Back to Journals » International Journal of Nanomedicine » Volume 14
Arsenic sulfide nanoformulation induces erythroid differentiation in chronic myeloid leukemia cells through degradation of BCR-ABL
Authors Wang T ![]() , Wen T, Li H, Han B, Hao S, Wang C, Ma Q, Meng J, Liu J
, Wen T, Li H, Han B, Hao S, Wang C, Ma Q, Meng J, Liu J ![]() , Xu H
, Xu H ![]()
Received 1 March 2019
Accepted for publication 17 June 2019
Published 22 July 2019 Volume 2019:14 Pages 5581—5594
DOI https://doi.org/10.2147/IJN.S207298
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Video abstract presented by Haiyan Xu.
Views: 276
Tao Wang,1 Tao Wen,1 Hongmin Li,2 Bing Han,2 Suisui Hao,1 Chuan Wang,1 Qiang Ma,1 Jie Meng,1 Jian Liu,1 Haiyan Xu1
1Department of Biomedical Engineering, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, People’s Republic of China; 2Department of Hematology, Peking Union Medical College Hospital, Beijing, People’s Republic of China
Background: Chronic myeloid leukemia (CML) is a myeloproliferative disorder due to the existence of BCR-ABL fusion protein that allows the cells to keep proliferating uncontrollably. Although tyrosine kinase inhibitors can inhibit the activity of BCR-ABL fusion protein to trigger the cells apoptosis, drug resistance or intolerance exists in part of CML patients. Arsenic sulfide in its raw form (r-As4S4) can be orally administrated and certain therapeutic effects have been found out in the treatment of hematologic malignancies through inducing cell apoptosis.
Methods: In this work, a water-dissolvable arsenic sulfide nanoformualtion (ee-As4S4) composed of As4S4 particulates with 470 nm in diameter and encapsulated by a kind of hydrophilic polymer was fabricated and applied to the CML cell line K562, K562/AO2 and primary cells from the bone marrow of CML patients.
Results: Results showed that instead of inhibiting the activity of BCR-ABL, ee-As4S4 induced direct degradation of BCR-ABL in K562 cells within 6 hr incubation, followed by the occurrence of erythroid differentiation in K562 after 72 hr incubation, evidenced by the significantly upregulated CD235a and benzidine staining, which was not detectable with r-As4S4. The ee-As4S4-induced erythroid differentiation was also observed in K562/AO2 cells and bone marrow mononuclear cells of CML patients. Mechanistic studies indicated that ee-As4S4 induced autophagy by downregulating the level of intracellular ROS and hypoxia-inducible factor-1α significantly, which led to the subsequent degradation of BCR-ABL. When the concentration was increased, ee-As4S4 induced much more significant apoptosis and cell cycle arrest than r-As4S4, and the cytotoxicity of the former was about 178 times of the latter.
Conclusion: ee-As4S4 was capable of inducing significant erythroid differentiation of CML cells by inducing the direct degradation of BCR-ABL; the new effect could improve hematopoietic function of CML patients as well as inhibit the leukemic cell proliferation.
Keywords: chronic myeloid leukemia, erythroid differentiation, As4S4, ROS
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disorder due to the existence of the BCR-ABL fusion protein, a constitutively active tyrosine kinase produced as a result of translocation of chromosomes 9 and 22.1 The BCR-ABL protects the cells from apoptosis, leading to the constant proliferation of undifferentiated stem cells in the absence of growth factors.2 It has been well known that tyrosine kinase inhibitors (TKIs) can trigger the apoptosis and differentiation of CML cells by specifically inhibiting the activity of BCR-ABL fusion protein;3,4 therefore the inhibitors have been used as first-line therapeutics for CML treatment.5,6 Nevertheless, TKIs resistance or intolerance happened sometimes;7,8 hence it is of significance to develop alternative or supplementary therapeutic options for CML therapy.
Arsenic sulfide is a kind of mineral drug that can be orally administrated and has shown certain therapeutic effects in the treatment of leukemia by inducing cell apoptosis,9 either in single10 or multiple content formulation.11–13 Nevertheless, arsenic sulfide in its raw form is poorly soluble in neutral or acidic aqueous solution; hence, only the leachable content is bioavailable, which was as low as about 4%.14 We previously reported that a solid dispersion formulation of arsenic sulfide could be dissolved in normal saline quickly, which significantly enhanced the bioavailability as well as the therapeutic efficacy when orally administrated in the mouse model of acute promyelocytic leukemia.15 Encouraged by those results, in this study, we fabricated and applied a water-dissolvable As4S4 nanoformulation (ee-As4S4) to the CML cell line K562, K562/AO2 and primary cells from CML patients, investigating whether ee-As4S4 could exert unknown therapeutic effect due to its largely increased bioavailability. We showed for the first time that ee-As4S4 induced direct degradation of BCR-ABL fusion protein instead of inhibiting the protein’s activity, which resulted in the remarkable erythroid differentiation in the CML cells, and the degradation of BCR-ABL resulted from the occurrence of autophagy following the effective downregulation of intracellular ROS by ee-As4S4.
Materials and methods
Preparation and characterization of ee-As4S4
ee-As4S4 was prepared according to the following steps. First, r-As4S4 (Alfa Aesar Co., Ward Hill, MA, USA, PubChem CID: 139298) and polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol copolymer (PVCL-PVAc-PEG, MW= 1.18 × 105 Da; BASF SE, Ludwigshafen, Germany) were mixed with a mass ratio of 1:15 and fed into a HAAKE MiniLab II co-rotating twin-screw extruder (ThermoFisher Scientific, Rockford, IL, USA). The processing conditions were set as described below: blending temperature in the mix chamber 120°C, screw rotation rate 10 rpm and cycling time 70 mins. The co-extruded product was ground in a coffee grinder at room temperature for preparing the stock solution of ee-As4S4. Next, 0.1 g As4S4 was added in 100 mL saline or complete medium, followed by the centrifugation for 5 mins in a speed of 1,000 rpm. The supernatant was taken out as the stock solution, and the concentration of As4S4 was determined with XSERIES 2 Quadrupole ICP-MS instrument (ThermoFisher Scientific Inc., Franklin, MA, USA). The hydrodynamic diameter of ee-As4S4 particles was measured using dynamic light scattering (DLS, Nano ZS90 Zetasizer, Malvern Instruments, and Malvern, UK).
Cell culture
Cell lines
K562 cells were purchased from the Cell Resource Center of the Chinese Academy of Medical Sciences (Beijing, China). The cell line was checked free of mycoplasma contamination by PCR and culture, and its species origin was confirmed with PCR. Cells were cultured in modified RPMI medium (Hyclone, GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% FBS (Gibco, Life Technologies, Carlsbad, CA, USA), 100 U/mL penicillin (Hyclone), and 100 μg/mL streptomycin (Hyclone) in a humidified atmosphere of 5% CO2 at 37°C. Adriamycin-resistant K562/AO2 cells were purchased from Institute of Hematology, Chinese Academy of Medical Sciences (Tianjin, China) and cultured in the same condition as K562. Cells of passage number between P3 and P10 were used for experiments.
CML patients and bone marrow mononuclear cells (BMMNCs) isolation
This study was performed in accordance with the Declaration of Helsinki and was approved by the Peking Union Medical College Hospital ethics committee, and written informed consent was obtained from all subjects. CML patients were recruited from Peking Union Medical College Hospital, confirmed by central review and classified in accordance with International Prognostic Scoring System. BMMNCs were isolated by Ficoll (Dakewe Biotech Co., Ltd., Shenzhen, China) gradient centrifugation (1.077±0.001 g/mL), and then cultured in modified RPMI medium supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin in a humidified atmosphere of 5% CO2 at 37°C.
Cellular uptake of As4S4
K562 cells were incubated with or without ee-As4S4 for different time. After incubation, viable single cells were collected by a MoFlo XDP flow sorter (BD Biosciences, San Jose, CA, USA). The arsenic content in the collected cells was measured using hydride generation atomic fluorescence spectrometry (AFS-8230 HG-AFS, Beijing Titan Instrument Co. Ltd., Beijing, China).
Cell viability assay
Cell viability was measured using CCK-8 (Dojindo, Kumamoto, Japan) according to the manufacturer’s protocol. In brief, after incubation with r-As4S4 or ee-As4S4 for 72 hrs at 37°C, 10 μL CCK-8 reagent was added to each well and incubated at 37°C for 1 hr. The absorbance was read at 450 nm with a Synergy H1 microplate spectrophotometer (BioTek Instruments, Winooski, VT, USA) and normalized with the untreated group.
Apoptosis assay
K562 cells were seeded in 24-well plate and incubated with ee-As4S4 for 72 hrs, and then stained with FITC-Annexin V and PI (eBioscience, Vienna, Austria) following the manufacturer instruction before subjected to flow cytometer.
Western blotting
Cells were harvested, lysed with RIPA (Beyotime Biotechnology, Haimen, China) supplement with Phenylmethanesulfonyl fluoride (PMSF) (Sigma-Aldrich, St Louis, MO, USA) and protein phosphatase inhibitor (Applygen, Beijing, China) and centrifuged. Protein samples were solubilized in protein-loading buffer (Applygen) and denatured by boiling. The samples were electrophoresed in 12% SDS/PAGE gels (Applygen) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA). The membranes were incubated with 5% BSA at room temperature for 1 hr, and then the following primary antibodies were used: anti LC3B (Cell Signaling Technology, CST; Danvers, MA, USA; CAT#: 3868S, LOT#: 11), anti AKT (CST, CAT#: 9272S, LOT#: 27), anti-phosp-AKT (CST, CAT#: 4060S, LOT#: 23), anti β-ACTIN (CST, CAT#: 3700S, LOT#: 17), anti-p38 MAPK (CST, CAT#: 8690S, LOT#: 8), anti-phosp-p38 MAPK (CST, CAT#: 9215S, LOT#: 7), anti-ERK1/2 (CST, CAT#: 4695S, LOT#: 21), anti-phosp-ERK1/2 (CST, CAT#: 4370S, LOT#: 17), anti-p21 (CST, CAT#: 2947T, LOT#: 9), anti-c-CBL (CST, CAT#: 2179T, LOT#: 1), anti-GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA, CAT#: sc25778, LOT#: H0612) and anti-Hypoxia-inducible factor-1 α (HIF-1α) (CST, CAT#: ab179483, LOT#: GR3228170-4). BCR-ABL was blotted by anti-c-ABL (CST, CAT#: 2862S, LOT#: 16). To develop immunostaining, we used Horseradish Peroxidase (HRP)-conjugated secondary antibodies against rabbit IgG and mouse IgG (Jackson ImmunoResearch, CAT#: 111-035-003, LOT#: 129736). The immune-complex on the membrane was visualized using an automatic chemoluminescence image analysis system (Tanon, Shanghai, China) with HRP substrate luminol reagent and peroxide solution (Millipore).
Quantitative real-time PCR
Total RNA was extracted by TRIzol reagent (Sigma-Aldrich) and reverse transcribed into cDNA via RT-PCR. Quantitative real-time PCR was performed using SRBY probe (Takara Bio, Shiga, Japan) and GAPDH was used as an internal control. The following primers were used:
BCR-ABL forward primer 5ʹ-TCCACTCAGCCACTGGATTTAA-3ʹ,
BCR-ABL reverse primer 5ʹ-TGAGGCTCAAAGTCAGATGCTACT-3ʹ,
GAPDH forward primer 5ʹ-CCAGCAAGAGCACAAGAGGAAGAG-3ʹ,
GAPDH reverse primer 5ʹ-AGCACAGGGATACTTTATTAGATG-3ʹ.
Cell differentiation assay
The hemoglobin content of K562 cells was assessed by benzidine staining. In brief, 2% (w/v) benzidine (Aladdin, Shanghai, China) solution in 3% HAc was prepared in prior. 30 μL H2O2 (wt%=30%, Aladdin) was added into the mixture before use. K562 cells were incubated with ee-As4S4 for 72 hrs and then collected and washed with PBS and then suspended in 50 μL PBS. 5 μL benzidine working solution was added in cell suspension and incubated at room temperature for 30 mins in dark. Smears of cells were observed under a microscope (Olympus BX53, Tokyo, Japan). Take photos of 5 fields of vision and count blue-colored cells. The cells were also collected and stained with PE-conjugated antibodies against CD235a (ebioscience, ThermoFisher Scientific, CAT#: 12–9987-82, LOT#: 4329624). The antibody-labeled cells were subsequently analyzed by flow cytometer (Accuri C6 flow cytometer; BD Biosciences).
Transmission electron microscope (TEM) observation of cells
Cells were incubated with or without ee-As4S4 and then collected and fixed with 2.5% glutaraldehyde overnight. After being washed and post-fixed in 1% OsO4 for 30 mins, the specimens were dehydrated gradually by alcohol and embedded in epon. Sections were then cut with an ultra-microtome and placed on copper grids for TEM observation using a JEM-1010 transmission electron microscope (JEOL Ltd., Tokyo, Japan).
ROS detection
Cells were incubated with ee-As4S4 for 0.5–72 hrs. Following incubation, cells were collected and washed with PBS, incubated in 300 μL 10 μM 2ʹ, 7ʹ-dichlorodihydrofluorescein diacetate (Sigma-Aldrich) for 30 mins at 37°C. Afterward, cells were washed by PBS and suspended in 100 μL PBS for flow cytometer analysis.
Electron spin resonance (ESR) spectroscopic measurements
All ESR measurements were carried out using a Bruker EMX ESR spectrometer (Billerica, MA) at ambient temperature with 20 mW microwave power, 1 G field modulation. Fifty microliter aliquots of sample solution was put in glass capillary tubes with internal diameters of 1 mm and sealed. The spin trap, 5-Tert-Butoxycarbonyl-5-Methyl-1-Pyrroline N-oxide (BMPO), was used to identify superoxide anion during the ESR measurements. The chemical KO2 system (1O2) was generated by dissolving KO2 in DMSO solvent in the presence of crown ether to verify the ability of scavenging 1O2.
Cell cycle analysis
Cells were incubated with ee-As4S4 for 72 hrs, washed with PBS, fixed and permeabilized with 70% cold ethanol overnight at 4°C. Cells were washed and incubated with 20 mg/L RNase (Beyotime Biotechnology) for 20 mins at 37°C and then stained with 50 mg/L PI (Sigma-Aldrich) for 10 mins at room temperature before being subjected to flow cytometer analysis of DNA content. The percentage of cell cycle distribution was calculated by FlowJo software.
Statistical analysis
All data were expressed as mean and SEM. The data shown were obtained from at least 3 independent experiments. One-way ANOVA was used for statistical analysis and performed in SPSS 22.0. P-value <0.05 was considered statistically significant.
Results
ee-As4S4 induced direct elimination of BCR-ABL in K562 cells
The ee-As4S4 was dissolvable in saline quickly (Figure 1A), and the average hydrodynamic diameter of the particulates was 470 nm (Figure 1B). When incubated with K562 cells, ee-As4S4 could be taken up and reached the highest accumulation in the cells after 12 hr incubation, while r-As4S4 was hardly taken up (Figure 1C). The cytotoxicity of ee-As4S4 to K562 cells was concentration dependent (Figure 1D); the IC50 of 72 hr incubation was 2.4 mg/L. The apoptosis occurred significantly when ee-As4S4 was incubated at 2.0 mg/L (Figure 1E). Next, the expression of BCR-ABL was examined in K562 cells incubated with ee-As4S4 at 4.0 mg/L or imatinib at 1 μM for 6 hrs, and within this short incubation the cell viability was about 80% (Figure S1). It was shown that the ee-As4S4 incubation led to a significant reduction of the total amount of BCR-ABL protein that stayed unchanged after the incubation of imatinib (Figure 1F), therefore, strongly suggesting that ee-As4S4 interacted with BCR-ABL in a different mechanism from imatinib. The mRNA of BCR-ABL was not changed after the same incubation (Figure 1G), indicating that the ee-As4S4-induced BCR-ABL elimination did not take place at the transcriptional level but at post-translational modification. In addition, different from the mechanism of As4S4 dissolved in NaOH aqueous solution, ee-As4S4 did not eliminate BCR-ABL through the enhancement of ubiquitination and subsequent proteolysis of its substrate BCR-ABL,16 evidenced by the unchanged expression of c-CBL after the treatment (Figure 1H).
 |
Figure 1 ee-As4S4 induced direct elimination of BCR-ABL in K562 cells. (A) The powder (insets) and solution of r-As4S4 and ee-As4S4 in ddH2O. (B) The size distribution of ee-As4S4 dissolved in ddH2O. (C) Cellular uptake of As4S4 in K562 cells incubated with 4.0 mg/L ee-As4S4 or r-As4S4. (D) The viability of K562 cells incubated with ee-As4S4 for 72 hrs. Data represent the mean ± SEM (n=4). **P<0.01, ***P<0.001 compared with the untreated group by Dunnett-T3 test. (E) The percentages of Annexin V- and PI-positive cells. Data represent the mean ± SEM (n=3). *P<0.05, **P<0.01 compared with untreated group by Dunnett-T3 test. (F) The expression of BCR-ABL in K562 cells incubated with imatinib and ee-As4S4 for 6 hrs. β-actin served as loading control. (G) Relative level of BCR-ABL mRNA in K562 cells incubated with ee-As4S4. Data represent the mean ± SEM (n=3). (H) The expression of c-CBL in K562 cells incubated with ee-As4S4 for 72 hrs. GAPDH served as loading control. |
ee-As4S4 induced erythroid differentiation through phosphorylation of p38 MAPK
The elimination of BCR-ABL protein implied the status of the cells changed, because CML cell differentiation is largely involved with BCR-ABL blockade.2 K562 cells are of the erythroleukemia type, which was derived from a 53-year-old female CML blast crisis, and used as a model cell line for erythroid differentiation. Therefore, we next examined if ee-As4S4 induced erythroid differentiation in K562 cells. When K562 cells were incubated with ee-As4S4 at 1.0, 2.0 or 4.0 mg/L for 72 hrs followed by benzidine staining, the number of blue-colored cells increased remarkably in the population referred to the untreated group (Figure 2A); the result of polymer control group is given in the supporting information (Figure S2). The statistical counting ratio of blue-colored cells in the K562 incubated with ee-As4S4 at 1.0, 2.0 and 4.0 mg/L was 14.41%, 19.96% or 11.51%, respectively (Figure 2B), clearly indicating the occurrence of erythroid differentiation in the cells. In addition, ee-As4S4 of 2.0 mg/L also induced significant erythroid differentiation in adriamycin-resistant K562 cells (K562/AO2) (Figure 2C). It should be addressed that erythroid differentiation was also observed in adriamycin-resistant K562 cells, which suggested that there was no cross-resistance between adriamycin and ee-As4S4, and ee-As4S4 could be an alternative medicine for those adriamycin-resistant cells. At the same time, the level of CD235a was upregulated significantly in the cells incubated with ee-As4S4 at 0.5–4.0 mg/L for 72 hrs (Figure 2D, S3). In accordance with benzidine staining, 2.0 mg/L induced the highest ratio of differentiation after 48 hr and 72 hr incubation, and the level of CD235a after 72 hr incubation was increased to 2.54- and 4.04-folds of the untreated group, respectively, while 6 hr and 24 hr incubation did not induce appreciable differentiation (Figure 2D), indicating that ee-As4S4-induced the differentiation was time-consuming and the internalized ee-As4S4 underwent some chemical reactions inside the cells. Different from ee-As4S4, r-As4S4 did not induce any erythroid differentiation in K562 cells even at 200 mg/L (Figure S4), which was possibly due to the extreme low bioavailability, thus clearly indicating that the induction of erythroid differentiation was one unique function of ee-As4S4. When the concentration of ee-As4S4 increased to 3.0 mg/L and 4.0 mg/L, and thereby the toxicity, the percentage of differentiation went down.
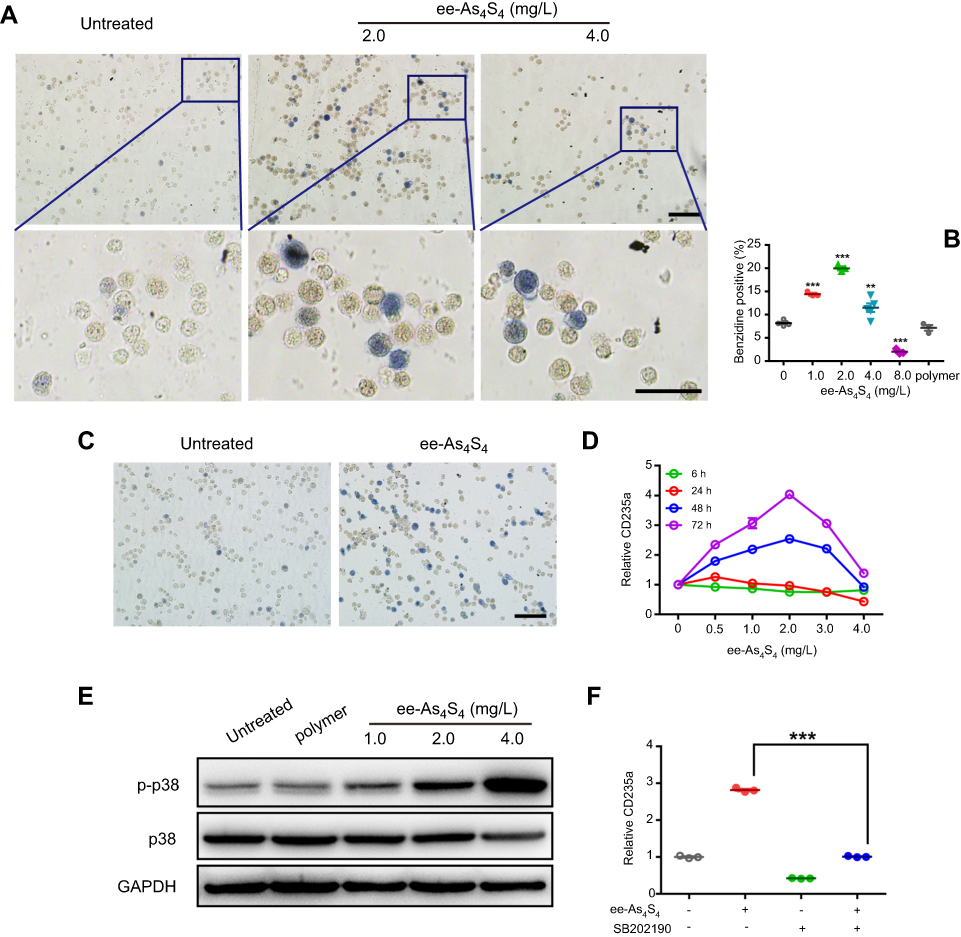 |
Figure 2 ee-As4S4 induced erythroid differentiation of CML cells through phosphorylation of p38 MAPK. (A) Representative benzidine staining images of K562 cells incubated with 0, 2.0 and 4.0 mg/L ee-As4S4, Bar represents 100 μm. (B) Quantification of benzidine-positive K562 cells according to the statistically counting. Data represent mean ± SEM (untreated group and 4.0 mg/L ee-As4S4 group, n=5; 2.0 mg/L ee-As4S4 group, n=3). **P<0.01 and ***P<0.001 compared with untreated group by Dunnett t-test. (C) Representative benzidine staining images of K562/AO2 cells incubated with or without ee-As4S4, Bar represents 100 μm. (D) The expression of CD235a for K562 cells incubated with ee-As4S4 for different time. (E) Phosphorylation of p38 MAPK in K562 cells incubated with ee-As4S4 for 72 h. (F) The expression of CD235a in K562 cells after incubation with ee-As4S4 together with SB202190. Data represent the mean ± SEM (n=3). ***P<0.001, by LSD post-hoc test. |
It is well-documented that p38 MAPK was involved in the erythroid differentiation of K562 cells;2,17,18 here consistently we observed that ee-As4S4 increased the expression of p-p38 MAPK in K562 cells after 72 hr incubation (Figure 2E). When a p-38 inhibitor SB202190 was supplemented in the culture medium, the expression level of CD235a went down (Figure 2F), demonstrating that ee-As4S4 induced erythroid differentiation through the phosphorylation of p38 MAPK.
Importantly, the ee-As4S4-induced erythroid differentiation was also detected in BMMNCs isolated from CML patients diagnosed by central review and classified in accordance with International Prognostic Scoring System. The proportion of primary cells in the bone marrow of Patient 1# was 7.5%, and the proliferation of bone marrow was active (Figure 3A); the proportion of BCR-ABL-positive cells was 95% determined by FISH. In Patient 2#, after treatment with imatinib for 6 months, IS BCR-ABL was 36.9% compared to 35.2% determined by PCR before imatinib treatment. In the sample of 1# CML patient who was newly diagnosed and had not received any treatment, the proportion of erythroid cells was only about 0.43%, while that of granulocyte cells was 97.5% under the optical microscope (Figure 3A), which meant the situation was extremely severe. In this worse status, the treatment with 0.5, 1.0 and 2.0 mg/L of ee-As4S4 for 7 days could raise the proportion of CD235a to 1.07%, 1.86% and 2.36%, respectively (Figure 3B), which was equivalent as 2.49-folds, 4.33-folds and 5.49-folds, respectively. This is a significant improvement for Patient 1#. As for Patient 2#, who was post-treated with imatinib for 6 months, the proportion of granulocyte cells was 37.5% and that of erythroid cells was 31.5% (Figure 3C), ee-As4S4 was also capable of further increasing the proportion of CD235a+ cells in the BMMNCs (Figure 3D). These results provided valuable clinical evidence that ee-As4S4 could induce effective erythroid differentiation and in our opinion, the occurrence of erythroid differentiation is likely to lead the increase of erythrocyte in blood; therefore it may improve hematopoietic function. Therefore, we suggested that ee-As4S4 is a supplementary therapy option for CML patients.
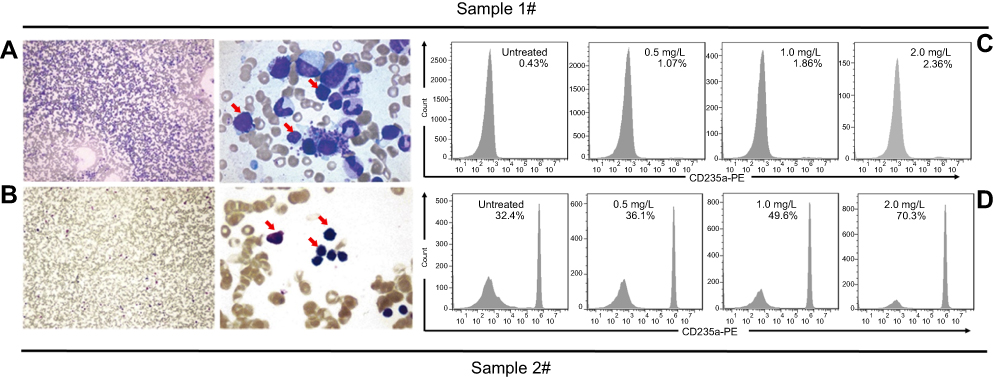 |
Figure 3 ee-As4S4 induced erythroid differentiation in BMMNCs derived from two CML patients. (A and C) Bone marrow examination images of the patients. (B and D) The percentage of CD235a+ cells of the CML patients before and after incubation with ee-As4S4. |
ee-As4S4 decreased BCR-ABL by inducing autophagy
As ee-As4S4-induced BCR-ABL elimination did not take place at the transcriptional level, we hypothesized that the BCR-ABL was degraded in the autophagy-dependent pathway,19 because autophagy is intracellular lysosomal degradation and recycling of proteins and organelles.20 In this study, the autophagosomes accumulation was observed in the cells incubated with ee-As4S4, which was few in the untreated group (Figure 4A). It was also found out that the expression of LC3B-II began to increase only after 1 hr of ee-As4S4 incubation, while BCR-ABL remained unchanged at that time point (Figure 4B), indicating ee-As4S4 triggered the autophagy prior to the BCR-ABL elimination. Correspondingly, ee-As4S4 at 2.0 mg/L, 4.0 mg/L, and 8.0 mg/L downregulated both total and the phosphorylation of AKT that is largely involved in the autophagy process (Figure 4C).
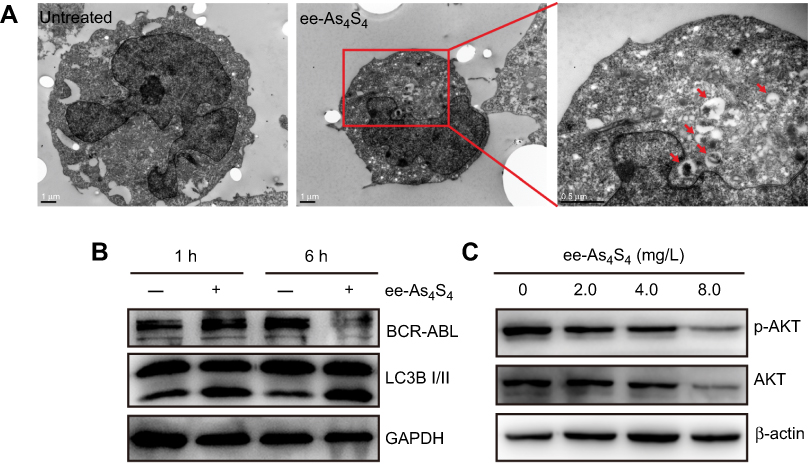 |
Figure 4 ee-As4S4 decreased BCR-ABL through autophagy. (A) TEM images of K562 cells incubated with ee-As4S4. The autophagosomes were pointed by red arrows.The scale bar represents 1 μm. (B) The expression of LC3B and BCR-ABL in K562 cells incubated with ee-As4S4 for 1 or 6 hrs. (C) Phosphorylation and total of AKT in K562 cells after incubation with ee-As4S4 for 72 hrs. GAPDH and β-ACTIN served as loading control. |
ee-As4S4 induced autophagy closely associated with downregulation of intracellular ROS and HIF-1α
Autophagy has been demonstrated to interact with the intracellular ROS21–23 that has crucial effects on the destiny of tumor cells.24 Accumulating evidence indicates that tumor cells usually have abnormal high level of ROS, which benefits their survival and resistance to drugs,25 and particularly in CML cells, the constitutively activated BCR-ABL produces more ROS.26 Considering As4S4 is a reducible compound, therefore we hypothesized that As4S4 is likely to react with the intracellular ROS. Strikingly it was observed that the ROS level in K562 cells was kept on decreasing over the incubation time when ee-As4S4 was supplemented in the culture medium (Figure 5A), the effect was detected even at the very beginning of the incubation of 30 mins, which was much earlier than the occurrence of autophagy. When exposed to 2.0 mg/L of ee-As4S4 for 48 hrs, the ROS level in K562 cells was decreased to about 20% of the untreated group. As a control, the polymer only increased the ROS level in K562 cells slightly (Figure S5), confirming the downregulated ROS level was contributed by the incubation of ee-As4S4. Interestingly, ESR analysis showed that As4S4 could scavenge superoxide anion in the KO2 solution system (Figure 5B), providing evidence that interacting with ROS was the intrinsic character of As4S4. It is well known that the accumulated intracellular ROS can stabilize HIF-1α, which activates downstream signals to enhance the survival of the cells.27 Our results showed that with the downregulation of intracellular ROS by ee-As4S4, the expression of HIF-1α in K562 cells was decreased correspondingly (Figure 5C).
 |
Figure 5 ee-As4S4 decreased intracellular ROS and HIF-1α in K562 cells. (A) Relative ROS level determined using 2ʹ, 7ʹ-dichlorodihydrofluorescein diacetate in K562 cells incubated with ee-As4S4. Data represent mean ± SEM (n=3). (B) ESR spectra of BMPO/·OOH adducts obtained from samples containing 25 mM BMPO, 20% DMSO, 2.5 mM KO2 and 0.35 mM 18-crown-6 in the absence or presence of 0.1mg/mL of As4S4. (C) The expression of HIF-1α incubated with ee-As4S4 for 6 and 12 hrs. GAPDH served as loading control. |
ee-As4S4 induced cell cycle arrest through p-erk1/2-dependent pathway
The cell cycle arrest was detected in K562 cells after ee-As4S4 incubation, showing the cells were arrested in G2/M phases. When incubated with ee-As4S4 at 1.0, 2.0 or 4.0 mg/L, the percentage of G2/M was 16.8%, 28.7% or 36.8%, respectively (Figure 6A, B). At the same time, the cell cycle-related protein p21 Waf1/Cip1 was upregulated in an incubation time-dependent manner (Figure 6C). In particular, after 6 hr incubation with ee-As4S4 at 2.0 or 4.0 mg/L, p21 Waf1/Cip1 was upregulated remarkably. The tumor-suppressor protein p53 is one of the most important upstream proteins in the regulation of p21expression. However, K562 cells do not express p53 in mRNA or protein level,28 and we would suggest that the downregulation of p21 in K562 cells induced by ee-As4S4 was not mediated through p53. Therefore, it was rational to consider that ee-As4S4 might increase p21 through other pathways.
 |
Figure 6 ee-As4S4 induced cell cycle arrest in G2/M phases through p-ERK1/2 dependent pathway. (A) Representative cell cycle analysis performed by flow cytometry of K562 cells incubated with ee-As4S4 at 1.0–4.0 mg/L for 72 hrs. (B) The percentage of cells in each phase. Data represent mean ± SEM (n=3) (C) The expression of cell cycle-dependent protein p21, GAPDH served as loading control. (D) The phosphorylation of ERK1/2 in K562 cells incubated with ee-As4S4. GAPDH served as loading control. (E) Cell viability of K562 cells treated with 4.0 mg/L of ee-As4S4, 5 μM of UO126 or both. (F) Representative cell cycle analysis of K562 cells incubated with 4.0 mg/L of ee-As4S4, 5 μM of UO126 or both. |
It was shown that the phosphorylation of ERK1/2 was increased in K562 cells incubated with ee-As4S4 at 2.0, 4.0 or 8.0 mg/L for 72 hrs (Figure 6D). UO126 is MEK inhibitor, which can inhibit the phosphorylation of ERK1/2. The cell viability of K562 treated with UO126 alone was 84.6% (Figure 6E). UO126 treatment decreased the cell cycle arrest induced by ee-As4S4 (Figure 6F). Therefore, we would suggest that phosphorylation of ERK1/2 participated in the G2/M phase arrest induced by ee-As4S4.
Discussion
It is well documented that the leukemogenic BCR-ABL fusion protein blocks the differentiation of CML cells and protects the cells from apoptosis, leading to the constant proliferation of undifferentiated stem cells. Therefore, inducing CML cells to cause differentiation represents a promising therapeutic strategy in the treatment of CML.4,29 Small-molecule BCR-ABL TKIs have fundamentally improved the treatment of CML and have become a paradigm for molecularly targeted therapy.8 However, single-point mutations within the BCR-ABL tyrosine kinase domain that interfere with drug-binding always cause BCR-ABL-dependent resistance to currently approved TKIs.8 Here we found out that ee-As4S4 acted as an anti-CML agent not through the way of inhibiting the activity of BCR-ABL, instead, through eliminating BCR-ABL directly, which makes ee-As4S4 a supplementary drug for CML patients. The function of inducing erythroid differentiation for ee-As4S4 may also decrease the fraction of cells endowed with self-renewal and/or accelerate the maturation of nonclonogenic leukemic cells30 and sensitize CML stem/progenitor cells to anticancer agents.18 Therefore, ee-As4S4 may improve the hematopoietic function of CML patients as well as inhibit the tumor cels proliferation. Additionally, these results are of implications to treatments of other hematopoietic disorders caused by myeloid diseases. It is shown that arsenic trioxide together with IFN decreases CML leukemia-initiating cells, therefore giving a chance to inhibit minimal residual disease and providing a curative approach for CML treatment.31 As4S4 showed anti-CML effect through degradation of BCR-ABL oncoprotein in K562 cells, make it holding a curative potential as well for the treatment for CML.
Although several studies have reported the enhanced cytotoxicity of nanoscaled As4S4,32–35 the function of inducing erythroid differentiation for As4S4 has not been observed. In this study, the water-dissolvable As4S4 nanoformulation (ee-As4S4) was applied to the CML cell lines and primary cells from CML patients, and the therapeutic effect of ee-As4S4 inducing erythroid differentiation in the CML cells was observed, which was attributable to the reductive property of As4S4 and the water-dissolvable nanoformulation. The ee-As4S4 could react with intracellular ROS effectively due to the large specific area of nanoparticles, downregulating the ROS level (Figure 5). ROS has been recognized as an upstream signal in the autophagic promotion of hematopoietic stem cell differentiation.23 For examples, an antioxidant resveratrol was reported to induce autophagy by scavenging intracellular ROS.21 In our work, ee-As4S4 induced ROS reduction from the very beginning of incubation (much less than 1 hr), autophagy was observed after 1 hr of incubation, and the differentiation was observed at 48 hrs of incubation (Figures 1, 4, 5). Therefore, we would suggest that ee-As4S4 induced erythroid differentiation of CML cells through downregulating the intracellular ROS level to trigger autophagy, which led to the autophagic elimination of BCR-ABL. In addition, the decrease of intracellular ROS destabilized HIF-1α that is closely associated with stemness of stem cells,36 which could enhance the ee-As4S4-induced erythroid differentiation.
Conclusion
In conclusion, ee-As4S4 could be taken up by K562 cells and induce effective erythroid differentiation both in the CML cell lines and in the CML patient-derived BMMNCs through BCR-ABL elimination that was resulted from the autophagy triggered by the intracellular ROS downregulation, in addition to inducing much more cytotoxicity to the cells than r-As4S4.
Ethics approval and consent to participate
This study was performed in accordance with the Declaration of Helsinki and was approved by the Peking Union Medical College Hospital ethics committee, and written informed consent was obtained from all subjects.
Abbreviation list
CML, chronic myeloid leukemia; BMMNCs, bone marrow mononuclear cells; ESR, electron spin resonance; TEM, transmission electron microscope; TKIs, tyrosine kinase inhibitors.
Acknowledgments
We thank Dr. Jun-jie Yin for his expertise support on the ESR experiment. This work was supported by National Key R&D Program of China (2017YFA0205504) and CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-3-004).
Author contributions
H.X. designed the experiments, discussed the results with the authors and revised the manuscript; T. Wang designed part of and performed the main experiments, conducted the data analysis, prepared the figures and wrote the main manuscript; T. Wen performed the ESR experiment and discussed the results; S.H. performed the q-PCR experiment; C.W. and Q.M. performed the cellular uptake assay; H.L. and B.H. provided the bone marrow samples and discussed the results; J.M. and J.L. reviewed the manuscript and discussed the results. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work
Disclosure
The authors report no conflicts of interest in this work.
References
1. Faderl S, Talpaz M, Estrov Z, O’Brien S, Kurzrock R, Kantarjian HM. Mechanisms of disease - the biology of chronic myeloid leukemia. New Engl J Med. 1999;341(3):164–172. doi:10.1056/NEJM199907153410306
2. Pierce A, Spooncer E, Ainsworth S, Whetton AD. BCR-ABL alters the proliferation and differentiation response of multipotent hematopoietic cells to stem cell factor. Oncogene. 2002;21(19):3068–3075. doi:10.1038/sj.onc.1205424
3. Fang GF, Kim CN, Perkins CL, et al. CGP57148B (STI-571) induces differentiation and apoptosis and sensitizes Bcr-Abl-positive human leukemia cells to apoptosis due to antileukemic drugs. Blood. 2000;96(6):2246–2253.
4. Jacquel A, Colosetti P, Grosso S, et al. Apoptosis and erythroid differentiation triggered by Bcr-Abl inhibitors in CML cell lines are fully distinguishable processes that exhibit different sensitivity to caspase inhibition. Oncogene. 2007;26(17):2445–2458. doi:10.1038/sj.onc.1210034
5. Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. New Engl J Med. 2006;355(23):2408–2417. doi:10.1056/NEJMoa062867
6. O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. New Engl J Med. 2003;348(11):994–1004. doi:10.1056/NEJMoa022457
7. Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376(10):917–927. doi:10.1056/NEJMoa1609324
8. O’Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12(8):513–526. doi:10.1038/nrc3317
9. Tong Y, Wu YL, Sun HP, et al. Combined effects of As4S4 and imatinib on chronic myeloid leukemia cells and BCR-ABL oncoprotein. Blood. 2004;104(13):4219–4225. doi:10.1182/blood-2004-04-1433
10. Lu DP, Qiu JY, Jiang B, et al. Tetra-arsenic tetra-sulfide for the treatment of acute promyelocytic leukemia: a pilot report. Blood. 2002;99(9):3136–3143. doi:10.1182/blood.v99.9.3136
11. Zhu HH, Huang XJ, Arsenic O. Retinoic acid for non-high-risk acute promyelocytic leukemia. New Engl J Med. 2014;371(23):2239–2241. doi:10.1056/NEJMc1412035
12. Zhu HH, Liu YR, Jia JS, Qin YZ, Zhao XS, Lai YY. Oral arsenic and all-trans retinoic acid for high-risk acute promyelocytic leukemia. Blood. 2018;131(26):2987–2989. doi:10.1182/blood-2018-02-834051
13. Zhu HH, Wu DP, Du X, et al. Oral arsenic plus retinoic acid versus intravenous arsenic plus retinoic acid for non-high-risk acute promyelocytic leukaemia: a non-inferiority, randomised phase 3 trial. Lancet Oncol. 2018;19(7):871–879. doi:10.1016/S1470-2045(18)30295-X
14. Kwan SY, Tsui SK, Man TO. Release of soluble arsenic from Realgar in simulated gastric juice. Anal Lett. 2001;34(9):1431–1436. doi:10.1081/AL-100104917
15. Ma Q, Wang C, Li X, et al. Fabrication of water-soluble polymer-encapsulated As4S4 to increase oral bioavailability and chemotherapeutic efficacy in AML mice. Sci Rep. 2016;6:29348. doi:10.1038/srep29348
16. Mao JH, Sun XY, Liu JX, et al. As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia. P Natl Acad Sci USA. 2010;107(50):21683–21688. doi:10.1073/pnas.1016311108
17. Huang YW, Lee WH, Tsai YH, Huang HM. Activin A induction of erythroid differentiation sensitizes K562 chronic myeloid leukemia cells to a subtoxic concentration of imatinib. Am J Physiol Cell Physiol. 2014;306(1):C37–C44. doi:10.1152/ajpcell.00130.2013
18. Lee YL, Chen CW, Liu FH, Huang YW, Huang HM. Aclacinomycin A sensitizes K562 chronic myeloid leukemia cells to imatinib through p38MAPK-mediated erythroid differentiation. PLoS One. 2013;8(4):e61939. doi:10.1371/journal.pone.0061939
19. Goussetis DJ, Gounaris E, Wu EJ, et al. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012;120(17):3555–3562. doi:10.1182/blood-2012-01-402578
20. White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. 2015;21(22):5037–5046. doi:10.1158/1078-0432.CCR-15-0490
21. Cao Y, Fang YX, Cai JY, et al. ROS functions as an upstream trigger for autophagy to drive hematopoietic stem cell differentiation. Hematology. 2016;21(10):613–618. doi:10.1080/10245332.2016.1165446
22. Liang Q, Wang XP, Chen TS. Resveratrol protects rabbit articular chondrocyte against sodium nitroprusside-induced apoptosis via scavenging ROS. Apoptosis. 2014;19(9):1354–1363. doi:10.1007/s10495-014-1012-1
23. Moosavi MA, Sharifi M, Ghafary SM, et al. Photodynamic N-TiO2 nanoparticle treatment induces controlled ros-mediated autophagy and terminal differentiation of leukemia cells. Sci Rep. 2016;6:34413.
24. Chen YF, Liu H, Luo XJ, et al. The roles of reactive oxygen species (ROS) and autophagy in the survival and death of leukemia cells. Crit Rev Oncol Hemat. 2017;112:21–30. doi:10.1016/j.critrevonc.2017.02.004
25. Irwin ME, Rivera-Del Valle N, Chandra J. Redox control of leukemia: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Sign. 2013;18(11):1349–1383. doi:10.1089/ars.2011.4258
26. Sattler M, Verma S, Shrikhande G, et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000;275(32):24273–24278. doi:10.1074/jbc.M002094200
27. Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1 alpha during hypoxia - A mechanism of O-2 sensing. J Biol Chem. 2000;275(33):25130–25138. doi:10.1074/jbc.M001914200
28. Woo S-M, Choi YK, Kim AJ, Cho S-G, Ko S-G. p53 causes butein‑mediated apoptosis of chronic myeloid leukemia cells. Mol Med Rep. 2016;13(2):1091–1096. doi:10.3892/mmr.2015.4672
29. Mazharian A, Ghevaert C, Zhang L, Massberg S, Watson SP. Dasatinib enhances megakaryocyte differentiation but inhibits platelet formation. Blood. 2011;117(19):5198–5206. doi:10.1182/blood-2010-12-326850
30. de Thé H. Differentiation therapy revisited. Nat Rev Cancer. 2018;18(2):117–127. doi:10.1038/nrc.2017.103
31. El Eit RM, Iskandarani AN, Saliba JL, et al. Effective targeting of chronic myeloid leukemia initiating activity with the combination of arsenic trioxide and interferon alpha. Int J Cancer. 2014;134(4):988–996. doi:10.1002/ijc.28427
32. Cholujova D, Bujnakova Z, Dutkova E, et al. Realgar nanoparticles versus ATO arsenic compounds induce in vitro and in vivo activity against multiple myeloma. Br J Haematol. 2017;179(5):756–771. doi:10.1111/bjh.14974
33. Song P, Chen P, Wang D, et al. Realgar transforming solution displays anticancer potential against human hepatocellular carcinoma HepG2 cells by inducing ROS. Int J Oncol. 2017;50(2):660–670. doi:10.3892/ijo.2016.3831
34. Tian Y, Wang X, Xi R, et al. Enhanced antitumor activity of realgar mediated by milling it to nanosize. Int J Nanomedicine. 2014;9:745–757. doi:10.2147/IJN.S56391
35. Zhao W, Lu X, Yuan Y, et al. Effect of size and processing method on the cytotoxicity of realgar nanoparticles in cancer cell lines. Int J Nanomedicine. 2011;6:1569–1577. doi:10.2147/IJN.S21373
36. Takubo K, Goda N, Yamada W, et al. Regulation of the HIF-1 alpha Level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391–402. doi:10.1016/j.stem.2010.06.020
Supplementary materials
 |
Figure S1 Cell viability of K562 cells after treatment with ee-As4S4 and imatinib for 6 hrs. |
 |
Figure S2 Representative benzidine staining images of K562 cells incubated with 300 mg/L soluplus. Bar represents 100 μm. |
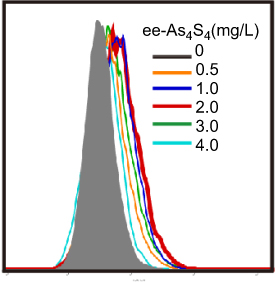 |
Figure S3 Overlapped distribution of CD235a expression of K562 cells after treatment with ee-As4S4 for 72 hrs in FACS. |
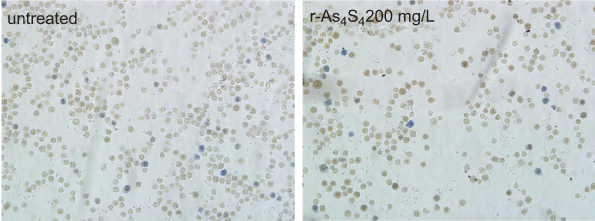 |
Figure S4 R-A4S4 did not induce any differentiation in K562 cells even when the concentration of As4S4 was up to 200 mg/L. Original magnification ×200. |
 |
Figure S5 Hydrophilic polymer increased the ROS in K562 cells slightly. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.