Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Application of a diagnostic algorithm for the rare deficient variant Mmalton of alpha-1-antitrypsin deficiency: a new approach
Authors Belmonte I ![]() , Barrecheguren M, López-Martínez RM
, Barrecheguren M, López-Martínez RM ![]() , Esquinas C, Rodríguez E, Miravitlles M
, Esquinas C, Rodríguez E, Miravitlles M ![]() , Rodríguez-Frias F
, Rodríguez-Frias F ![]()
Received 27 June 2016
Accepted for publication 18 August 2016
Published 11 October 2016 Volume 2016:11(1) Pages 2535—2541
DOI https://doi.org/10.2147/COPD.S115940
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Video abstract presented by Irene Belmonte.
Views: 2032
Irene Belmonte,1,2 Miriam Barrecheguren,3 Rosa M López-Martínez,4 Cristina Esquinas,3 Esther Rodríguez,3,5 Marc Miravitlles,3,5 Francisco Rodríguez-Frías1,6
1Liver Pathology Unit, Department of Biochemistry and Microbiology, Hospital Universitari Vall d´Hebron, Universitat Autònoma de Barcelona (UAB), Barcelona, Spain; 2Vall d´Hebron Institut de Recerca (VHIR), Barcelona, Spain; 3Pneumology Department, Hospital Universitari Vall d’Hebron, Barcelona, Spain; 4Biochemistry Department, Hospital Universitari Vall d’Hebron, Barcelona, Spain; 5CIBER of Respiratory Diseases, Barcelona, Spain; 6CIBER of Liver and Digestive Diseases, Instituto Nacional de Salud Carlos III, Madrid, Spain
Background and objectives: Alpha-1-antitrypsin deficiency (AATD) is associated with a high risk for the development of early-onset emphysema and liver disease. A large majority of subjects with severe AATD carry the ZZ genotype, which can be easily detected. Another rare pathologic variant, the Mmalton allele, causes a deficiency similar to that of the Z variant, but it is not easily recognizable and its detection seems to be underestimated. Therefore, we have included a rapid allele-specific genotyping assay for the detection of the Mmalton variant in the diagnostic algorithm of AATD used in our laboratory. The objective of this study was to test the usefulness of this new algorithm for Mmalton detection.
Materials and methods: We performed a retrospective revision of all AATD determinations carried out in our laboratory over 2 years using the new diagnostic algorithm. Samples with a phenotype showing one or two M alleles and AAT levels discordant with that phenotype were analyzed using the Mmalton allele-specific genotyping assay.
Results: We detected 49 samples with discordant AAT levels; 44 had the MM and five the MS phenotype. In nine of these samples, a single rare Mmalton variant was detected. During the study period, two family screenings were performed and four additional Mmalton variants were identified.
Conclusion: The incorporation of the Mmalton allele-specific genotyping assay in the diagnostic algorithm of AATD resulted in a faster and cheaper method to detect this allele and avoided a significant delay in diagnosis when a sequencing assay was required. This methodology can be adapted to other rare variants. Standardized algorithms are required to obtain conclusive data of the real incidence of rare AAT alleles in each region.
Keywords: rare variant, emphysema, genotyping, phenotyping, serum levels
Introduction
Alpha-1-antitrypsin deficiency (AATD) is an autosomal recessive disorder caused by several mutations in the alpha-1-antitrypsin (AAT) gene (SERPINA1). The most important clinical manifestations of AATD include COPD in adults and liver disorders in children as well as in adults.1 Liver disease is mediated by aggregation of misfolded AAT molecules to polymers that accumulate within the hepatocytes.2 This results in a reduction in the circulating levels of AAT, an excess of neutrophil elastase (the main substrate for AAT), and therefore, pulmonary tissue destruction. The SERPINA1 gene is located on the long arm of chromosome 14 and is organized in three nonencoding (Ia, Ib, and Ic) exons and four (II, III, IV, and V) encoding exons.3 At least 120 variants of the SERPINA1 gene have been identified, and these variants are codominantly inherited and closely associated with specific serum AAT levels.
The most common normal AAT allele is the M variant, and the most frequent deficient variants in Caucasian populations are Z (G/A, Glu342Lys) and S (A/T, Glu264Val).4,5 Nevertheless, several AAT alleles, other than the Z and S variants, are associated with significantly reduced or absent serum AAT levels. These deficient variants are called “rare”, with little being known about their epidemiology and even less about the associated clinical phenotypes.6–8 The Mmalton variant is a rare deficiency variant that differs from the normal allele by deletion of the entire codon (TTC) for the residue Phe at position 51/52 (exon II). It is associated with severe AATD and hepatic inclusions due to the formation of polymers9,10 and is characterized by a normal isoelectrophoretic pattern,11 which may, at first, be confounded with an M protein.
In a retrospective review of AATD studies performed in our laboratory, a remarkably high frequency of the deficient I and Mmalton alleles was observed, which together account for 54% of all the rare AAT variants in Spain (34% for I and 20% for Mmalton).7 While the I variant causes moderate AATD (60%–70% of normal level), the Mmalton causes a deficiency similar to the Z allele and is also considered the most common cause of severe AATD in Sardinia (Italy)8 and Tunisia,12,13 where it accounts for 60% and 35% of the rare variants, respectively. Mmalton represents 8% of the rare variants in Switzerland.7
The current laboratory testing for AATD involves the determination of a combination of serum AAT levels, AAT phenotyping by isoelectrofocusing (IEF), and an allele-specific genotyping assay for detecting the most prevalent deficiency alleles (Z and S).14 Nevertheless, most rare variants can only be detected by molecular biology techniques, such as genome sequencing, which is not available in all routine laboratories.15 Moreover, since Mmalton and other rare variants are not easily recognizable, this may contribute to misclassification of many of these cases, with the subsequent underestimation of their true frequency.16 For this reason, in our laboratory, we have recently developed a rapid real-time polymerase chain reaction (PCR) and melting curves assay designed for the detection of the Mmalton allele.17 This method detects the Mmalton mutation by analysis of the melting point of one of the two adjacent fluorescently labeled probes. This last probe hybridizes over the mutation position and matches perfectly with the mutated sequence (deletion of an entire TTC codon). This technology is optimal for working with small amounts of DNA, such as dried blood spot (DBS) samples and even with residual DNA present in serum samples. This technology has been included in the diagnostic algorithm of AATD used in our laboratory, as follows: AAT levels are determined, and in cases with concentrations <1.16 g/L,18 the phenotype is determined by IEF. A specific range of AAT levels was established for each of the major phenotypes;19 therefore, if AAT values are in accordance with the phenotype observed, the laboratory results are considered definitive. If AAT levels are not within the range established for their phenotype and the phenotype contains one or two M alleles, the Mmalton genotype is determined by the allele-specific genotyping assay. In addition, S and Z alleles are determined by the allele-specific genotyping method in order to test possible S and Z variants not correctly characterized by phenotyping. If these variants are not detected, exonic sequencing of the SERPINA1 gene is performed.17 This method consists of amplification of the four encoding exons of the SERPINA1 gene by PCR followed by Sanger automated sequencing.
Serum samples are needed for the determination of AAT levels and IEF, while DNA extracted from whole blood samples is necessary for sequencing. Currently, DNA is usually obtained from DBS samples14,20 or even from serum samples (residual DNA). Nevertheless, when there is discordance between AAT levels and the IEF phenotype pattern, an additional whole blood or DBS sample is usually required for exonic sequencing of the SERPINA1 gene. This sample requirement represents an additional venopuncture or finger puncture resulting in a significant delay in AATD diagnosis. Besides, this additional sample can sometimes not be obtained for different reasons: the patient does not return to the doctor’s office for the results, delay between results and new blood extraction, or disabled individuals. Therefore, we have proposed a fast allele-specific genotyping assay for the detection of Mmalton, the most prevalent deficient rare variant in our population. For this purpose, the same serum sample used for the determination of AAT levels and phenotyping was processed. This methodology is applicable to all deficient alleles and can be adapted to other populations and their more prevalent rare variants.21
The aim of this study was to test the usefulness of this new algorithm for AATD Mmalton detection used in our laboratory. This is a retrospective review of all AATD determinations performed at the Clinical Laboratory of Hospital Vall d´Hebron (Barcelona) over the 2 years after this Mmalton allele-specific methodology became available.
Materials and methods
Clinical samples
We performed a retrospective revision of the results obtained in all the AATD studies carried out in our laboratory over a 2-year period. All samples were processed according to the following algorithm: AAT serum levels were determined and samples with AAT levels below the established cutoff (1.16 g/L) were characterized by phenotyping. In case of discrepancy, the Mmalton allele-specific genotyping assay was performed. All discordant AATD cases were analyzed by sequencing of the entire encoding region of the SERPINA1 gene.
Serum samples were used to complete the diagnostic algorithm except in the case of SERPINA1 sequencing in which whole blood samples were required.
The study was approved by the Ethics Committee of the Vall d’Hebron Hospital (Barcelona, Spain); as this was a retrospective study, and all samples were anonymized, the committee did not require informed consent. The study was conducted in accordance with the principles of the Declaration of Helsinki, Guidelines for Good Clinical Practice, and in full conformity with relevant national regulations.
Detection of Mmalton variants
AAT levels were determined in serum samples by nephelometry (Siemens, BNII Instrument, Munich, Germany). In order to facilitate the interpretation of the results, each laboratory has reference values for AAT concentrations in serum samples obtained from a healthy population.22
Phenotyping was performed using the semiautomatic Hydrasys System and the Hydragel 18 A1AT Isofocusing kit (Sebia, Evry, France),23 designed for the qualitative detection and characterization of the different AAT phenotypes.
Genotyping was performed when there was a discrepancy between AAT levels and the phenotype observed. In this study, we focused on the search for Mmalton variants. Therefore, the candidate samples to be analyzed by the Mmalton allele-specific genotyping assay were patients with a phenotype showing one or two M alleles, deficient AAT serum levels, and without other deficiency alleles detectable by phenotyping.
The protocol for Mmalton allele-specific genotyping has been described in detail elsewhere.17 In cases in which the Mmalton allele was not found, direct sequencing was performed after PCR amplification of all the encoding exons (II–V) using a whole blood sample.
Results
During the 24 months from January 2014 to December 2015, 654 subjects were screened. A total of 273 AAT deficiency studies were performed in our clinical laboratory in samples with serum AAT levels <1.16 g/L (range: 0.18–1.15 g/L) following the algorithm described earlier. All these samples were characterized by phenotyping. Samples with one or two M alleles and AAT levels discordant with that phenotype were analyzed using the Mmalton allele-specific genotyping assay. We detected 49 samples with these characteristics; 7.5% of the total samples and 18% of those with low AAT concentrations. Of these, 44 had the MM and five the MS phenotype. The AAT concentrations ranged from 0.46 g/L to 1.02 g/L. In nine of these samples, the rare Mmalton variant was detected in a heterozygous fashion (AAT levels ranging from 0.46 g/L to 0.9 g/L). Of these nine Mmalton heterozygote samples, eight also carried a normal M variant (0.6–0.9 g/L) and one sample carried an S allele (0.46 g/L). During the study period, family screenings of previously detected M/Mmalton and S/Mmalton individuals were performed. In these studies, two relatives of each patient were included in the diagnostic algorithm. This allowed the identification of four additional Mmalton variants in heterozygosity: three M/Mmalton and one S/Mmalton (value range: 0.56–0.86 g/L).
Of the 40 remaining deficient samples with the MM and MS phenotypes and without Mmalton alleles, we analyzed 24 by SERPINA1 exonic sequencing. The results showed 22 cases of MM without exonic mutations, with a range of serum concentrations of 0.64–1.02 g/L. Nineteen of these 22 samples had AAT serum levels close to the limit established (range of values: 0.96–1.02 g/L). We also detected two M/Null Mattawa (range of values: 0.68 g/L and 0.94 g/L). In the remaining 16 cases, we could not obtain a second sample to complete the study. The results are shown in Tables 1 and 2.
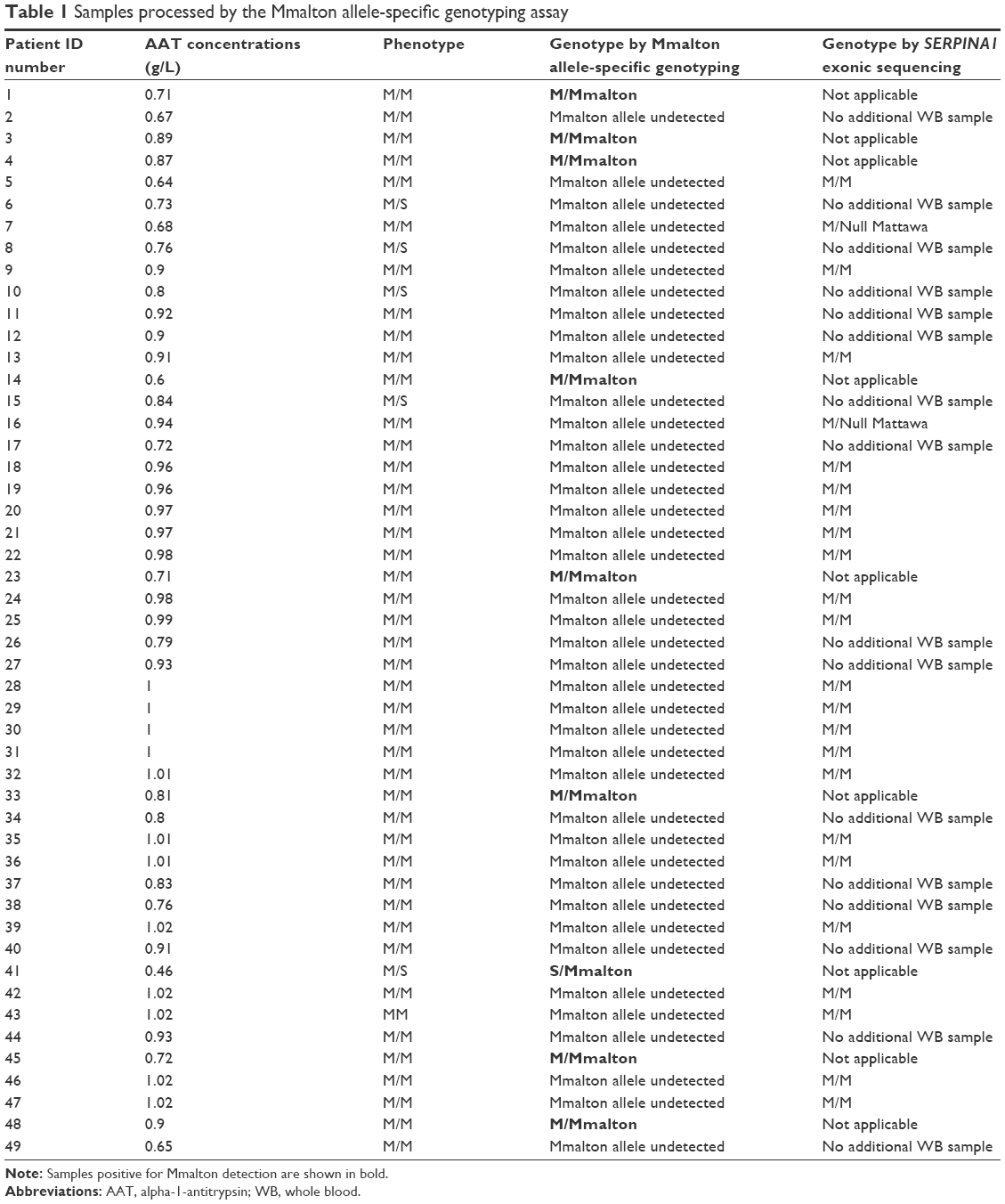 | Table 1 Samples processed by the Mmalton allele-specific genotyping assay |
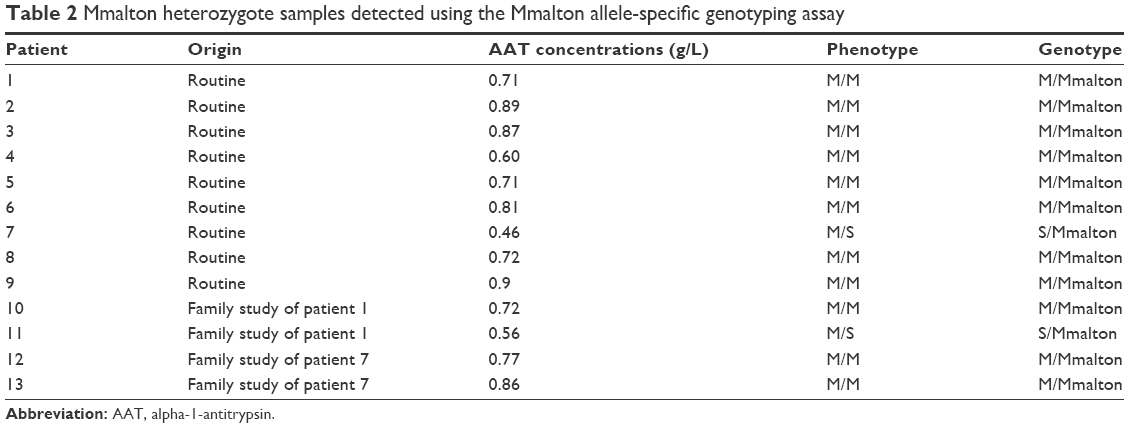 | Table 2 Mmalton heterozygote samples detected using the Mmalton allele-specific genotyping assay |
Discussion
We have introduced a new algorithm of AATD diagnosis including the Mmalton allele-specific genotyping assay. We determined the serum AAT concentrations and phenotypes in all the samples attended in the hospital with a request for AATD diagnosis over a period of 2 years. The discrepancy between the IEF phenotype and AAT serum levels led to suspicion of the presence of rare alleles in several cases.24,25 The Mmalton allele-specific genotyping assay was applied in samples that showed one or two M alleles and discordant AAT serum levels, and we detected this rare variant in 18% of the cases. Using this method, we avoided the sequencing technique, which is quite labor intensive and time consuming.
Little is known about the epidemiology of the non-S- and non-Z-deficient AAT variants, due to their low prevalence. A retrospective study of samples obtained in Barcelona from 1998 to 2010 showed that among these variants, Mmalton represented 20% of cases.7 It must be kept in mind that the Mmalton variant is difficult to interpret without molecular studies.26 For this reason, it is important to complement the diagnostic algorithm of AATD with simple molecular biology methods such as real-time PCR melting curves. This technique is considered one of the fastest and most sensitive and reproducible methods.20,27 Indeed, the extremely high sensitivity of this technology allows the analysis to be performed with the very low amount of DNA recovered from DBS samples or with the residual DNA that remains in standard clinical serum samples. The incorporation of the allele-specific genotyping assay for the Mmalton variant in the diagnostic algorithm of AATD results in a faster and cheaper approach to diagnose this rare allele. In addition, the use of the same serum samples available for the determination of AAT levels and IEF avoids a new appointment by the patient and an additional venopuncture. This practice allows a faster diagnosis and avoids the significant delay in AATD diagnosis when the SERPINA1 gene sequencing method is needed.
Population studies indicate that AATD is an underdiagnosed condition, and prolonged delays in diagnosis are common.28 Recommendations of the World Health Organization and the European Respiratory Society advocate screening programs for the detection of patients with AATD.29 In the Spanish population, there is a high frequency of I and rare Mmalton AAT variants,7 Mmalton being the more deficient than the I variant. In this case, the combination of this technique with the habitually used allele-specific genotyping assay for detecting the S and Z variants20 in large-scale screening programs may increase the number of individuals with AATD who are actually diagnosed with this disease in Spain. Genotyping using real-time PCR has the advantage that a definitive diagnosis is given, and it can be adapted for automation. Moreover, routine genotyping procedures may be cheaper than other methods for large screening programs. For this reason, it would be of interest to implement a simple allele-specific genotyping assay to study the most prevalent rare variants in each region. This could be done with the I variant in Ireland, in which this allele accounts for 90% of the total of rare variants,30 or in Finland, where the Mmalton variant has not been found, but the F allele is the most prevalent rare variant in the Finnish population.31,32 Other cases are certain regions of Italy, where Mmalton and Mprocida are more prevalent than Z.8 Bornhorst et al33 reported the M/I phenotype to be associated with AAT levels ranging from 0.80 g/L to 1.99 g/L, with this variant causing a moderate deficiency. However, the F variant presents AAT levels similar to the M variant but has reduced functional ability to inhibit neutrophil elastase compared to the normal allele.34 In our case, we found the Mmalton variant in combination with the M allele with AAT levels ranging from 0.6 g/L to 0.9 g/L. Most of the M/Mmalton individuals described have normal pulmonary functions, while Z/Mmalton patients often have a high risk of developing emphysema, especially smokers.35,36 Mprocida is another rare deficiency variant with IEF mobility similar to the M allele, which has been reported to produce 3%–4% of the normal amount of AAT.37,38
In this study, we have described a new diagnostic algorithm that has been used for 24 months with the samples received for AAT study. This new algorithm was computerized and provided definitive results even using serum samples. In this sense, it should be kept in mind that serum samples are used for routine determination of AAT levels and phenotyping. Therefore, the use of these samples for specific genotyping avoids the significant delay in diagnosis when a new whole blood sample is required for further molecular characterizations. A new appointment with the patient requires weeks or even months, while the use of the same serum sample significantly accelerates the routine diagnostic process. In fact, a significant number of patients did not provide a second sample for analysis in our study.
The detection of Mmalton variants is relevant because patients with homozygote Mmalton or Z/Mmalton genotypes with evidence of emphysema are candidates to receive augmentation therapy. Even some cases of S/Mmalton or in combination with other deficient alleles that show serum AAT levels <11 μM would fulfill the internationally accepted criteria for initiation of this therapy.29
Conclusion
The incorporation of the Mmalton allele-specific genotyping assay in the diagnostic algorithm of AATD could allow the clinical characterization of Mmalton individuals who, to date, are not well characterized. Our results show the strong need to develop standardized algorithms to obtain conclusive data of the real incidence of rare AAT variants in each country or region.
Acknowledgments
This study was supported in part by a grant from the Fundación Catalana de Pneumología (FUCAP 2014) and through funding from Grifols to the Catalan Center for Research in AATD of the Vall d’Hebron Research Institute in the Vall d’Hebron University Hospital, Barcelona, Spain.
Disclosure
The authors report no conflicts of interest in this work.
References
Carrell RW, Lomas DA, Sidhar S, Foreman R. Alpha 1-antitrypsin deficiency. A conformational disease. Chest. 1996;110(suppl 6):S243–S247. | ||
Lomas DA. Twenty years of polymers: a personal perspective on alpha-1 antitrypsin deficiency. COPD. 2013;10(suppl 1):17–25. | ||
Stoller JK, Aboussouan LS. Alpha1-antitrypsin deficiency. Lancet. 2005;365(9478):2225–2236. | ||
Janciauskiene S, Ferrarotti I, Laenger F, Jonigk D, Luisetti M. Clinical utility gene card for: α-1-antitrypsin deficiency. Eur J Hum Genet. 2011;19(5):1–3. | ||
de Serres FJ. Worldwide racial and ethnic distribution of alpha1-antitrypsin deficiency: summary of an analysis of published genetic epidemiologic surveys. Chest. 2002;122(5):1818–1829. | ||
Luisetti M, Ferrarotti I, Corda L, et al. Italian registry of patients with alpha-1 antitrypsin deficiency: general data and quality of life evaluation. COPD. 2015;12(suppl 1):52–57. | ||
Rodriguez-Frias F, Miravitlles M, Vidal R, Camos S, Jardi R. Rare alpha-1-antitrypsin variants: are they really so rare? Ther Adv Respir Dis. 2012;6(2):79–85. | ||
Ferrarotti I, Baccheschi J, Zorzetto M, et al. Prevalence and phenotype of subjects carrying rare variants in the Italian registry for alpha1-antitrypsin deficiency. J Med Genet. 2005;42(3):282–287. | ||
Joly P, Guillaud O, Hervieu V, Francina A, Mornex J-F, Chapuis-Cellier C. Clinical heterogeneity and potential high pathogenicity of the Mmalton Alpha 1 antitrypsin allele at the homozygous, compound heterozygous and heterozygous states. Orphanet J Rare Dis. 2015;10:130. | ||
Carroll TP, Connor CAO, Reeves EP, Mcelvaney NG. Alpha-1 Antitrypsin Deficiency – A Genetic Risk Factor for COPD. In: Ong KC, editor. Chronic Obstructive Pulmonary Disease – Current Concepts and Practice. Rijeka: InTech; 2012:179–198. | ||
Curiel DT, Holmes MD, Okayama H, et al. Molecular basis of the liver and lung disease associated with the alpha 1-antitrypsin deficiency allele Mmalton. J Biol Chem. 1989;264(23):13938–13945. | ||
Denden S, Zorzetto M, Amri F, et al. Screening for Alpha 1 antitrypsin deficiency in Tunisian subjects with obstructive lung disease: a feasibility report. Orphanet J Rare Dis. 2009;4:12. | ||
Denden S, Lakhdar R, Keskes NB, Hamdaoui MH, Chibani JB, Khelil AH. PCR-based screening for the most prevalent alpha 1 antitrypsin deficiency mutations (PI S, Z, and Mmalton) in COPD patients from Eastern Tunisia. Biochem Genet. 2013;51(9–10):677–685. | ||
Ferrarotti I, Scabini R, Campo I, et al. Laboratory diagnosis of alpha1-antitrypsin deficiency. Transl Res. 2007;150:267–274. | ||
Miravitlles M, Herr C, Ferrarotti I, et al. Laboratory testing of individuals with severe alpha1-antitrypsin deficiency in three European centres. Eur Respir J. 2010;35(5):960–968. | ||
Denden S, Lakhdar R, Leban N, Ben Chibani J, Haj Khelil A. Rapid genotyping of alpha 1 antitrypsin deletion mutation (PI*Mmalton) using bi-directional PCR allele-specific amplification. Mol Biotechnol. 2010;45(2):111–115. | ||
Belmonte I, Montoto L, Miravitlles M, et al. Rapid detection of Mmalton α1-antitrypsin deficiency allele by real-time PCR and melting curves in whole blood, serum and dried blood spot samples. Clin Chem Lab Med. 2016;54:241–248. | ||
Vidal R, Miravitlles M, Jardí R, et al. Estudio de la frecuencia de los diferentes fenotipos de la alfa-1-antitripsina en una población de Barcelona [Study of the frequency of different phenotypes of alpha-1-antitrypsin in a population of Barcelona]. Med Clin (Barc). 1996;107(6):211–214. | ||
Casas F, Blanco I, Martínez MT, et al. Indications for active case searches and intravenous alpha-1 antitrypsin treatment for patients with alpha-1 antitrypsin deficiency chronic pulmonary obstructive disease: an update. Arch Bronconeumol. 2015;51:185–192. | ||
Rodriguez F, Jardí R, Costa X, et al. Rapid screening for alpha1-antitrypsin deficiency in patients with chronic obstructive pulmonary disease using dried blood specimens. Am J Respir Crit Care Med. 2002;166(6):814–817. | ||
Seyama K, Nukiwa T, Takabe K, Takahashi H, Miyake K, Kira S. Siiyama (serine 53 (TCC) to phenylalanine 53 (TTC)). A new alpha 1-antitrypsin-deficient variant with mutation on a predicted conserved residue of the serpin backbone. J Biol Chem. 1991;266(19):12627–12632. | ||
Lara B, Miravitlles M. Spanish registry of patients with alpha-1 antitrypsin deficiency; comparison of the characteristics of PISZ and PIZZ individuals. COPD. 2015;12(suppl 1):S27–S31. | ||
Zerimech F, Hennache G, Bellon F, et al. Evaluation of a new Sebia isoelectrofocusing kit for alpha 1-antitrypsin phenotyping with the Hydrasys System. Clin Chem Lab Med. 2008;46(2):260–263. | ||
Lara B, Martínez-Delgado B, Torres ML, Marín-Arguedas S, Bustamante A, Miravitlles M. Alpha-1-antitrypsin deficiency associated with the Mattawa variant. Arch Bronconeumol. 2013;49(12):548–550. | ||
Rodríguez-Frías F, Vila-Auli B, Homs-Riba M, Vidal-Pla R, Calpe-Calpe JL, Jardi-margalef R. Diagnóstico del déficit de alfa 1-antitripsina: limitaciones de las pruebas de laboratorio de diagnóstico rápido [Diagnosis of alpha-1 antitrypsin deficiency: limitations of rapid diagnostic laboratory tests]. Arch Bronconeumol. 2011;47(8):415–417. | ||
Orru G, Faa G, Montaldo C, Pusceddu G, Piras V, Coni P. Rapid PCR real-time genotyping of M-malton a 1-antitrypsin deficiency alleles by molecular beacons. Diagnostic Mol Pathol. 2005;14(4):237–242. | ||
Kaczor MP, Sanak M, Szczeklik A. Rapid and inexpensive detection of alpha1-antitrypsin deficiency-related alleles S and Z by a real-time polymerase chain reaction suitable for a large-scale population-based screening. J Mol Diagn. 2007;9(1):99–104. | ||
Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest. 2005;128(4):1989–1994. | ||
American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900. | ||
Carroll TP, O’Connor CA, Floyd O, et al. The prevalence of alpha-1 antitrypsin deficiency in Ireland. Respir Res. 2011;12:91. | ||
Häggblom J, Kettunen K, Karjalainen J, Heliövaara M, Jousilahti P, Saarelainen S. Prevalence of PI*Z and PI*S alleles of alpha-1-antitrypsin deficiency in Finland. Eur Clin Respir J. 2015;2:28829. | ||
Arnaud P, Koistinen J, Wilson GB, Fudenberg HH. alpha-1-antitrypsin (Pi) phenotypes in a Finnish population. Scand J Clin Lab Invest. 1977;37(4):339–343. | ||
Bornhorst JA, Greene DN, Ashwood ER, Grenache GD. α1-Antitrypsin phenotypes and associated serum protein concentrations in a large clinical population. Chest. 2013;143(4):1000–1008. | ||
Sinden NJ, Koura F, Stockley RA. The significance of the F variant of alpha-1-antitrypsin and unique case report of a PiFF homozygote. BMC Pulm Med. 2014;14:132. | ||
Allen MB, Ward AM, Perks WH. Alpha 1 antitrypsin deficiency due to MMaltonZ phenotype: case report and family study. Thorax. 1986;41(7):568–570. | ||
Sproule BJ, Cox DW, Hsu K, Salkie ML, Herbert FA. Pulmonary function associated with the Mmalton deficient variant of alpha 1-antitrypsin. Am Rev Respir Dis. 1983;127(2):237–240. | ||
Takahashi H, Nukiwa T, Satoh K, et al. Characterization of the gene and protein of the alpha 1-antitrypsin deficiency allele Mprocida. J Biol Chem. 1988;263(30):15528–15534. | ||
Cox DW, Billingsley GD. Rare deficiency types of alpha 1-antitrypsin: electrophoretic variation and DNA haplotypes. Am J Hum Genet. 1989;44(6):844–854. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.