Back to Journals » Lung Cancer: Targets and Therapy » Volume 10
Approaches to Tumor Classification in Pulmonary Sarcomatoid Carcinoma
Authors Baldovini C, Rossi G ![]() , Ciarrocchi A
, Ciarrocchi A ![]()
Received 27 July 2019
Accepted for publication 26 November 2019
Published 5 December 2019 Volume 2019:10 Pages 131—149
DOI https://doi.org/10.2147/LCTT.S186779
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ou
Chiara Baldovini,1 Giulio Rossi,1 Alessia Ciarrocchi2
1Operative Unit of Pathologic Anatomy, Azienda USL della Romagna, Hospital S. Maria delle Croci, Ravenna, Italy; 2Laboratory of Translational Research, Azienda Unità Sanitaria Locale - IRCCS Reggio Emilia, Reggio Emilia 42123, Italy
Correspondence: Giulio Rossi
Operative Unit of Pathologic Anatomy, Azienda USL della Romagna, Hospital S. Maria delle Croci, Viale Randi 5, Ravenna 48121, Italy
Tel +39 0544 285368
Fax +39 0544 285758
Email [email protected]
Abstract: Pulmonary sarcomatoid carcinoma (PSC) is a heterogeneous category of primary lung cancer accounting from 0.3% to 3% of all primary lung malignancies. According to the most recent 2015 World Health Organization (WHO) classification, PSC includes several different variants of malignant epithelial tumors (carcinomas) histologically mimicking sarcomas showing or entirely lacking a conventional component of non-small cell lung cancer (NSCLC). Thus, this rare subheading of lung neoplasms includes pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, pulmonary blastoma, and carcinosarcoma. A diagnosis of PSC may be suspected on small biopsy or cytology, but commonly requires a surgical resection to reach a conclusive definition. The majority of patients with PSC consists of elderly, smoking men with a large, peripheral mass characterized by well-defined margins. However, presentation with a central, polypoid endobronchial lesion is well-documented, particularly in pleomorphic carcinoma and carcinosarcoma showing a squamous cell carcinoma component. As expected, PSC may pose diagnostic problems and immunohistochemistry is largely used when pathologists deal these tumors in routine practice. Indeed, PSC tends to overexpress molecules associated with the epithelial-to-mesenchymal transition, such as vimentin, but the panel of immunostains also includes epithelial markers (cytokeratins, EMA), TTF-1, p40 and negative markers (e.g., melanocytic, mesothelial and sarcoma-related primary antibodies). Although rare, PSC has increased their interest among oncologist community for different reasons: a. identification of the epithelial-to-mesenchymal phenomenon as a major mechanism of secondary resistance to tyrosine kinase inhibitors; b. over-expression of PD-L1 and effective treatment with immunotherapy; c. identification of c-MET exon 14 skipping mutation representing an effective target to crizotinib and other specific inhibitors. In this review, the feasibility of the diagnosis of PSC, its differential diagnosis and novel molecular findings characterizing this group of lung tumor are discussed.
Keywords: sarcomatoid, carcinoma, c-MET, PD-L1, cytokeratins, immunohistochemistry, WHO
Basic Concepts of Pulmonary Sarcomatoid Carcinoma
The occurrence of pulmonary carcinomas showing a sarcomatoid or sarcomatous cell component is a well-wknown phenomenon indicating a divergent tumor cell dedifferentiation from epithelial to mesenchymal phenotype in conventional non-small-cell lung cancer (NSCLC).1 The term pleomorphic (spindle/giant cell) carcinoma was first introduced in 1994 by Fishback et al2 to recognize and better categorize a specific subset of high-grade carcinomas, including unusual variants previously defined inflammatory-type sarcomatoid carcinoma and pseudoangiosarcomatous carcinoma (so-called pseudovascular adenoid squamous cell carcinoma).3–10
The seminal paper by Fishback et al,2 subsequently supported by other similar series,11–23 provided the basis of the 2004 World Health Organization (WHO) classification of lung cancers under the designation of “sarcomatoid carcinoma”, then including five main variants, namely pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma and pulmonary blastoma.24
According to this statement, overall sarcomatoid carcinoma is a group of poorly differentiated NSCLC (squamous cell carcinoma, large-cell carcinoma and/or adenocarcinoma) containing a component of sarcoma-like elements or true sarcomatous areas. Of note, the occurrence of small-cell carcinoma (SCLC) or large-cell neuroendocrine carcinoma (LCNEC) with sarcoma or sarcoma-like component is considered a combined variant into the category of neuroendocrine neoplasms.25,26
Pleomorphic carcinoma is the most common type of PSC (>50%), followed by spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and pulmonary blastoma.2,5,13–16,24
Several review articles have been performed on this topic and there is general agreement on the pathogenesis of this intriguing tumor entity. Indeed, previous works have consistently evidenced the clonal origin of PSC demonstrating identical mutations of KRAS and p53 in either carcinoma and sarcoma/sarcomatous component,27–35 as also disclosed in sarcomatoid carcinoma arising in other sites.36–38
Basically, the appearance of an additional sarcomatoid/sarcomatous component in an otherwise conventional NSCLC is due to up-regulation of the epithelial-to-mesenchymal transition (EMT) secondary to activation of genetic mechanisms generally associated with resistance to chemotherapy and tyrosine kinase inhibitors, such as KRAS mutations, c-MET gene alterations, overexpression of vimentin, ZEB1, Snail, MiR-34 coupled to down-regulation of E-cadherin and expression of epithelial markers, miR-200, mutations of EGFR (Figure 1).39
 |
Figure 1 A concise scheme illustrating the balance of epithelial-to-mesenchymal transition markers in sarcomatoid carcinoma sustained by up- and down-regulation of different molecules and gene alterations governing the tumor cell cellular program. |
Epidemiology, Imaging Studies, Clinical Features
Several series of PSC have been published in literature using the current criteria of the WHO classification.2,11–23 Overall, the incidence of PSC ranges from 0.3% to 3% of all lung malignancies with a significant higher prevalence in male gender, current/former smokers. The median age at diagnosis is about 65 years, although pulmonary blastoma usually affects younger smokers without gender predilection.
Pelosi et al7 in a series of 92 PSC reported a male/female ratio of about 4:1, with 90% of smoking patients. As in conventional NSCLC, PSC may occur also in patients under the age of 40 years and non-smokers,40 while even asbestos exposure seems to have a promoting role in PSC.41
At imaging studies, PSC may present as either a central or peripheral location.2,5,24,42 Pleomorphic carcinomas with squamous cell carcinoma component and carcinosarcoma may show a peculiar endobronchial, polypoid appearance (Figure 2), while peripheral PSC typically manifests as a large mass (over 10 cm) with rounded, well-defined margins (Figures 3 and 4) with necrotic and/or hemorrhagic areas and soft surface with/without cavitation (Figure 5).
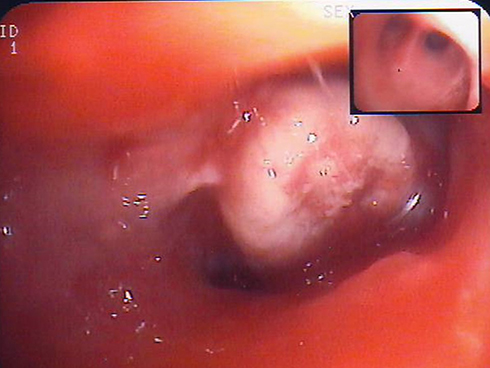 |
Figure 2 Bronchoscopy showing a whitish polypoid lesion consistent with carcinosarcoma at histology. |
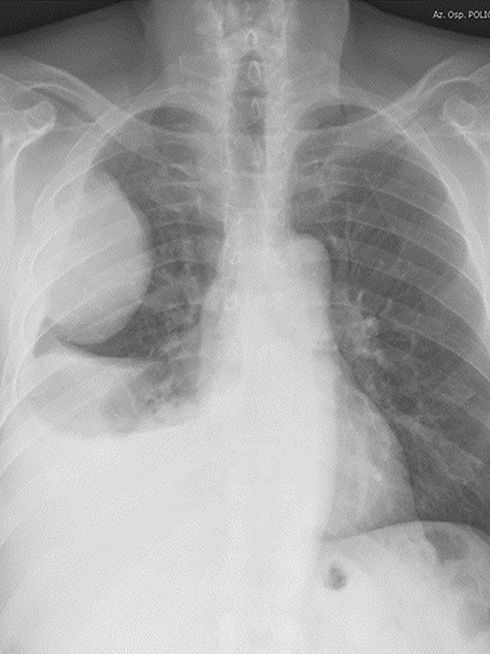 |
Figure 3 Sarcomatoid carcinoma of the right lung presenting as a large, rounded mass at chest X-rays. |
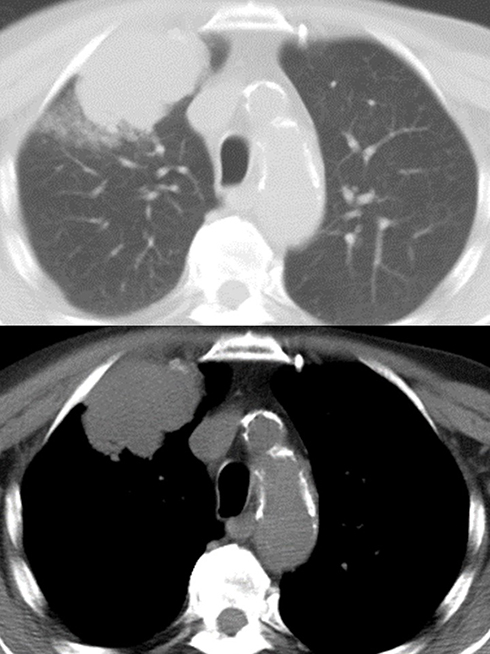 |
Figure 4 Sarcomatoid carcinoma appearing as a large nodular lesion of the right upper lobe with relatively well-defined margins at computed tomography of the thorax. |
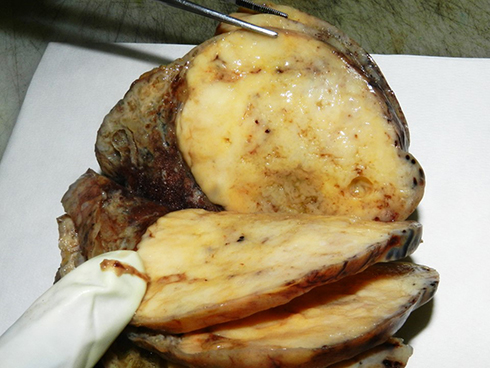 |
Figure 5 Macroscopic appearance of a pulmonary carcinosarcoma consisting of a large (>10 cm), whitish and fleshy mass with well-demarcated tumor border and heterogenous cut surface. |
Clinical symptoms are generally non-specific and basically related to the tumor location and involved structures, such as main bronchi, mediastinal infiltration, and chest wall.5,7
Of note, giant cell carcinoma is often characterized by peripheral neutrophilia and fever due to production of granulocyte colony-stimulating factor (G-CSF) or other paraneoplastic symptoms.43,44
A summary of the most characteristic features of patients with PSC is reported in Table 1. As conventional NSCLC, PSC may metastasize through lymphatic channels and blood vessels leading to usual distant metastases (i.e., brain, bone, adrenal gland, liver), but unusual sites (i.e., pancreas, and kidney) and involvement of the digestive tract are not infrequent.5,7,45,46
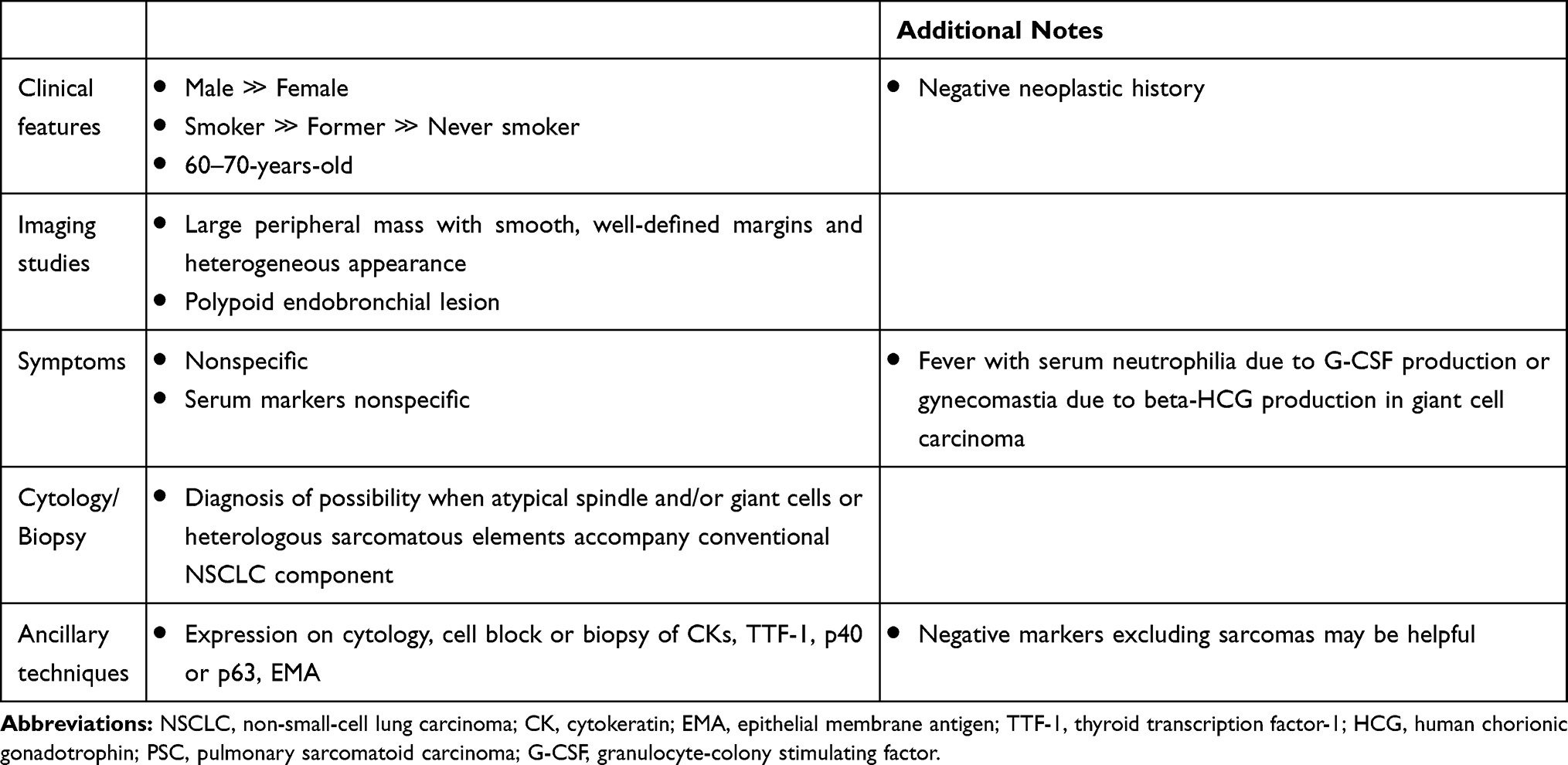 |
Table 1 Characteristics Possibly Indicating a Patient with Pulmonary Sarcomatoid Carcinoma |
The overall survival of stage-matched, surgically resected PSC is generally worse than that observed in conventional NSCLC. This finding has been further confirmed by some large series of PSC.15,16
The median survival is about 10 months and the estimated 5-year survival rate 15%.15,16 In the recent study of the Mayo Clinic, multivariate analysis showed that T and M stage, and surgery plus neoadjuvant or adjuvant therapy were significant prognostic parameters.15,16 At matched analysis, PSC confirmed a worse survival when compared with adenocarcinoma and squamous cell carcinoma.
PSCs are staged as for other NSCLC, according to the most recent edition of the AJCC/UICC/IASLC staging system.
Diagnosis of Pulmonary Sarcomatoid Carcinoma According to the WHO Classification
The classification of sarcomatoid carcinomas of the lung has posed significant challenges over the years, mainly based on their rare occurrence, heterogeneous histology, and unclear histogenesis. This led to multiple changes along their subsequent WHO classifications, both in terms of diagnostic criteria and nomenclature. Moreover, alternative descriptive classifications have been formulated, which encompass the following: monophasic and biphasic lesions, the latter further subdivided into homologous or heterologous neoplasms.4,47
The 2004 WHO classification established the unification of a spectrum of histological subtypes under the generic term of sarcomatoid carcinoma, which includes pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and blastoma.48 Since then, no significant changes have been made as the current 2015 WHO classification basically adopts the same terminology and diagnostic criteria with few recommendations, mainly regarding molecular tests for predictive gene alterations.24,49 Importantly, most of the entities encompassed under the definition of PSC can be recognized from their morphological features, with immunohistochemical stains being necessary only in a subset of cases (Table 2).
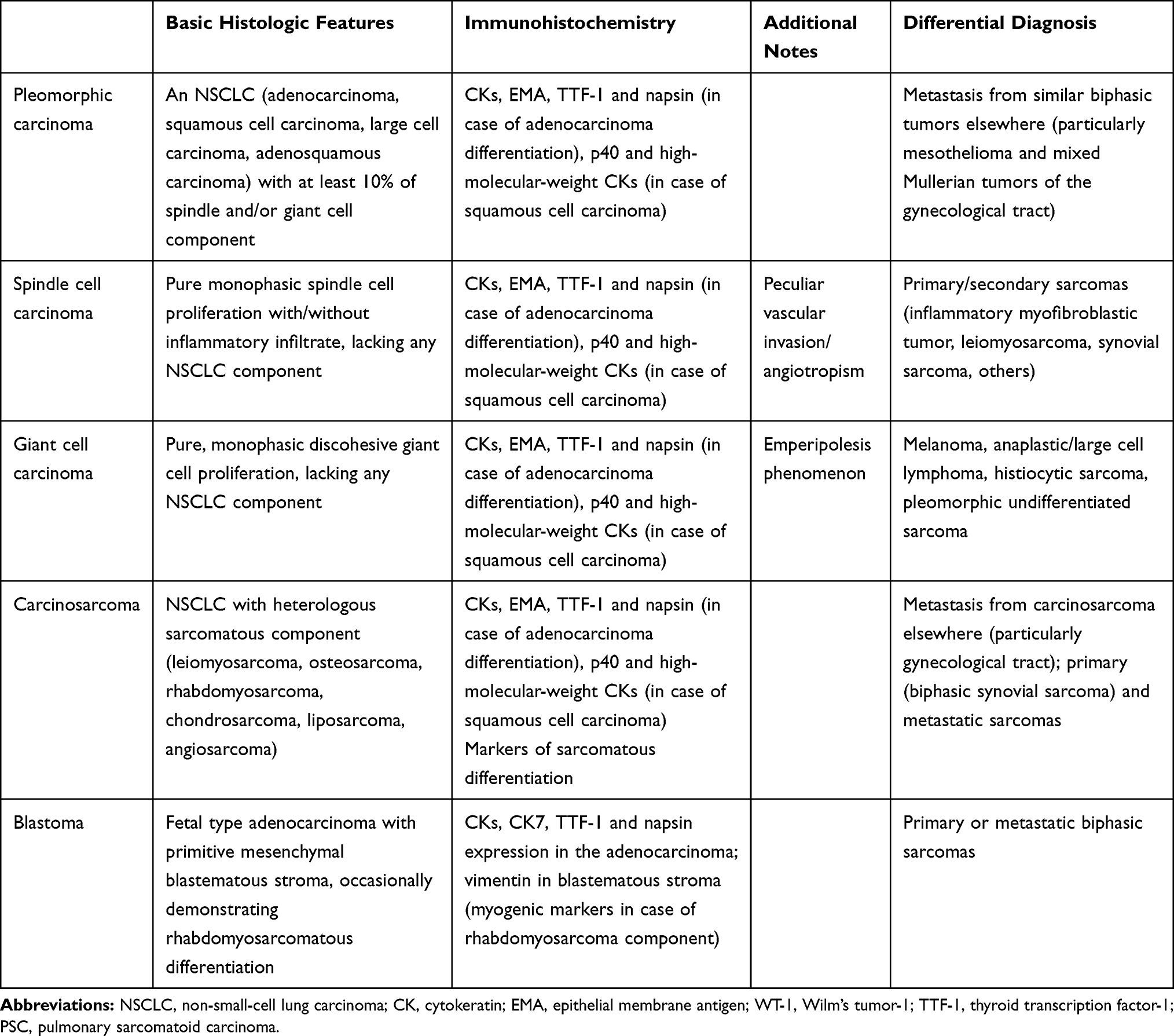 |
Table 2 Diagnostic Criteria of Pulmonary Sarcomatoid Carcinomas |
More in details, in the current WHO classification, pleomorphic carcinoma is defined as a poorly differentiated NSCLC (squamous cell carcinoma, adenocarcinoma, or large cell carcinoma) containing at least 10% of spindle cell and/or giant cell component, or a carcinoma consisting exclusively of spindle and giant cells (Figure 6). The occurrence of different histotypes of carcinoma varies widely across studies, with adenocarcinoma representing the most frequent finding, up to 73% of the cases.2,50,51 Spindle cells and/or neoplastic giant cells can be found intimately admixed with differentiated carcinomatous elements. Once identified, the NSCLC component should be mentioned in the report. Malignant spindle cells generally consist of fusiform elements with eosinophilic cytoplasm, arranged in sheets and fascicles. However, a certain degree of morphological variability exists: slender cells with fibroblastic appearance can be seen admixed with frankly epithelioid elements, displaying large nuclei and prominent nucleoli (Figure 7). Vascular invasion is very common in spindle cell carcinoma even in the bland-looking inflammatory type. A meticulous examination of the vessel wall, also using a pan-cytokeratin stain, is crucial to diagnose spindle cell carcinoma (Figure 8). Giant tumour cells are often discohesive, with multiple nuclei or a single pleomorphic nucleus, which can assume anaplastic and bizarre features; their cytoplasm is typically abundant and eosinophilic, sometimes characterized by neutrophil emperipolesis (Figure 9). These cells are embedded in a variably fibrous to myxoid stroma.
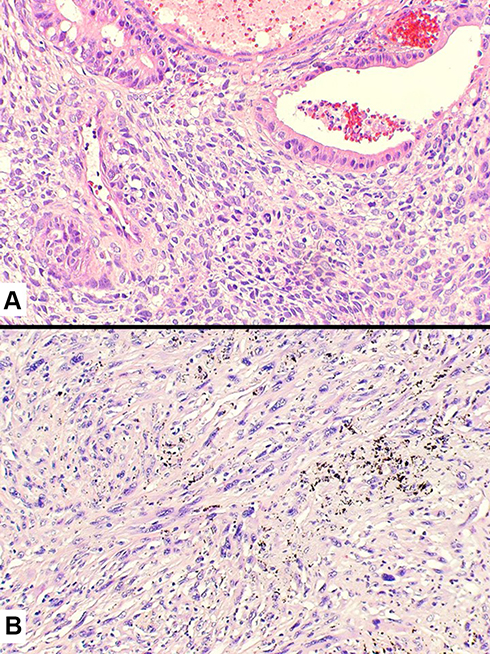 |
Figure 6 Histology of pleomorphic carcinoma revealing an adenocarcinoma (at the top) intermingled with neoplastic spindle cell component (A). Pure spindle cell carcinoma (B) consisting of a dense and irregular proliferation of atypical spindle cells with marked nuclear atypia and eosinophilic cytoplasm. |
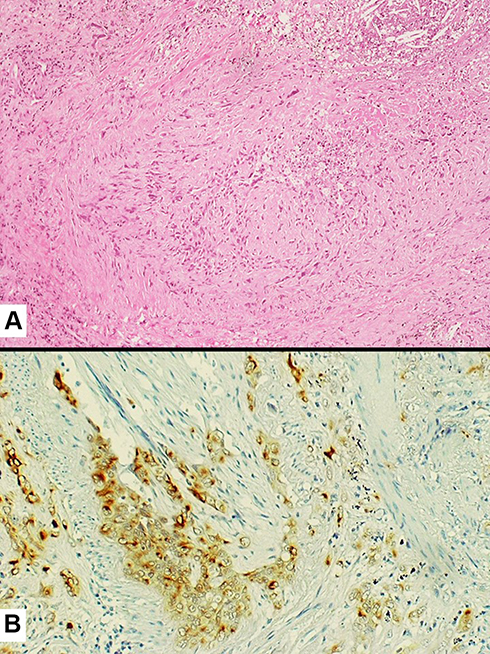 |
Figure 7 Tumor cell infiltration of a medium-sized vessel wall by spindle cell carcinoma (A) also highlighted by the expression of pan-cytokeratins (clone CAM5.2) (B). |
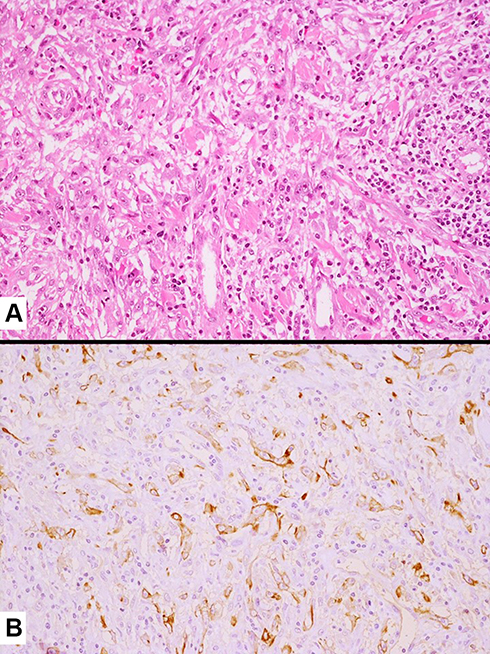 |
Figure 8 Inflammatory-type spindle cell carcinoma showing a chaotic proliferation of spindle cells with prominent nucleoli into collagenized stroma with a mixed inflammatory infiltrate (A). Positive staining with pan-cytokeratins (clone AE1/AE3) revealing the epithelial/carcinoma differentiation of spindle elements (B). |
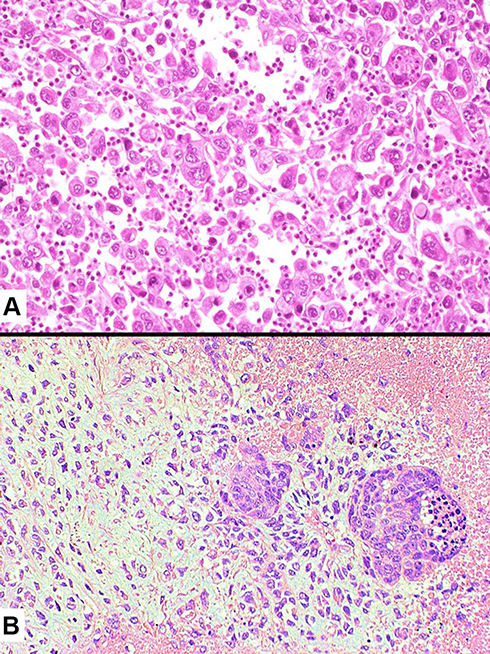 |
Figure 9 Histology of giant cell carcinoma showing a discohesive proliferation of bizarre, large tumor cells and the presence of sparse inflammatory infiltrates focally involving the cytoplasm of tumor cells (emperipolesis) (A). Carcinosarcoma consisting of a poorly differentiated squamous cell carcinoma (right) with a heterologous chondrosarcomatous component (left) (B). |
Spindle cell carcinoma and giant cell carcinoma are almost entirely composed of spindle-shaped and malignant giant cells, respectively, without differentiated carcinomatous elements. Histological characteristics are superimposable to those previously described. Caution should be used to distinguish between spindle cell carcinoma and desmoplastic stroma. In this regard, cytological atypia and mitotic activity favour the diagnosis of carcinoma. However, immunohistochemistry may be of help in difficult cases. In addition, an inflammatory background is frequently found in association with these variants.
Carcinosarcoma is a mixture of NSCLC and true sarcomatous elements (rhabdomyosarcoma, chondrosarcoma, osteosarcoma, liposarcoma, leiomyosarcoma, angiosarcoma or combinations of these elements) (Figure 8). Again, pathological report should list all histological types. In a classical study, the most frequent epithelial component was squamous cell carcinoma, followed by adenocarcinoma, adenosquamous carcinoma, and large-cell carcinoma.17 Nonetheless, a component of high-grade in well-differentiated fetal adenocarcinoma/clear cell carcinoma represents a non-negligible finding; in such cases, the term carcinosarcoma should be used instead of the traditional expression “blastomatoid variant of carcinosarcoma”, according to the WHO scheme.24,49 Combinations of neuroendocrine components and sarcoma-containing heterologous elements are exceedingly rare, and the WHO recommends to classify these cases as “small-cell lung carcinoma or large-cell neuroendocrine carcinoma with associated sarcomatous elements”. Along with the most common sarcomatous elements, cases displaying liposarcoma or angiosarcoma components have been described, although rarely.52,53 Less differentiated sarcomatous components with spindle or round cell features can also be observed in the background of more differentiated sarcomatous elements.
Finally, pulmonary blastoma is defined as a biphasic tumour that contains both fetal adenocarcinoma (low-grade/well differentiated) and a primitive mesenchymal stroma, which occasionally has foci of differentiated sarcoma (Figure 10). The epithelial component is characterized by irregularly branching glandular structures, lined by pseudostratified columnar cells with clear cytoplasm and little nuclear atypia, thus resembling the fetal lung in the pseudoglandular phase. Despite the generally bland appearance, foci of cellular atypia consistent with fetal or conventional adenocarcinoma can be found in up to a third of cases.54 In most cases, solid cell aggregates (morules) can be noted. The occurrence of a neuroendocrine epithelial component such as small-cell carcinoma is overtly rare in pulmonary blastoma, whereas sparse neuroendocrine cells represent a common finding. An embryonic stroma composed of oval cells with high nuclear-to- cytoplasmic ratio is present by definition, but in a subset of cases (up to 25%) heterologous elements such as osteosarcoma, chondrosarcoma and rhabdomyosarcoma can be identified.
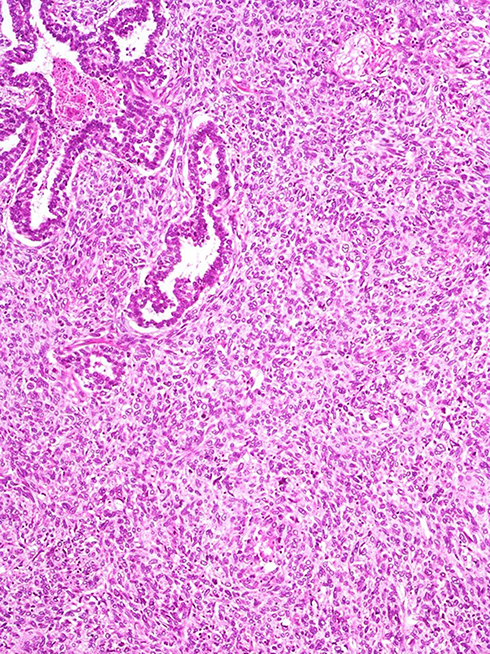 |
Figure 10 Pulmonary blastoma showing a minor component of well-differentiated adenocarcinoma (top) admixed with an undifferentiated blastematous component. |
Immunohistochemistry
The diagnosis of PSC is mainly based on morphological features, although immunohistochemical stains can be helpful to highlight the different cell components that characterize these tumours (Table 3). This is particularly useful to demonstrate carcinomatous differentiation in small biopsies or in those cases lacking an unambiguous epithelial component despite thorough sampling, thus prompting differential diagnosis with true sarcoma. As expected, the differentiated epithelial component stains with cytokeratins and other markers including epithelial membrane antigen (EMA) and carcinoembryonic antigen (CEA), whereas the sarcomatoid component often displays immunoreactivity with vimentin and fascin.7 In most cases, a single pancytokeratin antibody is sufficient to confirm the carcinomatous differentiation of spindle cell/giant cell components, while the use of AE1AE3 alone or in combination with Oscar is highly recommended compared with other pools that proved less sensitive.55 Of note, different cytokeratin filaments can be detected in PSC according to the subtypes of epithelial cells: high-molecular-weight cytokeratins (i.e. 34βE12 and 5/6) are found in squamous and adenosquamous carcinoma and low-weight cytokeratins (i.e. 7, 8 and 18) in adenocarcinoma and large-cell carcinoma.7,56 In particular, CK7 represents a sensible marker to highlight the spindle/giant cell component and was identified in up to 78% of cases in a recent study,57 in keeping with previously reported rates of 60-70%.14
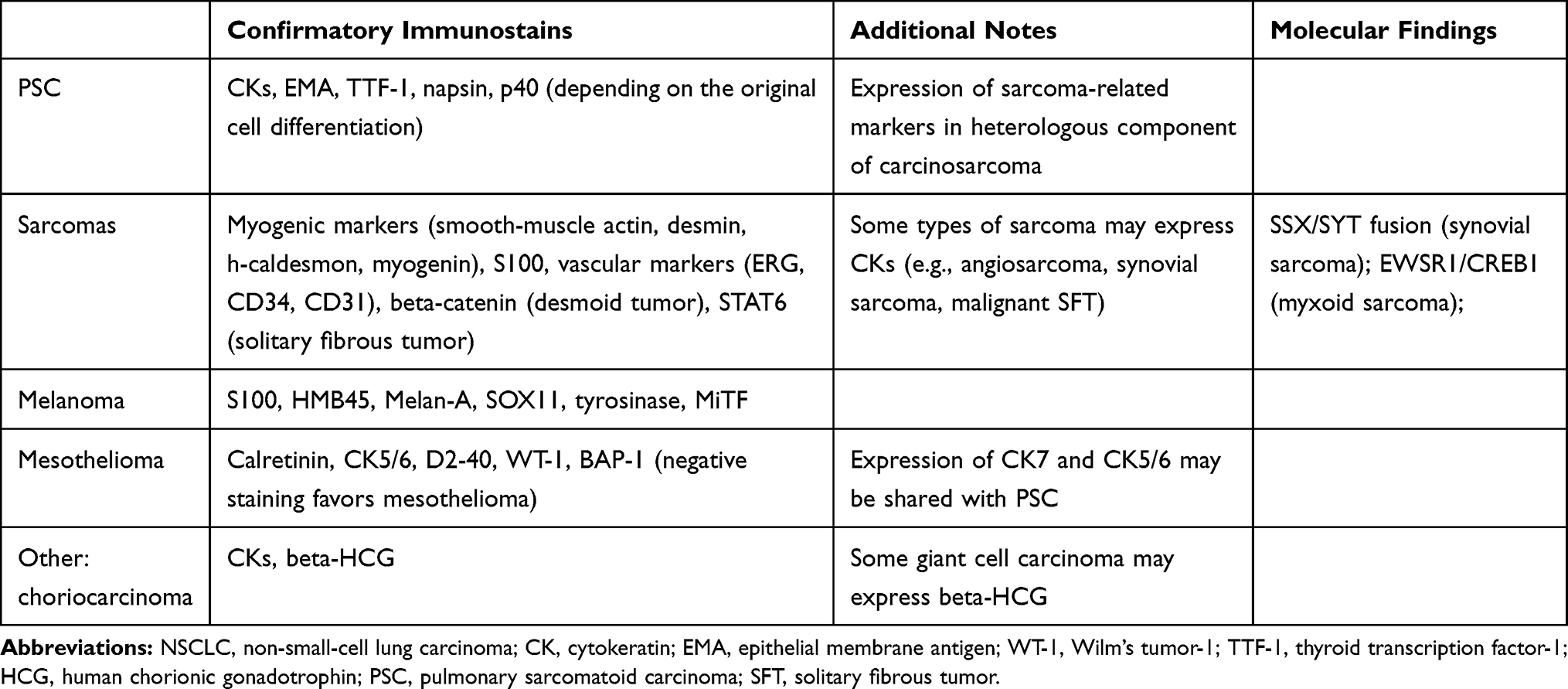 |
Table 3 Ancillary Techniques in Differential Diagnosis Between Pulmonary Sarcomatoid Carcinoma and Mimicking Tumors |
Although cytokeratins have significant diagnostic value in sarcomatoid carcinomas, staining is often focal and weak, and some cases can even be completely negative with a number of antibodies. In these cases, additional markers can be helpful, with thyroid transcription factor-1 (TTF-1) demonstrating higher sensitivity than Napsin A for the detection of sarcomatoid carcinoma.55,57 Markers associated with squamous differentiation, including p63, p40, and Desmocollin-3, have been repeatedly tested in sarcomatoid carcinomas of the lung. Among these, p63 showed the highest sensitivity (50%) and diagnostic utility, being positive in the spindle cell component of 2/4 cases, where staining for pan-cytokeratins and EMA was completely negative.58 On the other hand, p40 and Desmocollin-3 were expressed in the spindle/giant cell elements in 8% and 3% of cases, respectively.57 Nevertheless, a word of caution on p63 expression is necessary, since this marker is characterized by low specificity (i.e., positivity in high-grade lymphomas).59 A modified histological score for Vimentin (M-VHS), an intermediate filament involved in the EMT, has also proved a valuable tool for diagnosing sarcoma-like lesions with spindle and/or giant cells changes, especially in small biopsies. Indeed, an intense and diffuse expression of vimentin strongly supports the diagnosis of pulmonary sarcoma.60 Finally, the observation of patchy reactivity with mesothelioma-associated markers (Calretinin and D2-40) in a subset of PSC suggests to include sarcomatoid mesothelioma in the differential diagnosis.61
In carcinosarcoma, immunohistochemistry is required to demonstrate a poorly differentiated sarcomatous component which cannot be recognized from morphology. Most of these cases harbour a rhabdomyosarcomatous component which can be highlighted with skeletal muscle markers such as myogenin and desmin. In the same way, S100 will confirm a chondrosarcomatous component. On the other hand, cytokeratins allow to demonstrate the epithelial component, with TTF-1 and p63 being particularly useful in cases of adenocarcinoma and squamous cell carcinoma, respectively.14,17
In pulmonary blastoma, the epithelial component stains with cytokeratins, EMA, CEA, TTF-1. In particular, TTF-1 and GATA-6, a fetal lung transcription factor, proved useful to highlight the morules.62 The mesenchymal component can express smooth muscle actin and vimentin, whereas desmin and myogenin are found in cases showing rhabdomyosarcoma elements; pancytokeratins (AE1AE3) can also show focal positivity.63 The occurrence of neuroendocrine differentiation, with consistently positive chromogranin, neurone-specific enolase (NSE) and polypeptide hormones, is seen mainly within morules in up to two-third of cases.54 Finally, nuclear and cytoplasmic staining of β-catenin in both tubular structures and blastematous component of pulmonary blastoma prompts differential diagnosis with the blastomatoid variant of carcinosarcoma.64
Diagnosis of Sarcomatoid Carcinoma in Selected Conditions: Cytology/Small Biopsy and Intraoperative Examination
Some entities in lung cancer require a large amount of tumor tissue to achieve a correct and reliable diagnosis, including PSC. By definition, definitive diagnosis is not possible on small biopsy or cytology in light of the requirement for a sarcomatoid/sarcomatous component in at least 10% of the neoplasm.24,49 Nevertheless, a diagnosis of “NSCLC with sarcomatoid/sarcomatous component, possible sarcomatoid carcinoma” is reasonable.7
Indeed, in an adequate sample, smeared cytology, cell-block or small biopsy may contain either malignant epithelial components (adenocarcinoma or squamous cell carcinoma) and sarcomatoid elements, namely spindled and/or giant neoplastic cells. Akin to neoplastic cells, the background may also contain a significant rate of inflammatory cells (mainly neutrophils and lymphocytes).64–66 The spindle and giant cells usually show loss of intercellular cohesion, mitotic figures and necrotic debris. In small biopsy/cell block, the finding of epithelial component thorough serial haematoxylin–eosin sections and/or immunostains with pan-cytokeratins (low to high-molecular-weight, such as AE1/AE3, MNF116, CAM5.2), EMA, p40, TTF-1, CEA is extremely helpful (Figure 11).
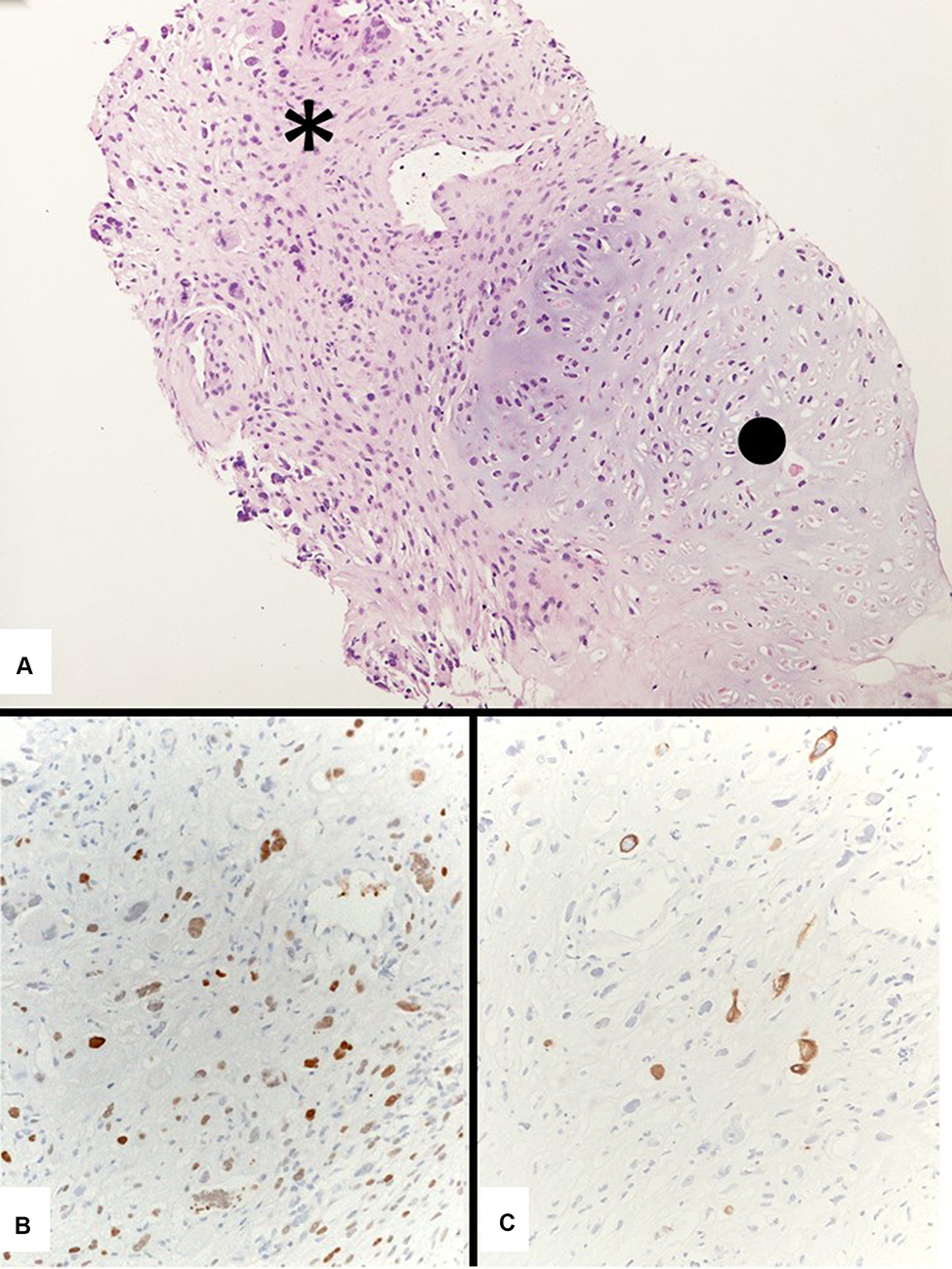 |
Figure 11 A generous transthoracic biopsy consistent with a carcinosarcoma showing an undifferentiated spindle cell carcinoma (asterisk) and a heterologous chondrosarcoma (dot) (A). Immunostains with pan-cytokeratins (clone MNF116) (B) and TTF-1 (clone 8G7G3/1) (C) demonstrate the epithelial adenocarcinomatous differentiation of the spindle cell component. |
A prompt diagnosis of PSC during frozen section examination may be a real challenge, even in the hands of expert pathologists. Knowledge of the clinical history of previous neoplasms and careful search for an NSCLC component using serial sections and/or analysis of more samples are mandatory. In any case, a provisional diagnosis of NSCLC is entirely adequate for practical purposes to guide the surgical procedure.7
At intraoperative examination or definitive gross sampling, abundant tumor sampling is crucial in the diagnosis of PSC, particularly when facing with a huge mass (at least 1 block per centimeter of the entire tumor size is necessary).
The differential diagnosis may include several tumors with various cell differentiations and non-neoplastic conditions. Primary or metastatic monophasic and biphasic sarcomas (i.e., synovial sarcoma, malignant solitary fibrous tumor, inflammatory myofibroblastic tumor), sarcomatoid mesothelioma, spindle cell melanoma, sarcomatoid thymic carcinoma, choriocarcinoma are the main neoplastic lesions entering in differential with PSC (Table 2).5,7,65–75
Even benign spindle cell lesions (i.e., desmoid tumor, inflammatory pseudotumor, organizing pneumonia) may mimic malignant spindle cell carcinoma.76 This is particularly true at frozen section or in small biopsy when extensive immunostains and molecular analysis (i.e., SYT-SSX fusion gene indicating synovial sarcoma) are not accessible or have several limitations, respectively. Nevertheless, pathologists should keep all these diagnostic options in differential diagnosis with PSC.
Among sarcomatoid/sarcomatous lesions in the thoracic region, PSC and sarcomatoid mesothelioma may overlap clinical presentation (i.e., localized malignant mesothelioma) and morphology. Pathologists should keep in mind that immunostains indicating mesothelial and epithelial phenotype may be aberrantly expressed either in PSC and mesothelioma.77 A careful evaluation of clinicopathologic and imaging information with histology, immunohistochemistry and molecular biology has been advised to render the correct final diagnosis. Of note, BAP-1 negativity at immunohistochemistry may be very helpful in ruling out PSC and favouring mesothelioma.78
Recent Insights into PSC Genetic and Transcriptional Features
PSCs are a highly peculiar and biologically fascinating forms of NSCLC,2,5,14 characterized by highly aggressive clinical behaviour with dismal outcome and variable response to chemo-radiotherapy.79,80 Owing to their rarity, the genetic events and molecular pathways that account for PSC development and progression have been eluded for a long time and recently, began to emerge.
The first clues about the genetic landscape of PSC became available only in the most recent years.81–86 The raise and rapid expansion of the new sequencing techniques, together with the collection of relatively large series of PSC samples available for the analysis have represented major turning points in getting clues into the genetic assets of these tumors.
In 2015 Fallet and colleagues described the first multi-targeted genetic analysis on a large cohort of PSCs.81 Using the LungCarta Panel on the Sequenom platform these Authors investigated the mutational assets of 26 oncogenes in 114 surgically resected PSCs. They reported mutations in TP53 (22.6%) KRAS (27.2%), EGFR (22.2%) and STK1 (7.4%) as the most common genetic alterations in these lesions. Additional but less frequent alterations were found in NOTCH1, NRAS, and PI3KCA. In 2016, Schrock et al used a deep sequencing approach to map whole-exome mutations in a cohort of 125 PSCs.82 While confirming that mutations in KRAS (34.4%) and TP53 (73.6%) are the most common mutations in PSCs, the results of these study refined the frequency of mutations and called into questions other relevant oncogenes that were not present in the Sequenom panel. In particular, these authors showed that PSCs are characterized by a quite high tumor mutational burden (TMB, average of 8.1 mutations/Mb) and that both TP53 mutated tumors and KRAS mutated tumors were characterized by a higher TMB than WT samples.
TP53 mutations were found in a consistent part of PSCs analyzed in this study and were reported to be widely heterogeneous as subsequentially confirmed by other reports.83 Furthermore, the occurrence of TP53 mutations in PSCs seemed to underpin a specific genetic subtype. For instance, mutation in NF1 and RB1 were reported to preferentially occur in TP53 mutated vs wild-type samples while NF2 mutations were more frequent in TP53 wild-type samples, thus hypothesizing distinct evolution pathways for PSCs with different genetic assets. Besides TP53 and KRAS mutations, additional frequent alterations were found, including those in CDKN2B (23.2%), CDKN2A (37.6%), MET (13.6%) and NF1 (17.6%). Noticeably, mutations in EGFR (8.8%) were detected at a lower frequency than previously reported and BRAF alterations (7.3%) were limited to a reduced number of PSC samples. Even if providing new important clues into the genetic event of PSCs, these data did not highlight genetic specificity that separates PSCs from NSCLCs. In this regard, the work by Liu et al83 has marked a major point. By means of whole-exome sequencing, the Authors identified mutations in the exon 14 of MET as preferentially associated with PSC.
MET exon 14 skipping mutations already described in other tumors including NSCLC affect either donor or acceptor splice site between exon 13 and exon 14, causing a 47 amino acid deletion in the juxta-membrane domain of the MET proteins. This region is crucial for the correct homeostasis of MET signaling being the docking site for the c-CBL ubiquitin E3 ligase (Y1003).87,88 Loss of c-CBL binding either by point mutations in the accepting site or by exon 14 skipping, leads to MET stabilization and constitutive activation of MET-dependent signaling.88 As in NSCLC, in PSC MET exon 14 mutations were mutually exclusive with other oncogene mutations like KRAS, BRAF, EGFR or ALK rearrangement, but co-occurrence with PI3K mutations has been reported.82,83 Furthermore, at least in the original report, MET exon 14 mutations were exclusively found in pure PSCs or PSCs with an ADK component.83,89 Noticeably, while MET exon 14 mutations occur in about 3% of NSCLCs (according to TCGA data), up to 32% of PSCs are reported to carry this alteration. Indeed, according to a recent systematic meta-analysis,90 the incidence of MET exon 14 mutations in PSC is variable. Overall, its frequency in NSCLC is 2% in adenocarcinoma, 1% in squamous cell carcinoma, 6% in adenosquamous carcinoma and 13% in PSC. Despite the different characteristics of analyzed case series, MET exon 14 mutations seem to occur particularly in PSC, ranging from 3% to 31.8%.81–83,87-93 The particular attention focused on MET exon 14 skipping mutations is then related to the relatively high frequency of this genetic alteration in PSC histology when compared with other conventional NSCLC, also predicting a good clinical response to MET inhibitors. The rate of MET exon 14 mutations is significantly higher in case series including PSC without common targetable mutations in EGFR, KRAS, ALK, ROS1, and RET.92,93 Nevertheless, whenever possible all histological types of NSCLC should be investigated for this mutation.
Activation of MET signaling is frequently associated and functionally supports the Epithelial to Mesenchymal Transition (EMT),90,91 which is believed to be a driver mechanism in the progression of PSC from well-differentiated lesions.
The frequent coexistence of both well-differentiated and sarcomatoid components within the same lesions led to the common assumption that PSCs originate from the progressive de-differentiation of well-differentiated NSCLC into sarcomatous like entities.
This hypothesis has found supporting evidence in recently released studies in which genetic and molecular profiling were used to investigate similarities and differences between the different components of PSCs. A recent study used exome sequencing to compare the mutational landscape of well-differentiated and sarcomatoid components in a small series of four PSCs.92 Even if limited by the number of cases analyzed, the authors concluded that during evolution, the sarcomatoid component branches very early from the well-differentiated epithelioid component and independently accumulates mutations establishing a profound difference with the original lesions.
We took further these observations showing in vivo that the transcriptional activation of an EMT program drives PSC phylogeny.93 Using Nanostring technology, we performed a deep transcriptional analysis of a series of 32 biphasic PSCs (17 training sets, 15 validation sets). We identified a list of 146 genes whose expression largely marks the phenotypic differences between epithelioid and sarcomatoid components of PSC. Gene Ontology (GO) enrichment analysis demonstrated loss of cell-adhesion and cell-junction properties as well as other epithelial specific features in the sarcomatous components and identified TGF-beta and many TGF-beta-induced transcription factors as upstream regulators of this phenotype. In accordance with previous reports, ECAD, a typical marker of epithelial cancers, emerged as differentially expressed gene and was consistently lost in sarcomatoid cells. Noticeably, from our analysis strongly emerged that the molecular reorganization that sustains the acquisition of the sarcomatoid phenotype relies on a profound rewiring of the gene expression program. This is driven by a functional switch between epithelial-associated TFs and mesenchymal-associated TFs, thus providing new clues into the molecular events that underline PSCs evolution but also the first formal proves that the activation of the EMT program drives this process.
Clinical Impact of PSC Molecular Profiling
Given the discouraging results with conventional chemotherapy, new therapies are needed to improve the management of PSCs. The recent identification of MET exon 14 mutations shed new lights on the unique biology of PSCs and opened the first prospective on PSC-oriented therapy. Here, we discuss on how the recently gained molecular insights into their biology may overturn PSCs therapy in the next years.
While providing confirmation of the close genetic proximity between PSC and NSCLC, these studies also indicate how these lesions harbor precise differences that at least in part may account for the failure of previously attempted targeted therapy in PSC.94 For instance, EGFR actionable mutations are quite frequent in NSCLC and treatment with TKI is a first-line therapy for EGFR mutated patients. Few reports of PSC patients treated with EGFR inhibitors are currently available but thus far a lack of significant and stable response has been observed.50,95 EGFR mutations in PSC is controversial (0–28% depending on series) but surely inferior to NSCLC.81–84,96–98 Furthermore, according to the available information, the number of actionable EGFR mutations (p.L858R) is even lower further reducing the rationale for the employment of these drugs in this setting. Besides, the absence of evident response to EGFR inhibitors even in EGFR mutated PSCs, seems to indicate that EGFR mutations in these tumors may not be an essential driver but rather a secondary event.
KRAS mutations are among the most common alterations detected in non-squamous NSCLC. Together with alterations in TP53, KRAS mutations are also quite frequent in PSCs with a reported occurrence that range between 15% and 39% depending on series.81–85 Mutations in 12 codon are the most frequent even if mutation in codon 61 has also been reported. KRAS mutations have been found significantly associated with pure sarcomatoid or with PSC with an adenocarcinoma component.82,84 KRAS mutations are predominantly found in PSCs from smokers, and the majority of the alteration detected are transversions, the typical DNA alterations found in smokers’ adenocarcinomas.84 Prognostic significance of KRAS mutations in PSC has been a matter of debate, in particular due to the limited number of cases in the analyzed series that precluded conclusive results. However, we recently showed that KRAS mutations alone or in combination with TP53 mutations were associated with local metastases at recurrence and with a significantly decreased survival probability in a cohort of surgically resected PSCs.84 Similarly, Mehrad and colleagues reported a significant correlation of KRAS mutations with worse patients’ outcome.86 We also showed that the presence of KRAS mutations significantly correlates with increased PD-L1 expression suggesting a possible correlation, to be further investigated, between these mutations and response to immunotherapy.99 These results together with the recent emerging MEK inhibitors may imply a potential value of KRAS mutations as relevant predictive markers in orienting PSC tailored treatment. Last but not least, the encouraging preclinical success of KRAS-specific inhibitors further consolidate the relevance of these mutations in the managing of these tumors.100,101
MET exon 14 skipping mutations cause stabilization of MET protein leading to the constitutive activation of its signaling.87 NSCLCs with activating alterations of MET (including MET locus amplification) have shown remarkable response to small molecules like crizotinib, cabozantinib or capmatinib that target MET activity.102 For the same reason, the presence of MET activating mutations in PSCs would qualify a good portion of patients (about 30%) based on the reported mutations incidence82,83,87,103 for treatment with these drugs. Preclinical evidence shows that tumor cells harboring MET ex14 mutations are responsive to MET inhibition, even if co-occurrence of PI3KCA concomitant gain of function alterations may partially reduce effectiveness of MET inhibition.83 Preliminary clinical data on small series or single case confirmed that in vivo, PSC harboring MET exon 14 mutations show effective response to MET targeting drugs.83,104–107
Even if revolutionary in the desolated PSC treatments landscape, the discovery of MET exon 14 mutations and the consequential possibility of directing patients to MET inhibitors is limited to a reduced number of patients. Besides, MET inhibitors as well as other selective targeting drugs are limited by innate or adaptive resistance keeping the need of new PSC-oriented therapies. The identification of effective therapies for rare cancer like PSCs poses many challenges and does not fit within the standard design of clinical trials. Thus, alternative strategies like the one of drug re-purposing offer obvious appeal (reduced costs and time) and represent a sustainable strategy for rare cancers. Coherently with this reasoning, we used gene expression profiling data based on human samples as the basis for searching the cMap database108 containing information of over one million FDA-approved drugs and identified ready-to-use compounds potentially matching the ability of counteracting the PSC-associated gene program. Dasatinib was found as the top scoring drug in this analysis. In vitro studies confirmed the effectiveness of this drug in PSC-like cancer cells and its preferential activity for PSC vs well-differentiated NSCLC model.96 Dasatinib is a broad-spectrum TKI designed to target among the others pro-oncogenic kinases like SRC, DDR1 and DDR2. It has obtained positive results in NSCLC trials and has been registered for clinical use in this setting.109 Furthermore, consistent evidence indicates that Dasatinib suppresses TGF-beta-mediated EMT in both lung normal and cancer cells preventing insurgence of pulmonary fibrosis and EMT-mediated drug resistance.110
Targetable mutations in PSC are less frequent than in NSCLC with adenocarcinoma histology. However, Fallet et al81 reported uncommon/rare EGFR mutations in 22%, NRAS and PI3KCA in 5%. Li et al85 performed an NGS study evidencing hot spot druggable gene alterations involving EGFR (exon 19 deletion) and EML4-ALK fusion in sporadic cases. Mehrad et al86 evidenced actionable genetic mutations in EGFR in 2 out of 23 PSCs (8.7%) and Schrock et al82 observed targetable genomic alteration in EGFR (8.8%), BRAF (7.2%), erb-b2 receptor tyrosine kinase 2 gene (HER2) (1.6%), and ret proto-oncogene (RET) (0.8%). Of note, no rearrangements in ALK, ROS1 and NTRKs were found in the last series.
Finally, Alì et al111 disclosed sporadic mutations in EGFR (1 case), MET (2 cases) and BRAF (1 case), KRAS (2 cases), PI3KCA (2 cases) and 1 ALK-rearranged case among 14 PSC using a combined approach with Sequenom Mass-Array and Sanger sequencing and NanoString technology.
By contrast, Nakagomi et al95 evidenced prevalent mutations in TP53 and KRAS coupled to Microsatellite instability/Mismatch repair system alteration in one case among four PSCs deeply investigated by NGS.
Several reasons may explain the wide range of incidence of druggable gene alterations in PSC, as follows: a) a selection bias related to the accuracy in diagnosing this unusual histology (i.e., application of morphologic criteria in biopsy versus resections, lack of specific diagnostic primary antibodies at immunohistochemistry); b) various methodologies in detecting gene alterations, including techniques with a significantly large range of sensitivity; and c) different ethnicities of the reported case series.
Overall, targetable gene alterations are less frequent in PSC than in non-squamous NSCLC, although MET and BRAF oncogene mutations are relatively more common in this unusual histology.82,94
Interestingly, among acquired resistance mechanisms during treatments with tyrosine kinase inhibitors (i.e., EGFR and ALK inhibitors) or chemotherapy, histologic “change” from adenocarcinoma to small cell or squamous cell carcinomas have been well demonstrated in EGFR mutated (about 10% of cases) and ALK rearranged adenocarcinomas. However, activation of epithelial-to-mesenchymal (EMT) transition leading to sarcomatoid “transformation” has been recently reported by Hsieh et al112 in 6 cases of adenocarcinoma (5 EGFR mutated, 1 ROS1 rearranged). The change from adenocarcinoma histology to PSC was often associated with PD-L1 over-expression (83%), c-MET gene alterations or MET overexpression (5 out of 6 cases), further supporting the activation of EMT phenomenon in tumor cells.
This histologic change has been previously observed in EGFR-mutated cell lines of lung adenocarcinoma by Sequist et al,113 demonstrating that acquired resistance to EGFR TKI was sustained by EMT activation with sarcomatoid spindle cell modification at histology and increased expression of vimentin. EMT as mechanism of resistance to TKI is observed in 1% to 10% of cases112–114 and sarcomatoid change in the experience of Hsieh et al is observed in 5% of EGFR-mutated NSCLC.112
Xu et al115 reported a case of lung adenocarcinoma resistant to EGFR TKI with concurrent acquired T790M secondary EGFR mutation and sarcomatoid spindle cell occurrence.
Finally, a case of exon 21 L858R EGFR-mutated sarcomatoid carcinoma of the lung resistant to icotinib was recently reported.116 Coexistence of Nkx2-4 mutation was considered responsible of its intrinsic resistance to EGFR TKI.116
PSC and Immunotherapy
Immunotherapy by blockade of immune checkpoint pathways like the PD1/PD-L1 signaling has dramatically impacted on therapy of many chemo-resistant cancer patients.117,118 Rational exists for the use of immune checkpoints inhibitors also in PSCs, relying on at least two different assumptions. First, immunotherapy has shown remarkable results in NCSLC improving progression-free and overall survival, causing fewer adverse events than standard chemotherapy in a significant portion of patients.119 Second, recent evidence indicates that the strength of immunotherapy is higher in tumors that like PSC or, more in general tobacco-associated tumors, are characterized by high degree of genetic damage.120 Up to now only sporadic reports of single cases on the use of immunotherapy in PSCs are available, but these preliminary data are encouraging and seem to corroborate the employment of immune checkpoint blockade in this setting.121
Currently, no definitive predictive marker to anticipate response to anti-PD1 or anti-PD-L1 antibody is available. PD-L1 expression in either tumor or lymphocyte infiltrate positive122 and tumor mutational burden120 have been reported to be positively associated with the strength of immunotherapy response and investigated as potential predictive markers.
In PSCs, several reports have investigated the expression of PD1 and/or PD-L1 to envisage a possible use of these therapies in this setting.101,123–133 Although widely employed and approved from different national drug agencies,123 PD-L1 expression is an imperfect predictive biomarker for immunotherapy in lung cancer. PSC tends to show high levels of immunohistochemical expression with PD-L1, even if it is quite difficult to compare the data so far reported in literature in this histology. Indeed, the main problem is related to technical clues related to numerous cut-offs of expression to quote positivity, different primary antibody clones and automated platforms as well as laboratory-developed tests used in scientific works.
A recent paper by Alì et al111 showed PD-L1 expression ≥1% in 84% out of 44 PSC and 47% of these cases revealed staining ≥50% (clone SP263 using BenchMark XT/Ventana platform). Similarly, Nakagomi et al95 demonstrated high PD-L1 expression ≥50% in all four PSCs analyzed (clone 28–8 using ULTRA/Ventana platform). Wu et al124 detected PD-L1 expression in 21 cases (87.5%) of PSC and SUVmax of 17 lesions with PD-L1 expression degree ≥50% was higher than that <50% and related to KRAS mutations. Chan et al125 assessed the PD-L1 expression in 713 consecutive NSCLC by four commercially available PD-L1 immunohistochemical assays (22C3, 28–8, SP142 and SP263) evidencing a high PD-L1 expression ≥50% significantly associated with PSC (p < 0.001) and mutant KRAS (p = 0.005) also reporting good agreement between 22C2, 28–8 and SP263 primary antibodies. Sim et al126 showed high levels of PD-L1 in 8 out of 13 PSC (61.5%) and Yvorel et al127 analogously reported high expression of PD-L1 (clone E1L3N, Cell Signaling Technology) in 75% of 36 PSC. In the series of Kim et al129 PD-L1 and PD-L2 were highly expressed in PSC (90.2%; 37/41 and 87.8%; 36/41), respectively. Of note, PD-L1 expression in pulmonary PCs was significantly higher in sarcomatous areas than in the carcinomatous component (p=0.006). Vieira et al130 reported high PD-L1 (clone 5H1) immunoreactivity in 40 out of 75 (53%) PSC, also confirming a significant association with the presence of KRAS mutations. High expression of PD-L1 in PSC has been similarly noted in other studies.101,131
The frequency of PD-L1 positive PSC samples is significantly higher than the one observed in NSCLC, coherently with higher presence of CD3+ or CD163+ infiltrating lymphocytes or macrophages observed in these tumors.130 Together, these observations encourage the use of immunotherapy in the treatment of PSCs.131,133
Take-Home Messages
- Patients with PSC are generally elderly smoking men with large peripheral mass with rounded margins or endobronchial fleshy polyp. Symptoms are non-specific, as well as serum markers, but giant cell carcinoma may produce G-CSF or beta-HCG leading to paraneoplastic conditions (i.e., fever and gynecomastia, respectively).
- The diagnosis of PSC is based on careful examination of morphologic features on hematoxylin–eosin-stained slides using a light microscope, but ancillary techniques (immunohistochemistry, molecular morphology or even ultrastructure analysis) are very helpful in challenging cases. Extensive sampling and serial consecutive sections may be required to disclose the NSCLC component, especially on small biopsy.
- A diagnosis of possible PSC is acceptable on cytology, cell-block and small biopsy, but definitive confirmation should be made on surgical specimen, according to the last 2015 WHO classification of lung tumors.
- In agreement with the 2015 WHO scheme, PSC encompasses five different histological subtypes, namely pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma and blastoma.24
- Sarcomatoid/sarcomatous component in PSC has a monoclonal origin driving from metaplastic changes of the carcinoma, secondary to the activation of a genetic program of epithelial-to-mesenchymal transition.39
- Prognosis of PSC is dismal; surgery remains the mainstay of treatment and the role of neoadjuvant/adjuvant chemo-radiotherapy is controversial and rarely effective.125
- PD-L1 over-expression and MET exon 14 skipping mutation are the most important predictive biomarkers for effective targeted therapies in PSC.134–136
Disclosure
The authors declared no conflicts of interest with respect to the authorship and/or publication of this article.
References
1. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi:10.1038/nrc822
2. Fishback NF, Travis WD, Moran CA, Guinee DG
3. Humphrey PA, Scroggs MW, Roggli VL, Shelburne JD. Pulmonary carcinomas with a sarcomatoid element: an immunocytochemical and ultrastructural analysis. Hum Pathol. 1988;19:155–165. doi:10.1016/S0046-8177(88)80343-5
4. Nappi O, Glasner SD, Swanson PE, Wick MR. Biphasic and monophasic sarcomatoid carcinomas of the lung. A reappraisal of “carcinosarcomas” and “spindle-cell carcinomas”. Am J Clin Pathol. 1994;102:331–340. doi:10.1093/ajcp/102.3.331
5. Colby T, Koss M, Travis W. Tumors of the Lower Respiratory Tract. Washington, DC: Armed Forces Institute of Pathology; 1995.
6. Wick MR, Ritter JH, Humphrey PA. Sarcomatoid carcinomas of the lung: a clinicopathological review. Am J Clin Pathol. 1997;108:40–53. doi:10.1093/ajcp/108.1.40
7. Pelosi G, Sonzogni A, De Pas T, et al. Pulmonary Sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18(2):103–120. doi:10.1177/1066896908330049
8. Wick MR, Ritter JH, Nappi O. Inflammatory sarcomatoid carcinoma of the lung: report of three cases and clinicopathologic comparison with inflammatory pseudotumors in adult patients. Hum Pathol. 1995;26:1014–1021. doi:10.1016/0046-8177(95)90092-6
9. Antic T, Kapur U, Vigneswaran WT, Oshima K. Inflammatory sarcomatoid carcinoma: a case report and discussion of a malignant tumor with benign appearance. Arch Pathol Lab Med. 2005;129:1334–1337. doi:10.1043/1543-2165(2005)129[1334:ISCACR]2.0.CO;2
10. Nappi O, Swanson PE, Wick MR. Pseudovascular adenoid squamous cell carcinoma of the lung: clinicopathologic study of three cases and comparison with true pleuropulmonary angiosarcoma. Hum Pathol. 1994;25:373–378. doi:10.1016/0046-8177(94)90145-7
11. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84:973–980. doi:10.1016/j.athoracsur.2007.03.099
12. Venissac N, Pop D, Lassalle S, Berthier F, Hofman P, Mouroux J. Sarcomatoid lung cancer (spindle/giant cells): an aggressive disease? J Thorac Cardiovasc Surg. 2007;134:619–623. doi:10.1016/j.jtcvs.2007.05.031
13. Pelosi G, Fraggetta F, Nappi O, et al. Pleomorphic carcinomas of the lung show a selective distribution of gene products involved in cell differentiation, cell cycle control, tumor growth and tumor cell motility: a clinicopathological and immunohistochemical study of 31 cases. Am J Surg Pathol. 2003;9:1203–1215. doi:10.1097/00000478-200309000-00003
14. Rossi G, Cavazza A, Sturm N, et al. Pulmonary carcinomas with pleomorphic, sarcomatoid, or sarcomatous elements: a clinicopathologic and immunohistochemical study of 75 cases. Am J Surg Pathol. 2003;27:311–324. doi:10.1097/00000478-200303000-00004
15. Maneenil K, Xue Z, Liu M, et al. Sarcomatoid carcinoma of the lung: the Mayo clinic experience in 127 patients. Clin Lung Cancer. 2017;19:e323–e333. doi:10.1016/j.cllc.2017.12.008
16. Ung M, Roquette I, Filleron T, et al. Characteristics and clinical outcomes of sarcomatoid carcinoma of the lung. Clin Lung Cancer. 2016;17:391–397. doi:10.1016/j.cllc.2016.03.001
17. Koss MN, Hochholzer L, Frommelt RA. Carcinosarcomas of the lung: a clinicopathologic study of 66 patients. Am J Surg Pathol. 1999;23:1514–1526. doi:10.1097/00000478-199912000-00009
18. Ro JY, Chen JL, Lee JS, Sahin AA, Ordonez N, Ayala AG. Sarcomatoid carcinoma of the lung. Immunohistochemical and ultrastructural studies of 14 cases. Cancer. 1992;69:376–386.
19. Nakajima M, Kasai T, Hashimoto H, Iwata Y, Manabe H. Sarcomatoid carcinoma of the lung: a clinicopathologic study of 37 cases. Cancer. 1999;86:608–616. doi:10.1002/(ISSN)1097-0142
20. Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34:91–97. doi:10.1016/S0169-5002(01)00224-0
21. Raveglia F, Mezzetti M, Panigalli T, et al. Personal experience in surgical management of pulmonary pleomorphic carcinoma. Ann Thorac Surg. 2004;78:1742–1747. doi:10.1016/j.athoracsur.2004.04.084
22. Davis MP, Eagan RR, Weiland LH, Pairolero PC. Carcinosarcoma of the lung: Mayo clinic experience and response to chemotherapy. Mayo Clin Proc. 1984;59:598–603. doi:10.1016/S0025-6196(12)62410-0
23. Mochizuki T, Ishii G, Nagai K, et al. Pleomorphic carcinoma of the lung: clinicopathologic characteristics of 70 cases. Am J Surg Pathol. 2008;32:1727–1735. doi:10.1097/PAS.0b013e3181804302
24. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10:1243–1260. doi:10.1097/JTO.0000000000000630
25. Tsubota YT, Kawaguchi T, Hoso T, Nishino E, Travis WD A combined small cell and spindle cell carcinoma of the lung. Report of a unique case with immunohistochemical and ultrastructural studies. Am J Surg Pathol. 1992;16:1108–1115. doi:10.1097/00000478-199211000-00010
26. Khalifa M, Hruby G, Ehrlich L, Danjoux C, Perez-Ordonez B. Combined large cell neuroendocrine carcinoma and spindle cell carcinoma of the lung. Ann Diagn Pathol. 2001;5:240–245. doi:10.1053/adpa.2001.27256
27. Berho M, Moran CA, Suster S. Malignant mixed epithelial/mesenchymal neoplasms of the lung. Semin Diagn Pathol. 1995;12:123–139.
28. Guarino M, Tricomi P, Giordano F, Cristofori E. Sarcomatoid carcinomas: pathological and histopathogenetic considerations. Pathology. 1996;28:298–305. doi:10.1080/00313029600169224
29. Holst VA, Finkelstein S, Colby TV, Myers JL, Yousem SA. p53 an K-ras mutational genotyping in pulmonary carcinosarcoma, spindle cell carcinoma, and pulmonary blastoma: implications for histogenesis. Am J Surg Pathol. 1997;21:801–8110. doi:10.1097/00000478-199707000-00008
30. Pelosi G, Scarpa A, Manzotti M, et al. K-ras gene mutational analysis supports a monoclonal origin of biphasic pleomorphic carcinoma of the lung. Mod Pathol. 2004;17:538–546. doi:10.1038/modpathol.3800058
31. Dacic S, Finkelstein SD, Sasatomi E, Swalsky PA, Yousem SA. Molecular pathogenesis of pulmonary carcinosarcoma as determined by microdissection-based allelotyping. Am J Surg Pathol. 2002;26:510–516. doi:10.1097/00000478-200204000-00015
32. Wick MR, Swanson PE. Carcinosarcomas: current perspectives and an historical review of nosological concepts. Semin Diagn Pathol. 1993;10:118–127.
33. Ansari-Lari MA, Hoque MO, Califano J, Westra WH. Immunohistochemical p53 expression patterns in sarcomatoid carcinomas of the upper respiratory tract. Am J Surg Pathol. 2002;26:1024–1031. doi:10.1097/00000478-200208000-00007
34. Bodner SM, Koss MN. Mutations in the p53 gene in pulmonary blastomas: immunohistochemical and molecular studies. Hum Pathol. 1996;27:1117–1123. doi:10.1016/S0046-8177(96)90302-0
35. Przygodzki RM, Koss MN, Moran CA, et al. Pleomorphic (giant and spindle cell) carcinoma is genetically distinct from adenocarcinoma and squamous cell carcinoma by K-ras-2 and p53 analysis. Am J Clin Pathol. 1996;106:487–492. doi:10.1093/ajcp/106.4.487
36. Thompson LD, Chang B, Barsky SH. Monoclonal origins of malignant mixed tumors (carcinosarcomas). Am J Surg Pathol. 1996;20:277–285. doi:10.1097/00000478-199603000-00003
37. Sreenan JJ, Hart WR. Carcinosarcomas of the female genital tract. A pathological study of 29 metastatic tumors: further evidence for the dominant role of the epithelial component and the conversion theory of histogenesis. Am J Surg Pathol. 1995;19:666–674. doi:10.1097/00000478-199506000-00007
38. Gotzmann J, Mikula M, Eger A, et al. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res. 2004;566:9–20. doi:10.1016/S1383-5742(03)00033-4
39. Pang A, Carbini M, Moreira AL, Maki RG. Carcinosarcomas and related cancers: tumors caught in the act of epithelial-Mesenchymal transition. J Clin Oncol. 2018;36(2):210–216. doi:10.1200/JCO.2017.74.9523
40. Aketa A, Yamada G, Aketa K, et al. Two younger male patients with rapidly progressing pulmonary pleomorphic carcinoma. Nihon Kokyuki Gakkai Zasshi. 2004;42:164–169.
41. Farrell DJ, Cooper PN, Malcolm AJ. Carcinosarcoma of lung associated with asbestosis. Histopathology. 1995;27:484–486. doi:10.1111/his.1995.27.issue-5
42. Rapicetta C, Lococo F, Stefani A, et al. Primary sarcomatoid carcinoma of the lung: radiometabolic (18)F-FDG PET/CT findings and correlation with clinic-pathological and survival results. Lung. 2016;194:653–657. doi:10.1007/s00408-016-9904-1
43. Yaturu S, Harrara E, Nopajaroonsri C, Singal R, Gill S. Gynecomastia attributable to human chorionic gonadotropin-secreting giant cell carcinoma of lung. Endocr Pract. 2003;9:233–235. doi:10.4158/EP.9.3.233
44. Nonami Y, Yamamoto M, Sasaguri S. G-CSF producing giant tumor in the lung. J Cardiovasc Surg (Torino). 2005;46(3):313–314.
45. Aokage K, Yoshida J, Ishii G, et al. Long-term survival in two cases of resected gastric metastasis of pulmonary pleomorphic carcinoma. J Thorac Oncol. 2008;3:796–799. doi:10.1097/JTO.0b013e31817c925c
46. Rossi G, Marchioni A, Romagnani E, et al. Primary lung cancer presenting with gastrointestinal tract involvement: clinicopathologic and immunohistochemical features in a series of 18 cases. J Thorac Oncol. 2018;2:115–120. doi:10.1016/S1556-0864(15)30037-X
47. Nappi O, Wick MR. Sarcomatoid neoplasms of the respiratory tract. Semin Diagn Pathol. 1993;10(2):137–147.
48. Corrin B, Chang YL, Rossi G, et al. Pathology and genetics of tumours of the lung, pleura, thymus and heart. In: Travis WD, Brambilla E, Mu¨ller-Hermelink HK, Harris CC, editors. World Health Organization Classification of Tumours. Vol. 10. Lyon, France: IARC Press; 2004;53–58.
49. Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. Lyon: International Agency for Research on Cancer; 2015.
50. Kaira K, Horie Y, Ayabe E, et al. Pulmonary pleomorphic carcinoma: a clinicopathological study including EGFR mutation analysis. J Thorac Oncol. 2010;5(4):460–465. doi:10.1097/JTO.0b013e3181ce3e3c
51. Lee S, Kim Y, Sun JM, et al. Molecular profiles of EGFR, K-ras, c-met, and FGFR in pulmonary pleomorphic carcinoma, a rare lung malignancy. J Cancer Res Clin Oncol. 2011;137(8):1203–1211. doi:10.1007/s00432-011-0986-0
52. Bull JC, Grimes OF. Pulmonary carcinosarcoma. Chest. 1974;65(1):9–12. doi:10.1378/chest.65.1.9
53. Kitazawa R, Kitazawa S, Nishimura Y, Kondo T, Obayashi C. Lung carcinosarcoma with liposarcoma element: autopsy case. Pathol Int. 2006;56(8):449–452.
54. Koss MN, Hochholzer L, O’Leary T. Pulmonary blastomas. Cancer. 1991;67:2368–2381. doi:10.1002/(ISSN)1097-0142
55. Terra SB, Aubry MC, Yi ES, Boland JM. Immunohistochemical study of 36 cases of pulmonary sarcomatoid carcinoma–sensitivity of TTF-1 is superior to napsin. Hum Pathol. 2014;45(2):294–302. doi:10.1016/j.humpath.2013.09.005
56. Yatabe Y, Dacic S, Borczuk AC, et al. Best practices recommendations for diagnostic immunohistochemistry in lung cancer. J Thorac Oncol. 2019;14(3):377–407. doi:10.1016/j.jtho.2018.12.005
57. Weissferdt A, Kalhor N, Rodriguez Canales J, Fujimoto J, Wistuba II, Moran CA. Spindle cell and pleomorphic (“sarcomatoid”) carcinomas of the lung: an immunohistochemical analysis of 86 cases. Hum Pathol. 2017;59:1–9. doi:10.1016/j.humpath.2016.08.003
58. Lewis JS, Ritter JH, El-Mofty S. Alternative epithelial markers in sarcomatoid carcinomas of the head and neck, lung, and bladder-p63, MOC-31, and TTF-1. Mod Pathol. 2005;18(11):1471–1481. doi:10.1038/modpathol.3800451
59. Bishop JA, Teruya-Feldstein J, Westra WH, Pelosi G, Travis WD, Rekhtman N. p40 (ΔNp63) is superior to p63 for the diagnosis of pulmonary squamous cell carcinoma. Mod Pathol. 2012;25(3):405–415. doi:10.1038/modpathol.2011.173
60. Pelosi G, Melotti F, Cavazza A, et al. A modified vimentin histological score helps recognize pulmonary sarcomatoid carcinoma in small biopsy samples. Anticancer Res. 2012;32(4):1463–1473.
61. Marchevsky AM, LeStang N, Hiroshima K, et al. The differential diagnosis between pleural sarcomatoid mesothelioma and spindle cell/pleomorphic (sarcomatoid) carcinomas of the lung: evidence-based guidelines from the International Mesothelioma Panel and the MESOPATH National Reference Center. Hum Pathol. 2017;67:160–168. doi:10.1016/j.humpath.2017.07.015
62. Yamazaki K. Pulmonary well-differentiated fetal adenocarcinoma expressing lineage-specific transcription factors (TTF-1 and GATA-6) to respiratory epithelial differentiation: an immunohistochemical and ultrastructural study. Virchows Arch. 2003;442(4):393–399. (). doi:10.1007/s00428-003-0798-y
63. Yousem SA, Wick MR, Randhawa P, Manivel JC. Pulmonary blastoma. An immunohistochemical analysis with comparison with fetal lung in its pseudoglandular stage. Am J Clin Pathol. 1990;93(2):167–175.
64. Nakatani Y, Miyagi Y, Takemura T, et al. Aberrant nuclear/cytoplasmic localization and gene mutation of beta-catenin in classic pulmonary blastoma: beta-catenin immunostaining is useful for distinguishing between classic pulmonary blastoma and a blastomatoid variant of carcinosarcoma. Am J Surg Pathol. 2004;28(7):921–927. doi:10.1097/00000478-200407000-00012
65. Choi HS, Seol H, Heo IY, et al. Fine-needle aspiration cytology of pleomorphic carcinomas of the lung. Korean J Pathol. 2012;46(6):576–578. doi:10.4132/KoreanJPathol.2012.46.6.576
66. Hummel P, Cangiarella JF, Cohen JM, Yang G, Waisman J, Chhieng DC. Transthoracic fine-needle aspiration biopsy of pulmonary spindle cell and mesenchymal lesions: a study of 61 cases. Cancer. 2001;93(3):187–198. doi:10.1002/cncr.9028
67. Ito K, Oizumi S, Fukumoto S; Hokkaido Lung Cancer Clinical Study Group, et al. Clinical characteristics of pleomorphic carcinoma of the lung. Lung Cancer. 2010;68(2):204–210.
68. Attanoos RL, Appleton MA, Gibbs AR. Primary sarcomas of the lung: a clinicopathological and immunohistochemical study of 14 cases. Histopathology. 1996;29:29–36. doi:10.1046/j.1365-2559.1996.d01-481.x
69. Etienne-Mastroianni B, Falchero L, Chalabreysse L, et al. Primary sarcomas of the lung: a clinicopathologic study of 12 cases. Lung Cancer. 2002;38:283–289. doi:10.1016/S0169-5002(02)00303-3
70. Suster S. Primary sarcomas of the lung. Semin Diagn Pathol. 1995;12:140–157.
71. Gebauer C. Primary pulmonary sarcomas: etiology, clinical assessment and prognosis with a comparison to pulmonary carcinomas—a review of 41 cases and 394 other cases of the literature. Jpn J Surg. 1982;12:148–159. doi:10.1007/BF02469384
72. Zeren H, Moran CA, Suster S, Fishback NF, Koss MN. Primary pulmonary sarcomas with features of monophasic synovial sarcoma: a clinicopathological, immunohistochemical, and ultrastructural study of 25 cases. Hum Pathol. 1995;26:474–480. doi:10.1016/0046-8177(95)90242-2
73. Begueret H, Galateau-Salle F, Guillou L, et al. Primary intrathoracic synovial sarcoma: a clinicopathologic study of 40 t(X;18)-positive cases from the French Sarcoma Group and the Mesopath Group. Am J Surg Pathol. 2005;29:339–346. doi:10.1097/01.pas.0000147401.95391.9a
74. Lucas DR, Pass HI, Madan SK, et al. Sarcomatoid mesothelioma and its histological mimics: a comparative immunohistochemical study. Histopathology. 2003;42:270–279. doi:10.1046/j.1365-2559.2003.01583.x
75. Knuuttila A, Jee KJ, Taskinen E, Wolff H, Knuutila S, Anttila S. Spindle cell tumours of the pleura: a clinical, histological and comparative genomic hybridization analysis of 14 cases. Virchows Arch. 2006;448:135–141. doi:10.1007/s00428-005-0059-3
76. Colby TV. Malignancies in the lung and pleura mimicking benign processes. Semin Diagn Pathol. 1995;12(1):30–44.
77. Comin CE, Novelli L, Cavazza A, Rotellini M, Cianchi F, Messerini L. Expression of thrombomodulin, calretinin, cytokeratin 5/6, D2-40 and WT-1 in a series of primary carcinomas of the lung: an immunohistochemical study in comparison with epithelioid pleural mesothelioma. Tumori. 2014;100(5):559–567. doi:10.1700/1660.18182
78. Carbone M, Shimizu D, Napolitano A, et al. Positive nuclear BAP1 immunostaining helps differentiate non-small cell lung carcinomas from malignant mesothelioma. Oncotarget. 2016;7(37):59314–59321. doi:10.18632/oncotarget.v7i37
79. Vieira T, Girard N, Ung M, et al. Efficacy of first-line chemotherapy in patients with advanced lung sarcomatoid carcinoma. J Thorac Oncol. 2013;8:1574–1577. doi:10.1097/01.JTO.0000437008.00554.90
80. Bae HM, Min HS, Lee SH, et al. Palliative chemotherapy for pulmonary pleomorphic carcinoma. Lung Cancer. 2007;58:112–115. doi:10.1016/j.lungcan.2007.05.006
81. Fallet V, Saffroy R, Girard N, et al. High-throughput somatic mutation profiling in pulmonary sarcomatoid carcinomas using the LungCarta TM panel: exploring therapeutic targets. Ann Oncol. 2015;26:1748–1753. doi:10.1093/annonc/mdv232
82. Schrock AB, Li SD, Frampton GM, et al. Pulmonary sarcomatoid carcinomas commonly harbor either potentially targetable genomic alterations or high tumor mutational burden as observed by comprehensive genomic profiling. J Thorac Oncol. 2017;12:932–942. doi:10.1016/j.jtho.2017.03.005
83. Liu X, Jia Y, Shen YF, et al. Detection of frequent MET Exon 14 skipping events in pulmonary sarcomatoid carcinoma and response to targeted inhibition. J Clin Oncol. 2016;34:794–802. doi:10.1200/JCO.2015.62.0674
84. Lococo F, Gandolfi G, Rossi G, et al. Deep sequencing analysis reveals that KRAS mutation is a marker of poor prognosis in patients with pulmonary Sarcomatoid carcinoma. J Thorac Oncol. 2016;11:1282–1292. doi:10.1016/j.jtho.2016.04.020
85. Li X, Wang D, Zhao Q, et al. Cinical significance of next-generation sequencing of Chinese pulmonary sarcomatoid carcinoma. Sci Rep. 2017;7:3947.
86. Mehard M, Rosy S, LaFramboise WA, et al. KRAS mutation is predictive of outcome in patients with pulmonary sarcomatoid carcinoma. Histopathology. 2018;73:207–214. doi:10.1111/his.13505
87. Awad MM, Oxnard GR, Jackman DM, et al. MET exon 14 mutations in non-small cell lung cancer are associated with advanced age and stage dependent MET genomic amplification and c-MET overexpression. J Clin Oncol. 2016;34:721–730. doi:10.1200/JCO.2015.63.4600
88. Awad MM. Impaired c-MET receptor degradation mediated by MET exon 14 mutations in non-small cell lung cancer. J Clin Oncol. 2016;34:879–881. doi:10.1200/JCO.2015.64.2777
89. Mengoli MC, Bertolini F, Tiseo M, et al. MET DNA alterations in NSCLC. Clin Cancer Res. 2016;22:3697–3698. doi:10.1158/1078-0432.CCR-16-0351
90. Vuong HG, Ho ATN, Altibi AMA, et al. Clinicopathological implications of MET exon 14 mutations in non-small cell lung cancer. A systematic review and meta-analysis. Lung Cancer. 2018;123:76–82.
91. Kim EK, Kim KA, Lee CY, et al. Molecular diagnostic assays and clinicopathologic implications of MET exon 14 skipping mutation in non-small cell lung cancer. Clin Lung Cancer. 2019;20:e123–e132. doi:10.1016/j.cllc.2018.10.004
92. Yu Y, Zhang Q, Zhang J, Lu S. Prevalence of MET exon 14 skipping mutation in pulmonary sarcomatoid carcinoma patients without common targetable mutations: a single-institute study. J Cancer Res Ther. 2019;15:909–913. doi:10.4103/jcrt.JCRT_591_18
93. Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013;31(8):1089–1096. doi:10.1200/JCO.2012.43.9422
94. Shum E, Stuart M, Borczuk A, Wang F, Cheng H, Halmos B. Recent advances in the management of pulmonary sarcomatoid carcinoma. Expert Rev Respir Med. 2016;10(4):407–416. doi:10.1586/17476348.2016.1157475
95. Nakagomi T, Goto T, Hirotsu Y, et al. New therapeutic targets for pulmonary sarcomatoid carcinomas based on their genomic and phylogenetic profiles. Oncotarget. 2018;9(12):10635–10649. doi:10.18632/oncotarget.v9i12
96. Manzotti G, Torricelli F, Benedetta D, et al. An epithelial-to-mesenchymal transcriptional switch triggers evolution of Pulmonary Sarcomatoid Carcinoma (PSC) and identifies dasatinib as new therapeutic option. Clin Cancer Res. 2019;25:2348–2360. doi:10.1158/1078-0432.CCR-18-2364
97. Ushiki A, Koizumi T, Kobayashi N, et al. Genetic heterogeneity of EGFR mutation in pleomorphic carcinoma of the lung: response to gefitinib and clinical outcome. Jpn J Clin Oncol. 2009;39(4):267–270. doi:10.1093/jjco/hyn155
98. Italiano A, Cortot AB, Ilie M, et al. EGFR and KRAS status of primary sarcomatoid carcinomas of the lung: implications for anti-EGFR treatment of a rare lung malignancy. Int J Cancer. 2009;125:2479–2482. doi:10.1002/ijc.v125:10
99. Lin Y, Yang H, Cai Q, et al. Characteristics and prognostic analysis of 69 patients with pulmonary sarcomatoid carcinoma. Am J Clin Pathol. 2016;39:215–222.
100. Chang YL, Wu CT, Shih JY, Lee YC. EGFR and p53 status of pulmonary pleomorphic carcinoma: implications for EGFR tyrosine kinase inhibitors therapy of an aggressive lung malignancy. Ann Surg Oncol. 2011;18(10):2952–2960. doi:10.1245/s10434-011-1621-7
101. Lococo F, Torricelli F, Rossi G, et al. Inter-relationship between PD-L1 expression and clinic-pathological features and driver gene mutations in pulmonary sarcomatoid carcinomas. Lung Cancer. 2017;113:93–101. doi:10.1016/j.lungcan.2017.09.009
102. O’Bryan JP. Pharmacological targeting of RAS: recent success with direct inhibitors. Pharmacol Res. 2019;139:503–511. doi:10.1016/j.phrs.2018.10.021
103. Janes MR, Zhang J, Li LS, et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. 2018;172(3):578–589.e17. doi:10.1016/j.cell.2018.01.006
104. Ou SH, Kwak EL, Siwak-Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6(5):942–946. doi:10.1097/JTO.0b013e31821528d3
105. Tong JH, Yeung SF, Chan AW, et al. MET amplification and Exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res. 2016;22(12):3048–3056. doi:10.1158/1078-0432.CCR-15-2061
106. Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:850–859. doi:10.1158/2159-8290.CD-15-0285
107. Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5(8):842–849. doi:10.1158/2159-8290.CD-14-1467
108. Lee C, Usenko D, Frampton GM, McMahon C, Ali SM. Deletion in Sarcomatoid non-small-cell lung cancer detected by next-generation sequencing and successfully treated with a MET inhibitor. J Thorac Oncol. 2015;10(12):e113–4. doi:10.1097/JTO.0000000000000645
109. Subramanian A, Narayan R, Corsello SM, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171(6):1437–1452.e17. doi:10.1016/j.cell.2017.10.049
110. Johnson FM, Bekele BN, Feng L, et al. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(30):4609–4615. doi:10.1200/JCO.2010.30.5474
111. Alì G, Bruno R, Poma AM, et al. Whole transcriptome targeted gene quantification provides new insights on pulmonary sarcomatoid carcinomas. Sci Rep. 2019;9:3536.
112. Hsieh MS, Lin MW, Lee YH. Lung adenocarcinoma with sarcomatoid transformation after tyrosine kinase treatment and chemotherapy. Lung Cancer. 2019;137:76–84. doi:10.1016/j.lungcan.2019.08.029
113. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi:10.1126/scitranslmed.3002003
114. Chung JH, Rho JK, Xu X, et al. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer. 2011;73(2):176–182. doi:10.1016/j.lungcan.2010.11.011
115. Xu S, Liu X, Liu R, et al. Concurrent epidermal growth factor receptor T790M secondary mutation and epithelial-mesenchymal transition in a lung adenocarcinoma patient with EGFR-TKI drug resistance. Thorac Cancer. 2017;8(6):693–697. doi:10.1111/1759-7714.12484
116. Xia L, Shao YW, Xia Y. Nkx2-4 mutation confers resistance to EGFR-tyrosine kinase inhibitors in EGFR-mutant lung sarcomatoid carcinoma. J Thorac Oncol. 2019;14(6):e125–e126. doi:10.1016/j.jtho.2019.01.027
117. Sesumi Y, Suda K, Mizuuchi H, et al. Effect of dasatinib on EMT-mediated-mechanism of resistance against EGFR inhibitors in lung cancer cells. Lung Cancer. 2017;104:85–90. doi:10.1016/j.lungcan.2016.12.012
118. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi:10.1056/NEJMoa1200690
119. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi:10.1056/NEJMoa1200694
120. Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi:10.1056/NEJMoa1606774
121. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128.
122. Kotlowska MP, Rueda AG, Olmedo ME, et al. Efficacy of immunotherapy in sarcomatoid lung cancer, a case report and literature review. Respir Med Case Rep. 2019;26:310–314. doi:10.1016/j.rmcr.2019.02.017
123. Lantuejoul S, Damotte D, Hofman V, Adam J. Programmed death ligand 1 immunohistochemistry in non-small cell lung carcinoma. J Thorac Dis. 2019;11(Suppl 1):S89–S101. doi:10.21037/jtd.2018.12.103
124. Wu X, Huang Y, Li Y, et al. 18F-FDG PET/CT imaging in pulmonary sarcomatoid carcinoma and correlation with clinical and genetic findings. Ann Nucl Med. 2019;33(9):647–656. doi:10.1007/s12149-019-01374-5
125. Chan AWH, Tong JHM, Kwan JSH, et al. Assessment of programmed cell death ligand-1 expression by 4 diagnostic assays and its clinicopathological correlation in a large cohort of surgical resected non-small cell lung carcinoma. Mod Pathol. 2018;31(9):1381–1390. doi:10.1038/s41379-018-0053-3
126. Sim JK, Chung SM, Choi JH, et al. Clinical and molecular characteristics of pulmonary sarcomatoid carcinoma. Korean J Intern Med. 2018;33(4):737–744. doi:10.3904/kjim.2017.245
127. Yvorel V, Patoir A, Casteillo F, et al. PD-L1 expression in pleomorphic, spindle cell and giant cell carcinoma of the lung is related to TTF-1, p40 expression and might indicate a worse prognosis. PLoS ONE. 2017;12(7):e0180346. doi:10.1371/journal.pone.0180346
128. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–1550.
129. Kim S, Kim MY, Koh J, et al. Programmed death-1 ligand 1 and 2 are highly expressed in pleomorphic carcinomas of the lung: comparison of sarcomatous and carcinomatous areas. Eur J Cancer. 2015;51(17):2698–2707. doi:10.1016/j.ejca.2015.08.013
130. Vieira T, Antoine M, Hamard C, et al. Sarcomatoid lung carcinomas show high levels of programmed death ligand-1 (PD-L1) and strong immune-cell infiltration by T CD3 cells and macrophages. Lung Cancer. 2016;98:51–58. doi:10.1016/j.lungcan.2016.05.013
131. Velcheti V, Rimm DL, Schalper KA. Sarcomatoid lung carcinomas show high levels of programmed death ligand-1 (PD-L1). J Thorac Oncol. 2013;8(6):803–805. doi:10.1097/JTO.0b013e318292be18
132. Salati M, Baldessari C, Calabrese F, et al. Nivolumab-induced impressive response of refractory pulmonary sarcomatoid carcinoma with brain metastasis. Case Rep Oncol. 2018;11(3):615–621. doi:10.1159/000492666
133. Vieira T, Antoine M, Ruppert A-M, et al. Blood vessel invasion is a major feature and a factor of poor prognosis in sarcomatoid carcinoma of the lung. Lung Cancer. 2014;85:276–281. doi:10.1016/j.lungcan.2014.06.004
134. Pecuchet N, Vieira T, Rabbe N, et al. Molecular classification of pulmonary sarcomatoid carcinomas suggests new therapeutic opportunities. Ann Oncol. 2017;28:1597–1604. doi:10.1093/annonc/mdx162
135. Saffroy R, Fallet V, Girard N, et al. MET exon 14 mutations as targets in routine molecular analysis of primary sarcomatoid carcinoma of the lung. Oncotarget. 2017;8:42428–42437. doi:10.18632/oncotarget.16403
136. Heist RS, Shim HS, Gingipally S, et al. MET exon 14 skipping in non-small cell lung cancer. Oncologist. 2016;21:481–486. doi:10.1634/theoncologist.2015-0510
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.