Back to Journals » Breast Cancer: Targets and Therapy » Volume 11
APR-246 alone and in combination with a phosphatidylserine-targeting antibody inhibits lung metastasis of human triple-negative breast cancer cells in nude mice
Authors Liang Y, Besch-Williford C ![]() , Cook MT, Belenchia A
, Cook MT, Belenchia A ![]() , Brekken RA
, Brekken RA ![]() , Hyder SM
, Hyder SM
Received 14 March 2019
Accepted for publication 18 July 2019
Published 31 July 2019 Volume 2019:11 Pages 249—259
DOI https://doi.org/10.2147/BCTT.S208706
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Pranela Rameshwar
Yayun Liang,1 Cynthia Besch-Williford,2 Matthew T Cook,3 Anthony Belenchia,4 Rolf A Brekken,5 Salman M Hyder1
1Department of Biomedical Sciences and Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO, USA; 2IDEXX BioAnalytics, Columbia, MO, USA; 3Department of Biology, Washburn University, Topeka, KS, USA; 4Department of Nutrition and Exercise Physiology and Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO, USA; 5Hamon Center for Therapeutic Oncology Research and Department of Surgery, University of Texas Southwestern Medical Center, Dallas, TX, USA
Background: Approximately 15–20% of all human breast cancers are classified as triple-negative because they lack estrogen and progesterone receptors and Her-2-neu, which are commonly targeted by chemotherapeutic drugs. New treatment strategies are therefore urgently needed to combat triple-negative breast cancers (TNBCs). Almost 80% of the triple-negative tumors express mutant p53 (mtp5), a functionally defective tumor suppressor protein. Whereas wild-type p53 (wtp53) promotes cell-cycle arrest and apoptosis and inhibits vascular endothelial growth factor-dependent angiogenesis, mtp53 fails to regulate these functions, resulting in tumor vascularization, growth, resistance to chemotherapy, and metastasis. Restoration of p53 function is therefore a promising drug-targeted strategy for suppressing TNBC metastasis.
Methods: APR-246 is a small-molecule drug that reactivates mtp53, thereby restoring p53 function. In this study, we sought to determine whether administration of APR-246, either alone or in combination with 2aG4, an antibody that targets phosphatidylserine residues on tumor blood vessels and disrupts tumor vasculature, effectively inhibits stem cell-like characteristics of tumor cells and migration in vitro, and metastasis of human mtp53-expressing TNBC cells to the lungs in mouse models.
Results: APR-246 reduced both the stem cell-like properties and migration of TNBC cells in vitro. In mouse models, administration of either APR-246 or 2aG4 reduced metastasis of TNBC cells to the lungs; a combination of the two diminished lung metastasis to the same extent as either agent alone. Combination treatment significantly reduced the incidence of lung metastasis compared either single agent alone.
Conclusion: Metastasis of human mtp53-expressing TNBC cells to the lungs of nude mice is inhibited by the treatment that combines activation of mtp53 with targeting of phosphatidylserine residues on tumor blood vessels. We contend therefore that our findings strongly support the use of combination treatment involving mtp53 activation and immunotherapy in patients with TNBC.
Keywords: breast cancer, metastasis, blood-vessel targeting agent, p53, APR-246
Introduction
Approximately 200,000 new cases of breast cancer are detected every year in the United States, and 40,000 American women die of the disease annually.1 Most deaths occur following the emergence of drug-resistant cancer cells and tumor metastasis.1 Consequently, more effective treatment strategies to reduce breast cancer-related mortality are urgently needed. Approximately 15–20% of all breast cancers are classified as triple-negative breast cancers (TNBC) because they do not express estrogen receptor, progesterone receptor, or Her-2-neu.2–4 Because such tumors lack these targets, currently used chemotherapeutic protocols are largely ineffective against TNBC, making this cancer virtually untreatable.
However, it has recently been shown that almost 80% of the TNBC tumors express a mutant form of the p53 tumor suppressor protein (mtp53) that is functionally defective.5 Wild-type p53 tumor suppressor protein (wtp53) promotes cell-cycle arrest and apoptosis and inhibits vascular endothelial growth factor-dependent angiogenesis, which, if left unchecked, leads to rapid tumor growth, metastasis, and patient death.6–12 Most p53 mutations occur in the DNA-binding domain, thereby preventing normal regulation of p53 target genes involved in apoptosis, cell-cycle arrest, and/or angiogenesis.13,14 Dysregulation of these processes promotes neovascularization, unconstrained tumor growth, and metastasis, and can lead to the development of resistance to chemotherapeutic drugs.15 Conversely, wtp53 suppresses both the self-renewal properties of stem cells and the epithelial-to-mesenchymal transition, the latter of which is vital for the initiation of tumor metastasis.16–18 We contend therefore that restoration of wtp53 functions in women with p53-defective TNBC may represent a viable alternative therapeutic approach to combat this aggressive type of cancer.
APR-246 is a recently developed small-molecule drug that reactivates mtp53 by covalently modifying the DNA-binding core domain of the mutant protein through alkylation of thiol groups, thereby restoring wild-type conformation and function.19,20 Functional studies show that APR-246 is converted to methylene quinuclidinone, which covalently binds to cysteine residues in p53 protein and thereby reactivates mtp53.20,21 APR-246–facilitated restoration of wtp53 renews its ability to promote cell-cycle arrest and apoptosis of tumor cells,22,23 though the capacity of APR-246 and reactivated mtp53 to control TNBC tumor growth, drug resistance, and metastasis, which are the leading causes of TNBC patient death, is not fully known. In human clinical trials, doses of up to 135 mg/kg of APR-246 have been administered, with doses between 60 and 100 mg/kg being well tolerated and found to be clinically useful against hematologic malignancies and prostate cancer.24,25
In this study, we examined the in vitro and in vivo effects of APR-246 alone, as well as its effectiveness in vivo in combination with 2aG4 (human equivalent bavituximab; US Food and Drug Administration-approved for clinical trials), a tumor blood-vessel-specific antibody that has been shown to reduce tumor angiogenesis26–28 and therefore may be able to inhibit tumor cell metastasis. In vitro, APR-246 reduced both TNBC cell mammosphere formation and aldehyde dehydrogenase isoform 1 (ALDH) activity, both of which are characteristics of cancer stem cells, the major cause of the emergence of drug-resistant tumors in individuals with TNBC,29,30 and inhibited migration of TNBC cells. These findings suggest that APR-246 has the capacity to destroy cancer stem cells, and could represent an innovative therapy to prevent the emergence of drug-resistant tumors in individuals with TNBC. APR-246 also effectively reduced the metastasis of TNBC cells to the lungs in two different mouse models, without any toxicity to experimental animals. Furthermore, a combination of APR-246 and 2aG4 treatment inhibited incidence of lung metastasis even more effectively than APR-246 or 2aG4 alone.
Materials and methods
Cell lines and culture
All cell culture studies were approved by the University of Missouri Institutional Environmental Health and Safety Board (Columbia, MO, USA). MDA-MB-231 and MDA-MB-435 TNBC cells were obtained from the American Type Culture Collection (Manassas, VA, USA). MDA-MB-231-4175 LM2 cells31 were obtained from Dr. Joan Massague from Memorial Sloan-Kettering Cancer Center (New York, NY). Cells were grown at 37°C in DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA) in a humidified atmosphere of 5% CO2 and harvested for various experiments with 0.05% trypsin-EDTA (ThermoFisher Scientific, Waltham, MA, USA). Prior to treatment in fresh medium containing 5% FBS, cells were washed with phosphate-buffered saline (PBS).
Reagents
APR-246 (PRIMA-1MET) was purchased from Tocris Bioscience (Bristol, UK). For in vivo studies, APR-246 was kindly provided by APREA AB (Solinka, Sweden). 2aG4, a mouse IgG2a monoclonal antibody that binds directly to phosphatidylserine on tumor blood vessels,32 was provided by Dr. Rolf Brekken from University of Texas Southwestern Medical Center (Dallas, TX, USA). Binding of 2aG4 to anionic phospholipids relies on a 50-kD bovine plasma glycoprotein, β2-glycoprotein 1. Consequently, we mixed 2aG4 at a 1:1 ratio with β2-glycoprotein 1 to enhance binding of 2aG4 to exposed anionic phospholipids on the endothelial cell surface.33 The IgG2a mouse anti-colchicine monoclonal antibody C44 (also provided by Dr. Brekken) was used as a negative control for 2aG4.
ALDH activity
TNBC cells were washed once with PBS and harvested using Accutase (BD Biosciences, Franklin Lakes, NJ, USA). ALDH activity was measured using the ALDEFLUORTM kit (STEMCELL Technologies, Vancouver, BC, Canada) and flow cytometry per the manufacturer’s protocol. Treatment with the ALDH inhibitor N,N-diethylaminobenzaldehyde was used as a negative control.
Mammosphere-formation assay
MDA-MB-435 and MDA-MB-231 cells were grown in 100 mm dishes in DMEM/F12 medium supplemented with 10% FBS to 60% confluence. Cells were then washed twice in PBS, harvested and counted. Cells (5×103) in 0.1 mL complete Mammocult™ medium (STEMCELL Technologies) were seeded onto ultra-low adherent six-well plates (STEMCELL Technologies) containing 1.9 mL/well of complete Mammocult medium supplemented with APR-246 at the final concentrations indicated. For controls, an equal volume of PBS (vehicle) was added to the medium. Incubations were carried out in triplicate. Every 48 hrs, cells were retreated with additional 1 mL of fresh APR-246 (or vehicle) in complete Mammocult medium. Images of mammospheres were captured by EVOS light microscopy (10x objective) (Waltham, MA, USA) on days 2, 4, and 6 of treatment. Mammospheres≥100 μm were quantified.
Cell-migration assay
Viable MDA-MB-231 cells were counted and plated (4×104 cells/well) on an Oris Pro Cell Migration Assay 96-well tissue culture-treated plate containing a bead of biocompatible gel (Cat # PROCMA1; Platypus Technologies; Fitchburg, WI, USA). Plates were incubated at 37°C for 1 hr to allow the gel to dissolve, leaving a circular zone of detection. Two hours later, once cells had properly adhered to the plates, they were treated with APR-246 (or PBS vehicle) for 24 or 48 hrs. Images were captured at 0-, 24-, and 48-hr intervals on an EVOS XL Cell Imaging System utilizing a 4x objective. Migration was analyzed by determining the difference in area of the detection zone between pre-migration (0 hr) and subsequent time points (closure), using the lasso and measurement tools in Adobe Photoshop CS 5.5 (San Jose, CA, USA). Inter-well coefficients of variability given by the manufacturer for the detection zone created by the biocompatible gel were ≤12%.
Animal studies
All animal studies were approved by the Animal Care and Use Committee at the University of Missouri (Columbia, MO, USA). The study adhered to the guidelines of the US Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training. Female athymic nude (nu/nu) mice, 6 weeks old and 20–22 g, were purchased from Harlan Sprague Dawley, Inc. (Indianapolis, IN, USA) and housed in a laminar air-flow cabinet under specific pathogen-free conditions. All facilities were approved by the American Association for Accreditation of Laboratory Animal Care in accordance with the current regulations and standards of the United States Department of Agriculture, the Department of Health and Human Services, and the National Institutes of Health.
An initial experiment was performed by injecting 2×105 MDA-MB-231–4175 (LM2) metastatic cells intravenously (iv) via tail vein.31 Five days later, animals received APR-246 (100 mg/kg) or PBS vehicle by intraperitoneal (ip) injection daily for 7 days, then every other day for an additional 15 treatments. Animal weights were recorded twice weekly throughout the study. At the termination of the experiment on day 42, animals were sacrificed and the lungs harvested and fixed in Bouin’s Fixative Solution (Cat # 1120–32; Ricca Chemical Company; Arlington, TX, USA). Superficial lung colonies were then counted using a stereoscopic dissection microscope and the results from two independent researchers averaged, then statistically analyzed using SigmaPlot software, version 12.5 (San Jose, CA, USA).
A subsequent study was performed using well-established metastatic MDA-MB-435 breast cancer cells34,35 that were harvested by trypsinization and washed twice with DMEM/F12 medium. Animals were inoculated iv with MDA-MB-435 cells (2×106) on day 0 and then randomly assigned to four groups, with 10–11 mice/group. Treatment began on day 5 (see Figure 5A for detailed treatment protocol and schedule). One group of animals received APR-246 alone (100 mg/kg/treatment) by iv tail-vein injection. A second group received 2aG4 (100 µg/mouse/treatment) by ip injection, while a third group was given both APR-246 and 2aG4 (100 mg/kg+100 µg) by iv and ip injection, respectively. The study lasted for a total of 57 days from initial inoculation of animals with the MDA-MB-435 cells to animal sacrifice. The control group received ip injections of control antibody C44 (100 µg/mouse/treatment). A total of 19 treatments were performed. Animal weights were recorded twice weekly throughout the study. At the termination of the experiment on day 57, animals were sacrificed, lungs harvested, and fixed in Bouin’s Fixative Solution (Cat # 1120-32; Ricca Chemical Company; Arlington, TX, USA). Superficial lung colonies were counted and statistically analyzed.
Statistical analysis
Differences among groups were tested using Student’s t-test or one-way analysis of variance (ANOVA). SigmaPlot software (version 14) was used for statistical analysis. Data are reported as mean±standard error of the mean (SEM). For all comparisons, P<0.05 was considered significant. The assumption of the ANOVA was examined, and if necessary, a nonparametric measure based on ranks was used. If normality failed, Kruskal–Wallis one-way ANOVA on ranks was used in place of regular ANOVA. In cases where a significant effect was shown by ANOVA (F-ratio, P<0.05), the Student–Newman–Keuls multiple comparison test was employed to compare the means of the individual groups. Differences in the incidence of lung metastasis were examined using logistic analysis of variances performed using the GENMOD procedure in the SAS program (Cary, NC, USA).
Results
APR-246 inhibits ALDH activity in TNBC cells
ALDH activity correlates with the presence of the stem cell phenotype and metastatic potential in TNBC cells.36 With this in mind, we exposed both MDA-MB-231 and MDA-MB-435 TNBC cells to different doses of APR-246 to ascertain its effect on ALDH activity. Treatment of cells with 10–50 µM APR-246 significantly reduced the percentage of ALDH-positive cells by up to 90% compared with controls (Figure 1A and B). A dose of 50 µM APR-246 effectively abolished the incidence of ALDH-positive cells in both cell lines examined.
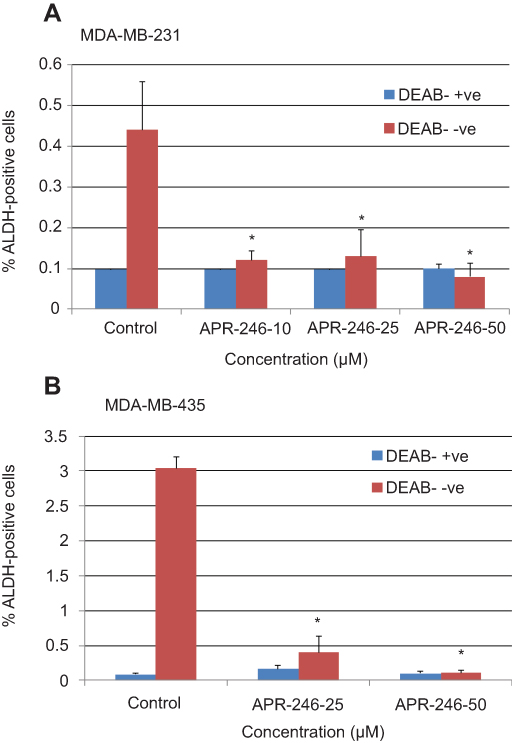 |
Figure 1 APR-246 inhibits ALDH activity in TNBC cells. Notes: MDA-MB-231 (A) or MDA-MB-435 (B) TNBC cells were treated in petri dishes with APR-246 or PBS vehicle (control) DMEM/F12 with 5% FBS for 24 hrs, then incubated with ALDEFLUOR substrate ±5 mM DEAB (an inhibitor of ALDH) as described by the ALDEFLUOR assay kit’s manufacturer (STEMCELL Technologies). Cells (2.5×104 cells/sample) were subsequently analyzed using flow cytometry. The ALDH gate was set using the negative control (+ DEAB). Viable cells were gated using propidium iodide. Results represent mean±SEM (n=3). *Significantly different from control (P<0.05, one-way ANOVA) followed by a Student–Newman–Keuls multiple comparison test.Abbreviations: ALDH, aldehyde dehydrogenase isoform 1; ANOVA, analysis of variance; DEAB, N,N-diethylaminobenzaldehyde; PBS, phosphate-buffered saline SEM, standard error of the mean; TNBC, triple-negative breast cancer. |
APR-246 inhibits formation of mammospheres by TNBC cells
Mammosphere-formation assays demonstrate anchorage-independent growth and are an excellent tool for examining cancer stem cells and progenitor cells.37 We next assessed whether APR-246 could interfere with cancer stem cell enrichment and thereby reduce the formation of mammospheres by TNBC cells. MDA-MB-231 and MDA-MB-435 TNBC cells were subjected to mammosphere-formation assays in the presence of different doses of APR-246 for up to 6 days. In PBS vehicle-treated wells, mammosphere formation increased steadily over the 6-day period (data not shown). However, lower concentration of APR-246 (1 μM) significantly reduced mammosphere formation in both cell lines and higher concentrations of APR-246 (5 or 10 µM) completely eradicated mammosphere formation at all time points in both cell lines examined (Figure 2A and B, data shown for day 4), suggesting that APR-246 effectively diminishes the cancer stem cell pool in TNBC cells.
 |
Figure 2 APR-246 inhibits mammosphere formation in TNBC cells. Notes: MDA-MB-231 and MDA-MB-435 cells were seeded into ultra-low adherent 6-well plates (5×103 cells/well) and treated for up to 6 days with 1–10 µM APR-246 or medium (control) in Complete Mammocult medium. Cells were retreated with fresh APR-246 or Complete Mammocult medium every 48 hrs. Representative light microscopic images of mammospheres formed on day 4 are shown in (A) and bar graph representing inhibition of mammospheres is shown in (B). Bar represents 100 μm. Inserts show an enlarged image of mammosphere. *Significantly different from control (P<0.05, one-way ANOVA) followed by a Student–Newman–Keuls multiple comparison test. **Significantly different from control and 1 μm APR-246 groups (P<0.05, one-way ANOVA) followed by a Student–Newman–Keuls multiple comparison test. Abbreviation: TNBC, triple-negative breast cancer. |
APR-246 inhibits migration of MDA-MB-231 TNBC cells
We used MDA-MB-231 cells to test whether APR-246 suppresses TNBC cell migration, which is an indicator that cells have acquired the ability to metastasize.38 Compared with controls, APR-246 significantly interrupted migration of MDA-MB-231 cells dose-dependently, with cell migration inhibited more than 50% in response to treatment with 25 µM APR-246 for 24 or 48 hrs (Figure 3).
 |
Figure 3 APR-246 inhibits migration of MDA-MB-231 TNBC cells. Notes: MDA-MB-231 cells (4×104 cells/well) were plated in a 96-well migration-assay plate and subjected to cell-migration assays in the presence of APR-246 (5 or 25 µM) or PBS (control) for 24–48 hrs. Pictures were taken just prior to initiation of treatment (0 hr; pre-migration) and after 24 and 48 hrs. Bar graph quantitates the difference in cell migration from pre-migration compared with control (value set at 100%) as described in Methods. Data represent mean±SEM (n=6). *Significantly different from controls; **Significantly different from APR-246–5 (P<0.05, one-way ANOVA) followed by a Student–Newman–Keuls multiple comparison test). Abbreviations: ANOVA, analysis of variance; PBS, phosphate-buffered saline; SEM, standard error of the mean; TNBC, triple-negative breast cancer. |
APR-246 inhibits metastasis of MDA-MB-231-4175 (LM2) TNBC cells to the lungs in nude mice
Using the MDA-MB-231–4175 (LM2) TNBC cell line, which has been selected for lung metastasis,31 we examined whether APR-246 could suppress metastasis of TNBC cells to the lungs. Compared with control-treated mice, APR-246 treatment significantly reduced the number of metastatic colonies in the lungs of nude mice (Figure 4, upper panel) with no loss of animal weight (Figure 4, lower panel), indicating that the treatment was not toxic.
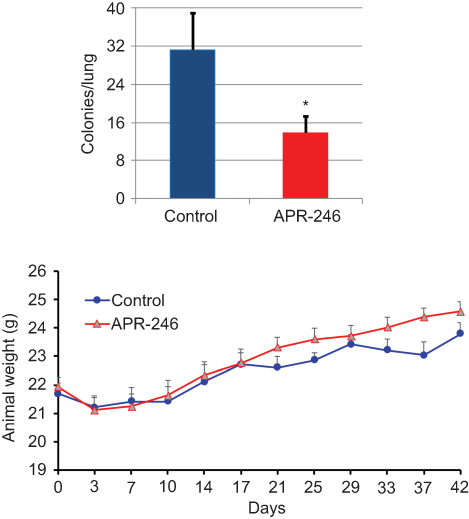 |
Figure 4 APR-246 inhibits metastasis of MDA-MB-231–4175 (LM2) TNBC cells to the lungs in nude mice. Notes: MDA-MB-231–4175 (LM2) cells (2×105) in 0.1 mL DMEM medium were injected into nude mice via tail vein and allowed to grow and circulate for 5 days. Animals were randomly assigned to two groups and treated with APR-246 (100 mg/kg; ip) or PBS (control) every day for 7 days, then every other day for an additional 15 treatments. Left, lungs were then removed and the number of metastatic colonies determined. Data represent mean±SEM (n=6 animals/group). *Significantly different from controls (P<0.05; Student’s t-test). Right, animals were weighed every 3–4 days throughout the experiment shown on the left. Data represent mean±SEM (n=6). No significant weight difference was found between the two groups using two-way repeated measures ANOVA (SAS Proc GLM). Abbreviations: i.p., intraperitoneal; SEM, standard error of the mean; TNBC, triple-negative breast cancer. |
APR-246 alone and in combination with antibody 2aG4 inhibits metastasis of MDA-MB-435 TNBC cells to the lungs in nude mice
We used a well-developed mouse model34,35 to study the effects of APR-246 when administered either alone or in combination with 2aG4, an antibody that targets tumor vasculature, on metastasis of MDA-MB-435 TNBC cells to the lungs (Figure 5A). Animals maintained their weight throughout the study (Figure 5B), showing that the treatments were non-toxic. The experiment was terminated on day 57 and lung tissues collected for photography and analysis. Compared with control-treated animals, those administered APR-246 or 2aG4 alone exhibited significantly fewer metastatic lung colonies (Figure 6A and B). The greatest effect was observed with a combination of the two agents, where the number of metastatic colonies was reduced more than 90% compared with control-treated animals. However, the number of lung colonies observed in mice treated with a combination of APR-246 and 2aG4 was not significantly different from the number seen with individual APR-246 or 2aG4 treatment alone.
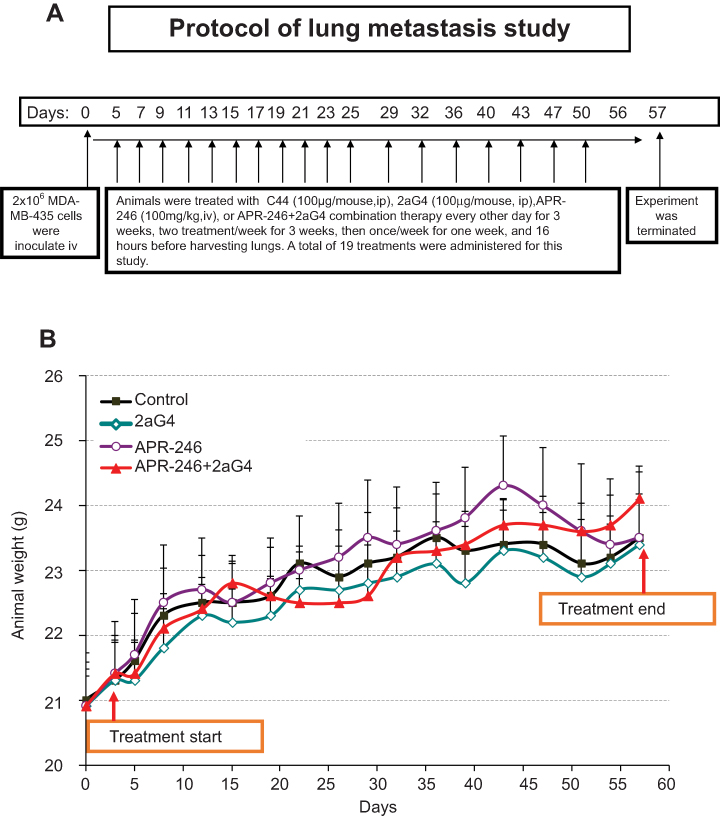 |
Figure 5 Protocol for lung metastasis study. Notes: (A) Protocol for examining the effect of APR-246 and 2aG4 antibody alone or in combination on lung metastasis of MDA-MB-435 TNBC cells in nude mice. (B) Animal weight during the treatment protocol shown in (A). Animals were weighed every 3–4 days throughout the experiment. Data represent mean±SEM (n=10–11/group). Control animals were treated with C44 antibody (ip). No significant weight differences were observed between control-treated animals and those administered APR-246 or 2aG4 alone or in combination using the one-way ANOVA on ranks (Dunn’s test). Abbreviations: ANOVA, analysis of variance; ip, intraperitoneal; SEM, standard error of the mean; TNBC, triple-negative breast cancer. |
 |
Figure 6 APR-246 and 2aG4 treatment inhibits metastasis of MDA-MB-435 TNBC cells to the lungs in nude mice. Notes: (A) Female nude mice were inoculated with MDA-MB-435 cells and then treated with C44 antibody (control), APR-246, 2aG4, or a combination of APR-246 and 2aG4 following the protocol described in Figure 5A. Data represent mean number of lung colonies±SEM (n=10–11/group) obtained at the end of the experiment. *Significantly different from control (P<0.05, ANOVA on ranks followed by Dunn’s test). (B) Representative images of lungs from four different animals from each group; colonies appear as off-white specks on the lungs (an example is circled). Abbreviations: ANOVA, analysis of variance; SEM, standard error of the mean; TNBC, triple-negative breast cancer. |
Compared with control-treated mice, administration of APR-246 and 2aG4 alone increased the number of animals exhibiting no metastatic lung colonies (ie, reduced incidence of metastasis), but this did not reach significance. However, the incidence of lung metastasis in animals receiving a combination of APR-246 and 2aG4 was significantly different from control-treated animals, and from the use of 2aG4 alone but not the use of APR-246 alone (Figure 7).
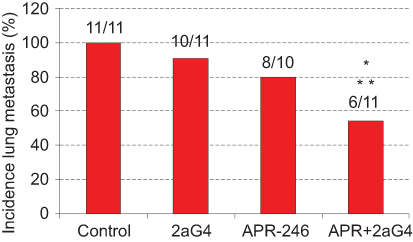 |
Figure 7 A combination of APR-246 and 2aG4 inhibits incidence of lung metastasis in animals inoculated with MDA-MB-435 TNBC cells. Notes: Animals (n=10–11) were inoculated with MDA-MB-435 cells. After treatment with C44 antibody (control), APR-246, 2aG4, or a combination of APR-246 and 2aG4 following the protocol described in Figure 5A, the number of animals with lung metastasis (incidence) among groups were compared using logistic analysis of variances performed with the GENMOD procedure in the SAS program. *Significantly different from control; **Significantly different from 2aG4 (P<0.05). Abbreviations: GENMOD, generalized linear models theory; SAS, statistical analysis software; TNBC, triple-negative breast cancer. |
Discussion
The presence, within a tumor, of stem-cell-like cells with the potential to metastasize is a major hallmark of metastatic breast cancer.39,40 Two important biomarkers of stem-cell-like cells in breast cancer are increased ALDH activity and the ability to form mammospheres.37 Our findings that the mtp53 activator APR-246 interfered with two such important components of metastatic cancer in two well-established mtp53-expressing TNBC cell lines suggest that it has the ability to reduce the population of stem-cell-like cells within a tumor and therefore inhibit metastasis. Using in vitro assays of MDA-MB-231 TNBC cells, we further found that APR-246 also suppressed cell migration, another hallmark of invasive breast cancer cells that go on to establish metastatic colonies at secondary sites.38
Initial studies in a mouse model of metastatic breast cancer using MDA-MB-231–4175 (LM2) TNBC cells31 demonstrated that APR-246 treatment reduced the number of metastatic colonies observed in the lungs of nude mice. Additional in vivo studies in a second mouse model of metastatic breast cancer indicated that administration of APR-246, either alone or in combination with 2aG4, an antibody that targets tumor vasculature, reduced the number of metastatic colonies derived from MDA-MB-435 TNBC cells in the lungs, and that a combination of APR-246 and 2aG4 also reduced incidence of lung metastasis in nude mice. APR-246’s ability to interrupt TNBC cell migration could underlie at least in part the reduced number of metastatic colonies in the lungs that we observed with APR-246 treatment in the two different mouse models of metastatic breast cancer employed in this report. However, in previous studies, we showed that APR-246 activated the mtp53 protein and induced apoptosis in hormone-dependent breast cancer cells.41,42 Consequently, in the studies presented here APR-246 may inhibit formation of metastatic lung colonies by activating mtp53 or by eradicating mtp53-containing tumor cells in the process of extravasation into the lungs since APR-246 treatment was started 5 days after injection of TNBC cells. These postulates remain to be tested.
Our finding that 2aG4 alone also reduced lung metastasis indicates that the antibody most likely prevented the growth of tumor-specific blood vessels that facilitate expansion of metastatic colonies following microscopic infiltration. However, we cannot rule out important recent observations that 2aG4 may also cause a heightened immune response and subsequent infiltration of immune cells to the site of metastasis in in vivo breast cancer metastasis models.43,44 This response may also contribute to reduced formation of metastatic lung colonies or prevent the cell migration and invasion necessary for the metastatic process, two possibilities that will be the subject of future studies. For example, the influence of 2aG4 on the infiltration of immune cells will be examined in syngeneic tumor models.
Conclusion
In conclusion, our study provides a strong rationale for using a therapy regimen that combines APR-246 and 2aG4 to treat and prevent metastatic breast cancer in lungs originating from mtp53-expressing TNBC cells. The former targets and activates mtp53, likely inducing apoptosis of metastatic cells, while the latter disrupts tumor blood vessels and may also activate an immune response in tissues. Our in vivo animal studies showed no signs of toxicity with any of the regimens tested, leading us to propose that such therapy could prove beneficial and safe for women with TNBC, who currently have few effective treatment options. Furthermore, because the p53 mutation is common in other types of cancer and angiogenesis is essential for the spread of all types of tumors,45,46 we contend that our findings support further study of APR-246 and 2aG4 for treating human TNBC, as well as other forms of cancer.47,48
Abbreviations
ALDH, aldehyde dehydrogenase isoform 1; ANOVA, analysis of variance; FBS, fetal bovine serum; ip, intraperitoneal; iv, intravenous; mtp53, mutant p53 tumor suppressor protein; PBS, phosphate-buffered saline; SEM, standard error of the mean; TNBC, triple-negative breast cancer; wtp53, wild-type p53 tumor suppressor protein.
Acknowledgments
Financial support was provided by a peer-reviewed faculty research grant from the College of Veterinary Medicine, University of Missouri and in part by APREA AB. APREA AB provided APR-246 without charge that was used in the in vivo experiments, they however, did not influence the experiments conducted or the reporting of results.
Disclosure
SMH is the Zalk Missouri Professor of Tumor Angiogenesis. RAB reports grants from Oncologie, during the conduct of the study and grants from Peregrine Pharmaceuticals, outside the submitted work. In addition, RAB has multiple patents issued related to PS targeting licensed to UT System. The authors report no other conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi:10.3322/caac.21442
2. Valentin MD, Da Silva SD, Privat M, Alaoui-Jamali M, Bignon YJ. Molecular insights on basal-like breast cancer. Breast Cancer Res Treat. 2012;134:21–30. doi:10.1007/s10549-011-1934-z
3. Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov. 2019;9:176–198. doi:10.1158/2159-8290.CD-18-1177
4. Engebraaten O, Vollan HK, Børresen-Dale AL. Triple-negative breast cancer and the need for new therapeutic targets. Am J Pathol. 2013;183:1064–1074. doi:10.1016/j.ajpath.2013.05.033
5. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi:10.1038/nature11412
6. Walerych D, Napoli M, Collavin L, Del Sal G. The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis. 2012;33:2007–2017. doi:10.1093/carcin/bgs232
7. Dhakal HP, Naume B, Synnestvedt M, et al. Expression of vascular endothelial growth factor and vascular endothelial growth factor receptors 1 and 2 in invasive breast carcinoma: prognostic significance and relationship with markers for aggressiveness. Histopathology. 2012;61:350–364. doi:10.1111/j.1365-2559.2012.04223.x
8. Berns EM, Klijn JG, Look MP, et al. Combined vascular endothelial growth factor and TP53 status predicts poor response to tamoxifen therapy in estrogen receptor-positive advanced breast cancer. Clin Cancer Res. 2003;9:1253–1258.
9. Nishizaki M, Fujiwara T, Tanida T, et al. Recombinant adenovirus expressing wild-type p53 is antiangiogenic: a proposed mechanism for bystander effect. Clin Cancer Res. 1995;5:1015–1023.
10. André F, Job B, Dessen P, et al. Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res. 2009;15:441–451. doi:10.1158/1078-0432.CCR-09-0547
11. Turner N, Moretti E, Siclari O, et al. Targeting triple negative breast cancer: is p53 the answer? Cancer Treat Rev. 2013;39:541–550. doi:10.1016/j.ctrv.2012.12.001
12. Mohammed RA, Ellis IO, Mahmmod AM, et al. Lymphatic and blood vessels in basal and triple-negative breast cancers: characteristics and prognostic significance. Mod Pathol. 2011;24:774–785. doi:10.1038/modpathol.2011.4
13. Bassett EA, Wang W, Rastinejad F, El-Deiry WS. Structural and functional basis for therapeutic modulation of p53 signaling. Clin Cancer Res. 2008;14:6376–6386. doi:10.1158/1078-0432.CCR-08-1526
14. Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer. 2011;2:466–474. doi:10.1177/1947601911408889
15. Rahko E, Blanco G, Soini Y, Bloigu R, Jukkola A. A mutant TP53 gene status is associated with a poor prognosis and anthracycline- resistance in breast cancer patients. Eur J Cancer. 2003;39:447–453. doi:10.1016/s0959-8049(02)00499-9
16. Spike BT, Wahl GM. p53, stem cells, and reprogramming: tumor suppression beyond guarding the genome. Genes Cancer. 2011;2:404–419. doi:10.1177/1947601911410224
17. Godar S, Ince TA, Bell GW, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi:10.1016/j.cell.2008.06.006
18. Chang CJ, Chao CH, Xia W, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–323. doi:10.1038/ncb2173
19. Bykov VJ, Wiman KG. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014;588:2622–2627. doi:10.1016/j.febslet.2014.04.017
20. Kaar JL, Basse N, Joerger AC, Stephens E, Rutherford TJ, Fersht AR. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010;19:2267–2278. doi:10.1002/pro.507
21. Lambert JM, Gorzov P, Veprintsev DB, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376–388. doi:10.1016/j.ccr.2009.03.003
22. Zandi R, Selivanova G, Christensen CL, Gerds TA, Willumsen BM, Poulsen HS. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin Cancer Res. 2011;17:2830–2841. doi:10.1158/1078-0432.CCR-10-3168
23. Synnott NC, Murray A, McGowan PM, et al. Mutant p53: a novel target for the treatment of patients with triple-negative breast cancer? Int J Cancer. 2017;140:234–246. doi:10.1002/ijc.30425
24. Lehmann S, Bykov VJ, Ali D, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol. 2012;30:3633–3639. doi:10.1200/JCO.2011.40.7783
25. Deneberg S, Cherif H, Lazarevic V, et al. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016;6:e447. doi:10.1038/bcj.2016.60
26. Ran S, He J, Huang X, Soares M, Scothorn D, Thorpe PE. Antitumor effects of a monoclonal antibody that binds anionic phospholipids on the surface of tumor blood vessels in mice. Clin Cancer Res. 2005;15:1551–1562. doi:10.1158/1078-0432.CCR-04-1645
27. He J, Luster TA, Thorpe PE. Radiation-enhanced vascular targeting of human lung cancers in mice with a monoclonal antibody that binds anionic phospholipids. Clin Cancer Res. 2007;13:5211–5218. doi:10.1158/1078-0432.CCR-07-0793
28. He J, Yin Y, Luster TA, Watkins L, Thorpe PE. Antiphosphatidylserine antibody combined with irradiation damages tumor blood vessels and induces tumor immunity in a rat model of glioblastoma. Clin Cancer Res. 2009;15:6871–6880. doi:10.1158/1078-0432.CCR-09-1499
29. Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugs. 2007;21:299–310. doi:10.2165/00063030-200721050-00002
30. Liu P, Kumar IS, Brown S, et al. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br J Cancer. 2013;109:1876–1885. doi:10.1038/bjc.2013.534
31. Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi:10.1038/nature03799
32. Luster TA, He J, Huang X, et al. Plasma protein beta-2-glycoprotein 1 mediates interaction between the anti- tumor monoclonal antibody 3G4 and anionic phospholipids on endothelial cells. J Biol Chem. 2006;281:29863–29871. doi:10.1074/jbc.M605252200
33. Huang X, Bennett M, Thorpe PE. A monoclonal antibody that binds anionic phospholipids on tumor blood vessels enhances the antitumor effect of docetaxel on human breast tumor in mice. Cancer Res. 2005;65:4408–4416. doi:10.1158/0008-5472.CAN-05-0031
34. Zhao X, Rezonzew G, Wang D, Siegal GP, Hardy RW. Diet modulation is an effective complementary agent in preventing and treating breast cancer lung metastasis. Clin Exp Metastasis. 2014;31:625–638.
35. Cook MT, Liang Y, Besch-Williford C, Hyder SM. Luteolin inhibits lung metastasis, cell migration, and viability of triple-negative breast cancer cells. Breast Cancer. 2016;9:9–19.
36. Marcato P, Dean CA, Pan D, et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 2011;29:32–45.
37. Cioce M, Gherardi S, Viglietto G, et al. Mammosphere-forming cells from breast cancer cell lines as a tool for the identification of CSC-like- and early progenitor- targeting drugs. Cell Cycle. 2010;9:2878–2887.
38. Zheng T, Lu M, Wang T, Zhang C, Du X. NRBE3 promotes metastasis of breast cancer by down-regulating E-cadherin expression. Biochim Biophys Acta Mol Cell Res. 2018;1865:1869–1877. doi:10.1016/j.bbamcr.2018.09.003
39. Xiang L, Semenza GL. Hypoxia-inducible factors promote breast cancer stem cell specification and maintenance in response to hypoxia or cytotoxic chemotherapy. Adv Cancer Res. 2019;141:175–212. doi:10.1016/bs.acr.2018.11.001
40. Tsang JY, Huang YH, Luo MH, et al. Cancer stem cell markers are associated with adverse biomarker profiles and molecular subtypes of breast cancer. Breast Cancer Res Treat. 2012;136:407–417. doi:10.1007/s10549-012-2271-6
41. Liang Y, Mafuvadze B, Besch-Williford C, Hyder SM. A combination of p53-activating APR-246 and phosphatidylserine-targeting antibody potently inhibits tumor development in hormone-dependent mutant p53-expressing breast cancer xenografts. Breast Cancer. 2018;10:53–67.
42. Liang Y, Besch-Williford C, Hyder SM. PRIMA-1 inhibits growth of breast cancer cells by re-activating mutant p53 protein. Int J Oncol. 2009;35:1015–1023. doi:10.3892/ijo_00000416
43. Yin Y, Huang X, Lynn KD, Thorpe PE. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res. 2013;1:256–268. doi:10.1158/2326-6066.CIR-13-0073
44. Cheng X, Li L, Thorpe PE, Yopp AC, Brekken RA, Huang X. Antibody-mediated blockade of phosphatidylserine enhances the antitumor effect of sorafenib in hepatocellular carcinomas xenografts. Ann Surg Oncol. 2016;23(Suppl 5):583–591. doi:10.1245/s10434-016-5107-5
45. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31.
46. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi:10.1038/nrc1093
47. Burrows FJ, Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004;10:415–427.
48. Denekamp J. Angiogenesis, neovascular proliferation and vascular pathophysiology as targets for cancer therapy. Br J Radiol. 1993;66:181–196. doi:10.1259/0007-1285-66-783-181
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.