Back to Journals » International Journal of Nanomedicine » Volume 11
Aptamer-functionalized peptide H3CR5C as a novel nanovehicle for codelivery of fasudil and miRNA-195 targeting hepatocellular carcinoma
Authors Liu Y, Wu X, Gao Y, Zhang J, Zhang D, Gu S, Zhu G, Liu G, Li X
Received 10 March 2016
Accepted for publication 23 May 2016
Published 12 August 2016 Volume 2016:11 Pages 3891—3905
DOI https://doi.org/10.2147/IJN.S108128
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Ying Liu,1,* Xin Wu,1,* Yuan Gao,2,* Jigang Zhang,1 Dandan Zhang,1 Shengying Gu,1 Guanhua Zhu,1 Gaolin Liu,1 Xiaoyu Li1
1Department of Clinical Pharmacy, Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, 2Department of Pharmaceutics, Changhai Hospital, Second Military Medical University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: Liver cancer is the fifth most commonly diagnosed malignancy, of which hepatocellular carcinoma (HCC) represents the dominating histological subtype. Antiangiogenic therapy aimed at vascular endothelial growth factor (VEGF) has shown promising but deficient clinical prospects on account of vasculogenic mimicry, a highly patterned vascular channel distinguished from the endothelium-dependent blood vessel, which may function as blood supply networks occurring in aggressive tumors including HCC. In this study, we used a new cationic peptide, disulfide cross-linked stearylated polyarginine peptide modified with histidine (H3R5), as a reducible vector, cell penetrating peptide-modified aptamer (ST21) with specific binding to HCC cells to conjugate to peptide H3R5 as the targeting probe, miRNA-195 (miR195)as a powerful gene drug to inhibit VEGF, and fasudil to suppress vasculogenic mimicry by blocking ROCK2, all of which were simultaneously encapsulated in the same nanoparticles. Fasudil was loaded by ammonium sulfate-induced transmembrane electrochemical gradient and miR195 was condensed through electrostatic interaction. ST21-H3R5-polyethylene glycol (PEG) exhibited excellent loading capacities for both fasudil and miR195 with adjustable dosing ratios. Western blot analysis showed that FasudilST21-H3R5-PEGmiR195 had strong silencing activity of ROCK2 and VEGF, as compared with FasudilH3R5-PEGmiR195. In vitro and in vivo experiments confirmed that ST21-modified nanoparticles showed significantly higher cellular uptake and therapeutic efficacy in tumor cells or tumor tissues than the unmodified counterparts. These findings suggest that aptamer-conjugated peptide holds great promise for delivering chemical drugs and gene drugs simultaneously to overcome HCC.
Keywords: aptamer, fasudil, miR195, combined therapy, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) represents the dominating histological subtype, accounting for 80%–90% of all primary liver cancers worldwide.1 The 5-year survival rate for HCC patients in People’s Republic of China is about 39% despite aggressive conventional therapy including surgery, radiotherapy, and chemotherapy. Generally, liver resection is restricted to early tumors without metastasis. The foremost impediments in cancer radiotherapy and chemotherapy are poor aqueous solubility, bioavailability, and adverse effects caused by nonspecific cytotoxicity of widely used anticancer drugs. Enhancing the potential of chemotherapeutic agents to discriminate between tumor cells and nonmalignant counterparts so as to improve their accumulation at the tumor site has become a major challenge of targeted therapeutics against cancer.2–4
As a typical hypervascular cancer, HCC can be inhibited by tumor angiogenesis inhibitors (TAI).5 Of many signaling-mediated angiogenesis pathways evaluated so far, vascular endothelial growth factor (VEGF) pathway is the most understood and studied.6 MicroRNAs (miRNAs) have the potential to regulate the posttranscriptional gene expression, and may simultaneously silence multiple genes involved in distinct tumor-related signaling networks, which is a distinguishing feature from small interfering RNA that specifically knocks down a single gene. Gene therapy based on miRNA has a great potential to become a more powerful tool in tumor treatment.7,8 In the tumor microenvironment, the downregulation of miRNA-195 (miR195) enhances VEGF levels, which subsequently activates VEGF receptor 2 signaling in endothelial cells and promotes angiogenesis. Some investigations have demonstrated the inhibitory role of miR195 in HCC.9 However, TAI cannot completely block the nutrient supply of the tumor tissue during treatment because blood is supplied to the tumor cells through a special way known as vasculogenic mimicry (VM). Through these pipes, similar to the blood vessels, HCC cells communicate with the host blood vessels and acquire blood supply for growth, invasion, and metastasis. To further increase the therapeutic effect in HCC, miR195, a powerful gene drug inhibiting VEGF, and fasudil, a serine/threonine protein kinase blocking Rho kinase, were simultaneously encapsulated in the same nanoparticles (NPs) in order to suppress VM.10–13
The cellular membrane plays the role of a selectively permeable barrier, which also presents a major barrier for intracellular delivery of cargo like plasmid DNA (pDNA), miRNA, small interfering RNA, and drugs. Although the small noncoding RNAs have been extensively studied as novel therapeutics for cancer treatment, large molecular weight, instability in blood circulation, and anionic surface charges have hindered the translation of these RNAs from bench to clinic.14–16 In our previous study, H3CR5C (HHHCRRRRRC, the peptide sequence), composed of arginine, histidine, and cysteine, has been found to be promising and efficient in gene delivery.17 Arginine-based cationic peptides in linear or branched polymeric configuration are good to mediate RNA condensation for effective RNA delivery. In order to achieve endosomal escape, arginine can be conjugated with histidine to generate a proton sponge effect. Cysteines can be oxidized to create cross-linking disulfide bonds in polyplexes, making them stable in the extracellular environment.18 Lack of selectivity and short biological life of the drug are the major deterrents for fasudil becoming a clinically viable therapeutic agent. The problem of short half-life can be overcome by mixing fasudil in H3CR5C (disulfide cross-linked stearylated polyarginine peptide modified with histidine [H3R5]), as it has an ability to extend the circulation time and control drug release. Meanwhile, the simultaneous loading of miR195 and fasudil in nanosized delivery systems is doubly difficult due to the different physicochemical properties of these molecules. To achieve maximal therapeutic efficiency from this dual delivery strategy, it is also important to ensure an optimal compromise between fasudil release and efficient miR195 expression, as the drug-induced toxicity may interfere with RNA expression.19 Particulate drug binding with homing devices has been used to deliver drugs for target drug delivery. The drawback of specificity can be addressed by means of homing device-empowered cell penetrating peptide-modified aptamer (ST21) (SFSIIHTPILPL-TAT-SFSIIHTPILPL, the peptide sequence).20 Aptamers are a class of DNA, RNA, and polypeptide fragments binding to different targets specifically, while cell penetrating peptides (CPP) can facilitate crossing of plasma membranes by poorly internalized biomacromolecules. Aptamer has been extensively used in basic research, pharmaceutical study, and clinical examinations due to its advantages of small molecular weight, good biocompatibility, and easy synthesis. ST21, a targeting probe, has a special configuration of Y and is composed of a novel peptide (SP94) that binds specifically to HCC cell lines and the cell penetrating peptides named TAT. A large number of aptamer-mediated nanocarriers have been developed for targeted cancer therapy because not only can they enhance water solubility and bioavailability, but they also have inherent properties of high specificity and affinity.21,22 Polyethylene glycol (PEG)-modified NPs have been used as a stable vehicle to carry drugs for the treatment of different aggressive cancers. PEGylation not only helps prevent rapid clearance of NPs by the reticuloendothelial system, but also prolongs their circulation time in blood.23,24 FasudilST21-H3R5-PEGmiR195 exhibits biodegradability, reduced aggregation, minimal drug loss, longer circulation time, and superior targeting efficiency. Moreover, the raw materials of methoxy polyethylene glycol-maleimide (mPEG-MAL), H3R5, and ST21 are nontoxic and biodegradable in vitro and in vivo.
In the present study, we perfected and fine-tuned the site specificity and pharmacological efficacy by loading weakly basic hydrophilic fasudil and negatively charged miR195 in H3R5-PEG, and modified them with aptamer that specifically recognized special structures that are not present in normal tissues but are expressed in HCC tissues (Figure 1). In addition, we characterized the particle size, surface zeta potential, morphology, condensation ability of carriers to miR195, particle stability in serum, fasudil encapsulation efficiency, and in vitro release. The targeting effects of FasudilH3R5-PEGmiR195 (FasudilHPmiR195) compared with FasudilST21-H3R5-PEGmiR195 (FasudilSHPmiR195) were further investigated by in vitro and in vivo models.
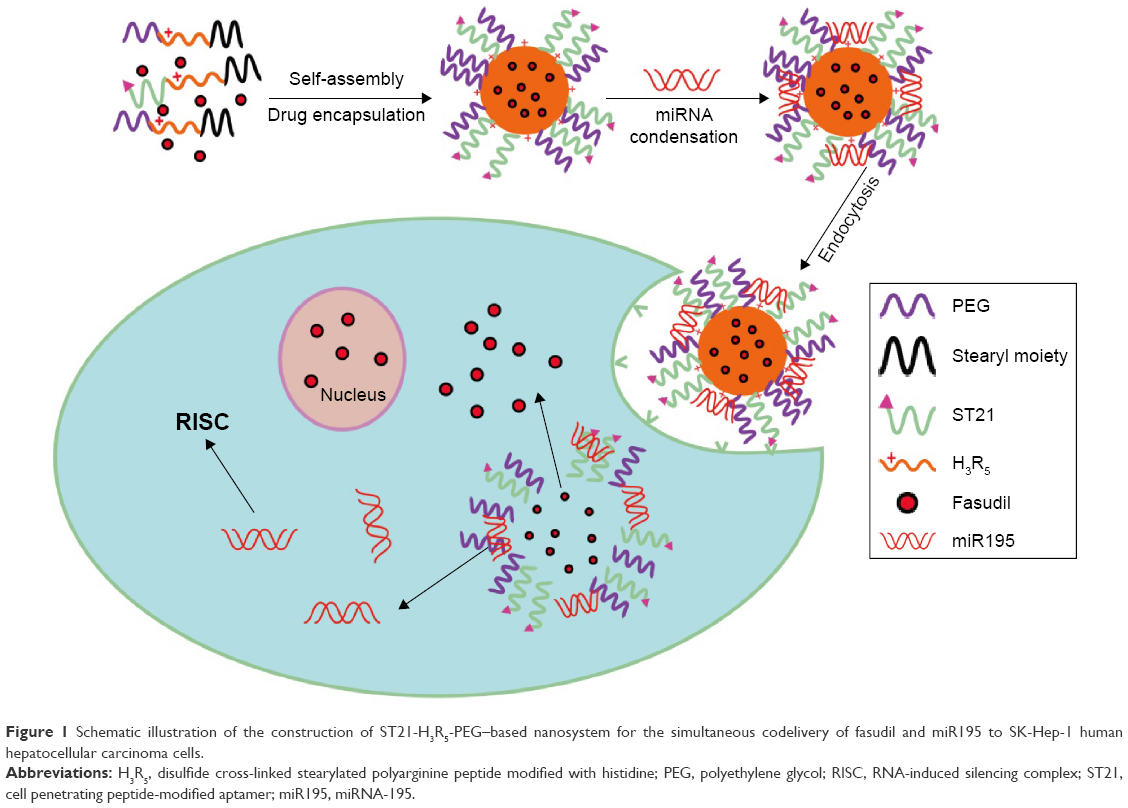 | Figure 1 Schematic illustration of the construction of ST21-H3R5-PEG–based nanosystem for the simultaneous codelivery of fasudil and miR195 to SK-Hep-1 human hepatocellular carcinoma cells. |
Materials and methods
Materials
Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), and penicillin–streptomycin solution (5 kU/mL) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Fasudil was obtained from Selleckchem (Houston, TX, USA). mPEG-MAL 3400 Da was purchased from Nektar Therapeutics (Huntsville, LA, USA). Dithiothreitol (DTT) was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Lipofectamine 2000 was purchased from Thermo Fisher Scientific. Enhanced bicinchoninic acid protein assay kit and 4′,6-diamidino-2-phenylindole were obtained from Beyotime (Nanjing, People’s Republic of China). Antibodies were purchased from Abcam (Cambridge, UK). Gelred was obtained from Biotium (Hayward, CA, USA). A Cell Counting Kit-8 (CCK8) was obtained from Dojindo Molecular Technologies Inc. (Tokyo, Japan). Fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit was obtained from BD Pharmingen (San Diego, CA, USA). Cy3 was obtained from LiTTLE-PA Sciences (Wuhan, People’s Republic of China). ST21 and H3R5 were custom synthesized from Guangzhou RiboBio Co., Ltd. (Guangzhou, People’s Republic of China). pGL3 and pEGFP were obtained from Shanghai Innovation Biotechnology Co., Ltd. (Shanghai, People’s Republic of China). Plasmid pre-miR195 (sequence 5′-UAGCAGCACAGAAAUAAUGGC-3′) was synthesized from YINGWEIXIN Information Technology (Shanghai, People’s Republic of China). Carboxylfluorescein (FAM)-labeled miR195 was obtained from GenePharma Co., Ltd. (Shanghai, People’s Republic of China). All other reagents and chemicals were of analytical grade. The human normal hepatic cells L02 and human HCC cells SK-Hep-1 (Cell Culture Center of Shanghai Institutes of the Chinese Academy of Sciences, Shanghai, People’s Republic of China) were cultured in DMEM supplemented with 10% FBS under a humidified atmosphere containing 5% CO2 at 37°C. Female Balb/c nude mice (4–6 weeks; ~20 g) were purchased from SLAC Laboratory Animal Co., Ltd. (Shanghai, People’s Republic of China). All animal experiments were performed in line with the protocols evaluated and approved by the Animal Care and Experiment Committee of Shanghai General Hospital affiliated to Shanghai Jiaotong University School of Medicine. All animal procedures were performed in line with the Guide for the Care and Use of Laboratory Animals of Shanghai General Hospital affiliated to Shanghai Jiaotong University School of Medicine and followed the guidelines of the Animal Welfare Act.
Synthesis of ST21-H3R5-PEG
ST21, H3R5, and PEG were successfully conjugated at a ratio of 1:5:2 (mol/mol/mol), as described previously with slight modification.17 Briefly, a 5:2 conjugate of mPEG-MAL and H3R5 was prepared through a specific reaction between the MAL groups and the –SH groups of H3R5 in phosphate-buffered saline (PBS; pH 7.0) for 2 hours at 25°C. Then, we introduced cysteine terminal to ST21 since the oxidation of the –SH groups of cysteine from ST21 and H3R5 generates disulfide bond. ST21 and H3R5-PEG were dissolved in PBS in a molar ratio of 1:1 (pH 7.0) and then added to H2O2 (0.1%–0.5%) under stirring at 25°C. After 24 hours, the resultant mixture was dialyzed in distilled water for 12 hours using a dialysis membrane (molecular weight cut-off 1,000) to remove residual reactants, and was lyophilized after that. To confirm conjugation, ST21-H3R5-PEG (SHP) was characterized by 1H nuclear magnetic resonance (NMR) spectra using deuterium oxide as the solvent with a concentration of 5 mg/mL.
Preparation of fasudil-loaded NPs
Fasudil hydrochloride was encapsulated either by passive or active loading.25,26 In the case of passive loading, SHP was rehydrated with 10 mg/mL fasudil in PBS and incubated for 30 minutes at 65°C. Free drug was removed by passing fasudil-loaded nanocomposites through Sephadex G-25 PD-10 column equilibrated with PBS. For active loading, we first rehydrated SHP with 200 mM (NH4)2SO4 solution at varying pH values (3.0, 5.4, 7.0 and 8.0) with an identical concentration of fasudil (10 mg/mL), exchanged external (NH4)2SO4 solution with PBS solution employing PD-10 column, loaded fasudil, and separated unentrapped drug using the same column. We then changed the drug-to-carrier ratio to optimize it. High performance liquid chromatography was used to measure unentrapped fasudil. The entrapment efficiency (EE%) was calculated by the following equation: T − U/T ×100%, where T represents the total fasudil added and U is the concentration of unentrapped fasudil in the NPs.
Preparation of miR195-condensed NPs
To format FasudilHPmiR195 and FasudilSHPmiR195, miRNA-195 in DEPC water was mixed with an equal volume of fasudil-loaded NPs (the molar ratio was 1.5:1 employing active loading at pH 7.0) at various N/P ratios, followed by gentle agitation using a vortex agitator for 5 seconds and incubation at 37°C for 30 minutes before use.
The condensation ability of FasudilSHPmiR195 to miR195 was determined by agarose gel electrophoresis. The polymeric NPs at various N/P ratios ranging from 0:1 to 15:1 were incubated for 30 minutes at room temperature. The samples were loaded onto 1.0% agarose gel containing Gelred at 100 V for 30 minutes in 1× TAE buffer solution.
Characterization of NPs in different formulations
Dynamic light scattering (DLS; Malvern, Westborough, MA, USA) was employed to analyze the particle size and zeta potential of coloaded NPs (FasudilHPmiR195, FasudilSHPmiR195) with fasudil-to-carrier molar ratio of 1.5:1. The particle morphology and size of the coloaded NPs at N/P =7.5 were observed on a transmission electron micrograph (TEM; Hitachi Ltd., Tokyo, Japan). Briefly, the sample was suspended in water and one drop of the sample solution was placed on a 200-mesh copper grid coated with carbon film. Images were recorded at an acceleration voltage of 75 kV.
Serum stability
For serum stability assay, naked miR195 and FasudilSHPmiR195 (N/P =7.5) were incubated in 50% FBS solution at 37°C. Aliquots measuring 20 μL were taken at 1, 2, 3, 4, 8, 16, 24, and 36 hours. To release the loaded miRNA, the mixtures were treated with 1% heparin solution and subsequently subjected to electrophoresis with 1.0% agarose gel.
In vitro drug release study
FasudilSHPmiR195 was incubated in PBS with different concentrations of DTT at 37°C with moderate stirring in dialysis cassettes (molecular weight cut-off 3,500; Thermo Fisher Scientific). The sample was withdrawn and immediately replenished with an equal volume of fresh PBS at predetermined time intervals. To evaluate whether the cassettes influenced drug release, plain fasudil was used as control. The concentration of fasudil in NPs was determined by high performance liquid chromatography (Shimazu, Tokyo, Japan).
Cellular uptake
The cellular uptake of targeted (FasudilSHPmiR195) and nontargeted (FasudilHPmiR195) NPs was investigated by confocal laser scanning microscopy (CLSM; Leica Microsystems, Wetzlar, Germany). Briefly, hydrophilic fluorescent probe CY-3 (red fluorescence) and FAM-labeled miR195 (green fluorescence) were coloaded into SHP and HP as described in the sections of preparation of fasudil-loaded NPs and preparation of miR195-condensed NPs, respectively, apart from the change of fasudil to Cy3 and miR195 to FAM-miR195. SK-Hep-1 (1×104 cells/well) and L02 (2×104 cells/well) were incubated for 24 hours, and then replaced in serum-free DMEM containing Cy3SHPFAM-miR195 or Cy3HPFAM-miR195. After incubation for prearranged time intervals (4 hours), cells were washed with PBS to remove the remaining NPs, fixed with 4% formaldehyde, and treated with 4′,6-diamidino-2-phenylindole for staining of nucleus.
Gene transfection assay
The gene transfection efficacy of HP/SHP was calculated in SK-Hep-1 cells using pDNA (pGL3 and pEGFP) as reporter genes. Briefly, SK-Hep-1 cells were cultured in 24-well plates at a density of 105 cells/well for 24 hours. Then, the culture medium was removed and the cells were incubated with serum-free DMEM containing HPpDNA/SHPpDNA at various N/P ratios. Lipofectamine 2000/pDNA complexes was used as control. After 3 hours of incubation, the medium was replaced by a fresh culture medium and the cells were incubated for another 24 hours. For qualitative analysis, cells were washed three times with PBS, incubated with lysis buffer for 0.5 hours, and centrifuged at 5,000 rpm for 3 minutes. The supernatant (20 μL) was added to substrate (100 μL) and analyzed by luminometer (Promega Corporation, Fitchburg, WI, USA). The relative luciferase light units were normalized to the amount of cellular protein, which was determined by Bradford colorimetric assay with bovine serum albumin. The luciferase protein expression was calculated as follows: (relative light units/mg protein) ×100%. The expression of enhanced green fluorescent protein was examined by fluorescent microscopy (Leica Microsystems).
Western blot assay
To verify the downregulation of VEGF and ROCK2 proteins, SK-Hep-1 cells were transfected with FasudilHPmiR195, FasudilSHPmiR195, FasudilSHP, and SHPmiR195. The untreated cells were used as control. Cultured cells were harvested after 48 hours, lysed with RIPA lysis buffer, and incubated for 60 minutes on ice. After centrifugation at 12,000 rpm for 10 minutes, the supernatant was analyzed for the protein concentration using bicinchoninic acid protein assay kit. An equal amount (20 μg) of protein was loaded on sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred onto polyvinylidene fluoride (EMD Millipore, Billerica, MA, USA), blocked, incubated overnight with primary antibodies at 4°C, washed with 1% TBST three times, and incubated in secondary antibody for 2 hours. After washing, the chemiluminescence signal was imaged using ChemiDoc XRS (Bio-Rad Laboratories Inc., Hercules, CA, USA).
In vitro cytotoxicity assay
In vitro cytotoxicity of different NPs against normal cells (L02) and HCC cells (SK-Hep-1) was determined by CCK8 assay. L02 and SK-Hep-1 were seeded in 96-well plates at 1.5×104 and 1×104 cells/well, respectively. After 24 hours, the culture medium was replaced by 100 μL of complete DMEM involving blank NPs (HP and SHP), plain fasudil, and coloaded NPs (FasudilHPmiR195, FasudilSHPmiR195) for 24 hours. Also, 100 nM miR195 was constantly used, and fasudil was loaded at three different drug concentrations (10, 30, and 60 μM) as low-, moderate-, and high-dose fasudil groups. Cells treated with the same amount of PBS were used as control. After 24 hours incubation, 10 μL of CCK8 solution was added to the culture medium and incubated for an additional 1.5 hours. Absorbance of each well at 450 nm was detected with a microplate reader (Thermo, Rockford, IL, USA). Cell viability (%) was calculated using the following equation: cell viability (%) = A − B/C − B ×100%, where A is the absorbance of the sample, B the absorbance of the blank (medium), and C is the absorbance of the control (cells). Compared with the untreated control, the dose of coloaded nanocomposites yielding 50% cell death was defined as IC50. All treatments were conducted in triplicate.
Cell apoptosis analysis
L02 and SK-Hep-1 (1×106 cells/well) were seeded in six-well plates and incubated at 37°C for 24 hours. After incubation with blank NPs (HP and SHP) or coloaded NPs (FasudilHPmiR195, FasudilSHPmiR195) for another 24 hours, apoptosis analysis was performed using the Annexin V-FITC (Annexin V) and propidium iodide detection kit (BD Pharmingen), according to the manufacturer’s instructions. The untreated cells were used as control.
In vivo fluorescence imaging for tumor-targeting analysis
For in vivo fluorescence imaging, 5×106 SK-Hep-1 cells were inoculated in nude mice by subcutaneous injection over their right flanks. The tumor volume was calculated according to the equation: tumor volume (mm3) =0.5× length × width2. The mice were injected with Cy7 coloaded NPs through the tail vein, when the tumor size reached ~500 mm3. The mice were anesthetized by 10% (w/v) chloral hydrate and images were taken at predetermined time intervals (1, 2, 6, 12, and 24 hours) on an in vivo imaging system. After 24 hours, the mice were sacrificed and different tissues including the tumor, heart, liver, spleen, lung, and kidney were excised for imaging.
Statistical analysis
Data were expressed as mean ± standard deviation. Statistical significance of differences in all measurements between control and treated groups was tested by two-tailed Student’s t-test. The differences were considered significant for *P<0.05 and highly significant for **P<0.01.
Results and discussion
Preparation and characterization of coloaded NPs
The chemical structure of SHP was confirmed by 1H NMR (Figure 2). The solvent peak of deuterium oxide was found at δ4.65 (signal e). The peak at δ0.75–1.30 (signal a) was assigned to the proton of the stearyl moiety. The peak at δ2.85 (signal b) was from –CH2– of cysteine, which generates disulfide bond to combine ST21 and H3R5. Signal c (δ3.10) was from -CH2- in arginine, except for -CH2- close to the tertiary carbon. The peak at δ3.6 (signal d) was attributed to PEG. Signal f (δ7.16) and signal g (δ8.56) were from the protons of imidazole in histidine. The 1H NMR results demonstrated that the SHP was synthesized successfully.
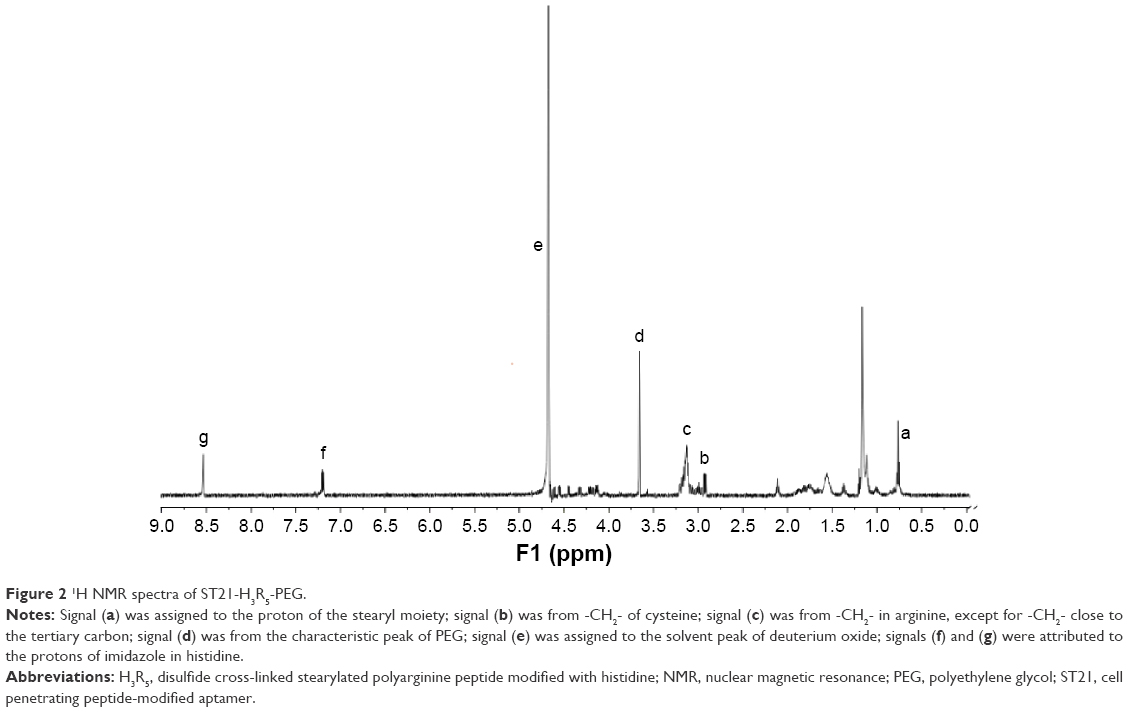 | Figure 2 1H NMR spectra of ST21-H3R5-PEG. |
As a weakly basic hydrophilic molecule, fasudil exhibits very low entrapment efficiency in nanocomposites.25 For this reason, we first optimized the EE% of fasudil employing passive and active loading methods, and then changed the drug-to-carrier ratio. FasudilSHPmiR195 prepared by the passive method at the ratio of 1.5:1 showed an EE% of 33.12% (Figure 3A). To increase the EE%, we adopted active loading using (NH4)2SO4 by changing the pH of the rehydrating medium. These results suggest that the drug entrapment was affected by the pH of the rehydrating medium, and the highest increase in entrapment was noticed at pH 7.0. In the active loading method, the dosing ratio affected the EE% of fasudil as well (Figure 3B). When the ratio progressed from 0.5:1 to 1.5:1, a slight change in EE% was observed, while an unappreciated decrease in EE% was noticed with an increase in molar ratio from 1.5:1 to 2.5:1. Along with the increase of the drug-to-carrier ratio, the decrease in EE% might be attributed to excessive drug, which hampered the overall stability of the formed NPs after H3R5 reached the saturation solubility in the polymer matrix. Irrespective of whether the drug was heavier or lighter than this certain critical value, the EE% could be reduced. Based on these data, we optimized the dosing ratio at 1.5:1 when the active loading method at pH =7.0 was used in the following experiments.
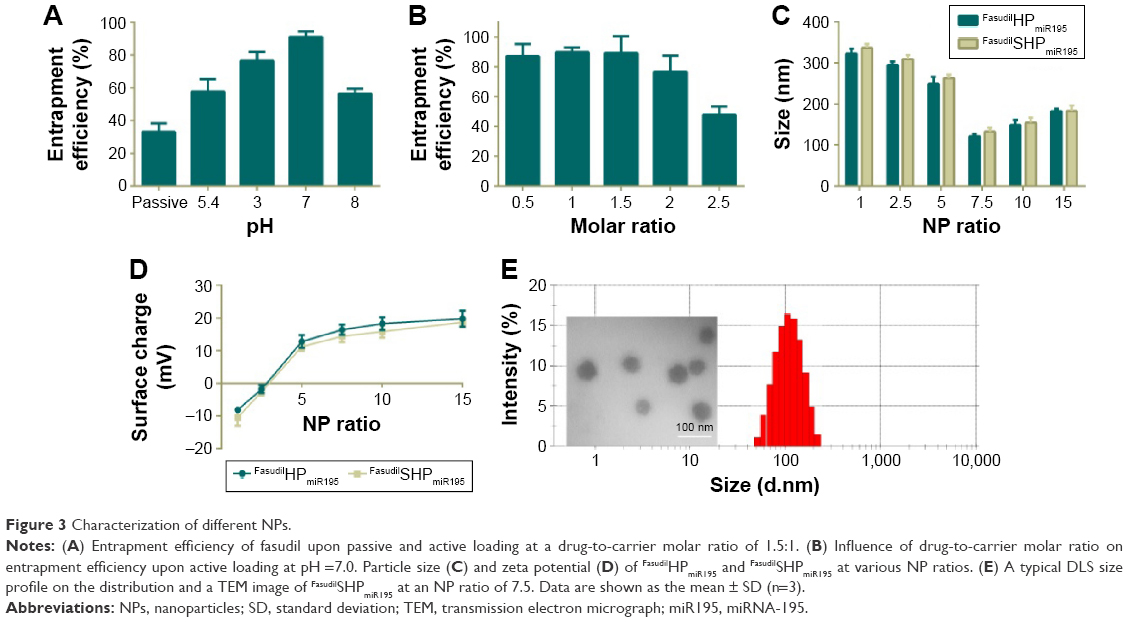 | Figure 3 Characterization of different NPs. |
This result is consistent with the finding of the previous studies that the (NH4)2SO4 gradient method could increase the EE% of hydrophilic drugs.27 Low encapsulation of hydrophilic drugs is known to have a limitation when NPs are applied to clinical therapy. In this gradient method, the presence of (NH4)2SO4 in the core of NPs produces an excess availability of protons owing to removal of (NH4)2SO4 from the external phase. Therefore, unprotonated fasudil can easily permeate through the membranes of NPs and can be protonated in the H+-rich environment and trapped in the aqueous core. The incubation time was limited to around 30 minutes because more neutral complexes that tend to effuse from NPs are produced between SO4−2 and protonated fasudil beyond 30 minutes.28 These EE% results suggest that (NH4)2SO4-based active loading is one of the most efficient methods in encapsulating fasudil in SHP/HP.
Particle size and zeta potential of different NPs are closely related to their gene condensing ability and in vivo profiles.29 When the N/P ratio increased from 1 to 7.5, the particle size of FasudilHPmiR195 reduced notably from ~320 nm to ~120 nm (Figure 3C), indicating that more compact complexes could be induced by electrostatic interactions. When the N/P ratio further increased to 15, the particle size began decreasing, indicating that the NPs had been compacted to the greatest extent at an N/P ratio of 7.5. The size of FasudilSHPmiR195 was slightly larger than that of FasudilHPmiR195, indicating that the presence of an additional hydrophilic molecule on the surface of NPs increased the hydrodynamic radius (P>0.05). With the N/P ratio increasing, the zeta potential of the NPs changed from negative to positive charges (Figure 3D). When the N/P ratio reached 15, the positive charge reached a plateau (~20 mV). Meanwhile, the negatively charged ST21 reduced the surface potential of the NPs (P>0.05). As NPs with positive charges are prone to interfere with negatively charged proteins in blood and disrupt the cell membrane resulting in decreased serum stability, we chose 7.5 as the optimal N/P ratio for the preparation of miR195-loaded NPs. The compact structure with the appropriate size and zeta potential of the complexes was expected to exhibit efficient cellular uptake. Since the DLS technique offers the hydrodynamic diameter of the NPs, the absolute particle size of the NPs was determined by TEM. It was found that the NPs had an apparent spherical shape with a mean particle size of about 60 nm (Figure 3E), which is smaller than that obtained in the DLS experiment because TEM measures the size of dried NPs while DLS measures the hydrodynamic diameter of NPs.
Agarose gel electrophoresis was carried out at different N/P ratios to confirm the condensation of miR195 with FasudilSHPmiR195. It was found that miR195 bands moved at an identical rate with the N/P ratio being from 0:1 to 2.5:1 and obviously more slowly at a ratio of 5:1 (Figure 4A). The miR195 was totally retarded when the N/P ratio was 7.5:1 or greater. Meanwhile, significant bands remained at the loading site. These results provide a suitable N/P ratio in accordance with the outcomes of size and potential measurement, which is an important reference for subsequent experiments.
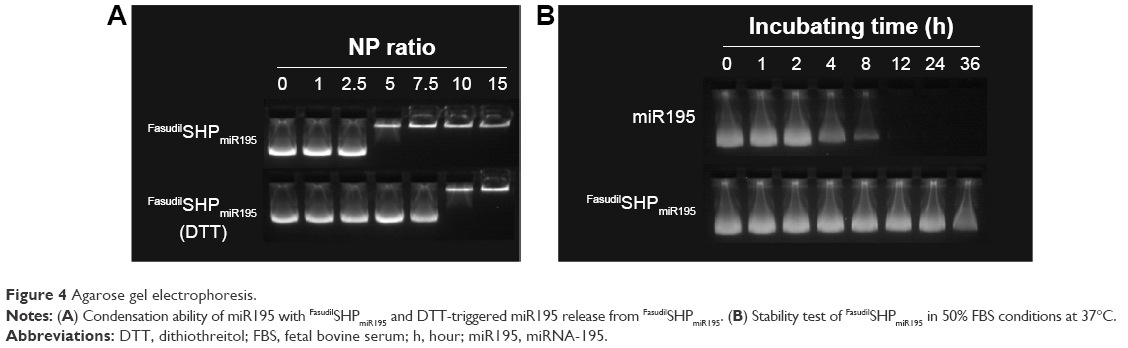 | Figure 4 Agarose gel electrophoresis. |
miR195 condensation and serum stability of miR195-loaded NPs
Disulfide bond has been regarded as an important linkage for constructing biologically responsive carriers owing to the rapid disulfide bond cleavage upon interaction with reducing molecules. DTT was applied as the reducing molecule to break the disulfide bonds.30,31 Figure 4A depicts the depolymerized FasudilSHPmiR195 formed due to DTT, showing a weaker binding of miR195. The stability of disulfide bonds is easily reversed from extracellular space to reductive intracellular homeostasis. Based on these findings, disulfide cross-linked SHP is often used as a carrier in enhancing complex formation and cytoplasm-sensitive dissociation.
In the serum-containing environment, the inefficient delivery of gene drugs by cationic polymeric carriers has largely limited their further therapeutic applications. FasudilSHPmiR195 prepared at an N/P ratio of 7.5 had an optimum positive surface charge, which is conducive to the enhanced serum stability of NPs on account of decreasing nonspecific interactions with anionic serum components. Naked miR195 was used as a control in this study. It was noticed that naked miR195 was completely degraded in 50% FBS between 8 and 12 hours, while miR195 formulated in SHP seemed to be less affected by serum, protecting the miR195 from complete degradation up to 32 hours (Figure 4B), implying that SHP NPs-encapsulated miR195 was stable and suitable for in vivo application.
In vitro release of fasudil in response to the reducing environment
Compared with normal cells, the cytoplasmic environment of tumor cells has a much higher reducing environment. Delivering and releasing the drugs actively into cancer cells using a redox-sensitive vehicle has been regarded as a potential method of delivery because it can rapidly degrade in the reductive intracellular cytoplasm.32 The in vitro drug release studies were performed to test the hypothesis that FasudilSHPmiR195 has the ability to quickly release drugs in the reducing environment. PBS with 20 mM DTT was set corresponding to the intracellular redox condition, while PBS with 10 μM DTT was regarded as the extracellular redox condition in tumor cells.33 FasudilSHPmiR195 showed sustained-release behavior of fasudil for 36 hours (Figure 5), while 100% of plain fasudil was released in 2–3 hours, indicating that the dialysis cassettes were not a rate-limiting factor for drug release. Figure 5 shows a burst in release of fasudil from FasudilSHPmiR195 in PBS with 20 mM DTT (58.49% of fasudil was released in 8 hours and 86.02% in 36 hours) and a dramatically decreased release rate of the drug in the presence of 10 μM DTT (16.24% of the drug was released in 8 hours and 26.02% in 36 hours) and no DTT (13.53% of fasudil was released in 8 hours and 25.02% in 36 hours), suggesting a concentration-dependent drug release of the reducing reagent. Based on these results, we postulated that FasudilSHPmiR195 could be an outstanding delivery system to provide sustained therapeutic efficacy and maintain stability in blood circulation for rapid release of encapsulated drugs upon targeting the cytoplasm of HCC cells.
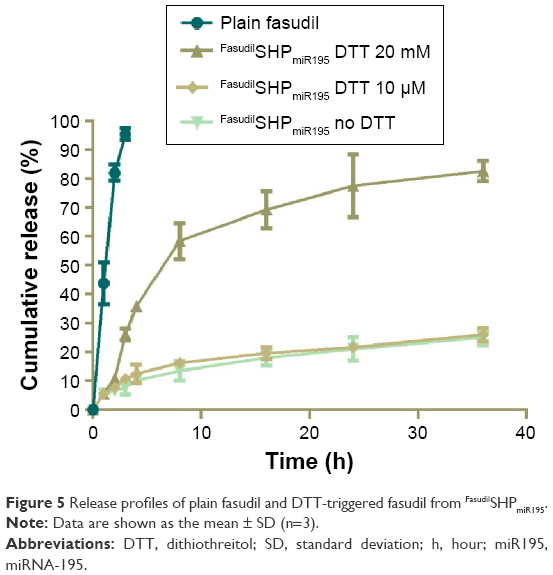 | Figure 5 Release profiles of plain fasudil and DTT-triggered fasudil from FasudilSHPmiR195. |
Cellular uptake
To demonstrate the intracellular dual delivery of fasudil and miR195 by redox-sensitive HP/SHP, a hydrophilic fluorescence probe (Cy3) and fluorescent-labeled miR195 (FAM-miR195) were coencapsulated into NPs. Cellular internalization of coloaded targeted and nontargeted NPs in L02 and SK-Hep-1 was studied using confocal microscopy. Figure 6 shows the confocal images of the normal and HCC cells after treatment for 4 hours. Both NPs could hardly be taken up by normal cells L02 (Figure 6A), while SK-Hep-1 showed the significantly stronger fluorescence signal after incubation with NPs (Figure 6B, C and D; P<0.01). In addition, SK-Hep-1 showed the strongest fluorescence signal after incubation with Cy3SHPFAM-miR195 than that of Cy3HPFAM-miR195 (P<0.05), which is consistent with the CCK8 finding and the flow cytometry result that are to be discussed subsequently. These results indicate the excellent role of ST21 in interacting with the HCC cell surface in cellular uptake.
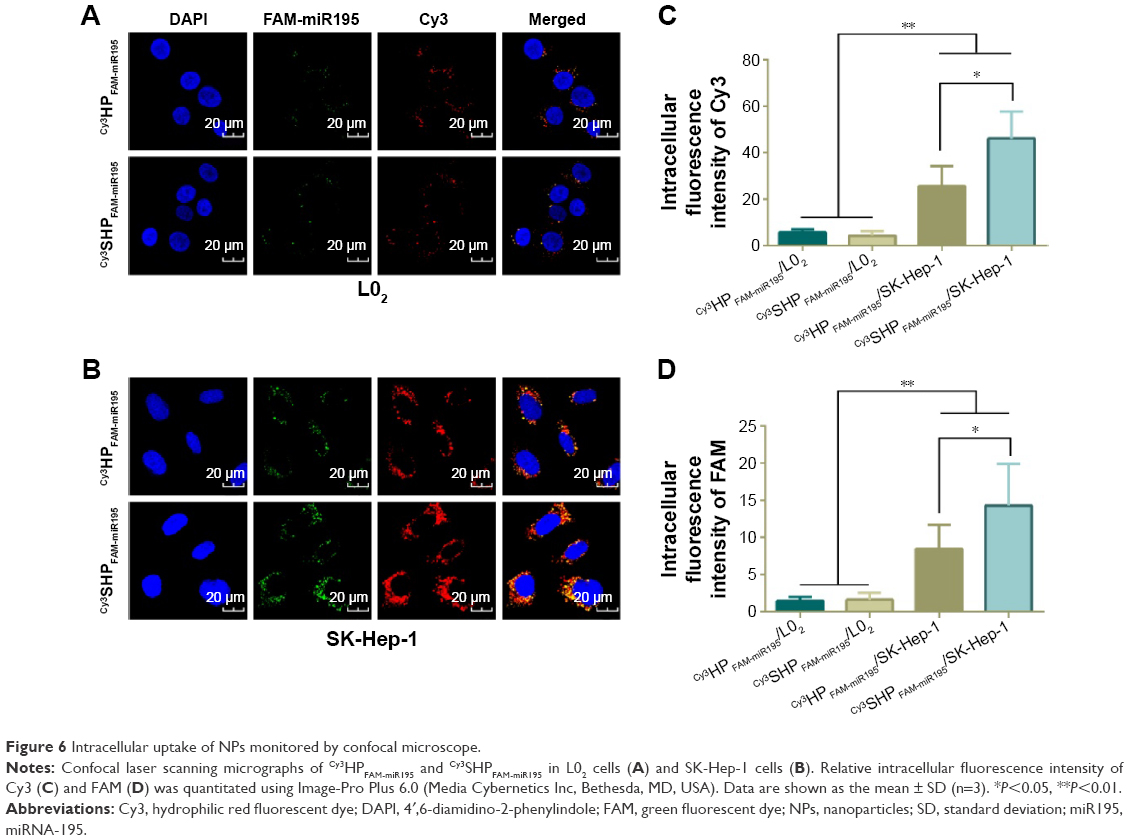 | Figure 6 Intracellular uptake of NPs monitored by confocal microscope. |
Gene transfection efficacy
To evaluate the surviving downregulation ability of HP/SHP, the in vitro gene transfection efficiency in SK-Hep-1 cells was assessed using pGL3 and pEGFP as the model plasmids to directly reflect the tendency. It seemed that the gene transfection efficacy was dependent on the N/P ratio (Figure 7A). The best transfection efficiency of HP/SHP existed at an N/P ratio of 7.5. The decreased enhanced green fluorescent protein intensity of HP/SHP at an N/P ratio larger than 7.5 could be due to the introduction of growth toxicity of the nanocarrier, because its positive charge contributed to the strong association and the consequent damage to the cell surface. Also, the pDNA transfection efficiency of the SHP complexes at an N/P ratio of 7.5 in SK-Hep-1 was comparable with that of lipofectamine 2000/pDNA (Figure 7B). Luciferase protein assays show that SHP could achieve more efficient transfection than HP. The higher transfection efficiency may be associated with the aptamer, resulting in more cellular internalization. As shown in Figure 7C, gene transfection efficiency in SHP group was significantly higher than that in HP group at an N/P ratio of 7.5 (P<0.01) or 10 (P<0.05). Since ST21 is a negatively charged molecule, its conjugation to HP may reduce the positive charge of the NPs and enlarge the diameter of the complexes, thus decreasing the transfection efficiency. However, the obtained result suggested that the attachment of ST21 increased the transfection ability dramatically with negligible adverse effects on charge and size.
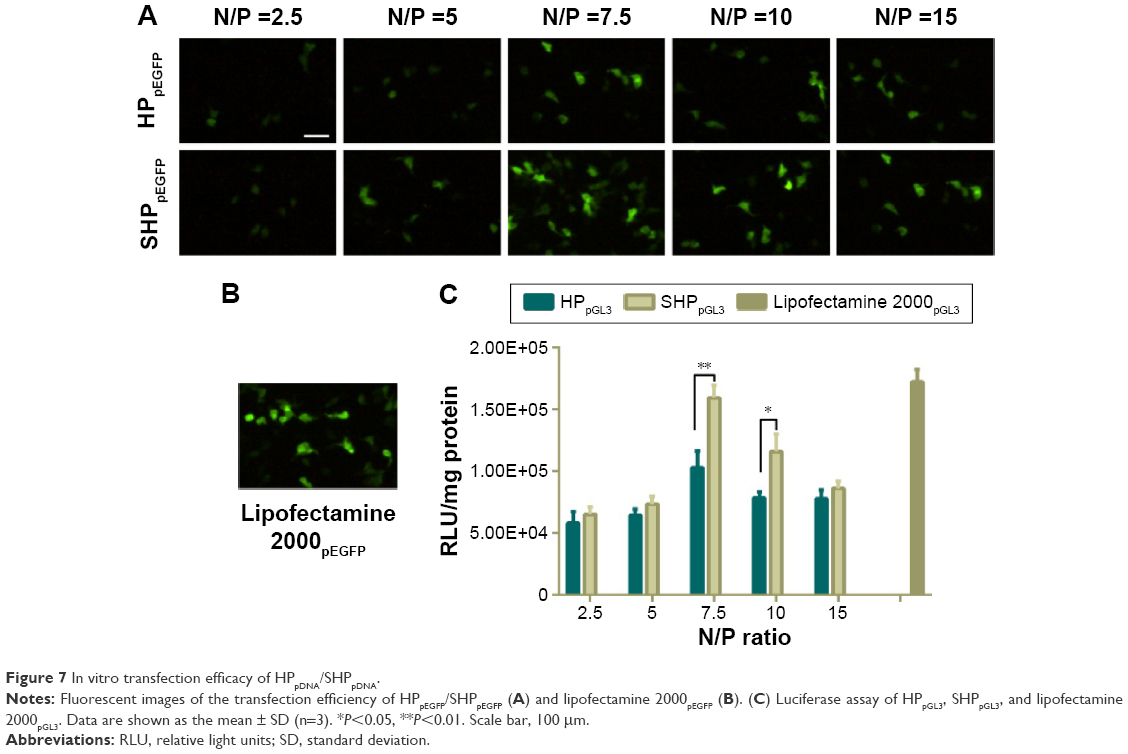 | Figure 7 In vitro transfection efficacy of HPpDNA/SHPpDNA. |
Downregulation of RhoA and VEGF protein expression
To confirm the gene silencing induced by fasudil and miR195 in targeted and nontargeted NPs, the expression of downstream protein (ROCK2 and VEGF) was further analyzed by Western blot. In the tumor microenvironment, the downregulation of miR195 enhanced the VEGF level and subsequently activated VEGF receptor 2 signaling in endothelial cells, thus promoting angiogenesis.9 The formation of tubular network structures suggested that high-level ROCK2 cells were more likely to form VM networks than low-level ROCK2 cells,34 implying that ROCK2 is positively correlated with the potential of VM formation in HCC cells. The expression levels of VEGF in FasudilSHP-treated cells and ROCK2 in SHPmiR195-treated cells are similar to those of control cells, implying that SHP had no regulatory effects on the expression of VEGF and ROCK2. Modest downregulation of VEGF and ROCK2 was observed in the cells treated with FasudilHPmiR195, while the expression decreased remarkably in FasudilSHPmiR195-treated cells (Figure 8), suggesting that FasudilSHPmiR195 was more capable of efficiently delivering fasudil and miR195 to cells than FasudilHPmiR195.These results also indirectly imply that compared with the nontargeted NPs, ST21-modified NPs possess increased tumor targeting.
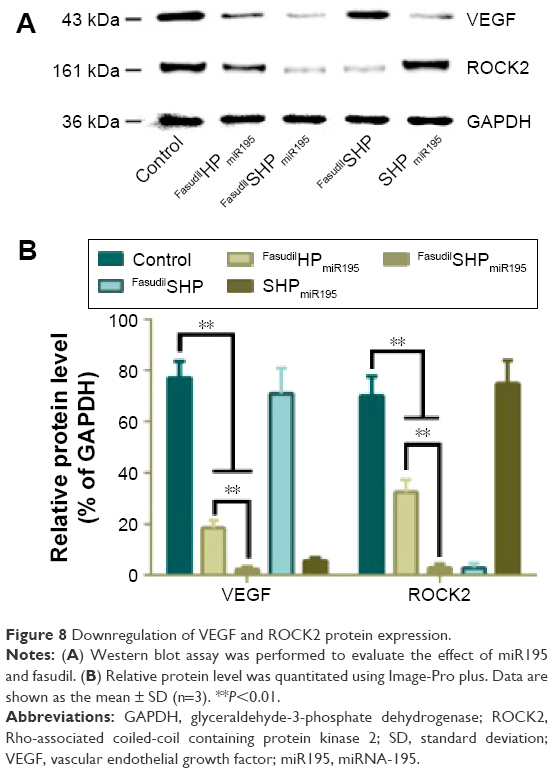 | Figure 8 Downregulation of VEGF and ROCK2 protein expression. |
In vitro cytotoxic efficacy and apoptosis assay
To investigate whether the aptamer-conjugated peptide had great therapeutic efficacy by actively targeting cell proliferation, cytotoxicity of different nanocomposites was evaluated by CCK8 assay. Cationic vehicles are closely related to significant cytotoxicity because their positive charges interact with glycocalyx that carries negative charges in blood and on the cell membranes.35 Hence, it is vital to assess the cytotoxicity of the cationic peptide used as a drug delivery carrier. In the case of L02 cells (Figure 9A), incubation with blank NPs showed cell viability above 90% in the concentration range of 5–80 μg/mL, while the largest dosage used in the experiment was under 20 μg/mL, indicating the biocompatibility of HP/SHP toward normal cells. The evaluated NPs did not cause significant damage to the L02, which can be attributed to the non-specific cellular uptake of coloaded NPs (Figure 9B).
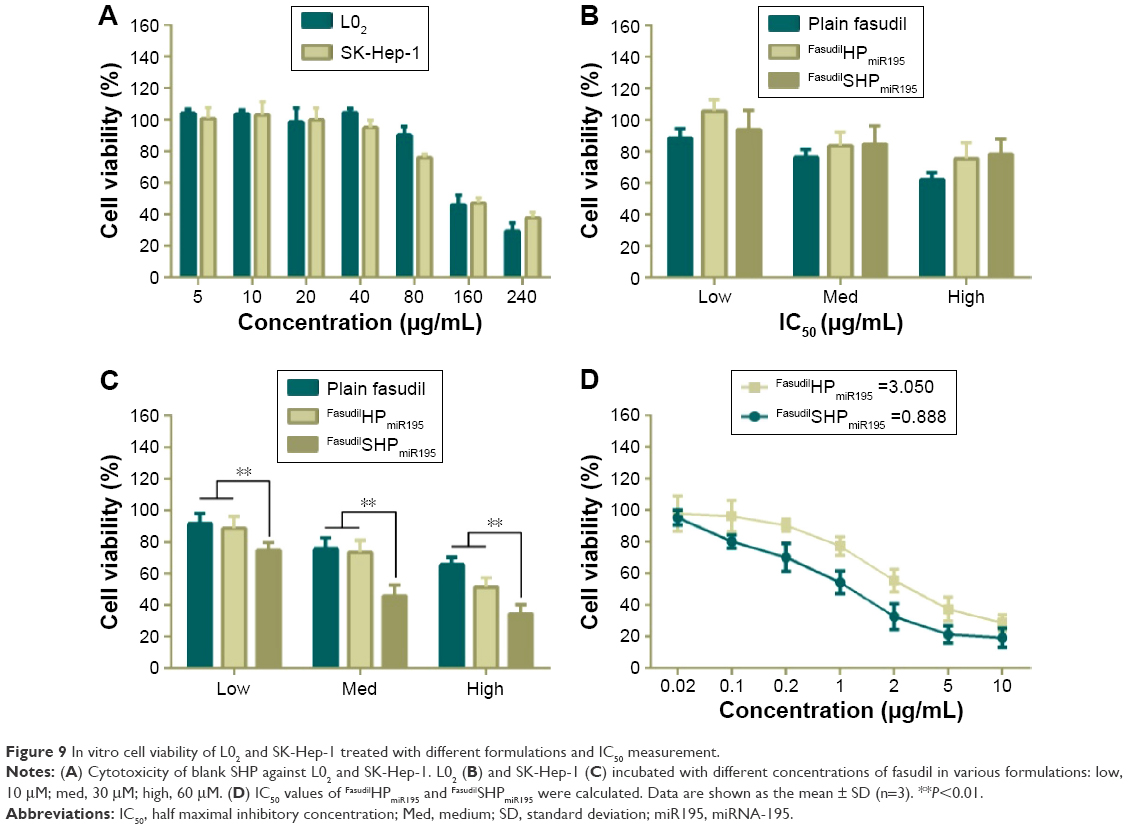 | Figure 9 In vitro cell viability of L02 and SK-Hep-1 treated with different formulations and IC50 measurement. |
Similar to normal cells, no obvious cell death was noticed in the drug-free NP-treated groups against SK-Hep-1 (Figure 9A), indicating the low cytotoxicity of blank HP/SHP. We further evaluated the cell inhibitory effect of plain fasudil and coencapsulated NPs at three different concentrations. Both FasudilHPmiR195 and FasudilSHPmiR195 showed remarkable cytotoxicity to HCC cells and exhibited a positive correlation with respect to the applied doses of fasudil in all tests. The viability of cells treated with FasudilSHPmiR195 at the same dose level was decreased significantly as compared with that of cells treated with plain fasudil and FasudilHPmiR195 (Figure 9C; P<0.01). As indicated in Figure 9D, the targeted NPs exhibited stronger cytotoxic activity to HCC cells with a significantly lower IC50 (0.888 μg/mL) than nontargeted NPs (3.050 μg/mL) at the same drug loading ratio. These results imply that FasudilSHPmiR195 has an increased cytotoxic activity to SK-Hep-1 cells by mediation of the additional active targeting.
Annexin V-FITC is used to quantitatively determine the percentage of cells within a population actively undergoing apoptosis. On the basis of the differential staining, the percentage of viable cells (lower left quadrant), damaged necrotic cells (upper left quadrant), early apoptotic cells (lower right quadrant), and necrotic cells (upper right quadrant) was determined by flow cytometry. Cells cultured in complete medium were used as negative control. In the case of L02 cells (Figure 10A), the control cells and most of the treated cells were healthy, indicating that the coloaded NPs had low toxicity to normal cells lacking overexpressed receptors of ST21 on their surfaces. In case of SK-Hep-1 cells (Figure 10B), few apoptotic cells were detected in the control group and no increase in apoptotic cells was observed in the cells treated with blank NPs (HP and SHP), suggesting that the cationic carrier is biocompatible and not able to induce cell apoptosis. The percentage of cell apoptosis (including the early apoptotic cells and the necrotic cells) induced by FasudilHPmiR195 (35.63%) was lower than that induced by FasudilSHPmiR195 (57.37%), which is a predictable result of targeting by ST21 (P<0.01).
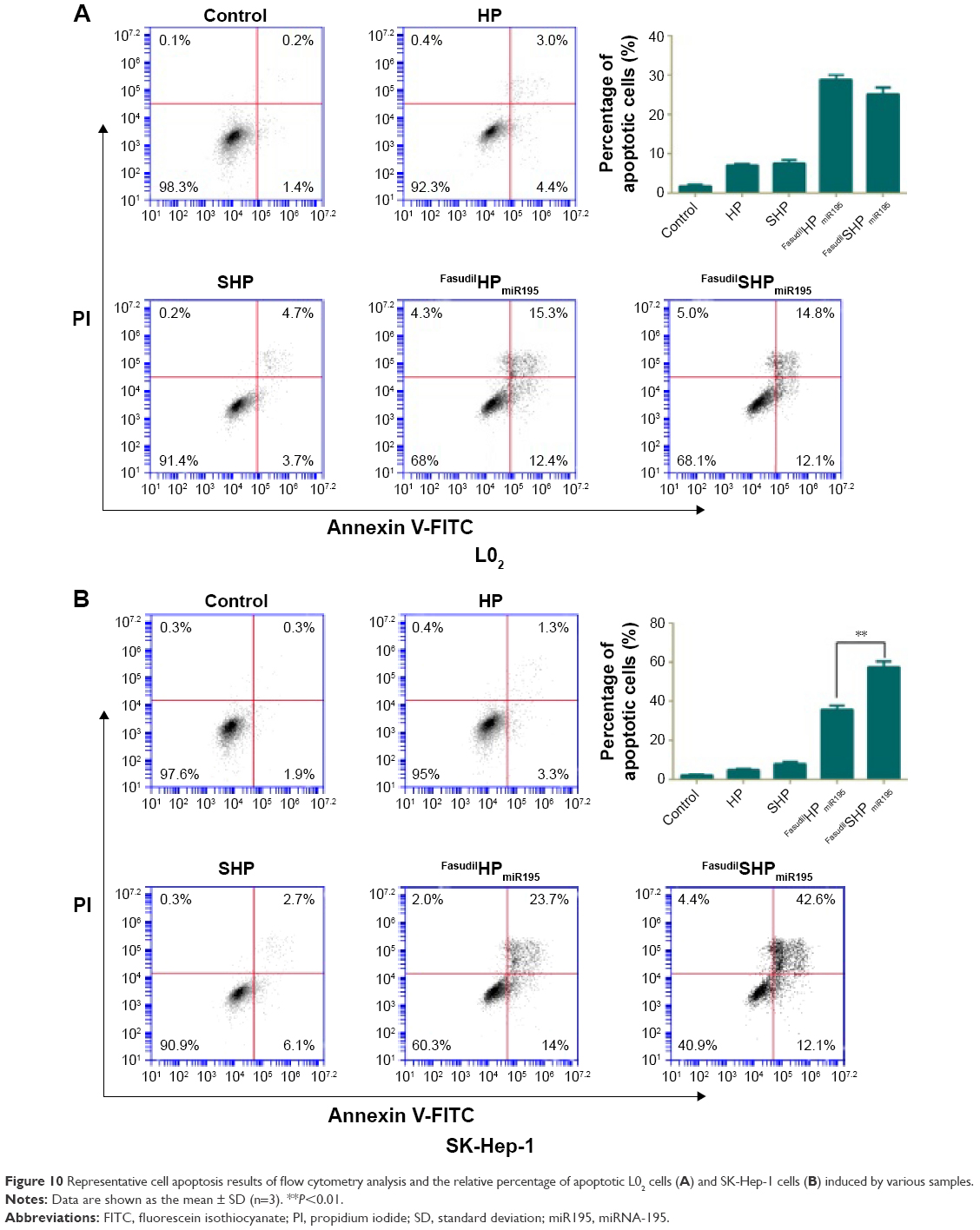 | Figure 10 Representative cell apoptosis results of flow cytometry analysis and the relative percentage of apoptotic L02 cells (A) and SK-Hep-1 cells (B) induced by various samples. |
In vivo tumor targeting
Fluorescence imaging showed that Cy7HP and Cy7SHP intravenously administered into the SK-Hep-1 tumor-bearing mice through the tail vein were distributed in a time-dependent manner. The intensity of their fluorescence was quantified using the Kodak in vivo imaging system equipped with a cooled charged coupled device camera at different time points. Figure 11A and B shows the presence of fluorescence as early as 1 hour postinjection. As time progressed, a preferential accumulation of fluorescence was detected in the tumor site other than the normal tissue 2 hours after injection, and then it decreased gradually 24 hours after injection. Compared with the nontargeted NPs, the fluorescence signal of Cy7SHP in the tumor site was dramatically stronger at any postinjection time point ranging from 1 to 24 hours.
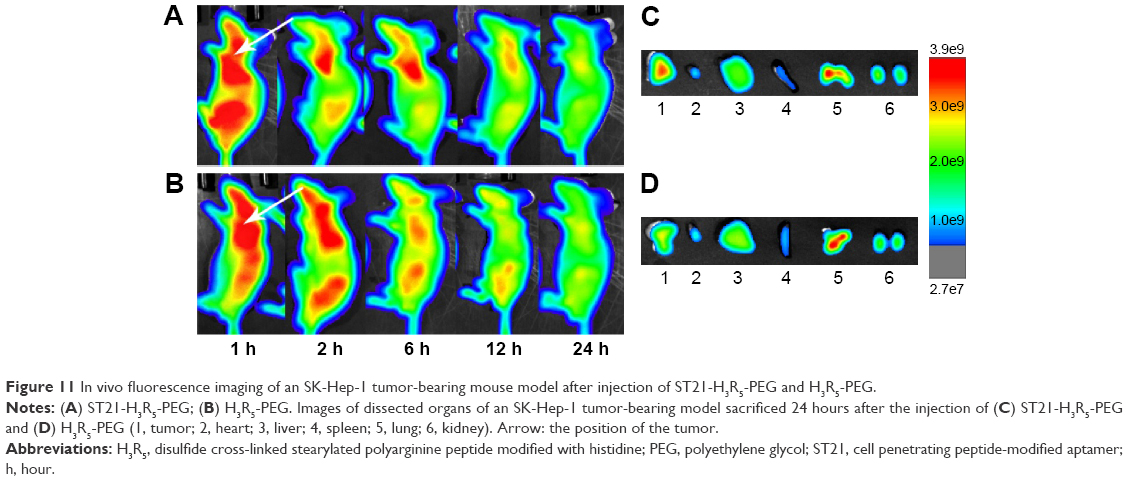 | Figure 11 In vivo fluorescence imaging of an SK-Hep-1 tumor-bearing mouse model after injection of ST21-H3R5-PEG and H3R5-PEG. |
Ex vivo optical imaging was performed where there was no impendence from soft tissues like the skin and blood vessels, which is similar to the intraoperative procedures in surgical oncology other than whole animal imaging. The images of the tumor and the tissues from the heart, liver, spleen, lung, and kidney were taken under the same conditions as those of whole body imaging. As shown in Figure 11C and D, the tumor possessed the strongest signal, while there were only background signals in the heart, liver, spleen, and kidneys. However, the lung displayed strong signals, perhaps reflecting that NPs underwent passive targeting. Compared with Cy7HP, more fluorescence of Cy7SHP was distributed in the tumor, indicating that targeted NPs not only accumulated in the tumor by enhanced permeability and retention effect but also were taken up in the HCC cells by aptamer receptor mediation. These results provide decisive evidence that the ST21-modified peptide could be a highly efficient codelivery vehicle for tumor-specific therapy.
Conclusion
In summary, we have developed a novel aptamer-functionalized cationic peptide for systemic delivery of a coloaded drug and gene. SHP exhibited a remarkable loading capacity for both fasudil and miR195, with an adjustable dosage ratio and ability to quickly release therapeutic agents in a reducing environment. The nontargeted and targeted NPs exhibited similar physicochemical properties, including an optimal particle size, zeta potential, and biocompatibility, while FasudilSHPmiR195 possessed significantly higher antitumor efficacy, which is attributed to aptamer-mediated active targeting. These findings indicate that the codelivery of fasudil and miR195 by the targeted peptide SHP may prove to be a promising treatment for targeted cancer therapy.
Acknowledgments
The authors acknowledge Professor Shen Gao from Changhai Hospital affiliated to Second Military Medical University for his kind guidance in this research. This research work was supported by the funds provided by the National Natural Science Foundation of China (number 81302212), the Natural Science Foundation of Shanghai (16ZR1428000), and the Crossing Program between Medicine and Industry supported by Shanghai Jiaotong University (numbers YG2014MS32, YG2015QN14).
Disclosure
The authors report no conflicts of interest in this work.
References
Romano M, De Francesco F, Pirozzi G, et al. Expression of cancer stem cell biomarkers as a tool for a correct therapeutic approach to hepatocellular carcinoma. Oncoscience. 2015;2(5):443–456. | ||
Lee KW, Yi NJ, Suh KS. Section 5. Further expanding the criteria for HCC in living donor liver transplantation: when not to transplant: SNUH experience. Transplantation. 2014;97(Suppl 8):S20–S23. | ||
Perez-Herrero E, Fernandez-Medarde A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015;93:52–79. | ||
Winkler GC, Barle EL, Galati G, Kluwe WM. Functional differentiation of cytotoxic cancer drugs and targeted cancer therapeutics. Regul Toxicol Pharmacol. 2014;70(1):46–53. | ||
Bishayee A, Darvesh AS. Angiogenesis in hepatocellular carcinoma: a potential target for chemoprevention and therapy. Curr Cancer Drug Targets. 2012;12(9):1095–1118. | ||
Sampat KR, O’Neil B. Antiangiogenic therapies for advanced hepatocellular carcinoma. Oncologist. 2013;18(4):430–438. | ||
Callegari E, Gramantieri L, Domenicali M, D’Abundo L, Sabbioni S, Negrini M. MicroRNAs in liver cancer: a model for investigating pathogenesis and novel therapeutic approaches. Cell Death Differ. 2015;22(1):46–57. | ||
Varshosaz J, Farzan M. Nanoparticles for targeted delivery of therapeutics and small interfering RNAs in hepatocellular carcinoma. World J Gastroenterol. 2015;21(42):12022–12041. | ||
Wang R, Zhao N, Li S, et al. MicroRNA-195 suppresses angiogenesis and metastasis of hepatocellular carcinoma by inhibiting the expression of VEGF, VAV2, and CDC42. Hepatology. 2013;58(2):642–653. | ||
Zhang JG, Zhang DD, Wu X, et al. Incarvine C suppresses proliferation and vasculogenic mimicry of hepatocellular carcinoma cells via targeting ROCK inhibition. BMC Cancer. 2015;15:814. | ||
Qiao L, Liang N, Zhang J, et al. Advanced research on vasculogenic mimicry in cancer. J Cell Mol Med. 2015;19(2):315–326. | ||
Zhang JG, Li XY, Wang YZ, et al. ROCK is involved in vasculogenic mimicry formation in hepatocellular carcinoma cell line. PLoS One. 2014;9(9):e107661. | ||
Pofali PA, Singh B, Dandekar P, et al. Drug-conjugated polymers as gene carriers for synergistic therapeutic effect. J Biomed Mater Res B Appl Biomater. 2016;104(4):698–711. | ||
Bakhtiar A, Sayyad M, Rosli R, Maruyama A, Chowdhury EH. Intracellular delivery of potential therapeutic genes: prospects in cancer gene therapy. Curr Gene Ther. 2014;14(4):247–257. | ||
Li L, Wei Y, Gong C. Polymeric nanocarriers for non-viral gene delivery. J Biomed Nanotechnol. 2015;11(5):739–770. | ||
Shukla RS, Qin B, Cheng K. Peptides used in the delivery of small noncoding RNA. Mol Pharm. 2014;11(10):3395–3408. | ||
Tai Z, Wang X, Tian J, et al. Biodegradable stearylated peptide with internal disulfide bonds for efficient delivery of siRNA in vitro and in vivo. Biomacromolecules. 2015;16(4):1119–1130. | ||
Mann A, Shukla V, Khanduri R, Dabral S, Singh H, Ganguli M. Linear short histidine and cysteine modified arginine peptides constitute a potential class of DNA delivery agents. Mol Pharm. 2014;11(3):683–696. | ||
Kang L, Gao Z, Huang W, Jin M, Wang Q. Nanocarrier-mediated co-delivery of chemotherapeutic drugs and gene agents for cancer treatment. Acta Pharm Sin B. 2015;5(3):169–175. | ||
Lo A, Lin CT, Wu HC. Hepatocellular carcinoma cell-specific peptide ligand for targeted drug delivery. Mol Cancer Ther. 2008;7(3):579–589. | ||
Jiang F, Liu B, Lu J, et al. Progress and challenges in developing aptamer-functionalized targeted drug delivery systems. Int J Mol Sci. 2015;16(10):23784–23822. | ||
Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew Chem Int Ed Engl. 2014;53(46):12320–12364. | ||
Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(pt A):28–51. | ||
Chen S, Yang K, Tuguntaev RG, et al. Targeting tumor microenvironment with PEG-based amphiphilic nanoparticles to overcome chemoresistance. Nanomedicine. 2016;12(2):269–286. | ||
Gupta V, Gupta N, Shaik IH, et al. Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary preferential vasodilation in pulmonary arterial hypertension. J Control Release. 2013;167(2):189–199. | ||
Nahar K, Absar S, Patel B, Ahsan F. Starch-coated magnetic liposomes as an inhalable carrier for accumulation of fasudil in the pulmonary vasculature. Int J Pharm. 2014;464(1–2):185–195. | ||
Zhou T, Llizo A, Wang C, Xu G, Yang Y. Nanostructure-induced DNA condensation. Nanoscale. 2013;5(18):8288–8306. | ||
Zhang W, Wang G, Falconer JR, et al. Strategies to maximize liposomal drug loading for a poorly water-soluble anticancer drug. Pharm Res. 2015;32(4):1451–1461. | ||
Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. | ||
Shirazi RS, Ewert KK, Leal C, Majzoub RN, Bouxsein NF, Safinya CR. Synthesis and characterization of degradable multivalent cationic lipids with disulfide-bond spacers for gene delivery. Biochim Biophys Acta. 2011;1808(9):2156–2166. | ||
Kim JS, Oh MH, Park JY, Park TG, Nam YS. Protein-resistant, reductively dissociable polyplexes for in vivo systemic delivery and tumor-targeting of siRNA. Biomaterials. 2013;34(9):2370–2379. | ||
Cui C, Xue YN, Wu M, et al. Cellular uptake, intracellular trafficking, and antitumor efficacy of doxorubicin-loaded reduction-sensitive micelles. Biomaterials. 2013;34(15):3858–3869. | ||
Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30(11):1191–1212. | ||
Xia Y, Cai XY, Fan JQ, et al. Rho kinase inhibitor fasudil suppresses the vasculogenic mimicry of B16 mouse melanoma cells both in vitro and in vivo. Mol Cancer Ther. 2015;14(7):1582–1590. | ||
Junquera E, Aicart E. Recent progress in gene therapy to deliver nucleic acids with multivalent cationic vectors. Adv Colloid Interface Sci. Epub 2015 Jul 26. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.