")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Application of nanoparticles for oral delivery of acid-labile lansoprazole in the treatment of gastric ulcer: in vitro and in vivo evaluations
Authors Alai M, Lin WJ, Yang HH
Received 6 February 2015
Accepted for publication 28 April 2015
Published 18 June 2015 Volume 2015:10(1) Pages 4029—4041
DOI https://doi.org/10.2147/IJN.S82366
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Milind Alai,1 Wen Jen Lin1,2
1Graduate Institute of Pharmaceutical Sciences, School of Pharmacy, 2Drug Research Center, College of Medicine, National Taiwan University, Taipei, Taiwan
Abstract: The aim of this study was to develop nanoparticles for oral delivery of an acid-labile drug, lansoprazole (LPZ), for gastric ulcer therapy. LPZ-loaded positively charged Eudragit® RS100 nanoparticles (ERSNPs-LPZ) and negatively charged poly(lactic-co-glycolic acid) nanoparticles (PLGANPs-LPZ) were prepared. The effect of charge on nanoparticle deposition in ulcerated and non-ulcerated regions of the stomach was investigated. The cellular uptake of nanoparticles in the intestine was evaluated in a Caco-2 cell model. The pharmacokinetic performance and ulcer healing response of LPZ-loaded nanoparticles following oral administration were evaluated in Wistar rats with induced ulcers. The prepared drug-loaded ERSNPs-LPZ and PLGANPs-LPZ possessed opposite surface charge (+38.5±0.3 mV versus -27.3±0.3 mV, respectively) and the particle size was around 200 nm with a narrow size distribution. The negatively charged PLGANPs adhered more readily to the ulcerated region (7.22%±1.21% per cm2), whereas the positively charged ERSNPs preferentially distributed in the non-ulcerated region (8.29%±0.35% per cm2). Both ERSNPs and PLGANPs were prominent uptake in Caco-2 cells, too. The nanoparticles sustained and prolonged LPZ concentrations up to 24 hours, and the half-life and mean residence time of LPZ were prolonged by 3.5-fold and 4.5-fold, respectively, as compared with LPZ solution. Oral administration of LPZ-loaded nanoparticles healed 92.6%–95.7% of gastric ulcers in Wistar rats within 7 days.
Keywords: nanoparticles, lansoprazole, Eudragit® RS100, PLGA
Introduction
Acid-related disorders, such as peptic ulcer and gastroesophageal reflux disease, frequently occur in elderly people and are associated with morbidity.1 Proton pump inhibitors are the most effective drugs for ulcer healing and achieve greater symptomatic relief in patients. The mechanism of action of proton pump inhibitors is via selective inhibition of H+/K+ ATPase (also known as a the proton pump), an enzyme system present in gastric parietal cells, to suppress secretion of gastric acid and promote ulcer healing activity in patients.2 Lansoprazole (LPZ) is a proton pump inhibitor. It is metabolized in the parietal cells to form an active sulfonamide metabolite that inactivates the sulfhydryl group of the proton pump, thereby reducing hydrogen ion secretion.3 LPZ induces onset of maximal acid suppression more rapidly than its analogs and provides earlier and better symptom relief.4
LPZ is an acid-labile and poorly water-soluble drug.5 It is necessary to protect LPZ from degradation in gastric acid when orally administered. The commercial available solid dosage forms of LPZ include enteric-coated granules, tablets, and capsules. The enteric delayed-release dosage form of LPZ has been reported to be unable to efficiently suppress nocturnal acid secretion in the event of gastroesophageal reflux disease.6,7 Nocturnal acid breakthrough frequently occurs in patients with gastroesophageal reflux disease during the first half of the sleeping period, and affects sleep quality and daytime functioning.
Eudragit® RS100 and poly(lactic-co-glycolic acid) (PLGA) polymers have been used as drug carriers. Eudragit RS100 is insoluble but swellable at physiological pH. It contains 4.5%–6.8% quaternary ammonium groups that impart a positive charge to the polymer and is responsible for the bioadhesive properties of this polymer.8 Eudragit RS100 has been used to prepare sustained-release nanoparticulate dosage forms. Adibkia et al developed Eudragit nanoparticles to sustain delivery of naproxen via ocular and intra-articular administration.9 Devarajan and Sonavane reported that Eudragit nanoparticles enabled a sustained-release profile for gliclazide after oral administration,10 and Dai et al used Eudragit nanoparticles as a carrier to enhance the oral bioavailability of cyclosporine.11
PLGA is a biocompatible and biodegradable copolymer. It contains carboxyl groups that impart a negative charge to the polymer. PLGA is approved by the US Food and Drug Administration as a biomedical material.12,13 An emulsification/solvent evaporation method is generally used to prepare PLGA microparticles and nanoparticles, which are used widely to control drug delivery.8
Nanoparticles are colloidal carriers with a size range of 10–200 nm and have the potential for uptake in inflamed tissues.14 It has been reported that the particle size and surface charge on polystyrene nanoparticles play a vital role in efficient uptake of these nanoparticles in stomach tissue.15 In a previous study, we combined the nanoparticle dosage form with an enteric-coating technique to protect LPZ from degradation in the stomach, and achieved a reliable gastric ulcer healing response in an animal model of induced ulcers.
In this study, we aimed to develop a non-enteric-coated nanoparticulate delivery system for acid-labile LPZ. Up until now, no non-enteric-coated nanoparticulate dosage form containing LPZ has been available. Such a formulation would be expected to provide reliable long-term acid suppression when administered orally once daily. LPZ-loaded positively charged Eudragit RS100 nanoparticles (ERSNPs-LPZ) and negatively charged PLGA nanoparticles (PLGANPs-LPZ) were prepared using a solvent evaporation method. The effect of charge on deposition of the nanoparticles in ulcerated and non-ulcerated regions of the stomach was investigated using fluorescent nanoparticles, and cellular uptake of the nanoparticles in the intestine was further evaluated in a monolayer of Caco-2 cells. Finally, the pharmacokinetic performance and ulcer healing response were demonstrated in Wistar rats with induced ulcers after oral administration of ERSNPs-LPZ and PLGANPs.
Materials and methods
Materials
LPZ was obtained from Alcon Biosciences Private Ltd (Mumbai, India). Eudragit RS100 (Degussa, Darmstadt, Germany) and PLGA (molecular weight 28,000 Da, copolymer ratio 50:50, Boehringer Ingelheim, Ingelheim, Germany) were also used. Coumarin-6 (Sigma-Aldrich, St Louis, MO, USA) was used as a fluorescence marker. A human colon adenocarcinoma (Caco-2) cell line was a gift from Li-Juan Shen (Graduate Institute of Pharmaceutical Sciences, National Taiwan University, Taipei, Taiwan) and originated from the American Type Culture Collection (Manassas, VA, USA). Dulbecco Modified Eagle’s Medium (with 4.5 g/L D-glucose and L-glutamine, without sodium pyruvate and sodium bicarbonate), non-essential amino acids, and mycoserum-tested fetal bovine serum were purchased from Biological Industries (Beit-Haemek, Israel). Hank’s balanced salt solution buffer (HBSS) and propidium iodide were purchased from Invitrogen Corporation (Carlsbad, CA, USA).
Preparation of LPZ-loaded Eudragit nanoparticles
The ERSNPs-LPZ were prepared by an oil-in-water (o/w) emulsion solvent evaporation/extraction method. Eudragit RS100 (200 mg) and LPZ (200 mg) were dissolved in 10 mL of a dichloromethane/methanol mixture (5/5, v/v). The organic phase was added to 100 mL of aqueous poly(vinyl alcohol) solution (0.25% w/v, pH 9.0) under sonication at 4°C for 20 minutes. The organic solvent was evaporated by magnetic stirring at room temperature for 3 hours followed by a rotavapor under reduced pressure at 35°C for 5 minutes. The nanoparticles were recovered after centrifugation at 17,000 rpm for 30 minutes (Avanti J26 XP centrifuge, Beckman Coulter, Miami, USA). The collected nanoparticles were washed with deionized water three times and resuspended in 1 mL of deionized water containing 5% w/v sucrose, and then freeze-dried.
Preparation of LPZ-loaded PLGA nanoparticles
PLGANPs-LPZ were prepared using a water-in-oil-in-water (w/o/w) emulsion method. PLGA (200 mg) and LPZ (100 mg) were dissolved in 10 mL of a dichloromethane/acetone mixture (5/5 v/v). An aqueous solution of NaHCO3 (1 mL, 0.2%) was added to PLGA solution and emulsified to obtain a primary water-in-oil emulsion using an ultrasonic probe at 4°C for 2 minutes. The primary emulsion was then added to 100 mL of aqueous poly(vinyl alcohol) solution (0.25% w/v, pH 9.0) and followed the same procedures as for preparation of ERSNPs-LPZ.
Characterization of nanoparticles
The particle size and zeta potential of the nanoparticles were determined by a Zetasizer (Nano ZS, Malvern Co Ltd, Malvern, UK). The morphology of the nanoparticles was examined using an H7100 transmission electron microscope (Hitachi High-technologies Corporation, Tokyo, Japan) where the freeze-dried nanoparticles were dispersed in deionized water and stained with 2% phosphotungstic acid for 30 seconds, then placed on copper grids for observation. For determination of LPZ content, about 5 mg each of ERSNPs-LPZ and PLGANPs-LPZ were dissolved in 5 mL of methanol and acetonitrile, respectively. Each sample was centrifuged at 14,000 rpm for 10 minutes, and 20 μL of the supernatant was injected into a high-performance liquid chromatography (HPLC) system (Jasco International Company Ltd, Tokyo, Japan). The HPLC system consisted of a pump (PU-2089) and an MD-2010 photo diode array detector at 285 nm. A reversed phase silica column (C-18, 4.6×250 mm, 5 μm, Phenomenex Inc, Torrance, CA, USA) was used. The mobile phase comprised water, acetonitrile, and triethylamine in a volume ratio of 50:50:0.1 (pH 7) at a flow rate of 1 mL per minute. The HPLC analytical method was validated prior to sample analysis. Drug loading (DL) and encapsulation efficiency (EE) were calculated using equations (1) and (2).
|
|
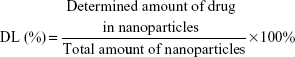
|
|
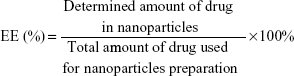
In vitro drug release
LPZ-loaded nanoparticles were suspended in 5 mL of phosphate-buffered solution (pH 7.4) which was placed in a dialysis bag (molecular weight cutoff 6,000–8,000 Da) followed by immersion in 100 mL of the same release medium at 37°C±0.5°C in a shaker bath at 75 rpm. Samples (1 mL) were collected at time intervals of 0.5, 1, 2, 4, 6, 8, 12, and 24 hours, and the same volume of fresh release medium was replaced. The amount of LPZ in each release sample was analyzed by the HPLC method. Release of LPZ from the nanoparticles was analyzed by equation (3) based on the Korsmeyer–Peppas model.16
Mt/M∞ = ktn | (3) |
where Mt/M∞ is the percentage of drug released, k is the release rate constant, n is the release exponent, and t is the release time. The n value of the Korsmeyer–Peppas equation was used to indicate the release mechanism.17 In addition, the similarity factor ( f2) was calculated to compare the release profiles of ERSNPs-LPZ and PLGANPs-LPZ.18
|
|
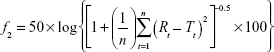
where n is the number of time points, Rt and Tt are the mean percent of drug released from the reference and test dissolution profiles, respectively, at each time point (t). Generally, an f2 value in the range of 50–100 indicates similarity between two comparative release profiles.
Preparation of coumarin-6-loaded fluorescence nanoparticles
Coumarin-6-loaded Eudragit RS100 (ERSNPs-C6) and PLGA (PLGANPs-C6) fluorescence nanoparticles were prepared by an o/w solvent evaporation method. Eudragit RS100 or PLGA (200 mg) and coumarin-6 (1 mg) were dissolved in 10 mL of dichloromethane/acetone mixture (5/5 v/v). The preparation procedures that followed were the same as those described for preparation of the LPZ nanoparticles. The amounts of coumarin-6 entrapped in ERSNPs-C6 and PLGANPs-C6 were determined using a fluorescence spectrophotometer (F4500, Hitachi Ltd, Tokyo, Japan). The nanoparticles were dissolved in acetone for measurement of fluorescence at an excitation wavelength of 430 nm and an emission wavelength of 490 nm. Leaching of coumarin-6 from the nanoparticles was also evaluated. ERSNPs-C6 and PLGANPs-C6 (10 mg) were dispersed in 1 mL of HBSS (pH 7.4) and then placed in the dialysis bag (molecular weight cutoff 6,000–8,000 Da) which was immersed in 10 mL of HBSS at 37°C±0.5°C in a mechanical shaker at 50 rpm for 24 hours. The sample was collected and diluted with acetone followed by fluorescence measurement.
Biodistribution of nanoparticles in stomach tissue
Male Wistar rats (250–300 g) were used in this study, and were obtained from the National Laboratory Animal Center (Taipei, Taiwan). All animal experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health, and all procedures were examined by the ethics committee on animal experimentation at National Taiwan University (Taipei, Taiwan). The rats were fasted but allowed free access to water overnight. Gastric ulceration was induced by oral administration of absolute ethanol (5 mL/kg).19 The ulcer-induced rats were divided into three groups (one control group and two treatment groups), and each group included four rats. Each treatment group received a hard gelatin capsule (#9, Torpac Inc, Fairfield, NJ, USA) filled with ERSNPs-C6 or PLGANPs-C6, and the control group received saline solution only. The formulations were administered orally 1 hour after administration of ethanol. The rats were sacrificed 4 hours after dose administration. The stomach was opened longitudinally and rinsed with saline solution. The ulcerated and non-ulcerated regions of stomach tissue were cut and the freshly excised tissues were cryofixed using Tissue-Tek®. The molded tissue sample was sectioned using a CM3050 S Cryostat (Leica Microsystems, Wetzlar, Germany) and observed under a fluorescence microscope combined with a digitally integrated photomicrography system (Axiophot 2, Carl Zeiss, Hamburg, Germany). In addition, the sectioned stomach tissues were stained with hematoxylin and eosin to illustrate the morphology of the healthy and ulcerated tissues. For quantitative determination, the total mucosal area and the ulcerated area were measured using Axio Vision software (version 4.8, Carl Zeiss International, New York, NY, USA). The ulcerated and non-ulcerated tissues were lyophilized separatedly in the dark. Acetone was added to each tissue sample followed by sonication for 15 minutes. The tissue samples were centrifuged at 2,000 rpm for 5 minutes and the supernatant was collected. The extraction procedure was repeated three times. Finally, the supernatant was diluted with acetone and analyzed using a fluorescence spectrophotometer (F4500, Hitachi Ltd) at an excitation wavelength of 430 nm and an emission wavelength of 490 nm. Uptake of the nanoparticles by stomach tissues was expressed as a percentage of nanoparticle uptake per cm2 of ulcerated or non-ulcerated stomach tissue.
Uptake of nanoparticles by Caco-2 cells
The Caco-2 cell monolayer was washed with HBSS (pH 7.4) twice followed by preincubation with HBSS at 37°C for 30 minutes. The HBSS (control), free coumarin-6 solution, ERSNPs-C6, and PLGANPs-C6 suspension in HBSS equivalent to 200 ng/mL of coumarin-6 were added in the apical side while 3 mL of HBSS was added in the basolateral side. The Caco-2 cell monolayer was incubated for 0.5 hours and 1 hours at 37°C in an atmosphere of 5% CO2 and 90% relative humidity. The cell monolayer was then washed with phosphate-buffered saline (PBS, pH 7.4) followed by trypsinization for 5 minutes (0.25% trypsin ethylenediaminetetraacetic acid). Cold PBS was added and the cells were centrifuged at 1,000 rpm for 5 minutes, resuspended in 2 mL of PBS, and analyzed using a fluorescence activated flow cytometer (FACSCalibur, BD Biosciences, Franklin Lakes, NJ, USA). For confocal microscopic observation, the cell monolayer was incubated at 37°C for 0.5 hours, washed with PBS, and then fixed with 3.7% paraformaldehyde solution in PBS for 30 minutes. The cells were washed again, then treated with RNase solution for 30 minutes. The nuclei were then stained with propidium iodide for 30 minutes. The cell monolayer was then mounted on a glass slide with Fluoromount™ medium (Sigma-Aldrich). The images were captured using a confocal laser scanning microscopy imaging system (TCS SP5, Leica).
Pharmacokinetic study
The Wistar rats (250–300 g) with induced ulcers were divided into three groups, with each group including four rats. The LPZ-NaHCO3 solution (5 mg LPZ/kg) and the hard gelatin capsules (#9, Torpac Inc) filled with sodium bicarbonate (20 mg/kg) and either ERSNPs-LPZ or PLGANPs-LPZ (5 mg LPZ/kg) were orally administered to the Wistar rats. The LPZ solution (1 mg/mL) was prepared by dissolving LPZ in a mixture of PEG 400 and 1% NaHCO3 (2:3 v/v).20 Blood samples were collected from the tail vein of each rat prior to drug administration and at time intervals of 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, and 24 hours after dosing. The blood samples were centrifuged at 12,000 rpm for 10 minutes at 4°C and the supernatant was collected. The LPZ in each sample was extracted using acetonitrile followed by centrifugation. The supernatant was collected, air-dried, and reconstituted with mobile phase for HPLC analysis. The recovery of LPZ from serum samples was in the range of 98.60%–101.78%. The pharmacokinetic parameters were obtained based on a noncompartmental pharmacokinetic analysis model (WinNonlin software, version 5.3, Pharsight Corporation, Mountain View, CA, USA). The statistical analysis was performed using the Student’s t-test, and a P-value of P<0.05 was considered to be statistically significant.
Pharmacodyamic study
The Wistar rats with induced ulcers were divided into four groups (n=4 each) to receive saline solution (control group), LPZ solution, or a capsule filled with ERSNPs-LPZ or PLGANPs-LPZ. The formulations were orally administered once daily for 7 days. The rats were euthanized 24 hours after the last dose. The stomach was cut along the greater curvature, and the mucosal surface of the stomach was washed with saline solution. Photographic images (Nikon E5000, Nikon Corporation, Tokyo, Japan) of the mucosal surface of the stomach were obtained, and the total mucosal area and the ulcerated area were measured using Axio Vision software (version 4.8, Carl Zeiss International). The ulcer index (UI) was calculated using equation (5).21
|
|

Results and discussion
Characterization of ERSNPs-LPZ and PLGANPs-LPZ
Figure 1A and 1B show a transmission electron micrograph of the ERSNPs-LPZ and PLGANPs-LPZ, indicating that these nanoparticles have a spherical shape and smooth surface. The mean particle size of ERSNPs-LPZ was 203.9±4.9 nm and the polydispersity index was 0.09±0.04, indicating a narrow size distribution (Figure 1C). The EE and DL were 79.3%±1.0% and 43.7%±0.5%, respectively. The ERSNPs-LPZ showed a zeta potential of +38.5±0.3 mV due to the quaternary ammonium groups of Eudragit RS100. The mean particle size of PLGANPs-LPZ was 219.2±2.9 nm and the polydispersity index was 0.13±0.07 (Figure 1D). The carboxyl end groups of the PLGA copolymer form an acidic microenvironment which may affect the stability of LPZ, so sodium bicarbonate was added as an alkaline stabilizer during preparation of PLGANPs-LPZ in order to maintain the stability of LPZ in the PLGA nanoparticles.22 The EE and DL of PLGANPs-LPZ were 79.6%±2.2% and 28.7%±1.2%, respectively. PLGANPs-LPZ showed a zeta potential of −27.3±0.3 mV due to the carboxyl end groups of PLGA.
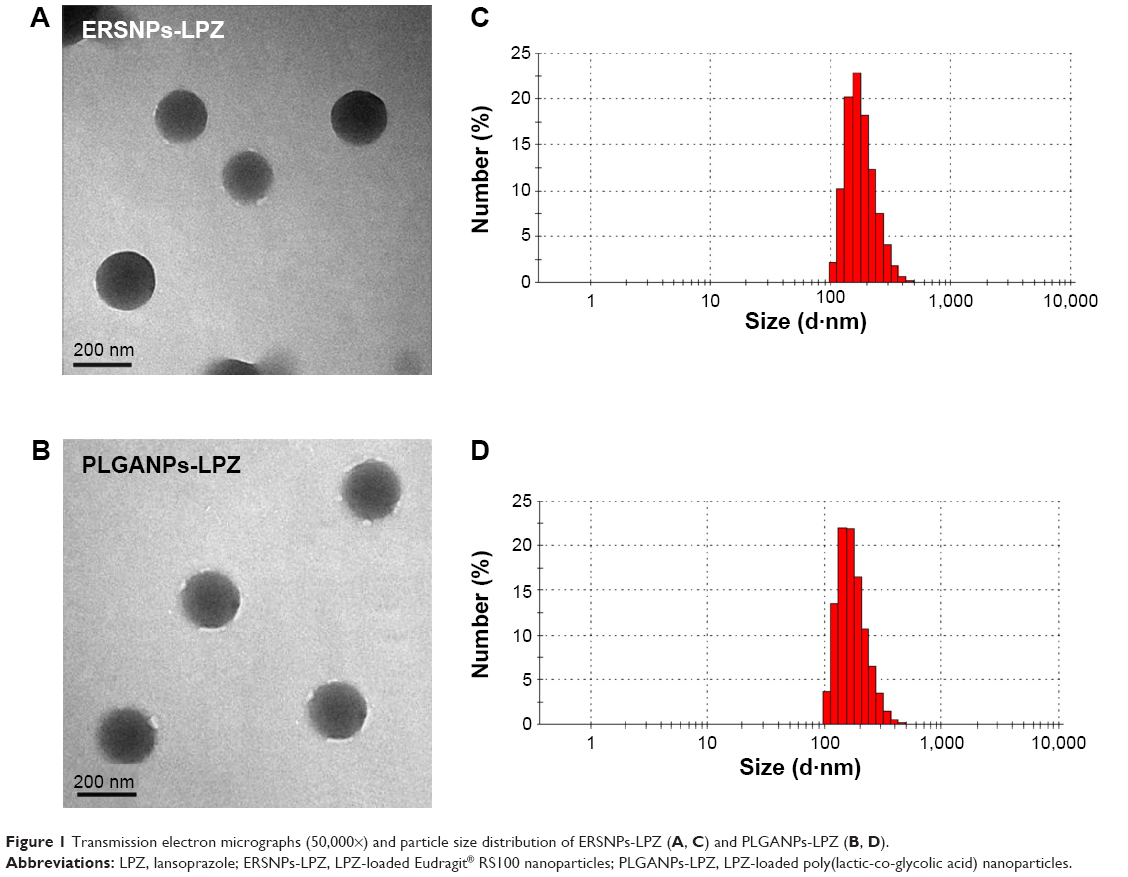 | Figure 1 Transmission electron micrographs (50,000×) and particle size distribution of ERSNPs-LPZ (A, C) and PLGANPs-LPZ (B, D). |
In vitro drug release
Figure 2 shows the in vitro release of LPZ from ERSNPs-LPZ and PLGANPs-LPZ in pH 7.4 release medium. ERSNPs-LPZ and PLGANPs-LPZ showed sustained-release behavior for up to 24 hours. The similarity factor, f2, was calculated to be 58, indicating no difference in drug release profile between ERSNPs-LPZ and PLGANPs-LPZ. Release of LPZ from the nanoparticles was analyzed using the Korsmeyer–Peppas model. The release rate constants (k) for ERSNPs-LPZ and PLGANPs-LPZ were 10.6%±0.4% h−n (R2=0.9305±0.015) and 11.0%±1.2% h−n (R2=0.9609±0.008), respectively, and the corresponding release exponent values (n) were 0.77±0.01 and 0.69±0.02, implying that the release of LPZ from both types of nanoparticles was dominated by non-Fickian diffusion. Eudragit RS100 is a water-insoluble but swellable polymer that behaves as a rate-limiting matrix to control drug release from ERSNPs-LPZ via a non-Fickian diffusion mechanism.23,24 The same release mechanism has been demonstrated for ERSNPs loaded with gatifloxacin or amphotericin B.25,26 PLGA is a water-insoluble and biodegradable polymer. Release of LPZ from PLGANPs-LPZ was dominated by polymer degradation and drug diffusion in terms of an n value in the range of 0.5–1.0. The same release mechanism has been demonstrated for bupivacaine-loaded PLGA nanoparticles.27
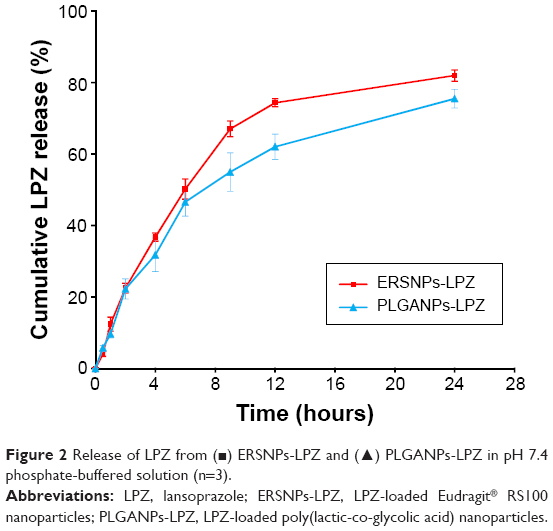 | Figure 2 Release of LPZ from (▪) ERSNPs-LPZ and (▴) PLGANPs-LPZ in pH 7.4 phosphate-buffered solution (n=3). |
Characterization of coumarin-6-loaded fluorescence nanoparticles
Coumarin-6 is widely used as a fluorescence marker for confocal microscopic investigation due to its high fluorescence sensitivity. It has been reported that about 0.05% w/w dye loading is sufficient to produce bright fluorescence nanoparticles for visualization.28 Coumarin-6-loaded fluorescence nanoparticles were prepared to demonstrate the biodistribution and cellular uptake of nanoparticles in the gastrointestinal tract. The mean particle size of ERSNPs-C6 and PLGANPs-C6 was 188.9±8.7 nm and 193.4±2.9 nm, respectively, and the zeta potential was +39.4±0.6 mV and −24.5±0.7 mV. The dye loadings of ERSNPs-C6 and PLGANPs-C6 were 0.35%±0.03% and 0.088%±0.003%, respectively. Use of a fluorescence marker for visualization of nanoparticles may lead to misinterpretation of results in the event of leaching or release of fluorescence marker into the release medium and subsequently into the cells.29 Less than 0.05% of the coumarin-6 was released from our ERSNPs-C6 and PLGANPs-C6 in the release medium within 24 hours, which was considered negligible. It was thus reasonable to assume that the coumarin-6 was stably retained in the nanoparticles ready for biodistribution and cellular uptake studies.
Biodistribution of fluorescence nanoparticles
Figure 3A illustrates the fluorescence microscopic images of ulcerated and non-ulcerated regions of the stomach after 4 hours of oral administration of ERSNPs-C6 and PLGANPs-C6 in Wistar rats with induced ulcers. Figure 3Ai and 3Aii show the pathology of the ulcerated and non-ulcerated regions of stomach tissue stained with hematoxylin and eosin before treatment with the nanoparticles. The nanoparticles were localized in both ulcerated (Figure 3Aiii and 3Av) and non-ulcerated (Figure 3Aiv and 3Avi) regions after oral administration of the nanoparticles, and the corresponding values are shown in Figure 3B. The PLGANPs-C6 (7.22%±1.21% per cm2) was deposited in the ulcerated region more than ERSNPs-C6 (4.28%±0.39% per cm2). In contrast, ERSNPs-C6 (8.29%±0.35% per cm2) was distributed more than PLGANPs-C6 (5.77%±0.66% per cm2) in the non-ulcerated region. These results indicate that the negatively charged PLGANPs-C6 adhered more readily to the ulcerated region, whereas the positively charged ERSNPs-C6 preferred localization in the non-ulcerated region of the stomach.
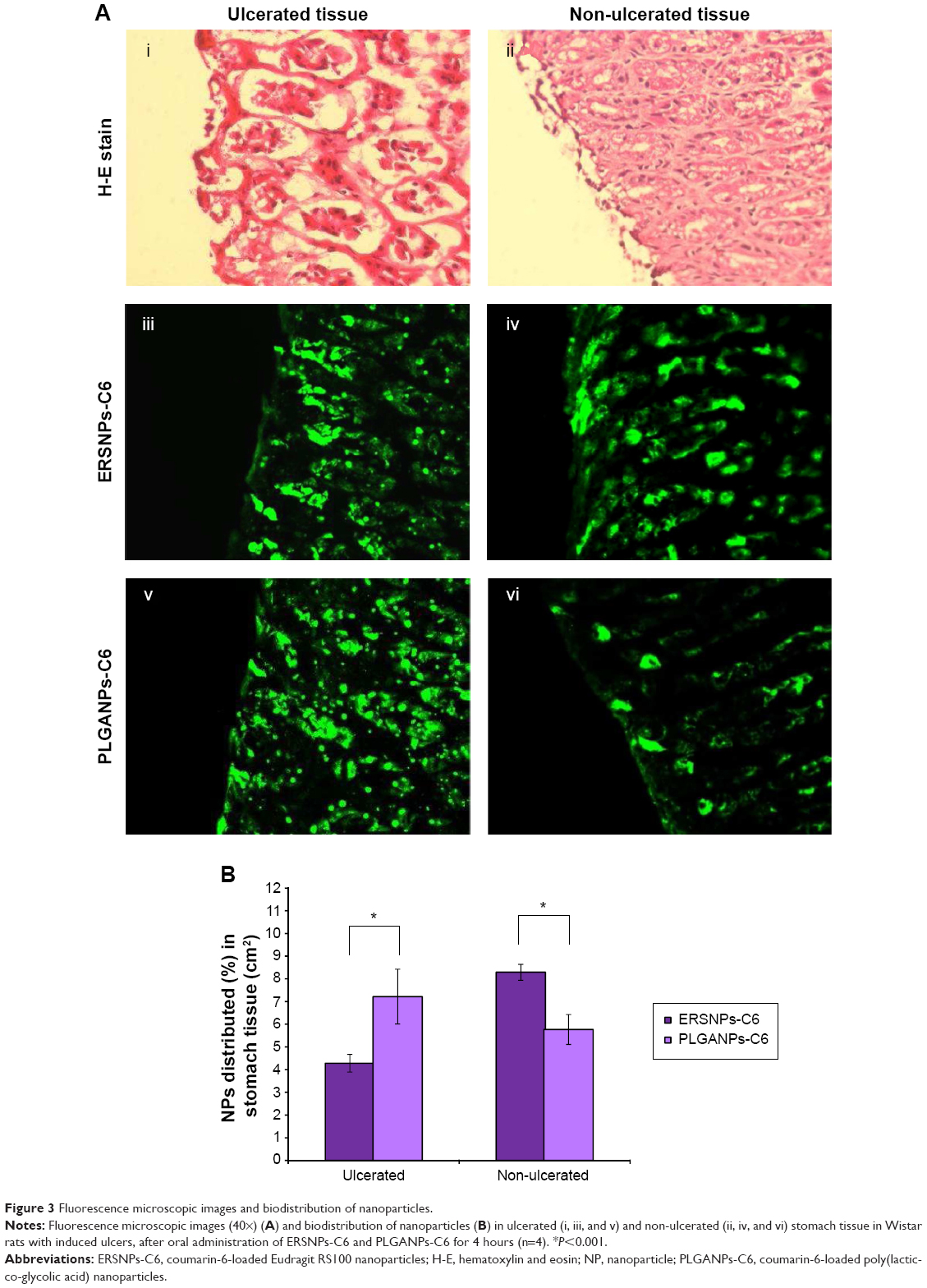 | Figure 3 Fluorescence microscopic images and biodistribution of nanoparticles. |
Different biodistribution of ERSNPs-C6 and PL GANPs-C6 in ulcerated and non-ulcerated regions of the stomach was observed. It has been reported that ulcerated tissue contains high concentrations of positively charged proteins which have a high affinity for negatively charged PLGANPs-C6 in terms of bioadhesion of these nanoparticles in the ulcerated region.15 In contrast, positively charged ERSNPs-C6 had a high affinity for the negatively charged cell membrane, resulting in bioadhesion in the non-ulcerated region. A similar result has been observed in negatively charged polystyrene nanoparticles which were taken up in the ulcerated region of stomach tissue more than in healthy stomach tissue.15
Cellular uptake of fluorescence nanoparticles
The uptake of coumarin-6-loaded ERSNPs-C6 and PLGANPs-C6 in the Caco-2 cell monolayer was monitored by flow cytometry, and the result is shown in Figure 4A. The cellular uptake of free coumarin-6 (1.27%±0.3%) was comparable with that in the control group (1.00%±0.03%). It has been suggested that it is not feasible for coumarin-6 to internalize into Caco-2 cells.30 However, a significant increase of fluorescence intensity in Caco-2 cells was observed when these cells were incubated with ERSNPs-C6 (78.4%±0.8%) and PLGANPs-C6 (45.3%±4.6%). The positively charged ERSNPs-C6 enhanced cellular uptake more prominently than negatively charged PLGANPs-C6 (P<0.05). The strong affinity of positively charged nanoparticles for negatively charged cell membranes accounted for this result. Figure 4B shows the confocal microscopic images of the Caco-2 cell monolayer after incubation with ERSNPs-C6 and PLGANPs-C6 for 0.5 hours. The strong green fluorescence in the cytoplasm indicated localization of nanoparticles in the cells. A similar charge effect has been reported by He et al whereby positively charged chitosan nanoparticles showed higher phagocytic uptake in LO2 and SMMC-7721 cells than negatively charged carboxymethyl-grafted chitosan nanoparticles.5 The uptake of nanoparticles into Caco-2 cells was further confirmed by three-dimensional analysis, where the fluorescence signal clearly appeared in the YZ plane, as shown in Figure 4B. All of these results confirm that both ERSNPs and PLGANPs were able to internalize into Caco-2 cells in a charge-dependent manner. This finding explored the possibility of nanoparticles being absorbed in the intestinal tract.
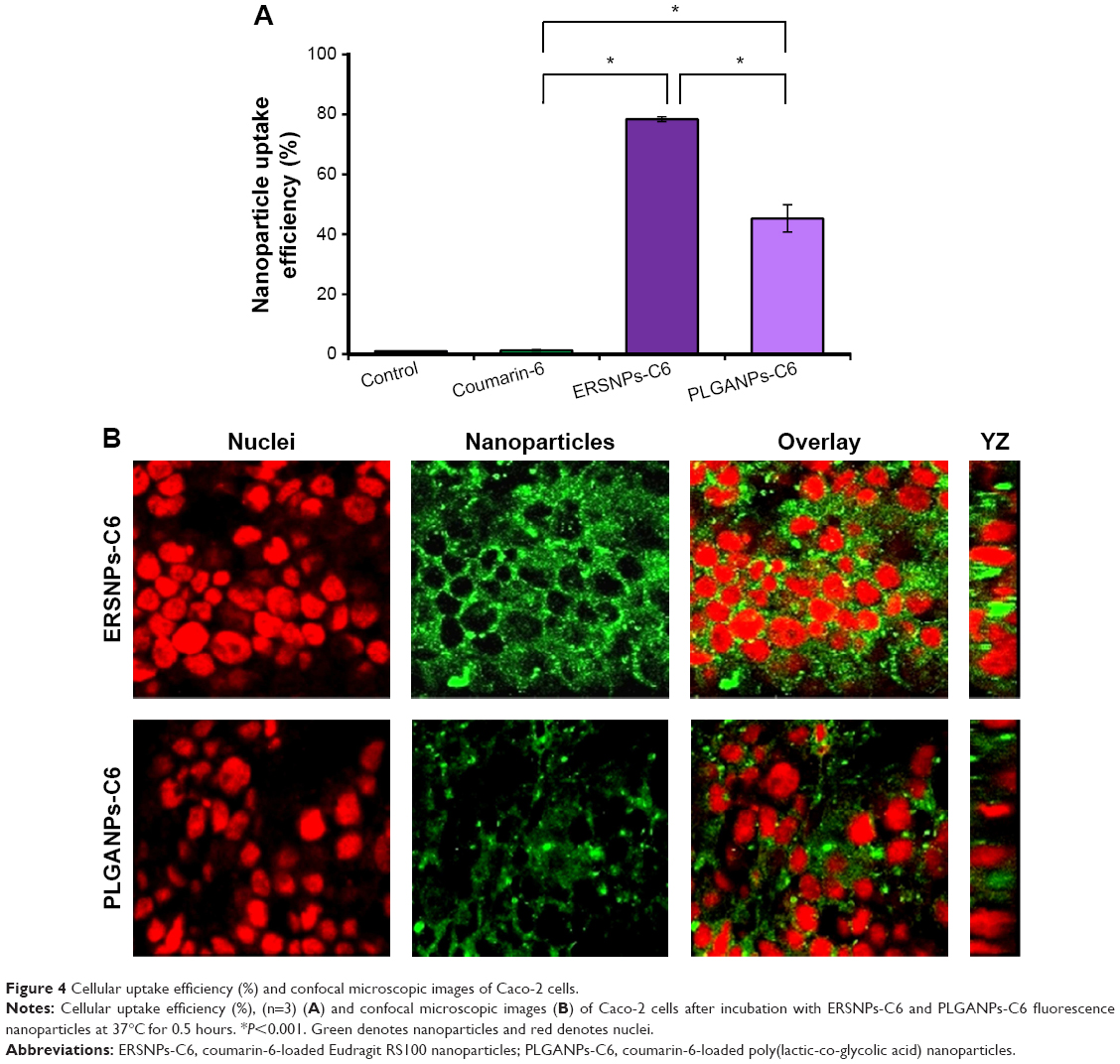 | Figure 4 Cellular uptake efficiency (%) and confocal microscopic images of Caco-2 cells. |
Pharmacokinetic study
Figure 5 shows the LPZ concentration versus time profiles after oral administration of ERSNPs-LPZ, PLGANPs-LPZ, and LPZ solution in Wistar rats with induced ulcers. The pharmacokinetic parameters are listed in Table 1. All LPZ was eliminated within 6 hours after oral administration of LPZ solution, while the nanoparticle formulations showed sustained and prolonged LPZ concentrations up to 24 hours. The elimination half-life and mean residence time for ERSNPs-LPZ (4.60±0.45 hours and 8.19±0.28 hours, respectively) and PLGANPs-LPZ (4.71±0.41 hours and 9.07±0.83 hours) were significantly longer than those for LPZ solution (1.36±0.24 hours and 1.94±0.44 hours; P<0.05). Meanwhile, both types of nanoparticles markedly reduced the peak plasma concentration of LPZ (475.3±37.5 ng/mL and 331.7±36.0 ng/mL) as compared with LPZ solution (2,338.3±255.3 ng/mL; P<0.05). Herein, ERSNPs-LPZ produced a comparable area under the curve from time zero to infinity (AUC0–∞) with LPZ solution. However, the AUC0–∞ of PLGANPs-LPZ was significantly lower than for LPZ solution and ERSNPs-LPZ (P<0.05). There were significant differences in AUC0–∞, peak plasma concentration, relative apparent volume of distribution (Vd/F), and relative clearance (CL/F) between the two types of nanoparticles (P<0.05). Since the AUC0–∞ of ERSNPs-LPZ was higher than that of PLGANPs-LPZ, the higher relative bioavailability of ERSNPs-LPZ resulted in the Vd/F and CL/F being significantly lower than for PLGANPs-LPZ.
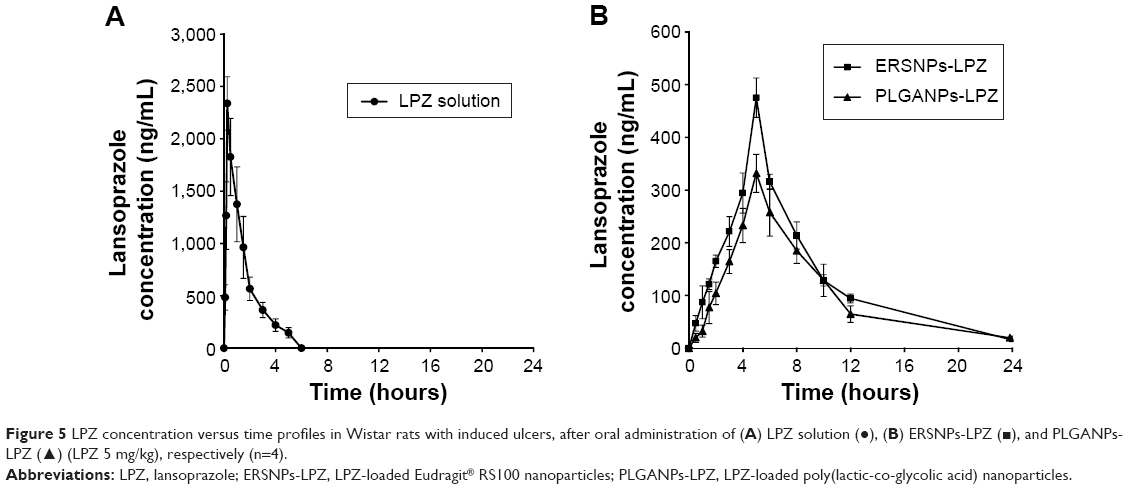 | Figure 5 LPZ concentration versus time profiles in Wistar rats with induced ulcers, after oral administration of (A) LPZ solution (•), (B) ERSNPs-LPZ (▪), and PLGANPs-LPZ (▴) (LPZ 5 mg/kg), respectively (n=4). |
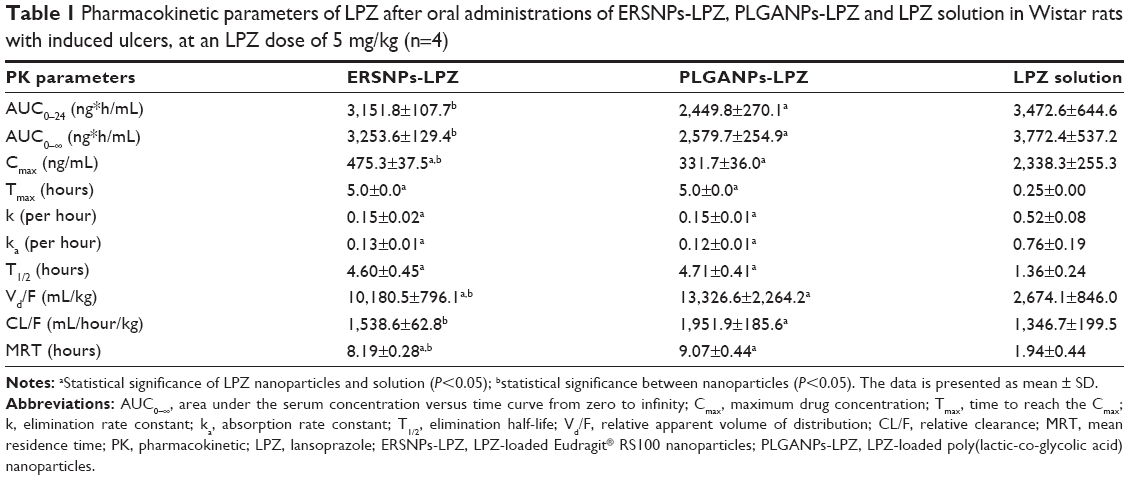 | Table 1 Pharmacokinetic parameters of LPZ after oral administrations of ERSNPs-LPZ, PLGANPs-LPZ and LPZ solution in Wistar rats with induced ulcers, at an LPZ dose of 5 mg/kg (n=4) |
It was reported that oral administration of Levant™ (an enteric-coated capsule containing LPZ 30 mg, Ranbaxy Ltd, London, UK) in healthy male subjects resulted in rapid LPZ absorption (mean peak plasma concentration 817±284 ng/mL; time taken to reach peak plasma concentration 2.72±0.87 hours; mean residence time 4.00±0.79 hours).31 A similar result was obtained following oral administration of the capsule filled with enteric-coated LPZ pellets in healthy Chinese male volunteers (peak plasma concentration 1,047±344 ng/mL; time taken to reach peak plasma concentration 2.0±0.7 hours; mean residence time 3.62±0.87 hours).32 Stefanič et al reported that oral absorption of dispersible tablets composed of enteric-coated LPZ pellets was completed in 6 hours in healthy subjects.33 Our nanoparticles provided sustained release of LPZ for up to 24 hours with a long mean residence time as compared with LPZ solution in an ulcer-induced Wistar rat model. In contrast with conventional enteric-coated LPZ formulations, the current nanoparticles have the potential to provide effective acid suppression, especially late in the day and throughout the night. It has been reported that higher LPZ blood levels are usually associated with a higher incidence of diarrhea as a side effect.34 The constant LPZ blood level delivered by these nanoparticles could also be of benefit in reducing such side effects.
Ulcer healing response
The ulcer healing efficacy of LPZ nanoparticles was further evaluated. Figure 6A shows photographic images of the stomach after oral administration of saline solution (control group), LPZ solution, ERSNPs-LPZ, and PLGANPs-LPZ, respectively, for 7 days in ulcer-induced Wistar rats. The corresponding gastric UI was 16.2±1.6, 4.5±1.0, 0.7±0.2, and 1.2±0.2 (Figure 6B). This result shows that the gastric ulceration gradually healed within 1 week after oral administration of LPZ solution and LPZ nanoparticles. Oral administration of LPZ solution for 7 days healed 72.2% of gastric ulcers as compared with the control group, while oral administration of ERSNPs-LPZ and PLGANPs-LPZ for 7 days healed 95.7% and 92.6% of gastric ulcers, respectively. The better ulcer healing efficacy of LPZ nanoparticles when compared to LPZ solution could be due to sustained release properties and the prolonged in vivo absorption characteristics of the nanoparticles which continuously suppressed acid secretion. Figure 7 shows the histopathology of healing ulcer tissue after oral administration of the four formulations. In the control group, the mucosal surface of the gastric ulcer was damaged and dilated gastric glands were spread across a large region of the ulcer. After oral administration of LPZ solution, granulation tissue developed during the ulcer healing process. Granulation tissue is an important component of healing which supplies connective tissue and microvessels in the ulcer scar.35 Oral administration of LPZ nanoparticles induced much more growth of granulation tissue than LPZ solution. All results confirmed the efficient ulcer healing effect of LPZ-loaded nanoparticles in an ulcer-induced animal model after 7 days of treatment. However, the possible absorption pathway(s) for oral administration of LPZ-loaded nanoparticles was/were not elucidated in this study, and should be investigated further.
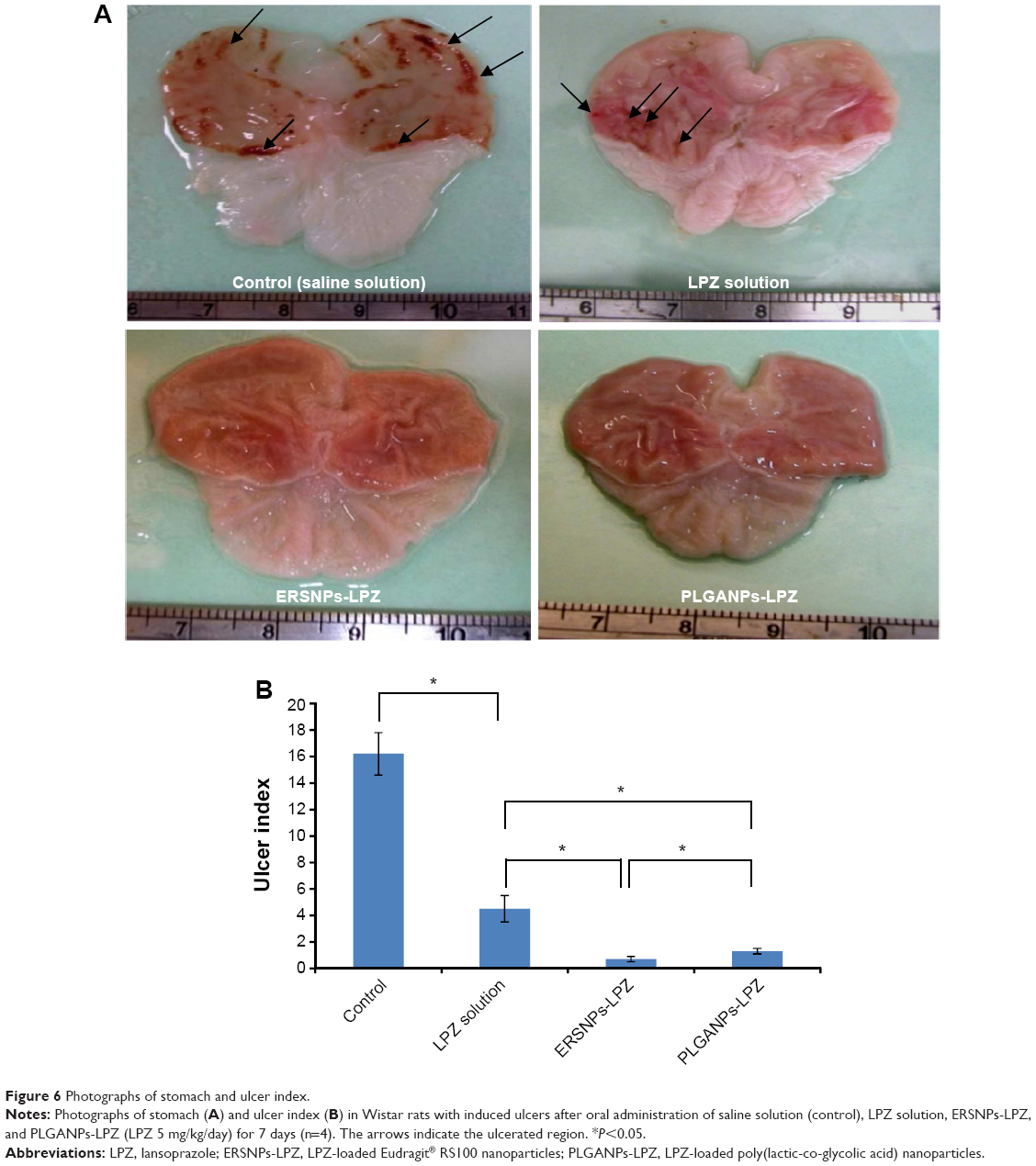 | Figure 6 Photographs of stomach and ulcer index. |
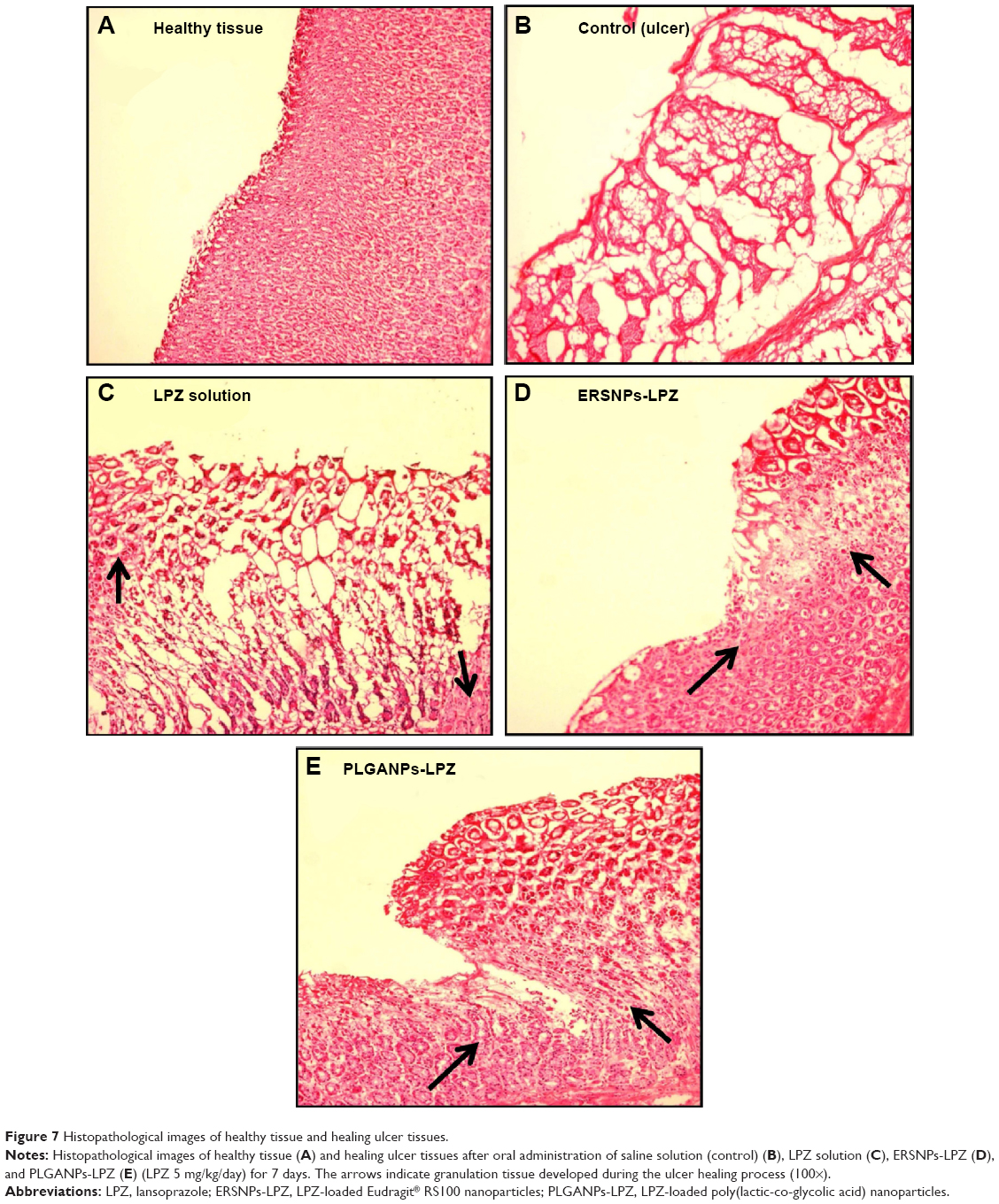 | Figure 7 Histopathological images of healthy tissue and healing ulcer tissues. |
Conclusion
Eudragit RS100 and PLGA nanoparticles were designed for oral delivery of LPZ as gastric ulcer therapy. The prepared LPZ-loaded nanoparticles had opposite surface charge and the particle size was around 200 nm with a narrow size distribution. The nanoparticles adhered and deposited in the stomach tissue were charge-dependent. The negatively charged PLGANPs adhered more readily to the ulcerated region whereas the positively charged ERSNPs preferred distribution in the non-ulcerated region of the stomach. Furthermore, the nanoparticles were able to internalize and localize in Caco-2 cells, where the positively charged ERSNPs enhanced cellular uptake more prominently than negatively charged PLGANPs. The nanoparticles allowed sustained and prolonged release of LPZ for up to 24 hours. Oral administration of LPZ-loaded nanoparticles for 7 days healed 92.6%–95.7% of gastric ulcers. Our results indicate that the nanoparticles developed in this study have the potential to release LPZ in a sustained manner and heal gastric ulcer effectively following oral administration.
Acknowledgments
This study was supported by the Ministry of Science and Technology in Taiwan (NSC 102-2320-B-002-007-MY3). The authors are grateful to Professor Wen Mei Fu (Department of Pharmacology, National Taiwan University, Taiwan) for lending the fluorescence microscope and to Dr Li-Juan Shen (School of Pharmacy, National Taiwan University, Taiwan) for donating the Caco-2 cells used in this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Matheson AJ, Jarvis B. Lansoprazole: an update of its place in the management of acid-related disorders. Drugs. 2001;61:1801–1833. | ||
Zhang D, Yang M, Liu M, et al. Pharmacokinetics of lansoprazole and its main metabolites after single intravenous doses in healthy Chinese subjects. Xenobiotica. 2012;42:1156–1162. | ||
Baldi F. Lansoprazole oro-dispersible tablet – pharmacokinetics and therapeutic use in acid-related disorders. Drugs. 2005;65(10): 1419–1426. | ||
Stedman CA, Barclay ML. Review article: comparison of the pharmacokinetics, acid suppression and efficacy of proton pump inhibitors. Aliment Pharmacol Ther. 2000;14:963–978. | ||
He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657–3666. | ||
Horn JR, Howden CW. Review article: similarities and differences among delayed-release proton-pump inhibitor formulations. Aliment Pharmacol Ther. 2005;22:20–24. | ||
Katz PO, Koch FK, Ballard ED, et al. Comparison of the effects of immediate-release omeprazole oral suspension, delayed-release lansoprazole capsules and delayed-release esomeprazole capsules on nocturnal gastric acidity after bedtime dosing in patients with night-time GERD symptoms. Aliment Pharmacol Ther. 2007;25:197–205. | ||
Wu ZM, Zhou LY, Guo XD, et al. HP55-coated capsule containing PLGA/RS nanoparticles for oral delivery of insulin. Int J Pharm. 2012;425:1–8. | ||
Adibkia K, Javadzadeh Y, Dastmalchi S, Mohammadi G, Niri FK, Alaei-Beirami M. Naproxen-Eudragit RS100 nanoparticles: preparation and physicochemical characterization. Colloids Surf B Biointerfaces. 2011;83:155–159. | ||
Devarajan PV, Sonavane GS. Preparation and in vitro/in vivo evaluation of gliclazide loaded Eudragit nanoparticles as a sustained release carriers. Drug Dev Ind Pharm. 2007;33:101–111. | ||
Dai J, Nagai T, Wang X, Zhang T, Meng M, Zhang Q. pH-sensitive nanoparticles for improving the oral bioavailability of cyclosporine A. Int J Pharm. 2004;280:229–240. | ||
Wang H, Jia Y, Hu W, Jiang H, Zhang J, Zhang L. Effect of preparation conditions on the size and encapsulation properties of mPEG-PLGA nanoparticles simultaneously loaded with vincristine sulfate and curcumin. Pharm Dev Technol. 2013;18:694–700. | ||
Cetin M, Atila A, Sahi S, Vural I. Preparation and characterization of metformin hydrochloride loaded-Eudragit®RSPO and Eudragit®RSPO/PLGA nanoparticles. Pharm Dev Technol. 2013;18:570–576. | ||
Collnot EM, Ali H, Lehr CM. Nano- and microparticulate drug carriers for targeting of the inflamed intestinal mucosa. J Control Release. 2012;161:235–246. | ||
Hassani S, Pellequer Y, Lamprecht A. Selective adhesion of nanoparticles to inflamed tissue in gastric ulcers. Pharm Res. 2009;26:1149–1154. | ||
Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35. | ||
Algieri C, Drioli E, Donato L. Development of mixed matrix membranes for controlled release of ibuprofen. J Appl Polym Sci. 2013;128:754–760. | ||
Alai MS, Lin WJ. A novel nanoparticulate system for sustained delivery of acid-labile lansoprazole. Colloids Surf B Biointerfaces. 2013;111:453–459. | ||
Raffin RP, Colome LM, Pohlmann AR, Guterres SS. Preparation, characterization, and in vivo anti-ulcer evaluation of pantoprazole-loaded microparticles. Eur J Pharm Biopharm. 2006;63:198–204. | ||
Ito Y, Arai H, Uchino K, Iwasaki K, Shibata N, Takada K. Effect of adsorbents on the absorption of lansoprazole with surfactant. Int J Pharm. 2005;289:69–77. | ||
Alai MS, Lin WJ. Novel lansoprazole-loaded nanoparticles for the treatment of gastric acid secretion-related ulcers: in vitro and in vivo pharmacokinetic pharmacodynamic evaluation. AAPS J. 2014;16:361–372. | ||
Johnson CE, Cober MP, Ludwig JL. Stability of partial doses of omeprazole-sodium bicarbonate oral suspension. Ann Pharmacother. 2007;41:1954–1961. | ||
Alai MS, Lin WJ. A novel once daily microparticulate dosage form comprising lansoprazole to prevent nocturnal acid breakthrough in the case of gastro-esophageal reflux disease: preparation, pharmacokinetic and pharmacodynamic evaluation. J Microencapsul. 2013;30: 519–529. | ||
Sun WZ, Lin WJ, Alai MS. Preparation of microparticles for acid-labile lansoprazole by solvent evaporation method combined with a spray drying process. J Food Drug Anal. 2012;20:438–445. | ||
Das S, Suresh PK, Desmukh R. Design of Eudragit RL 100 nanoparticles by nanoprecipitation method for ocular drug delivery. Nanomedicine. 2010;6:318–323. | ||
Ibrahim HK, El-Leithy IS, Makky AA. Mucoadhesive nanoparticles as carrier systems for prolonged ocular delivery of gatifloxacin/prednisolone bitherapy. Mol Pharm. 2010;7:576–585. | ||
Garcia X, Escribano E, Domenech J, Queralt J, Freixes J. In vitro characterization and in vivo analgesic and anti-allodynic activity of PLGA-bupivacaine nanoparticles. J Nanopart Res. 2011;13: 2213–2223. | ||
Sun B, Ranganathan B, Feng SS. Multifunctional poly(D,L-lactide-co-glycolide)/montmorillonite (PLGA/MMT) nanoparticles decorated by trastuzumab for targeted chemotherapy of breast cancer. Biomaterials. 2008;29:475–486. | ||
Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26:2713–2722. | ||
Dong Y, Feng SS. Poly(d,l-lactide-co-glycolide)/montmorillonite nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26:6068–6076. | ||
Vlase L, Popa A, Neag M, Muntean D, Leucuta SE. Effect of fluoxetine on the pharmacokinetics of lansoprazole: a two-treatment period study in healthy male subjects. Clin Drug Investig. 2011;31:727–733. | ||
Song M, Gao X, Hang TJ, Wen AD. Pharmacokinetic properties of lansoprazole (30-mg enteric-coated capsules) and its metabolites: a single-dose, open-label study in healthy Chinese male subjects. Curr Ther Res Clin Exp. 2009;70:228–239. | ||
Stefanič M, Locatelli I, Vrečer F, Sever T, Mrhar A, Bogataj M. The influence of gastric emptying kinetics on the drug release from enteric coated pellets in fasted state: an in vitro/in vivo correlation. Eur J Pharm Biopharm.2012;82:376–382. | ||
Capurso G, Marignani M, Attilia F, et al. Lansoprazole-induced microscopic colitis: an increasing problem? Results of a prospecive case-series and systematic review of the literature. Dig Liver Dis. 2011;43:380–385. | ||
Tarnawski AS. Cellular and molecular mechanisms of gastrointestinal ulcer healing. Dig Dis Sci. 2005;50:S24–S33. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.