Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Are 24-hour motor activity patterns associated with continued rapid response to ketamine?
Authors Duncan Jr WC, Slonena EE, Hejazi NS, Brutsche N, Park LT ![]() , Henter ID, Ballard ED, Zarate Jr CA
, Henter ID, Ballard ED, Zarate Jr CA
Received 24 April 2018
Accepted for publication 18 July 2018
Published 16 October 2018 Volume 2018:14 Pages 2739—2748
DOI https://doi.org/10.2147/NDT.S172089
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Wallace C Duncan Jr, Elizabeth E Slonena, Nadia S Hejazi, Nancy Brutsche, Lawrence T Park, Ioline D Henter, Elizabeth D Ballard, Carlos A Zarate Jr
Experimental Therapeutics and Pathophysiology Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD, USA
Purpose: This study examined the links between 24-hour activity patterns (specifically, amplitude and timing of wrist activity) and the persisting qualities of clinical antidepressant response to the glutamatergic modulator ketamine.
Methods: Twenty-four-hour activity patterns were compared across 5 days of 24-hour activity rhythms in patients with major depressive disorder who displayed either a brief antidepressant response (24–48 hours), a continued antidepressant response (>72 hours), or no antidepressant response to ketamine. These postinfusion-response profiles were then used retrospectively to examine cohort-specific fitted parameters at baseline, postinfusion day 1 (D1), and postinfusion D3.
Results: Relative to the nonresponders, the cohort experiencing a brief antidepressant response had blunted 24-hour amplitude that extended from baseline through D3 and postketamine phase advance of activity on D1 that reverted to baseline on D3. Relative to the nonresponders, the cohort experiencing a continued antidepressant response to ketamine had phase-advanced activity at both baseline and D1, as well as increased amplitude on D1 and D3.
Conclusion: Taken together, the results suggest that the time course of antidepressant response to ketamine is influenced by underlying biological differences in motor activity timekeeping. These differences may provide clues that link durable mood response with the molecular machinery of the circadian system, thus leading to more effective interventions. In addition, biomarkers of preinfusion motor activity (eg, amplitude, timing) may be useful for recommending future individualized treatment interventions, to the extent that they help identify patients who may relapse quickly after treatment.
Keywords: antidepressant, ketamine, glutamate, motor activity, sleep deprivation, clock-gene
Plain language summary
This study examined whether the 24-hour pattern of motor activity, ie, information that can be collected easily from wrist-worn activity monitors (such as Fitbit or other activity trackers), can be used to identify whether the effects of a promising new treatment for depression are both rapid and durable. The therapeutic drug ketamine produces rapid (within minutes to hours) antidepressant effects in up to 70% of patients with depression, including those with treatment-resistant depression, the more severe form of the disease. However, in patients who respond to ketamine, the duration of clinical benefit varies, typically ranging from a day to a week. Researchers are eager to identify easily obtained biomarkers, such as information obtained from activity trackers, that could predict which subjects are likely to respond to ketamine, as well as which responders are likely to have a continued antidepressant effect vs those who will quickly relapse. This study used easily worn activity watches to collect activity data from subjects with depression. We found differences between baseline patterns of 24-hour activity and initial response to ketamine (ie, response vs nonresponse) and differences between baseline patterns and continuing response to ketamine. The findings show that prior to treatment, 24-hour patterns of activity might be used to predict both response and relapse patterns in certain depressed individuals. While the results are preliminary, they offer a glimpse into the potential benefits of using simple, noninvasive technologies to identify and individualize personalized therapies for mood disorders.
Introduction
The molecular and cellular targets of rapid antidepressant interventions—such as sleep deprivation (SD) or the glutamatergic modulator ketamine—are the focus of intense research because they may provide clues for developing novel and improved rapidly acting antidepressant treatments. In addition to identifying the mechanisms underlying rapid antidepressant response, research is needed to understand the neurobiology of relapse and the durability of any such response. Studies have found that the timing of relapse following rapid interventions differs, suggesting that these therapies have different mediators. For instance, in SD, relapse rapidly follows recovery sleep. In contrast, relapse may extend for days (or weeks) after ketamine treatment.1
Numerous studies have indicated that chronotherapeutic interventions, such as SD, partial SD, sleep-phase advance, and bright-light therapy (BLT), can initiate an antidepressant response. For instance, previous studies found that 24-hour patterns of motor activity are advanced after SD and BLT and that different antidepressant treatment interventions are often associated with increased and decreased day and night motor activity.2 Furthermore, these rapid antidepressant responses can be extended using conventional drug therapies or used to augment antidepressant response to conventional therapies.2 The ability of electroconvulsive therapy and repetitive transcranial magnetic stimulation to extend the SD response have also been explored, with limited success.3,4 While this evidence suggests that the sleep–wake system can affect and prolong antidepressant response, its role in rapid antidepressant response to ketamine is complex and not well understood. For example, while lithium extended response to SD in a mixed population of subjects with either major depressive disorder (MDD) or bipolar depression,5 it did not extend duration of response to ketamine in subjects with bipolar depression vs MDD.1,6
At a system or cellular/molecular level, several factors have also been implicated in extending antidepressant response to SD. For instance, naps may shorten the duration of rapid antidepressant response,2,7 while repeated sleep-phase-advance episodes8 extend SD response rather than shorten response duration. Although pharmacological interventions that reduce microsleeps and naps (ie, modafinil, caffeine, and flumazenil) do not predictably enhance response to SD,9,10 the benzodiazepine receptor antagonist flumazenil has been found to extend response to SD,10 suggesting that sleep and circadian interactions could be involved. At the molecular level, a prolonged SD response is associated with the interaction between a variant of the circadian clock-associated GSK promoter (rs334558*C) and the long/short form of the serotonin transporter (5-HTTLPR) 5HT allele.11 Diurnal variation in brain derived neurotrophic factor (BDNF) levels predict mood response to SD,12 and increased BDNF levels at 240 minutes post-ketamine infusion predict continued mood response.13 The relationship between sleep, BDNF, and mood has a genetic basis that affects mood response to ketamine14 as well as slow-wave sleep (SWS) production.15
Unlike monoaminergic antidepressants, which require weeks to months to achieve antidepressant efficacy, ketamine acts within hours to produce a clinically significant antidepressant response.16,17 Indeed, numerous placebo-controlled, double-blind, and open-label studies have demonstrated that ketamine has rapid antidepressant effects; while often transient, these can nevertheless also be sustained in a significant proportion of responders.1,6,18–20 Ketamine’s rapid antidepressant effects have been associated with increased synaptic strength and plasticity following altered glutamatergic signaling and with associated changes in dendritic spines and protein synthesis, including BDNF.21 Enhanced synaptic plasticity and neuronal synchronization, especially in areas involved in mood and behavior, have rapid antidepressant effects22,23 that (along with BDNF) correlate with SWS.24 Ketamine also acts on mTOR and clock genes (Bmal1, Per2, Cry1) to reset circadian timing,25,26 and on BDNF and SWS to affect sleep quality.24 Taken together, the evidence suggests that numerous interactions between sleep homeostatic and circadian systems are possible.
Actigraphy-monitored activity–rest rhythms were recently examined as a paradigm to evaluate circadian rhythms as a core construct within the Arousal and Regulatory System domain of the National Institute of Mental Health (NIMH) Research Domain Criteria (https://www.nimh.nih.gov/research-priorities/rdoc/index.shtml). Wrist activity has been used as an adjunct measure of central timekeeping27,28 and may have diagnostic and treatment utility,29 especially combined with other measures (eg, dim light melatonin onset).30 A prior analysis from our laboratory examined how initial mood response to ketamine (occurring within 24 hours) altered motor activity patterns over the next few days.29 We found that an initial mood response to ketamine increased the amplitude of motor activity in responders vs nonresponders. In contrast, the current analysis examines how continued response is related to baseline motor activity patterns. The result may help identify at pretreatment which individuals may have short or extended treatment response to ketamine.
Methods
This study was conducted as part of several investigations exploring ketamine’s antidepressant mechanism of action in patients with treatment-resistant depression. Specifically, the study was part of protocols investigating the clinical effects of ketamine in MDD patients who subsequently received riluzole (another glutamatergic modulator) and the effects of ketamine in a double-blind study of MDD and bipolar depression patients, some of whom received maintenance mood stabilizers. The studies were conducted at the NIMH Clinical Research Center Mood Disorders Research Unit in Bethesda, Maryland (NCT00088699). Specific details have been reported previously.1,24 The studies were approved by the Combined Neuroscience Institutional Review Board of the National Institutes of Health. All subjects provided written informed consent before entry into the study and were assigned a clinical research advocate from the NIMH Human Subjects Protection Unit to monitor the consent process and research participation throughout the study.
Participants
A total of 51 inpatients (29 females) aged 20–65 (mean 42.6±1.6 years) with a DSM-IV-confirmed clinical diagnosis of MDD (n=30) or bipolar depression (n=21) were included in the analysis (Table S1). All patients were experiencing a current major depressive episode lasting at least 4 weeks and were required to have previously failed to respond to at least one adequate antidepressant trial (as assessed by the modified Antidepressant Treatment History Form).31 All patients had a Montgomery–Åsberg Depression Rating Scale (MADRS) score ≥20. Patients with bipolar depression were required to have not responded to a prospective open trial of a mood stabilizer while at the NIMH (lithium or valproate for at least 4 weeks at therapeutic levels [serum lithium 0.6–1.2 mEq/L or valproic acid 50–125 μg/mL]). Exclusion criteria included psychotic features, a DSM-IV diagnosis of drug or alcohol abuse or dependence in the last 3 months, or the presence of an unstable serious medical illness. Female subjects could not be pregnant or nursing. All participants had been free of psychotropic medications for 2–5 weeks prior to the study, with the exception of mood stabilizers among some patients with bipolar depression (lithium, n=14; valproate, n=5). Cigarette use was permitted during the clinical trial but alcohol use was not. Participants were not allowed to nap during the 3 days prior to and after the infusion procedure and selected a preferred sleep schedule between 10 pm and 7 am.
Experimental design
The 51 participants wore an Actiwatch (AW64; Philips, Amsterdam, the Netherlands) for 2–3 days before and 3 days after a scheduled ketamine infusion. The watch was removed during testing procedures and bathing. A diary was used to track watch removal and replacement. Ketamine infusion was conducted as previously described.19 Briefly, at about 10 am, MDD or bipolar depression patients received a single intravenous infusion of 0.5 mg/kg racemic ketamine hydrochloride over the course of 40 minutes. Depressive symptoms were examined via MADRS ratings conducted at baseline (60 minutes pre-ketamine infusion), 230 minutes postinfusion (D0), 1 day postinfusion (D1), and 3 days postinfusion (D3). At all time points, change in depressive symptoms was expressed as change in score from baseline.
Day 1 ketamine response
Patients showing <50% improvement on D0 or D1 were classified as D1 nonresponders (NRs). Patients exhibiting a 50% reduction in MADRS scores at 230 minutes postinfusion (D0) or at 9 am on D1 were classified as ketamine responders. This group of responders was subdivided into brief responders (RB) and continued responders (RC) based on the duration of their clinical response. Specifically, responders who did not meet the 50%-reduction response criteria on D3 were classified as RB. Patients who responded on D0 and/or D1 and maintained their response through D3 were classified as RC. The days defining the RB sample are of interest because, during this 2-day interval, patients typically exhibit the highest relapse rate during the 4-week postinfusion period.1
Data collection and analysis
Data collection methods have been previously described.29 Briefly, activity counts were collected in 1-minute bins and edited for activity data outliers and intervals of watch removal. One-minute averages were then calculated for each hour. Fitted parameter estimates of 24-hour activity pattern differences (amplitude, timing, and midline statistic of rhythm [MESOR]) were compared for the NR vs responder (RB, RC) groups. The clock time of the estimated peak activity (acrophase) was used as a reference marker of timing.
Statistics
GraphPad Prism (v.6.04) was used to compare 24-hour patterns of wrist-activity for baseline vs treatment on D1 (first day after infusion) and D3. Specifically, differences in 24-hour activity patterns were compared in the ketamine RB, RC, and NR cohorts at baseline, D1, and D3. Motor activity amplitude and timing were the primary focus of this analysis. Table 1 reports the clock time of the estimated peak activity time (acrophase).
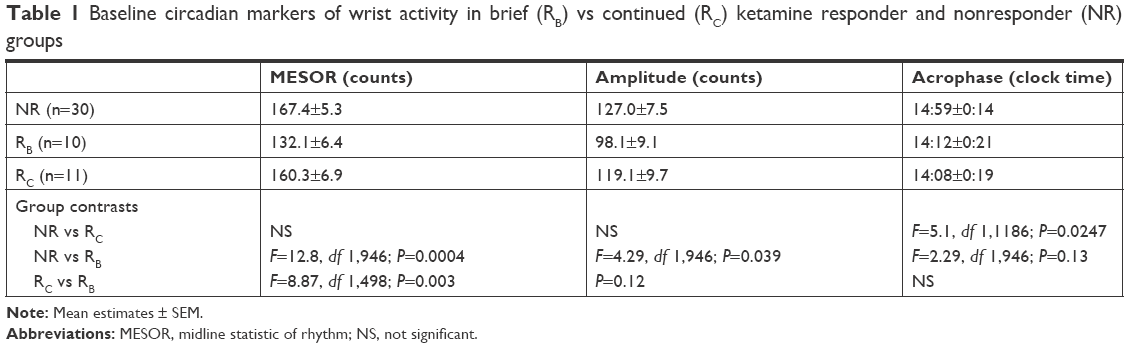 | Table 1 Baseline circadian markers of wrist activity in brief (RB) vs continued (RC) ketamine responder and nonresponder (NR) groups |
The analytical methods have been described previously.29 Briefly, the best-fit 24-hour curves for day (baseline, D1, and D3) and cohort differences (NR, RB, and RC) were characterized using a least squares sine-wave fit to activity scores. Period, amplitude, and phase estimates of wrist activity were derived using a minimal least squares algorithm to fit the 24-hour time series to a sinusoidal curve of the form (y[t] = M + Asin[2π·ft + P]) where M is MESOR (a “baseline” central value of the sinusoid), A amplitude, f frequency, and P phase. Frequency was constrained to be 6.28 (24 hours). Best-fit parameters for baseline, amplitude, and phase were derived for the aforementioned daily contrasts (eg, RB and RC vs NR). Group and treatment differences among 24-hour activity curves were evaluated using an F-test “loss of fit” when applying shared vs independently derived parameter estimates to group data. Bonferroni correction for multiple within-day curve comparisons was used with a corrected α-criterion significance level of P<0.0167, two-tailed.
Results
A post hoc analysis examined multiple demographic factors, including age, age of onset, and rating scale scores, and revealed no significant differences in these measures across the three cohorts (Table S1). D1 and D3 MADRS scores were used to classify the baseline patient sample as NR (MDD 17, bipolar depression 13), RB (MDD 7, bipolar depression 3), or RC (MDD 6, bipolar depression 5). Amplitude and peak activity estimates for the NR, RB, and RC cohorts at baseline, D1, and D3 are displayed in Figure 1.
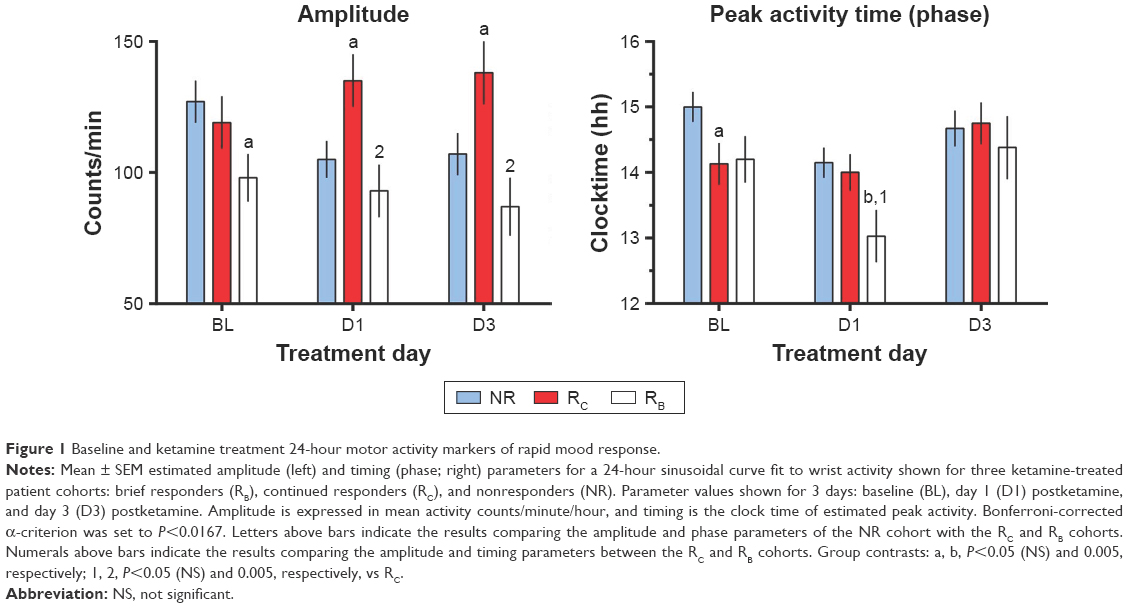 | Figure 1 Baseline and ketamine treatment 24-hour motor activity markers of rapid mood response. |
Rating scale scores at baseline, D1, and D3
At baseline, no significant difference between MADRS scores was observed across the three cohorts (Figure 2; Table S1).
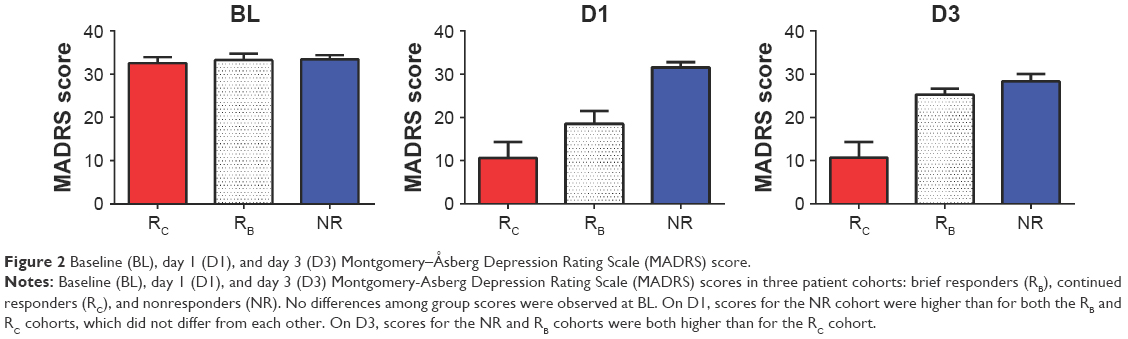 | Figure 2 Baseline (BL), day 1 (D1), and day 3 (D3) Montgomery–Åsberg Depression Rating Scale (MADRS) score. |
On D1, MADRS scores varied among the three cohorts (F=25.69, df 2,48; P<0.0001). MADRS scores for the RB (t=4.64, df 38; P<0.001) and RC cohorts (t=6.88, df 39; P<0.0001) were significantly lower than for the NR cohort. No significant difference in MADRS scores was observed between the RB and RC cohorts (Figure 2). On D3, MADRS scores also varied between the cohorts (F=15.51, df 2,46; P<0.0001). MADRS scores for the RB cohort were higher than for the RC cohort (t=3.648, df 19; P<0.0017), and MADRS scores for the RC cohort were lower than for the NR cohort (t=4.05, df 7; P<0.0001). No significant difference in MADRS scores was noted between the RB and NR cohorts (Figure 2).
24-Hour activity patterns in the NR, RB, and RC cohorts
The overall 24-hour baseline pattern of activity for the three cohorts was characterized by low values at night (mean <25 counts/minute) that began to rise from nighttime lows at about 6 am. Values continued to rise to peak values (200–300 counts/minute) until about 2 pm, then decreased. The lowest levels were observed at about 3 am. Mood-associated 24-hour activity pattern differences were found both pre- and postketamine (Figure 1).
Within-day cohort contrasts
Pre- and postketamine treatment-parameter estimates of 24-hour activity patterns are summarized in Table 1 and Figure 1. On each day, constraining the three cohorts to share MESOR, amplitude, and phase (timing) parameter values resulted in a significant loss of fit (baseline, F=3.996, df 6,1207, P=0.0006; D1, F=6.409, df 6,1185, P<0.0001; D3, F=4.375, df 6,1108, P<0.0002). Cohort differences between individual parameter tests were examined each day. At baseline, trends were observed for the amplitude of the RB cohort (but not the RC cohort) to be blunted relative to the NR cohort (Figure 1 top panel; Table 1) and for the timing (acrophase) of the RC cohort to be advanced relative to the NR cohort.
On D1, the timing of the RB cohort was advanced relative to the NR cohort (F=9.074, df 1,927; P=0.0027; Figure 1, middle panel) and trended toward an advance relative to the RC cohort (F=3.939, df 1,495; P=0.0477). Also on D1, a trend was observed for the amplitude of the RC cohort to be increased relative to the NR cohort (F=5.210, df 1,948; P=0.0227). Timing did not differ between the RC and NR cohorts. On D3, the amplitude of the RC cohort was greater than that of the RB cohort (F=10.02, df 1,462; P=0.0017; Figure 1, top panel), with a trend to be greater than the NR cohort (F=4.464, df 1,883; P=0.0349). No amplitude or timing differences were seen between the RB and NR cohorts. Timing also did not differ between the RC and NR cohorts.
Discussion
The current study sought to identify baseline and posttreatment indicators of rapid vs continuing antidepressant response to the glutamatergic modulator ketamine. We found that ketamine’s rapid antidepressant effects were associated with advanced timing and amplitude differences that varied with duration of antidepressant response. These effects were most pronounced when comparing brief (D1) and continued (D3) response activity patterns with the patterns of ketamine NRs. Notably, trends were observed for baseline 24-hour activity amplitude and timing that were associated with subsequent rapid antidepressant effects. Specifically, the timing of 24-hour activity (phase) in RB and RC subjects was earlier than in NR subjects. In addition, baseline activity amplitude trended lower in RB than NR subjects and in RB than RC subjects. At D3, the amplitude for RC subjects was increased relative to RB subjects, with a trend to increase relative to NR subjects. The current findings, which are specific to treatment-resistant depression, underscore the motor activity amplitude and timing effects of ketamine on rapid antidepressant response. On the one hand, this motor activity result may implicate central or noncentral circadian factors in moderating the durability of mood response to ketamine. On the other hand, homeostatic or environmental factors may contribute, and additional measures would need to be monitored to resolve the contribution of these factors to measured response. The fact that clinical response to ketamine was related to baseline differences in motor activity and, furthermore, that ketamine infusion altered 24-hour patterns of motor activity, suggests that underlying mechanisms linked to the generation of 24-hour activity patterns predict and contribute to the durable features of ketamine-mediated mood effects.
Baseline differences between responders and nonresponders
Baseline differences between 24-hour activity patterns for the NR, RB, and RC cohorts indicated that differences in circadian-related circuitry were linked to the acute and durable features of the mood response. These differences may occur at the genetic, cellular, systems, or behavioral level. Interestingly, baseline differences between ketamine responders and NRs with regard to both sleep–wake patterns and motor activity patterns have previously been described.24,32 These findings, as well as ketamine’s preclinical effects on altered circadian gene expression,26 suggest possible ketamine-associated interactions between the sleep homeostatic and circadian systems. However, it is beyond the scope of the current study to clearly separate their individual contributions.
This study extends the earlier finding that biomarkers of motor activity timing are important in examining the underlying mechanisms of ketamine’s rapid antidepressant effects. Specifically, such markers might be used to identify clinical interventions that could potentially augment, extend, or diminish antidepressant response, particularly because the present results suggest that ketamine responders tend to have advanced activity timing, and NRs tend to have both delayed and elevated activity patterns during pretreatment. While our findings do not provide evidence of ketamine’s effects on circadian clock-related molecules, if ketamine acts on central timekeeping, the results may suggest that ketamine-mediated mood effects are linked to markers of circadian timekeeping (specifically, phase and amplitude). Such markers have previously been shown to be altered by clock gene-related variants. It should be noted that while clock gene variants have been associated with increased motor activity33 – as was also observed here with ketamine NRs at baseline – the sample size in this study was not adequate to determine whether such variants contributed to cohort differences in activity patterns.
Differences in rapid antidepressant response for the NR, RB, and RC cohorts
The relationship between ketamine’s pharmacological effects and its effects on mood and activity level is complex. Activity amplitude and level often increase during effective antidepressant treatment,34 and in the current study amplitude effects were indeed greater for the RC cohort (on both D1 and D3) than for the RB cohort. Interestingly, rapid antidepressant response on D1 for the RB cohort was associated with a phase advance of activity but not with change in amplitude (Figure 1). The fact that the amplitude and timing parameters for the RB, RC, and NR cohorts differed at baseline, D1, and D3 (Figure 1) suggests biological differences in the organization and function of the sleep and circadian timekeeping systems. Delayed circadian phase, which was present in NRs relative to responder cohorts, has been linked to elevated glutamatergic levels in emerging depression.35 It is interesting to note that both delayed phase and increased amplitude may be markers of nonresponse in individuals with treatment-resistant depression. Delayed-phase phenotypes have previously been associated with increased circadian amplitude of body temperature36 as well as with increased risk of anxiety, depressive, or substance-use disorders.37
Baseline versus postinfusion effects between the NR, RB, and RC cohorts
Comparison of response maintenance features of the NR, RB, and RC cohorts demonstrated differences in motor activity amplitude, timing, and level of activity at baseline, with some effects persisting on D1 and D3. These differences indicate that timekeeping differences both mediate and moderate the duration of antidepressant response to ketamine. Specifically, blunted amplitude at baseline was associated with brief antidepressant response and elevated amplitude with more persistent response or nonresponse to ketamine. The observation that amplitude did not vary from baseline to D3 in the RB cohort suggests that low amplitude is a trait-like feature of brief response and rapid relapse. One might speculate that low amplitude could be used to identify which subjects would benefit from particular future treatment interventions, but this remains to be determined given that amplitude estimates varied across both the NR and responder groups. The fact that some interventions, such as BLT or conventional antidepressant treatments, increase 24-hour rhythm amplitude38–40 suggests that pretreatment or supplemental BLT might be used to prolong ketamine’s antidepressant effects.
In the RB and RC cohorts, but not in NRs, ketamine was associated with advanced timing (RB) and increased amplitude (RC) of 24-hour activity. This suggests that advanced timing and increased amplitude are specifically related to brief vs continued antidepressant response to ketamine. Interestingly, the advanced motor activity timing observed on D1 was not present in the RB cohort on D3, suggesting that the advance in the RB cohort was associated with positive mood response on D1, and that the delay was associated with relapse on D3. Altered timing and increased amplitude are associated with antidepressant response;40–44 therefore, the current results indicate possible linkage to duration of antidepressant response.
One might speculate that the relationship between 24-hour amplitude, clock gene variants, and circadian plasticity (eg, increased range of synchronization to environmental cues) might be associated with relapse and continued response, such that low amplitude (weak) circadian rhythms would allow for greater phase instability and larger phase shifts.45,46 These novel and as yet unexplored possibilities are discussed in the following section.
Circadian clock- and sleep-associated factors
Mutations in such canonical clock genes as CLOCK and PER and associated clock-related mutations may underlie the temporal variation in activity. These have been described in clinical and preclinical mood-disorder studies.33,47–51 Clock gene variants are often associated with diurnal preference, and are thus useful targets for exploring the pathogenesis of mood disorders.52 CLOCK gene variants affect timekeeping markers,53,54 and specific markers such as amplitude and phase vary with morning–evening chronotypes,36 suggesting both are useful for exploring rapid antidepressant response to ketamine. The extent to which response and relapse might be related to CLOCK gene- or sleep-related molecules remains to be determined.
Markers of circadian timekeeping—for instance, circadian amplitude and phase change—have been interpreted as markers of the intrinsic strength of the central circadian clock.55,56 In this regard, high-amplitude rhythms are often viewed as more stable and resistant to change by external perturbations, whereas those with low amplitude are more labile. Notably, other factors also influence amplitude and phase.36,57,58 At the gene level, circadian systems with mutant clock gene homologues have both lower activity amplitude and greater phase response to external challenges.56 Both may be related to continued ketamine mood response.
If increased circadian amplitude is related to a strong central clock, then clock strength would be expected to be related to continued mood response and greater resistance to external perturbations. Conversely, decreased amplitude and a weak clock would be consistent with D3 relapse in the RB cohort and less resistance to change from external cues and vulnerability to depressogenic factors. Further, because circadian amplitude is related to the genetic redundancy of clock genes,56 these results may indicate that the low-amplitude cohort that relapses rapidly has less clock gene redundancy than the high-amplitude cohort that exhibits continued antidepressant response. Differences in circadian amplitude and incidence of clock gene variants may thus contribute to the transient vs preserved mood-response patterns in these cohorts.
Conclusion
This clinical study found that markers of motor activity associated with timing and amplitude were important indicators and mediators of durable antidepressant response to ketamine. The current findings suggest that, prior to treatment: 1) a higher amplitude delayed 24-hour motor activity pattern is associated with nonresponse; 2) a low amplitude 24-hour activity pattern is associated with rapid relapse; and 3) a high amplitude advanced activity pattern is associated with continued response. Together, these observations suggest that biological elements underlying the expression of 24-hour motor activity rhythms contribute to durable antidepressant response and to relapse. In addition, postinfusion results suggested that increased 24-hour activity amplitude was associated with prolonged response. In contrast, in the RB cohort, rapid changes in motor activity timing were associated with rapid response followed by rapid relapse. Overall, the findings indicate that the amplitude and timing of 24-hour motor activity contribute to the durability of ketamine’s rapid antidepressant effects. It is important to recognize that while 24-hour patterns of motor activity might be associated with underlying central timekeeping effects, activity patterns are also influenced by other behavioral and environmental effects that mask the central effect. Therefore, studies using more established markers of central circadian timing (eg, dim light melatonin onset, 24-hour body temperature) are warranted to determine whether the strength and organization of the circadian system specifically contribute to preserving ketamine’s rapidly acting antidepressant effects.
Acknowledgments
The authors thank the 7SE research unit and staff for their support, and the patients for their invaluable contributions. This work was supported by the Intramural Research Program at the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH; ZIA MH002927; NCT00088699), by a NARSAD Independent Investigator Award to CAZ, and by a Brain and Behavior Mood Disorders Research Award to CAZ. These organizations had no further role in study design; in the collection, analysis, or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Disclosure
CAZ is listed as a coinventor on a patent for the use of ketamine in major depression and suicidal ideation; as a coinventor on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine, and other stereoisomeric “dehydro-” and hydroxylated metabolites of (R,S)-ketamine metabolites in the treatment of depression and neuropathic pain; and as a coinventor on a patent application for the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation, and posttraumatic stress disorders. He has assigned his patent rights to the US government, but will share a percentage of any royalties that may be received by the government. The other authors report no conflicts of interest in this work.
References
Ibrahim L, Diazgranados N, Franco-Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37(6):1526–1533. | ||
Dallaspezia S, Benedetti F. Chronobiological therapy for mood disorders. Expert Rev Neurother. 2011;11(7):961–970. | ||
Eichhammer P, Kharraz A, Wiegand R, et al. Sleep deprivation in depression stabilizing antidepressant effects by repetitive transcranial magnetic stimulation. Life Sci. 2002;70(15):1741–1749. | ||
Kreuzer PM, Langguth B, Schecklmann M, Eichhammer P, Hajak G, Landgrebe M. Can repetitive transcranial magnetic stimulation prolong the antidepressant effects of sleep deprivation? Brain Stimul. 2012;5(2):141–147. | ||
Szuba MP, Baxter LR, Altshuler LL, et al. Lithium sustains the acute antidepressant effects of sleep deprivation: preliminary findings from a controlled study. Psychiatry Res. 1994;51(3):283–295. | ||
Diazgranados N, Ibrahim L, Brutsche NE, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67(8):793–802. | ||
Benedetti F, Colombo C. Sleep deprivation in mood disorders. Neuropsychobiology. 2011;64(3):141–151. | ||
Berger M, Vollmann J, Hohagen F, et al. Sleep deprivation combined with consecutive sleep phase advance as a fast-acting therapy in depression: an open pilot trial in medicated and unmedicated patients. Am J Psychiatry. 1997;154(6):870–872. | ||
Beck J, Hemmeter U, Brand S, Muheim F, Hatzinger M, Holsboer-Trachsler E. Modafinil reduces microsleep during partial sleep deprivation in depressed patients. J Psychiatr Res. 2010;44(13):853–864. | ||
Hemmeter U, Hatzinger M, Brand S, Holsboer-Trachsler E. Effect of flumazenil-augmentation on microsleep and mood in depressed patients during partial sleep deprivation. J Psychiatr Res. 2007;41(10):876–884. | ||
Benedetti F, Dallaspezia S, Lorenzi C, et al. Gene-gene interaction of glycogen synthase kinase 3-β and serotonin transporter on human antidepressant response to sleep deprivation. J Affect Disord. 2012;136(3):514–519. | ||
Giese M, Beck J, Brand S, et al. Fast BDNF serum level increase and diurnal BDNF oscillations are associated with therapeutic response after partial sleep deprivation. J Psychiatr Res. 2014;59:1–7. | ||
Haile CN, Murrough JW, Iosifescu DV, et al. Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. Int J Neuropsychopharmacol. 2014;17(2):331–336. | ||
Laje G, Lally N, Mathews D, et al. Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biol Psychiatry. 2012;72(11):e27–e28. | ||
Bachmann V, Klein C, Bodenmann S, et al. The BDNF Val66Met polymorphism modulates sleep intensity: EEG frequency- and state-specificity. Sleep. 2012;35(3):335–344. | ||
Coyle CM, Laws KR. The use of ketamine as an antidepressant: a systematic review and meta-analysis. Hum Psychopharmacol. 2015;30(3):152–163. | ||
Kishimoto T, Chawla JM, Hagi K, et al. Single-dose infusion ketamine and non-ketamine N-methyl-d-aspartate receptor antagonists for unipolar and bipolar depression: a meta-analysis of efficacy, safety and time trajectories. Psychol Med. 2016;46(7):1459–1472. | ||
Zarate CA, Brutsche NE, Ibrahim L, et al. Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry. 2012;71(11):939–946. | ||
Zarate CA, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856–864. | ||
Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134–1142. | ||
Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338(6103):68–72. | ||
Machado-Vieira R, Ibrahim L, Henter ID, Zarate CA. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):678–687. | ||
Maeng S, Zarate CA. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467–474. | ||
Duncan WC, Sarasso S, Ferrarelli F, et al. Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. Int J Neuropsychopharmacol. 2013;16(2):301–311. | ||
Bunney BG, Bunney WE. Rapid-acting antidepressant strategies: mechanisms of action. Int J Neuropsychopharmacol. 2012;15(5):695–713. | ||
Bellet MM, Vawter MP, Bunney BG, Bunney WE, Sassone-Corsi P. Ketamine influences CLOCK:BMAL1 function leading to altered circadian gene expression. PLoS One. 2011;6(8):e23982. | ||
Ancoli-Israel S, Cole R, Alessi C, Chambers M, Moorcroft W, Pollak CP. The role of actigraphy in the study of sleep and circadian rhythms. Sleep. 2003;26(3):342–392. | ||
Teicher MH. Actigraphy and motion analysis: new tools for psychiatry. Harv Rev Psychiatry. 1995;3(1):18–35. | ||
Duncan WC, Slonena E, Hejazi NS, et al. Motor-Activity Markers of Circadian Timekeeping Are Related to Ketamine’s Rapid Antidepressant Properties. Biol Psychiatry. 2017;82(5):361–369. | ||
Benloucif S, Guico MJ, Reid KJ, Wolfe LF, L’Hermite-Balériaux M, Zee PC. Stability of melatonin and temperature as circadian phase markers and their relation to sleep times in humans. J Biol Rhythms. 2005;20(2):178–188. | ||
Sackeim HA. The definition and meaning of treatment-resistant depression. J Clin Psychiatry. 2001;62(Suppl 16):10–17. | ||
Duncan WC, Selter J, Brutsche N, Sarasso S, Zarate CA. Baseline delta sleep ratio predicts acute ketamine mood response in major depressive disorder. J Affect Disord. 2013;145(1):115–119. | ||
Mcclung CA. Circadian rhythms and mood regulation: insights from pre-clinical models. Eur Neuropsychopharmacol. 2011;21(Suppl 4):S683–S693. | ||
Burton C, Mckinstry B, Szentagotai Tătar A, Serrano-Blanco A, Pagliari C, Wolters M. Activity monitoring in patients with depression: a systematic review. J Affect Disord. 2013;145(1):21–28. | ||
Naismith SL, Lagopoulos J, Hermens DF, et al. Delayed circadian phase is linked to glutamatergic functions in young people with affective disorders: a proton magnetic resonance spectroscopy study. BMC Psychiatry. 2014;14(1):345. | ||
Baehr EK, Revelle W, Eastman CI. Individual differences in the phase and amplitude of the human circadian temperature rhythm: with an emphasis on morningness-eveningness. J Sleep Res. 2000;9(2):117–127. | ||
Reid KJ, Jaksa AA, Eisengart JB, et al. Systematic evaluation of Axis-I DSM diagnoses in delayed sleep phase disorder and evening-type circadian preference. Sleep Med. 2012;13(9):1171–1177. | ||
Kasper S, Hajak G, Wulff K, et al. Efficacy of the novel antidepressant agomelatine on the circadian rest-activity cycle and depressive and anxiety symptoms in patients with major depressive disorder: a randomized, double-blind comparison with sertraline. J Clin Psychiatry. 2010;71(2):109–120. | ||
Raoux N, Benoit O, Dantchev N, et al. Circadian pattern of motor activity in major depressed patients undergoing antidepressant therapy: relationship between actigraphic measures and clinical course. Psychiatry Res. 1994;52(1):85–98. | ||
Winkler D, Pjrek E, Praschak-Rieder N, et al. Actigraphy in patients with seasonal affective disorder and healthy control subjects treated with light therapy. Biol Psychiatry. 2005;58(4):331–336. | ||
Benedetti F, Dallaspezia S, Fulgosi MC, Barbini B, Colombo C, Smeraldi E. Phase advance is an actimetric correlate of antidepressant response to sleep deprivation and light therapy in bipolar depression. Chronobiol Int. 2007;24(5):921–937. | ||
Lewy AJ, Lefler BJ, Emens JS, Bauer VK. The circadian basis of winter depression. Proc Natl Acad Sci U S A. 2006;103(19):7414–7419. | ||
Wehr TA, Wirz-Justice A, Goodwin FK, Duncan W, Gillin JC. Phase advance of the circadian sleep-wake cycle as an antidepressant. Science. 1979;206(4419):710–713. | ||
Wirz-Justice A. Chronobiology and mood disorders. Dialogues Clin Neurosci. 2003;5(4):315–325. | ||
Soreca I. Circadian rhythms and sleep in bipolar disorder: implications for pathophysiology and treatment. Curr Opin Psychiatry. 2014;27(6):467–471. | ||
Vitaterna MH, Ko CH, Chang AM, et al. The mouse Clock mutation reduces circadian pacemaker amplitude and enhances efficacy of resetting stimuli and phase-response curve amplitude. Proc Natl Acad Sci U S A. 2006;103(24):9327–9332. | ||
Hampp G, Albrecht U. The circadian clock and mood-related behavior. Commun Integr Biol. 2008;1(1):1–3. | ||
Harvey AG. Sleep and circadian rhythms in bipolar disorder: seeking synchrony, harmony, and regulation. Am J Psychiatry. 2008;165(7):820–829. | ||
Milhiet V, Etain B, Boudebesse C, Bellivier F. Circadian biomarkers, circadian genes and bipolar disorders. J Physiol Paris. 2011;105(4–6):183–189. | ||
Roybal K, Theobold D, Graham A, et al. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci U S A. 2007;104(15):6406–6411. | ||
Schnell A, Albrecht U, Sandrelli F. Rhythm and mood: relationships between the circadian clock and mood-related behavior. Behav Neurosci. 2014;128(3):326–343. | ||
Partonen T. Clock gene variants in mood and anxiety disorders. J Neural Transm. 2012;119(10):1133–1145. | ||
Allebrandt KV, Teder-Laving M, Akyol M, et al. CLOCK gene variants associate with sleep duration in two independent populations. Biol Psychiatry. 2010;67(11):1040–1047. | ||
Nováková M, Sládek M, Sumová A. Human chronotype is determined in bodily cells under real-life conditions. Chronobiol Int. 2013;30(4):607–617. | ||
Aschoff J, Pohl H. Phase relations between a circadian rhythm and its zeitgeber within the range of entrainment. Naturwissenschaften. 1978;65(2):80–84. | ||
Erzberger A, Hampp G, Granada AE, Albrecht U, Herzel H. Genetic redundancy strengthens the circadian clock leading to a narrow entrainment range. J R Soc Interface. 2013;10(84):221. | ||
Dijk DJ, Duffy JF, Silva EJ, Shanahan TL, Boivin DB, Czeisler CA. Amplitude reduction and phase shifts of melatonin, cortisol and other circadian rhythms after a gradual advance of sleep and light exposure in humans. PLoS One. 2012;7(2):e30037. | ||
Ueda HR, Hayashi S, Chen W, et al. System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat Genet. 2005;37(2):187–192. |
Supplementary material
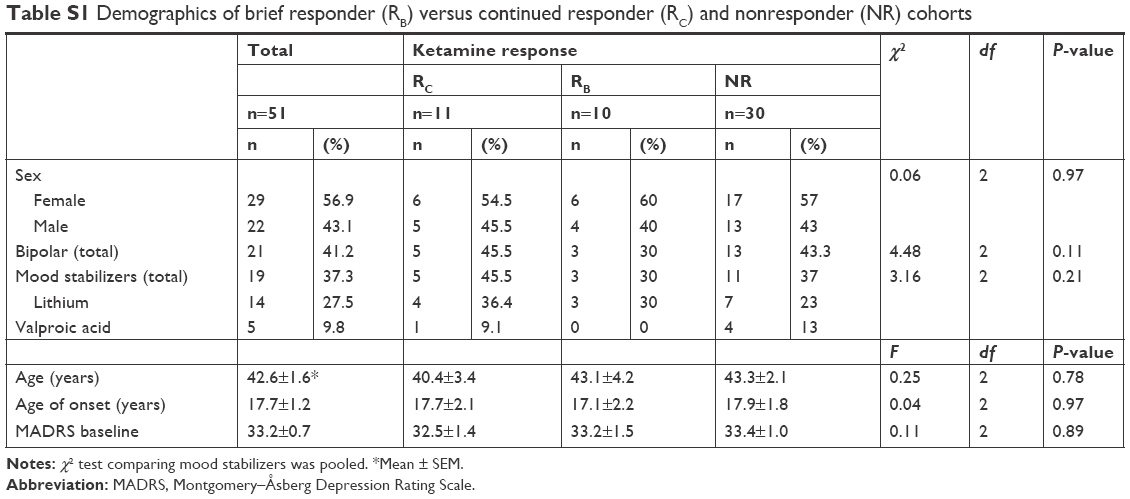 | Table S1 Demographics of brief responder (RB) versus continued responder (RC) and nonresponder (NR) cohorts |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.