")
Back to Journals » International Journal of Nanomedicine » Volume 13
Arginine, glycine, aspartic acid peptide-modified paclitaxel and curcumin co-loaded liposome for the treatment of lung cancer: in vitro/vivo evaluation
Received 22 November 2017
Accepted for publication 16 March 2018
Published 27 April 2018 Volume 2018:13 Pages 2561—2569
DOI https://doi.org/10.2147/IJN.S157746
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Kanqiu Jiang,* Mingjing Shen,* Weihua Xu
Department of Cardiothoracic Surgery, The Second Affiliated Hospital of Soochow University, Suzhou Shi, Jiangsu Sheng, People’s Republic of China
*These authors contributed equally to this work
Purpose: In this study, a novel arginine, glycine, aspartic acid peptide (RGD)-modified paclitaxel and curcumin co-loaded liposomes were developed to evaluate their antitumor activity in vitro and in vivo.
Materials and methods: Co-loaded liposomes were prepared using the solvent evaporation method. The particles had spherical shapes under electron microscopy with sizes <130 nm.
Results: By comparison with the free drug, RGD-modified paclitaxel and curcumin co-loaded liposomes and paclitaxel and curcumin co-loaded liposomes have sustained-release properties in vitro. In vivo, there was no significant difference in pharmacokinetic parameters between the RGD-modified paclitaxel and curcumin co-loaded liposomes and paclitaxel and curcumin co-loaded liposomes. A strong green fluorescence was observed in the cytoplasmic region after incubation of RGD-modified paclitaxel and curcumin co-loaded liposomes for 2 h. RGD-modified paclitaxel and curcumin co-loaded liposomes showed a superior antiproliferative effect on A549 cells with a possible mechanism that suppressed the multidrug resistance phenomenon and exhibited a clear synergistic effect.
Conclusion: The results indicate that RGD-modified paclitaxel and curcumin co-loaded liposomes had a better antitumor effect in vivo than the non-modified LPs. These results indicate that RGD-modified co-loaded liposomes are a promising candidate for antitumor drug delivery.
Keywords: arginine, glycine, aspartic acid peptide, paclitaxel, curcumin, liposome, cell uptake, cytotoxicity study, in vivo anti-tumor study
Introduction
Paclitaxel (Figure 1) (PTX), the prototype of taxanes agents, emerges from a natural source.1 Paclitaxel was effective in the treatment of a wide range of advanced human cancers, including non-small-cell lung cancer (NSCLC).2 The commercial PTX preparation (Taxol®) is formulated by Cremophor EL® (polyethoxylated castor oil used as a solubilizing agent) and anhydrous ethanol, which provides a homogeneous preparation. In clinical practice, PTX is usually administered as a 3-h and 24-h infusion representing a total dose of 135–175 mg/m2 of the body every 3 weeks.3 Although paclitaxel is the first choice for the treatment of lung cancer, its use is limited due to the limited uptake of the drug by p-gp.4
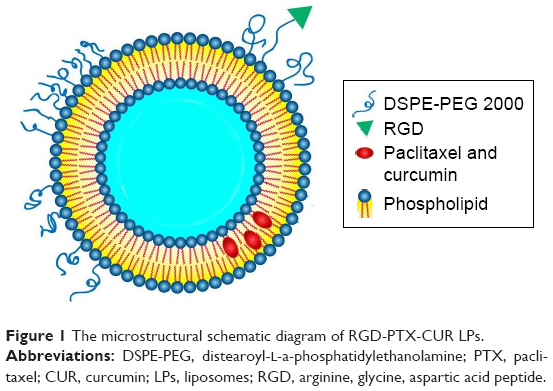 | Figure 1 The microstructural schematic diagram of RGD-PTX-CUR LPs. |
Curcumin (Figure 1B) (CUR) is a yellow polyphenol extracted from the rhizome of turmeric (Curcuma longa), a plant grown in tropical Southeast Asia.5 Enthusiasm for CUR as an anti-cancer agent was based on a larger number of epidemiological evidence, suggesting that dietary turmeric is associated with a lower incidence of gastrointestinal mucosal cancers.6,7 Numerous experimental data clearly indicate that free CUR induces cell cycle arrest and/or apoptosis in human cancer cell lines derived from a variety of solid tumors including colorectal, lung, breast, pancreatic and prostate carcinoma, and so on.8,9 In addition, some studies have shown that, after the reduction of PI3K/Akt and NF-kB pathways, CUR could independently display p-gp inhibitory activity.10,11 Therefore, CUR could be used as an effective p-gp inhibitor to maximize the cytotoxicity of anticancer drugs through combination with an anticancer drug on MDR-expressing cancer cells.
Despite PTX and CUR’s multiple medicinal benefits, low solubility and bioavailability of both continues to be highlighted as a major challenge in developing formulations for clinical efficacy. Current improvements are mainly focused on the development of formulations that are devoid of toxic solvent, surveys of the potential for large-scale preparation, and requirements for long-term stability. Liposomes represent versatile and advanced nanoscale delivery systems for a wide range of bioactive compounds.12
These relatively non-toxic systems have a considerable potential to entrap both hydrophobic and hydrophilic drugs. Traditional common delivery systems have some disadvantages, such as inappropriate release profiles, and the use of conventional p-gp inhibitors to be toxic and non-targeted to normal cells. Therefore, the ideal composite delivery system should have a target feature and an optimal release profile. In other words, the exposure of anticancer agents and p-gp inhibitors to normal cells or tissues should be minimized and limited to specific target cells or tissues.13,14 The integrin receptors, especially ανβ3, are often highly expressed in certain types of tumor cells and tumor vascular endothelial cells, but not in normal vessels.15 RGD is a short peptide containing arginine, glycine, and aspartic acid, which is widely found in vivo. RGD peptides, as the recognition site of integrin and its ligand, had adhesion between cells and extracellular matrix. The exogenous RGD peptide has a competitive inhibitory ligand to integrins and inhibits angiogenesis and migration of tumor cells. At the same time, the tumor can be targeted, labeled, and anticancer drugs can be target delivered.16–18
In this experiment, we used liposomes (LPs) as a carrier, PTX and CUR as model drugs to prepare RGD-modified PTX-CUR LPs. LPs have good biodegradability and biocompatibility, and can achieve a sustained-release effect by gradually degrading in vivo. RGD peptide-modified LPs could improve the targeting ability of LPs. In addition, through enhanced permeability and retention (EPR) effects, it can increase the number of residual LPs in the tumor.19 In order to verify these properties of the LPs, various properties of RGD modified PTX-CUR LPs were studied in detail. Although there have been many reports on the delivery system of loading both PTX and CUR recently,20,21 the strategy of RGD modified was reported for the first time.
Materials and methods
Materials
PTX and CUR was gifted from Yangzi River Pharmaceuticals Co Ltd (Jiangsu, People’s Republic of China). Distearoyl-L-a-phosphatidylethanolamine (DSPE)-PEG2000 was provided by the FanShuo Biopharma Ltd (Shanghai, People’s Republic of China). High purity cholesterol (CHOL) and hydrogenated soybean phosphatidyl choline (SPC) were purchased by Phospholipid Tech Ltd, Shanghai, People’s Republic of China. RGD peptide was purchased from Enzo Life Sciences (Farmingdale, NY, USA). A549 lung cancer cell lines were purchased from the Institute of Biochemistry and Cell Biology (Shanghai, People’s Republic of China). The chemical and solvents used were analytical or high performance liquid chromatography. Deionized water was prepared in the laboratory.
Preparation of RGD-modified PTX-CUR LPs
RGD-modified PTX and CUR co-loaded LPs were prepared using the solvent evaporation method.22 Briefly, PTX (5 mg), CUR (3 mg), CHOL (15 mg), DSPE-PEG 2000 (12.5 mg) and SPC (120 mg) were dissolved in 10 mL of chloroform to produce an oil phase. This was attached to a rotary evaporator and the organic phase was removed by evaporating at 45°C±2°C, which led to the formation of the film on the wall of the flask. The flask remained overnight to remove any trace in the solvent. The lipid membranes were hydrated with 5 mL of phosphate-buffered saline (PBS, pH 7.4, containing 50 mg RGD peptide) at 37°C for 30 min. The suspension was then subjected to ultrasonic treatment for 2 min at 500 W using a high-intensity probe ultrasonicator (JY92-2D; Xinzhi Equipment Instruction, Zhejiang, People’s Republic of China). The same method was used to prepare PTX and CUR co-loaded LPs without RGD peptide as the control samples.
Characterization
Morphological examination of LPs was performed using Philips CM120 TEM (FEI, Eindhoven, the Netherlands). In practice, the LPs solution containing 0.1% (w/v) phosphotungstic acid was placed on the carbon film, and was observed by electron microscope at 80 kV.
A NICOMP 380 Submicron Particle Sizer with dynamic light scattering (DLS, PSS company, Santa Barbara, USA) was used to determine the particle size distribution and mean diameter of the prepared RGD-modified PTX-CUR LPs. Sample solutions were transferred into the light scattering cells. The intensity autocorrelation was measured at a scattering angle of 90° at room temperature. Data was analyzed in terms of intensity-weighted NICOMP distributions. The experimental results of each report are obtained with an average autocorrelation function of at least three DH values accumulated at least 20 min from the analysis. Zeta potential was measured on the same samples prepared for size analysis.
Characterization of LPs
Drug-loading coefficient (DL%) and encapsulation ratio (ER%) were calculated as described previously.23,24 Firstly, PTX and CUR were extracted from the LPs with 1 mL 2% acetic acid: acetonitrile (1:1, v/v), and then the extract solution was properly diluted prior to HPLC analysis. The content of PTX and CUR in the LPs was determined by the HPLC method described below. Then, DL% and ER% were calculated according to Eq (1) and Eq (2):
|
|
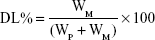
|
|
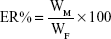
where WP is the weight of the initial feeding polymer, WM is the weight of the drug incorporated in LPs, and WF is the weight of the initial feeding drug.
In vitro drug release
In vitro release of CUR from the LPs was carried out by the dialysis membrane method. RGD-modified PTX and CUR co-loaded LPs (≈5 mg PTX and 3 mg CUR) were re-dispersed in 1 mL of deionized water in dialysis bags (sigma) with a molecular cut off of 10 kDa. The bags were suspended in the release of 18 mL medium (ethanol 50%, v/v) while shaking in a 37°C water bath at 75 rpm. The 0.1 mL sample was extracted at the predetermined time interval and replaced with fresh medium. Finally, the samples were analyzed by HPLC. In the release medium, ethanol was used to provide the sink condition because PTX and CUR were insoluble in water.
The control PTX and CUR co-loaded LPs without RGD peptide were prepared using the same method.
Stability study
Accelerated stability studies were conducted under the International Conference on Harmonisation (ICH) guidelines (1993). Sealed bottles of freshly prepared freeze-dried LPs were placed in a stability chamber maintained at 25°C, 60% relative humidity (RH). The physical appearance, size, physical and chemical properties of the samples were analyzed over a 3-month period and with a frequency of 1-month sampling.
Pharmacokinetics
All the in vivo experimental protocols were approved by the animal care committee of Soochow University and all experiments were conducted in strict accordance with the laboratory animal care and use guidelines adopted by the National Institutes of Health (Shanghai, People’s Republic of China). A total of 30 Sprague Daley rats (half male) weighing from 180 to 220 g were used for the pharmacokinetics study. Before the experiment commenced, the rats were kept in a state of fasting for 6 h. All animals were randomly divided into three groups with 10 in each group via iv injection of PTX and CUR suspension, conventional PTX/CUR LPs, and RGD-PTX/CUR LPs. The doses of PTX and CUR in the three groups were 10 and 6 mg/kg, respectively. Blood samples (2 mL) were collected from the tail vein into heparinized 5 mL polythene tubes just before administration and 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 h after dosing. The plasma obtained was stored at −20°C until analysis. The sample handling and determination process was referenced to previous literature by the LC-MS/MS method.25–28
Cell uptake
The cellular internalization of free PTX/CUR, PTX/CUR LPs and RGD-PTX/CUR LPs was observed with confocal microscopy by using coumarin-6 as a fluorescent probe. A549 cells were grown in RPMI 1640 medium with 10% (v/v) FBS and 5% antibiotics (100 IU/mL of penicillin G sodium and 100 μg/mL of streptomycin sulfate). A549 cells were inoculated at the initial density of 4×105 cells per dish. Cells were then incubated with coumarin-6-adsorbed free PTX/CUR, PTX/CUR LPs and RGD-PTX/CUR LPs (equivalent to 0.1 μg/mL of coumarin-6) for 2 h at 37°C±0.5°C.
Subsequently, the cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min under confocal microscopy. For PTX/CUR uptake quantitative estimates, the density of cells inoculated on 24-well plates was 3×104 cells. When reaching a confluence of 70%–80%, the cells were incubated with coumarin-6 adsorbed free PTX/CUR, PTX/CUR LPs and RGD-PTX/CUR LPs (equivalent to 0.1 μg/mL of coumarin-6). After 2 h of incubation, the cells were washed several times with cold PBS and then dissolved with Triton X-100 (0.1%). Multimode microplate reader was used to measure the fluorescence intensity and the excitation/emission wavelength was 440 and 520 nm, respectively.
Cytotoxicity study
The cytotoxicity of blank LPs, free PTX/CUR, PTX/CUR LPs and RGD-PTX/CUR LPs was evaluated in A549 cells by MTT assay. Briefly, A549 cells were seeded in a 96-well plate at a density of 3 to 4×103 cells per well. After 12 h, different PTX/CUR formulations (PTX/CUR concentrations ranging from 0.0025 to 25 μg/mL were added, and plates were incubated for 24 and 48 h, PTX:CUR=5:3). PTX/CUR standard solution was prepared with PTX/CUR dissolved in ethanol ranging from 0.25 to 2.5 mg/mL and then diluted 100 times with distilled water. Measurements were taken using a microplate reader.
In vivo anti-tumor study
The antitumor effect of RGD-PTX/CUR LPs was evaluated in vivo using BALB/c nude mice (aged 5–6 weeks, weight 20–22 g) that had been inoculated subcutaneously with 2×109 human lung adenocarcinoma (A549) cells. The treatments were started on the day when the tumor volume reached 100–150 mm3, which was designated as day 0. On the 8th day, the mice were randomly assigned to four groups (6 animals per group): group 1 was given a 5% glucose injection, group 2 was given free PTX and CUR, group 3 was given PTX/CUR LPs and group 4 was given RGD-PTX/CUR LPs. All the formulations were given via the tail vein on days 8, 10, 12, and 14, at a dose of 10 mg/kg (PTX) and 6 mg/kg (CUR). A digital caliper was used to measure the tumor diameters and tumor volumes (mm3) were calculated by the following formula: tumor volume=length×width2×0.5. Throughout the study, mice were weighed regularly to monitor the potential toxicities.
Statistical analysis
All experiments were expressed as mean ± SD and performed in triplicate. Statistical analysis was carried out by ANOVA analysis using GraphPad Prism 6.0. Statistically significant change is considered at p<0.05.
Results and discussion
Preparation and characterization
A solvent evaporation method was employed to prepare RGD-modified PTX and CUR co-loaded LPs and the method was simple and easy to scale up. In the process, lipid solution was first prepared by dissolving solid lipids with chloroform. The resulting lipid film was hydrated phosphate-buffered saline which was then filtered and dried. As shown in Figure 2, the surface morphology of PTX and CUR LPs was observed by TEM. The LPs were spherical in shape with a smooth surface and the size was uniform and appropriate for administration via intravenous injection. The mean diameter of RGD-PTX/CUR LPs was 120.6±10.8 nm and the poly disperse index (PDI) was 0.139±0.026. The other parameters are shown in Table 1. Obviously, there were no significant differences in the parameters between the modified liposomes and the unmodified liposomes. In addition, the zeta potential of liposomes indicates that there was a number of negative charges on the surface, which makes the whole system have good physical stability. There were no significant changes in the indexes after adding RGD into liposomes.
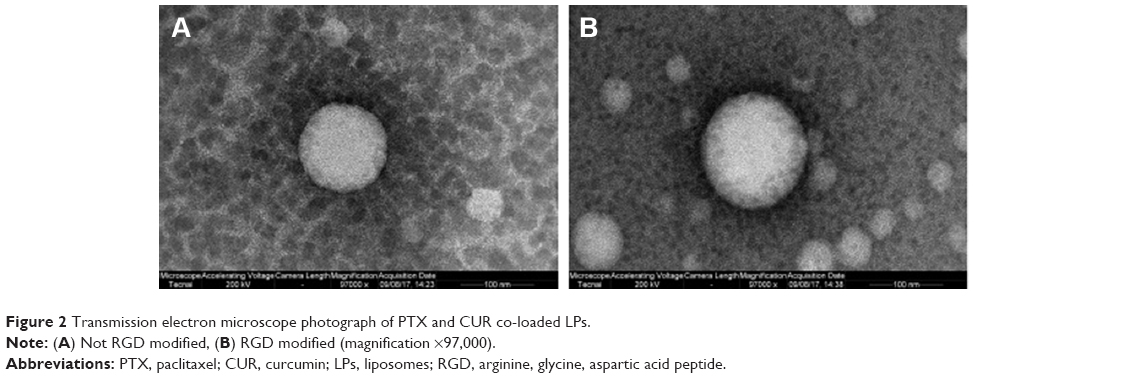 | Figure 2 Transmission electron microscope photograph of PTX and CUR co-loaded LPs. |
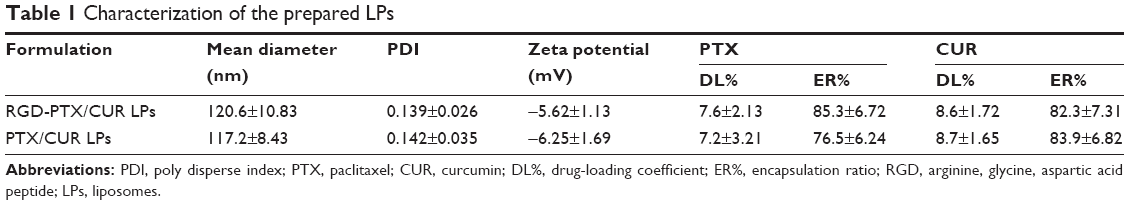 | Table 1 Characterization of the prepared LPs |
In vitro drug release
The in vitro cumulative drug-release profile is shown in Figure 3. Over time, both PTX and CUR in LPs was released much more slowly than the free drug. At 2 h, the cumulative release of the free drug (PTX) was about 90% but about 40% in LPs (CUR 86%, 30%). A biphasic release was observed in this case with a rapid release of about 40% in 2 h followed by sustained drug release of about 75% over 12 h (CUR 30%, 60%). This suggests that in comparison with the free drug, RGD-PTX-CUR LPs and PTX-CUR LPs have sustained-release properties. However, there was no significant difference between the two LPs in the release curve. Phospholipid, which is the main ingredient used in this experiment, is a corrosive substance. Because of the poor water solubility of this drug, it is difficult for it to penetrate into the matrix of the LPs. The sustained release of the drug might mainly contribute to the corrosion rates of the LPs prepared with the phospholipid. After calculation, the in vitro drug-release kinetic model of PTX and CUR (RGD-PTX-CUR LPs) in release medium fit well with the Higuchi equation: Q=6.595t1/2+0.652 (PTX, r=0.996) and Q=3.597t1/2−0.659 (CUR, r=0.995). Thus, it was speculated that the sustained-release property of LPs could enhance absorption of PTX and CUR.
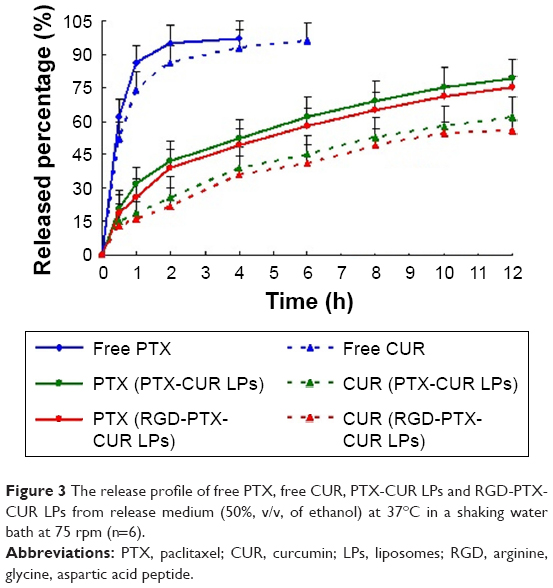 | Figure 3 The release profile of free PTX, free CUR, PTX-CUR LPs and RGD-PTX-CUR LPs from release medium (50%, v/v, of ethanol) at 37°C in a shaking water bath at 75 rpm (n=6). |
Stability study
RGD-modified PTX and CUR co-loaded LPs exhibited good stability over the period of 3 months. No significant change in physical appearance and particle aggregation was observed. Meanwhile LPs had a good physical stability. None of the indexes changed markedly during the observation period. Both PTX and CUR in the formulations were observed with no degradation (Table 2).
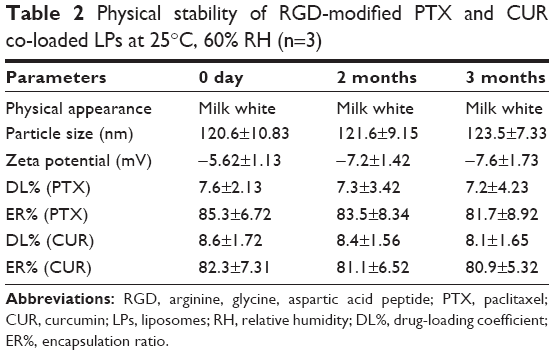 | Table 2 Physical stability of RGD-modified PTX and CUR co-loaded LPs at 25°C, 60% RH (n=3) |
Pharmacokinetics
The profiles of PTX and CUR concentration in plasma vs time are shown in Figure 4. As is already known, the liposomal PTX or CUR formulation had a much longer systemic circulation time when compared with the suspension. Both PTX and CUR from suspensions were eliminated in vivo in a short time, and the concentration in plasma was lower than 20 ng/mL at 6 h after administration. Based on the analysis of the models and parameters with DAS2.0, it was concluded that the in vivo pharmacokinetics of RGD-PTX/CUR LPs in blood could be described by a two-compartment model after iv administration. In this study, rats treated with RGD-PTX/CUR LPs had a slightly higher plasma concentration than those treated with PTX/CUR LPs. The pharmacokinetic parameters are reported in Table 3. As shown in Table 3, the pharmacokinetic parameters of RGD-PTX/CUR LPs in rats showed changes in comparison to the other two groups. The mean residence time was significantly higher than that of the suspension. The RGD-PTX/CUR LPs provided a higher AUC0–∞ (PTX:1.16-fold; CUR:1.23-fold), mean residence time (PTX:1.13-fold; CUR:1.39-fold), and biological half-life (PTX:1.17-fold; CUR:1.34-fold) when compared with PTX/CUR LPs. However, the RGD-PTX/CUR LPs showed decreased clearance when compared with the PTX/CUR LPs. There was no significant difference in pharmacokinetic parameters between the RGD-PTX/CUR LPs and PTX/CUR LPs. Usually, a hydrophilic surface-modified nano-carrier can be prevented by the reticuloendothelial system, have long circulating action, and change the pharmacokinetics. In the present study, hydrophobic RGD was coupled to PTX/CUR LPs and extended the circulation of PTX and CUR. The variation of AUC indicated that the RGD-PTX/CUR LPs provided higher bioavailability than the PTX/CUR suspension.
 | Figure 4 Plasma concentration-time profiles. |
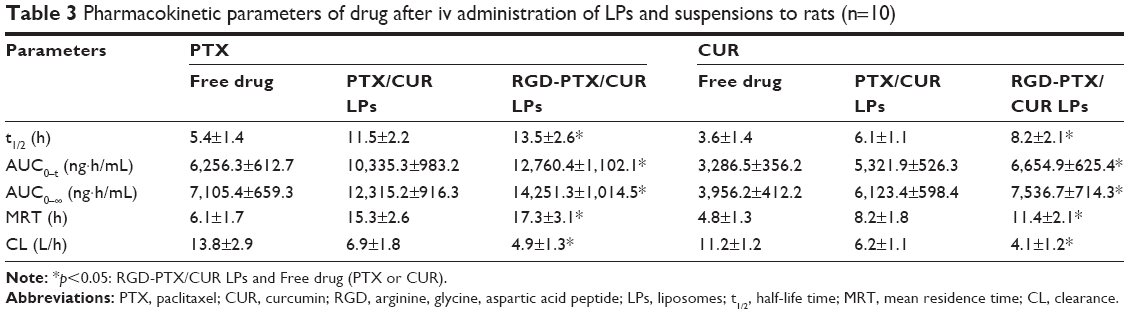 | Table 3 Pharmacokinetic parameters of drug after iv administration of LPs and suspensions to rats (n=10) |
Cell uptake
Confocal microscopy was employed to characterize the cellular internalization of free PTX/CUR, PTX/CUR LPs and RGD-PTX/CUR LPs in A549 cells. As shown in Figure 5, a strong green fluorescence was observed in the cytoplasmic region of RGD-PTX/CUR LPs after incubation for 2 h. In A549 cells, the cell internalization of RGD-PTX/CUR LPs was higher than others. The results showed that surface modification could indicate when cell internalization was changed and more drugs entered the cells successfully. In quantitative cell uptake studies, coumarin-6 in the three formulations was quantified by recovering the drug LPs from cells and measuring their fluorescence (normalized to per mg of the total cellular protein contents). Quantitative results were consistent with the confocal images (data not shown).
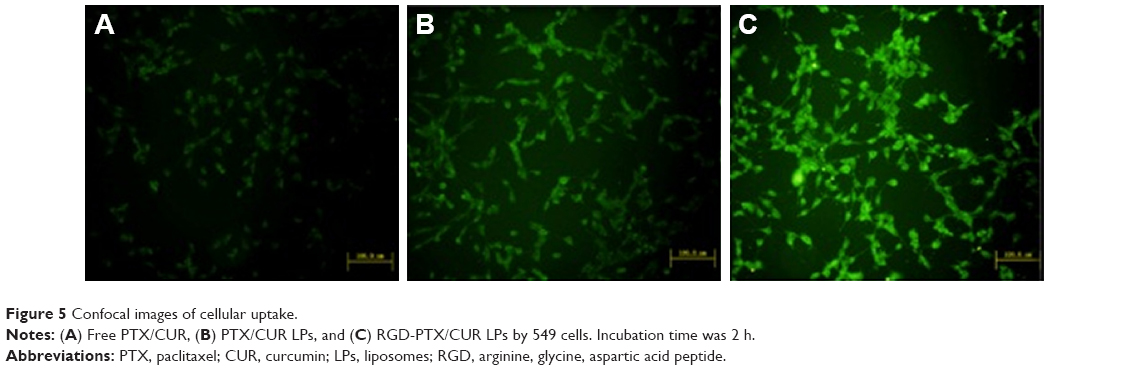 | Figure 5 Confocal images of cellular uptake. |
Cytotoxicity study
The effects of different PTX preparations on A549 cells were detected by MTT assay (Figure 6). It can be seen that free PTX/CUR, PTX/CUR LPs and RGD-PTX/CUR LPs exhibited a time-dependent and dose-dependent cytotoxicity in the cell lines tested. Although PTX and CUR are powerful antitumor drugs, they do not completely inhibit cell proliferation. Similarly, the combination of LPs and PTX/CUR has no superior effect. On the other hand, RGD coupled with PTX/CUR significantly reduces cell proliferation and improved therapeutic efficiency. Surprisingly, RGD-PTX/CUR LPs showed a higher antiproliferative effect on A549 cells, which may have a possible mechanism that inhibited multidrug resistance phenomenon and showed an obvious synergistic effect. All formulations values of half maximal inhibitory concentration (IC50) are listed in Table 4. The results showed that IC50 value decreased to different extents depending on incubation time, and RGD-PTX/CUR LPs have strong targeting ability, which can improve the cellular uptake and cytotoxic of PTX/CUR.
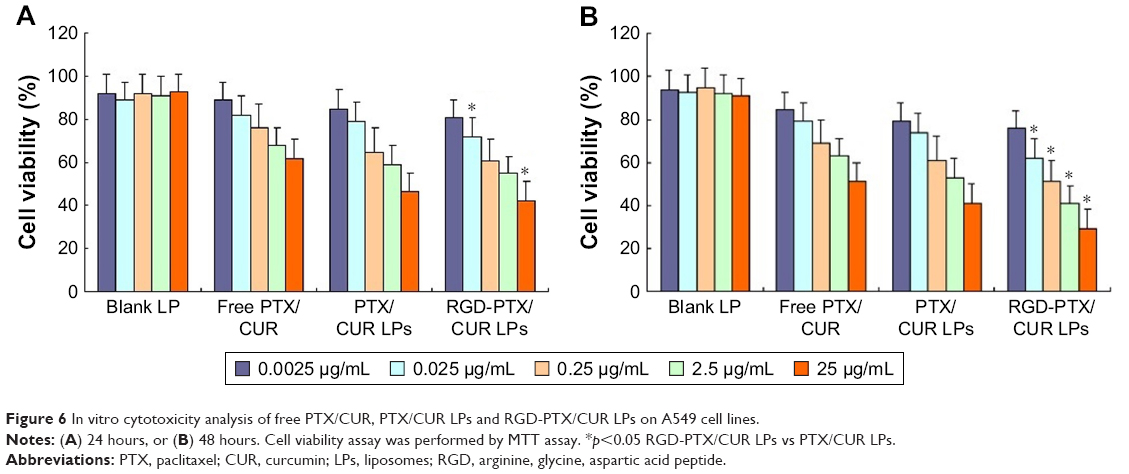 | Figure 6 In vitro cytotoxicity analysis of free PTX/CUR, PTX/CUR LPs and RGD-PTX/CUR LPs on A549 cell lines. |
 | Table 4 Effect of free PTX/CUR, PTX/CUR LPs and RGD-PTX/CUR LPs on A549 cells in nude mice (n=12) |
In vivo tumor growth inhibition
To test the antitumor activity of RGD-PTX/CUR LPs, mice bearing human lung adenocarcinoma (A549) tumors were treated with conventional LPs or RGD-modified LPs four times over 14 days. As shown in Figure 7, the two types of LPs inhibited growth of the A549 cells and reduced the size of the tumors compared with the control group. However, there was a significant decrease in tumor volume in the treatment group with RGD-PTX/CUR LPs and PTX/CUR LPs group with only a small decrease in tumor volume, when compared with the control group. The average weight and volume of the tumors are shown in Table 4. The appearance of tumors was consistent with the statistical analysis of tumor volume data. Compared with the control group, all the treatments were significant (p<0.05), and the tumor volume was inhibited, and the results were the most effective in the RGD modification group. Similarly, the weight of the tumor was significant (p<0.05), and was inhibited by both PTX/CUR treatments. Once again, the effect was most obvious in the RGD-PTX/CUR LPs group. Interestingly, the data show that the tumor weight was more sensitive to treatment than the tumor volume. In general, our results show that the antitumor effect of RGD-PTX/CUR LPs was better than that of unmodified LPs in vivo. No significant pathological changes were found in any of the treatment groups (data not shown).
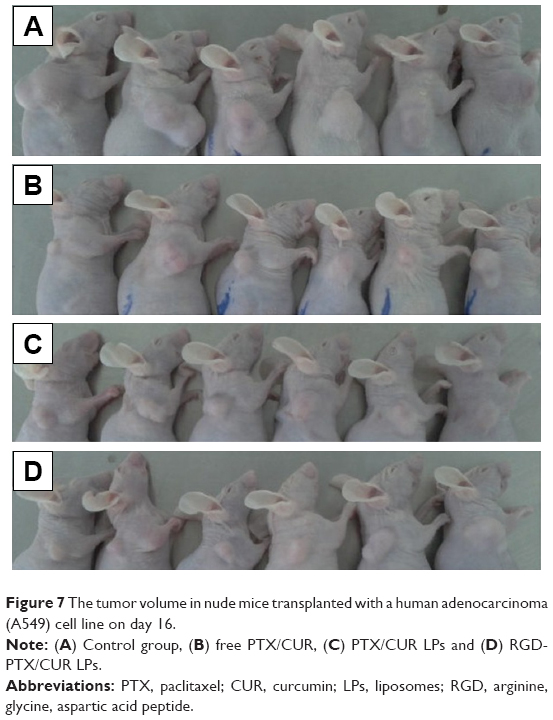 | Figure 7 The tumor volume in nude mice transplanted with a human adenocarcinoma (A549) cell line on day 16. |
Conclusion
A novel RGD-modified paclitaxel and curcumin co-loaded liposomes were developed by using the solvent evaporation method. In comparison with the free drug, RGD-PTX-CUR LPs and PTX-CUR LPs have sustained-release properties in vitro release. In vivo, there was no significant difference in pharmacokinetic parameters between the RGD-PTX/CUR LPs and PTX/CUR LPs. A strong green fluorescence was observed in the cytoplasmic region after incubation of RGD-PTX/CUR LPs for 2 h. RGD-PTX/CUR LPs showed a higher antiproliferative effect on A549 cells, which may be a possible mechanism that inhibited multidrug resistance phenomenon and showed an obvious synergistic effect. Finally, the results indicate that RGD-PTX/CUR LPs had a better antitumor effect in vivo than the non-modified LPs. These results indicate that RGD-modified co-loaded liposomes were a promising candidate for antitumor drug delivery.
Acknowledgments
This study was funded by Suzhou science and technology item (SYS201719) and (SYSD2016090).
Disclosure
The authors report no conflicts of interest in this work.
References
Stage TB, Bergmann TK, Kroetz DL. Clinical pharmacokinetics of paclitaxel monotherapy: an updated literature review. Clin Pharmacokinet. 2018;57(1):7–19. | ||
Lu H, Jiang Z. Advances in antiangiogenic treatment of small-cell lung cancer. Onco Targets Ther. 2017;10:353–359. | ||
Kramer I, Heuser A. Paclitaxel pharmaceutical and pharmacological issues. Eur Hosp Pharm. 1995;1:37–41. | ||
Zhao M, Lei C, Yang Y, et al. Abraxane, the nanoparticle formulation of paclitaxel can induce drug resistance by up-regulation of p-gp. PLoS One. 2015;10(7):e0131429. | ||
Shishodia S, Sethi G, Aggarwal BB. Curcumin: getting back to the roots. Ann N Y Acad Sci. 2005;1056:206–217. | ||
Mohandas KM, Desai DC. Epidemiology of digestive tract cancers in India. V. Large and small bowel. Indian J Gastroenterol. 1999;18(3):118–121. | ||
Sinha R, Anderson DE, McDonald SS, Greenwald P. Cancer risk and diet in India. J Postgrad Med. 2003;49(3):222–228. | ||
Khor TO, Keum YS, Lin W, et al. Combined inhibitory effects of curcumin and phenethyl isothiocyanate on the growth of human PC-3 prostate xenografts in immunodeficient mice. Cancer Res. 2006;66(2):613–621. | ||
Maheshwari RK, Singh AK, Gaddipati J, Srimal RC. Multiple biological activities of curcumin: a short review. Life Sci. 2006;78(18):2081–2087. | ||
Choi BH, Kim CG, Lim Y, Shin SY, Lee YH. Curcumin down-regulates the multidrug-resistance mdr1b gene by inhibiting the PI3K/Akt/NF kappa B pathway. Cancer Lett. 2008;259(1):111–118. | ||
Neerati P, Sudhakar YA, Kanwar JR. Curcumin regulates colon cancer by inhibiting p-glycoprotein in in-situ cancerous colon perfusion rat model. J Cancer Sci Ther. 2013;5:313–319. | ||
Hofheinz RD, Gnad-Vogt SU, Beyer U, Hochhaus A. Liposomal encapsulated anticancer drugs. Anticancer Drugs. 2005;16(7):691–707. | ||
Wessler JD, Grip LT, Mendell J, Giugliano RP. The P-glycoprotein transport system and cardiovascular drugs. J Am Coll Cardiol. 2013;61(25):2495–2502. | ||
Thadakapally R, Aafreen A, Aukunuru J, Habibuddin M, Joqala S. Preparation and characterization of PEG albumin-curcumin nanoparticles intended to treat breast cancer. Indian J Pharm Sci. 2016;78(1):65–72. | ||
Zetter BR. On target with tumor blood vessel markers. Nat Biotechnol. 1997;15(12):1243–1244. | ||
Danhier F, Le Breton A, Préat V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm. 2012;9(11):2961–2973. | ||
Garanger E, Boturyn D, Dumy P. Tumor targeting with RGD peptide ligands – design of new molecular conjugates for imaging and therapy of cancers. Anticancer Agents Med Chem. 2007;7(5):552–558. | ||
Zitzmann S, Ehemann V, Schwab M. Arginine-glycine-aspartic acid (RGD)-peptide binds to both tumor and tumor-endothelial cells in vivo. Cancer Res. 2002;62(18):5139–5143. | ||
Maeda H, Wu J, Sawa T, Matsumura Y Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Yao Q, Gutierrez DC, Hoang NH, et al. Efficient codelivery of paclitaxel and curcumin by novel bottlebrush copolymer-based micelles. Mol Pharm. 2017;14(7):2378–2389. | ||
Wei Y, Pu X, Zhao L. Preclinical studies for the combination of paclitaxel and curcumin in cancer therapy (Review). Oncol Rep. 2017;37(6):3159–3166. | ||
Trotta M, Debernardi F, Caputo O. Preparation of solid lipid nanoparticles by a solvent emulsification-diffusion technique. Int J Pharm. 2003;257(1–2):153–160. | ||
Kim SY, Lee YM. Taxol-loaded block copolymer nanospheres composed of methoxy poly(ethylene glycol) and poly(epsilon-caprolactone) as novel anticancer drug carriers. Biomaterials. 2001;22(13):1697–1704. | ||
Shi B, Fang C, You MX, et al. Stealth MePEG-PCL micelles: effects of polymer composition on micelle physicochemical characteristics, in vitro drug release, in vivo pharmacokinetics in rats and biodistribution in S180 tumor bearing mice. Colloid Polym Sci. 2005;283(9):954–967. | ||
Turner EA, Stenson AC, Yazdani SK. HPLC-MS/MS method for quantification of paclitaxel from keratin containing samples. J Pharm Biomed Anal. 2017;139:247–251. | ||
Malhi S, Stesco N, Alrushaid S, Lakowski TM, Davies NM, Gu X. Simultaneous quantification of reparixin and paclitaxel in plasma and urine using ultra performance liquid chromatography-tandem mass spectroscopy (UHPLC-MS/MS): application to a preclinical pharmacokinetic study in rats. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1046:165–171. | ||
Zheng Z, Sun Y, Liu Z, Zhang M, Li C, Cai H. The effect of curcumin and its nanoformulation on adjuvant-induced arthritis in rats. Drug Des Devel Ther. 2015;9:4931–4942. | ||
Cao Y, Xu RX, Liu Z. A high-throughput quantification method of curcuminoids and curcumin metabolites in human plasma via high-performance liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;949–950:70–78. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.