these two assays following OGD were both partially prevented by icFSP1. To confirm the role of FSP1 on perampanel-induced regulation of ferroptosis, Western blot was performed after icFSP1 treatment (Figure 8D). The results showed that the expression of FTH-1 (Figure 8E) and GPX-4 (Figure 8F) in perampanel treated group following OGD were significantly inhibited by icFSP1.
 |
Figure 8 Perampanel regulated neuronal ferroptosis via FSP1 in vitro. (A) Immunostaining with FSP1 showed the expression of FSP1 protein in neurons after OGD and perampanel treatment. (B) The decreased LDH release induced by perampanel after OGD was increased by icFSP1. (C) The increased Calcein signal induced by perampanel after OGD was decreased by icFSP1. (D–F) Western blot (D) and quantitative analysis (E and F) showed that icFSP1 decreased the expression of FTH-1 (E) and GPX-4 (F) after OGD and perampanel treatment. Scale bar = 50 μm. n=5 in each group. Data are shown as mean ± SD. *p < 0.05. |
Discussion
Neuroinflammation is helpful for repairing damaged tissues and homeostasis restore, but an excessive inflammatory response could exacerbate neuronal damage, disrupt blood-brain barrier integrity, and promote secondary injury mechanisms.15 In the present study, we identified perampanel as a neuroprotective agent against MCAO-induced neuroinflammation in rats. We found that (a) perampanel attenuates brain damage and preserves neurological function following brain ischemia; (b) perampanel inhibits the activation of microglia and astrocytes; (c) perampanel increases the expression of FTH-1 and GPX-4 following brain ischemia; (d) perampanel regulates FSP1 expression in cortical neurons; (e) the FSP1 inhibitor icFSP1 prevents the perampanel-induced effects on neuronal loss and ferroptosis; (f) perampanel protects against OGD-induced neuronal injury in a dose- and time-dependent manner in vitro; and (g) perampanel inhibits neuronal ferroptosis via activation of FSP1 after OGD in neurons.
Following ischemic stroke, the increased permeability of BBB promotes the infiltration of deleterious substances, facilitating the activation of microglia, astrocytes and release of inflammatory cytokines. Several drugs targeting inflammatory responses, including Etanercept and Fingolimod, have been demonstrated to exert clinical efficacy against ischemic stroke, but only Edaravone dexborneol has successfully completed the phase III clinical trial and received approval from the National Medical Products Administration (NMPA).15 In the present study, the MCAO-induced loss of neurons, as well as the increased number of Iba-1 and GFAP positive cells were significantly attenuated by perampanel, indicating its anti-inflammatory activity. Our data are consistent with previous published results in acute TBI mouse model,16 in subarachnoid hemorrhage,17 intracerebral hemorrhage18 and chronic pain models,19 making perampanel an ideal candidate for the treatment of ischemic stroke. As a neuroprotective agent, perampanel possesses some unique advantages. First, it exhibits no inhibitory effects on neurotransmission responses or calcium influx mediated by other glutamate receptors, rendering it a safer option with a reduced risk of psychotomimetic side effects commonly associated with NMDA receptor antagonists.20 In addition, perampanel is rapidly absorbed through the gastrointestinal tract, exhibiting a bioavailability of 100%.9 It has obtained approval in over 55 countries, including the USA and China, for use as either monotherapy or adjunctive therapy in treating certain types of epilepsy in patients aged 12 and older.21
Iron is a redox-active element that facilitates other redox reactions in nearly every biochemical pathway, including oxygen transport, DNA metabolism, oxidative phosphorylation and neurotransmitter synthesis.22 Brain iron homeostasis is largely dependent on the integrity of the BBB, and the increased BBB leakage following ischemic stroke leads to excess iron accumulation, lipid peroxidation, inactivation of endogenous antioxidative systems, as well as a newly characterized iron-dependent regulated cell death, ferroptosis.23 Unlike typical programmed cell death, ferroptosis exhibits a reduction in mitochondrial size, an increase in membrane density, a decrease in cristae number, and elevated levels of lipid peroxidation.24 Accumulating evidence showed that it is involved in neuronal death under various neurological disorders, ranging from acute and chronic brain injury, such as TBI, spinal cord injury (SCI), and Parkinson’s disease (PD), to brain tumors.25–27 Following ischemic stroke, ferroptosis commonly occurs during the reperfusion phase rather than ischemia phase, as evidenced by increased iron content, and the expression of transferrin, ferritin, and transferrin receptors.28,29 Accumulating evidence has shown that many neuroprotective agents and compounds with anti-oxidative or anti-inflammatory activities protect against ischemic brain damage via inhibiting ferroptosis related cascades.30–34 In addition, some neuroprotective treatments, such as electroacupuncture, could attenuate ferroptosis in both neurons and glial cells to suppress oxidative stress and lipid peroxidation following experimental ischemic stroke models.35–40 Our results showed that perampanel significantly attenuated the MCAO-induced decrease in the expression of FTH-1 and GPX-4, two essential factors for evaluating ferroptosis.41,42 Similar results have also been obtained in neuronal injury following OGD, the in vitro ischemia model. These data suggested that inhibition of neuronal ferroptosis might be involved in the perampanel-induced protection against ischemic stroke in our model.
Ferroptosis suppressor protein 1 (FSP1) was originally described as apoptosis inducing factor mitochondrial 2 (AIFM2), which regulates mitochondrial apoptotic cell death by affecting intracellular calcium homeostasis and mitochondrial membrane potential (MMP).43 It was renamed after its role in ferroptosis inhibition in a glutathione-independent manner was discovered.44 FSP1 is a polypeptide consisting of 373 amino acids with a molecular weight of approximately 40.5 kDa, which is encoded by the FSP1 gene located on human chromosome 10q21.3-q22.1 region.45 Utilizing N-myristoylation and NAD(P)H as substrates, FSP1 reduces oxidized CoQ10 (ubiquinone-10) to ubiquinol-10 at the plasma membrane independently of GPX-4 and GSH, diminishing the CoQ10 pool and halting lipid peroxide propagation to prevent ferroptosis.44,46 A recent study showed that overexpression of FSP1 via lentivirus transfection reduces ferroptosis by activating PI3K/AKT pathway in PC12 cells following OGD.47 Zou et al reported that Ginkgolide B protected against cerebral ischemia/reperfusion injury via binding GPX-4 and FSP1 in rats,48 indicating the protective activity of FSP1 against neuronal ferroptosis. Consistently, our results of immunostaining showed that MCAO-induced expression of FSP1 was enhanced by perampanel, accompanied by the preserved neurons in cortex. Thus, we speculated that FSP1 might be an endogenous protective mechanism following brain ischemia. To verify this hypothesis, we repeated experiments of NeuN staining and Western blot after using icFSP1, a newly designed drug with FSP1 inhibition activity.49 icFSP1 was shown to synergize with ferroptosis inducers to enhance ferroptosis and impair tumor growth in vivo, providing a rationale for targeting FSP1-dependent phase separation as an effective anticancer therapy. We found that perampanel-induced effects on neuronal loss and expression of FTH-1 and GPX-4 following MCAO were partially prevented by icFSP1, confirming the involvement of FSP1 in perampanel-induced protection.
There are a few limitations to the present study. Firstly, we did not perform infarct volume assessment using traditional methods such as TTC staining or MRI imaging, which are commonly employed to quantify the extent of ischemic injury. Future research could benefit from incorporating these techniques to provide a more comprehensive understanding of the neuroprotective effects of perampanel on ischemic brain damage. Secondly, the FSP1 inhibitor icFSP1 was used to investigate the involvement of FSP1 in perampanel-induced protection. While inhibitors offer the advantage of pharmacological modulation and potential reversibility, they are not devoid of constraints. One significant limitation lies in the specificity and selectivity of these inhibitors, which may cross-react with other proteins, thereby complicating the interpretation of experimental results.50 More experiments using siRNA-mediated knockdown of FSP1 might be helpful for further understanding the role of FSP1 in perampanel-induced protection. Additionally, while we focused on the biochemical markers of ferroptosis, further studies could investigate other forms of cell death, such as apoptosis and necroptosis, using appropriate inhibitors or knockdown strategies. Understanding the interplay between different cell death pathways in ischemic stroke will be crucial for developing targeted therapeutic interventions. Finally, long-term studies evaluating the functional recovery of animals after perampanel treatment and its potential effects on neuroplasticity and behavioral outcomes are needed to better understand the therapeutic potential of perampanel in stroke recovery.
Conclusion
In summary, our present study demonstrated that the anti-epileptic drug perampanel protects against the MCAO-induced brain damage and inflammation in rats. It also attenuates neuronal injury following OGD in cortical neurons. These protective effects were associated with the FSP1-mediated inhibition of neuronal ferroptosis.
Data Sharing Statement
All data sets and materials that support the conclusions of this article are provided with the manuscript.
Ethical Approval
The study was performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, and approved by the Laboratory Animal Ethics Committee of the Shaanxi Provincial People’s Hospital (Xi’an, China). Ethics No.2022-054.
Funding
This study has been funded by the Shaanxi Province Natural Science Foundation Research Program (No.2021JM-554 and No.2022JM-587).
Disclosure
The authors declare no conflicts of interest.
References
1. Martin SS, Aday AW, Allen NB, et al. 2025 heart disease and stroke statistics: a report of US and global data from the American Heart Association. Circulation. 2025;151:e41–660.
2. Li W, Cao F, Takase H, Arai K, Lo EH, Lok J. Blood-brain barrier mechanisms in stroke and trauma. Handb Exp Pharmacol. 2022;273:267–293.
3. Xie L, He M, Ying C, Chu H. Mechanisms of inflammation after ischemic stroke in brain-peripheral crosstalk. Front Mol Neurosci. 2024;17:1400808. doi:10.3389/fnmol.2024.1400808
4. Mellor IR. The AMPA receptor as a therapeutic target: current perspectives and emerging possibilities. Future Med Chem. 2010;2(5):877–891. doi:10.4155/fmc.10.27
5. Meden P, Overgaard K, Sereghy T, Boysen G. Enhancing the efficacy of thrombolysis by AMPA receptor blockade with NBQX in a rat embolic stroke model. J Neurol Sci. 1993;119(2):209–216. doi:10.1016/0022-510X(93)90136-M
6. Nakagawa T. The biochemistry, ultrastructure, and subunit assembly mechanism of AMPA receptors. Mol Neurobiol. 2010;42(3):161–184. doi:10.1007/s12035-010-8149-x
7. Fan PF, Zhuo C, Huang M. Efficacy and safety of perampanel for epilepsy: a systematic review and meta-analysis of real-world studies. Eur Rev Med Pharmacol Sci. 2023;27(13):6027–6039. doi:10.26355/eurrev_202307_32957
8. Chen T, Dai SH, Jiang ZQ, et al. The AMPAR antagonist perampanel attenuates traumatic brain injury through anti-oxidative and anti-inflammatory activity. Cell Mol Neurobiol. 2017;37(1):43–52. doi:10.1007/s10571-016-0341-8
9. Chen T, Liu WB, Qian X, Xie KL, Wang YH. The AMPAR antagonist perampanel protects the neurovascular unit against traumatic injury via regulating Sirt3. CNS Neurosci Ther. 2021;27(1):134–144. doi:10.1111/cns.13580
10. Chen T, Yang LK, Zhu J, Hang CH, Wang YH. The AMPAR antagonist perampanel regulates neuronal necroptosis via Akt/GSK3beta signaling after acute traumatic injury in cortical neuron. CNS Neurol Disord Drug Targets. 2021;20(3):266–272. doi:10.2174/1871527319666201001110937
11. Niu HX, Wang JZ, Wang DL, et al. The orally active noncompetitive AMPAR antagonist perampanel attenuates focal cerebral ischemia injury in rats. Cell Mol Neurobiol. 2018;38(2):459–466. doi:10.1007/s10571-017-0489-x
12. Nakajima M, Suda S, Sowa K, et al. AMPA receptor antagonist perampanel ameliorates post-stroke functional and cognitive impairments. Neuroscience. 2018;386:256–264. doi:10.1016/j.neuroscience.2018.06.043
13. Lv JM, Guo XM, Chen B, Lei Q, Pan YJ, Yang Q. The noncompetitive AMPAR antagonist perampanel abrogates brain endothelial cell permeability in response to ischemia: involvement of claudin-5. Cell Mol Neurobiol. 2016;36(5):745–753. doi:10.1007/s10571-015-0257-8
14. Dumbrava DA, Surugiu R, Borger V, et al. Mesenchymal stromal cell-derived small extracellular vesicles promote neurological recovery and brain remodeling after distal middle cerebral artery occlusion in aged rats. Geroscience. 2022;44(1):293–310. doi:10.1007/s11357-021-00483-2
15. Cao Y, Yue X, Jia M, Wang J. Neuroinflammation and anti-inflammatory therapy for ischemic stroke. Heliyon. 2023;9(7):e17986. doi:10.1016/j.heliyon.2023.e17986
16. Alqahtani F, Assiri MA, Mohany M, et al. Coadministration of ketamine and perampanel improves behavioral function and reduces inflammation in acute traumatic brain injury mouse model. Biomed Res Int. 2020;2020:3193725. doi:10.1155/2020/3193725
17. Kawakita F, Nakano F, Kanamaru H, Asada R, Suzuki H. Anti-apoptotic effects of AMPA receptor antagonist perampanel in early brain injury after subarachnoid hemorrhage in mice. Transl Stroke Res. 2024;15(2):462–475. doi:10.1007/s12975-023-01138-4
18. Yang L, Wang Y, Zhang C, Chen T, Cheng H. Perampanel, an AMPAR antagonist, alleviates experimental intracerebral hemorrhage‑induced brain injury via necroptosis and neuroinflammation. Mol Med Rep. 2021;24(2). doi:10.3892/mmr.2021.12183
19. De Caro C, Cristiano C, Avagliano C, et al. Analgesic and anti-inflammatory effects of perampanel in acute and chronic pain models in mice: interaction with the cannabinergic system. Front Pharmacol. 2020;11:620221. doi:10.3389/fphar.2020.620221
20. Olney JW, Labruyere J, Wang G, Wozniak DF, Price MT, Sesma MA. NMDA antagonist neurotoxicity: mechanism and prevention. Science. 1991;254(5037):1515–1518. doi:10.1126/science.1835799
21. Tsai JJ, Wu T, Leung H, et al. Perampanel, an AMPA receptor antagonist: from clinical research to practice in clinical settings. Acta Neurol Scand. 2018;137(4):378–391. doi:10.1111/ane.12879
22. Shi R, Hou W, Wang ZQ, Xu X. Biogenesis of iron-sulfur clusters and their role in DNA metabolism. Front Cell Dev Biol. 2021;9:735678. doi:10.3389/fcell.2021.735678
23. Wiegertjes K, Chan KS, Telgte AT, et al. Assessing cortical cerebral microinfarcts on iron-sensitive MRI in cerebral small vessel disease. J Cereb Blood Flow Metab. 2021;41(12):3391–3399. doi:10.1177/0271678X211039609
24. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
25. Wei Z, Yu H, Zhao H, et al. Broadening horizons: ferroptosis as a new target for traumatic brain injury. Burns Trauma. 2024;12:tkad051. doi:10.1093/burnst/tkad051
26. Zhang Y, Yang S, Liu C, Han X, Gu X, Zhou S. Deciphering glial scar after spinal cord injury. Burns Trauma. 2021;9:tkab035. doi:10.1093/burnst/tkab035
27. Wang Y, Wei Z, Pan K, Li J, Chen Q. The function and mechanism of ferroptosis in cancer. Apoptosis. 2020;25(11–12):786–798. doi:10.1007/s10495-020-01638-w
28. Chai Z, Zheng J, Shen J. Mechanism of ferroptosis regulating ischemic stroke and pharmacologically inhibiting ferroptosis in treatment of ischemic stroke. CNS Neurosci Ther. 2024;30(7):e14865. doi:10.1111/cns.14865
29. Tang LJ, Luo XJ, Tu H, et al. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol. 2021;394(2):401–410. doi:10.1007/s00210-020-01932-z
30. Abdul Y, Li W, Ward R, et al. Deferoxamine treatment prevents post-stroke vasoregression and neurovascular unit remodeling leading to improved functional outcomes in type 2 male diabetic rats: role of endothelial ferroptosis. Transl Stroke Res. 2021;12(4):615–630. doi:10.1007/s12975-020-00844-7
31. Jin Z, Gao W, Guo F, et al. Astragaloside IV alleviates neuronal ferroptosis in ischemic stroke by regulating fat mass and obesity-associated-N6-methyladenosine-acyl-CoA synthetase long-chain family member 4 axis. J Neurochem. 2023;166(2):328–345. doi:10.1111/jnc.15871
32. Liu H, Zhang TA, Zhang WY, Huang SR, Hu Y, Sun J. Rhein attenuates cerebral ischemia-reperfusion injury via inhibition of ferroptosis through NRF2/SLC7A11/GPX-4 pathway. Exp Neurol. 2023;369:114541. doi:10.1016/j.expneurol.2023.114541
33. Duan WL, Ma YP, Wang XJ, et al. N6022 attenuates cerebral ischemia/reperfusion injury-induced microglia ferroptosis by promoting Nrf2 nuclear translocation and inhibiting the GSNOR/GSTP1 axis. Eur J Pharmacol. 2024;972:176553. doi:10.1016/j.ejphar.2024.176553
34. Liu Z, Liu Q, Zhang X, Li G. Dexmedetomidine inhibits ferroptosis by regulating the SRY-box transcription factor 9/divalent metal transporter-1 axis to alleviate cerebral ischemia/reperfusion injury. Chem Biol Drug Des. 2025;105(1):e70022. doi:10.1111/cbdd.70022
35. Wang GL, Xu SY, Lv HQ, Zhang C, Peng YJ. Electroacupuncture inhibits ferroptosis induced by cerebral ischemiareperfusion. Curr Neurovasc Res. 2023;20(3):346–353. doi:10.2174/1567202620666230623153728
36. Lang J, Luo J, Wang L, et al. Electroacupuncture suppresses oxidative stress and ferroptosis by activating the mTOR/SREBP1 pathway in ischemic stroke. Crit Rev Immunol. 2024;44(6):99–110. doi:10.1615/CritRevImmunol.2024051934
37. Pu Y, Cheng J, Wang Z, et al. Electroacupuncture pretreatment inhibits ferroptosis and inflammation after middle cerebral artery occlusion in rats by activating Nrf2. Histol Histopathol. 2024;18780.
38. Ye T, Zhang N, Zhang A, Sun X, Pang B, Wu X. Electroacupuncture pretreatment alleviates rats cerebral ischemia-reperfusion injury by inhibiting ferroptosis. Heliyon. 2024;10(9):e30418. doi:10.1016/j.heliyon.2024.e30418
39. Zhu W, Dong J, Han Y. Electroacupuncture downregulating neuronal ferroptosis in mcao/r rats by activating Nrf2/SLC7A11/GPX-4 axis. Neurochem Res. 2024;49(8):2105–2119. doi:10.1007/s11064-024-04185-x
40. Yang XC, Jin YJ, Ning R, et al. Electroacupuncture attenuates ferroptosis by promoting Nrf2 nuclear translocation and activating Nrf2/SLC7A11/GPX-4 pathway in ischemic stroke. Chin Med. 2025;20(1):4. doi:10.1186/s13020-024-01047-0
41. Di Sanzo M, Quaresima B, Biamonte F, Palmieri C, Faniello MC. FTH1 pseudogenes in cancer and cell metabolism. Cells. 2020;9(12):2554. doi:10.3390/cells9122554
42. Seibt TM, Proneth B, Conrad M. Role of GPX-4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–152. doi:10.1016/j.freeradbiomed.2018.09.014
43. Xie K, Liu L, Wang M, et al. IMPA2 blocks cervical cancer cell apoptosis and induces paclitaxel resistance through p53-mediated AIFM2 regulation. Acta Biochim Biophys Sin. 2023;55(4):623–632. doi:10.3724/abbs.2023069
44. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX-4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. doi:10.1038/s41586-019-1705-2
45. Ohiro Y, Garkavtsev I, Kobayashi S, et al. A novel p53-inducible apoptogenic gene, PRG3, encodes a homologue of the apoptosis-inducing factor (AIF). FEBS Lett. 2002;524(1–3):163–171. doi:10.1016/S0014-5793(02)03049-1
46. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. doi:10.1038/s41586-019-1707-0
47. Wu Y, Shi H, Zheng J, et al. Overexpression of FSP1 ameliorates ferroptosis via PI3K/ AKT /GSK3beta pathway in PC12 cells with oxygen-glucose deprivation/reoxygenation. Heliyon. 2023;9(8):e18449. doi:10.1016/j.heliyon.2023.e18449
48. Zou R, Liu Z, Wang P, Liu Y. Ginkgolide B binds to GPX-4 and FSP1 to alleviate cerebral ischemia/reperfusion injury in rats. Toxicol Appl Pharmacol. 2025;495:117237. doi:10.1016/j.taap.2025.117237
49. Nakamura T, Hipp C, Santos Dias Mourao A, et al. Phase separation of FSP1 promotes ferroptosis. Nature. 2023;619(7969):371–377. doi:10.1038/s41586-023-06255-6
50. Martin KJ, Arthur JS. Selective kinase inhibitors as tools for neuroscience research. Neuropharmacology. 2012;63(7):1227–1237. doi:10.1016/j.neuropharm.2012.07.024
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.