")
Back to Journals » International Journal of Nanomedicine » Volume 12
Apoferritin as an ubiquitous nanocarrier with excellent shelf life
Authors Dostalova S, Vasickova K , Hynek D, Krizkova S, Richtera L , Vaculovicova M, Eckschlager T, Stiborova M, Heger Z , Adam V
Received 15 December 2016
Accepted for publication 21 January 2017
Published 24 March 2017 Volume 2017:12 Pages 2265—2278
DOI https://doi.org/10.2147/IJN.S130267
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Simona Dostalova,1,2 Katerina Vasickova,1 David Hynek,1,2 Sona Krizkova,1,2 Lukas Richtera,1,2 Marketa Vaculovicova,1,2 Tomas Eckschlager,3 Marie Stiborova,4 Zbynek Heger,1,2 Vojtech Adam1,2
1Department of Chemistry and Biochemistry, Mendel University in Brno, 2Central European Institute of Technology, Brno University of Technology, Brno, 3Department of Pediatric Hematology and Oncology, Second Faculty of Medicine, University Hospital Motol, Charles University, 4Department of Biochemistry, Faculty of Science, Charles University, Prague, Czech Republic
Abstract: Due to many adverse effects of conventional chemotherapy, novel methods of targeting drugs to cancer cells are being investigated. Nanosize carriers are a suitable platform for this specific delivery. Herein, we evaluated the long-term stability of the naturally found protein nanocarrier apoferritin (Apo) with encapsulated doxorubicin (Dox). The encapsulation was performed using Apo’s ability to disassemble reversibly into its subunits at low pH (2.7) and reassemble in neutral pH (7.2), physically entrapping drug molecules in its cavity (creating ApoDox). In this study, ApoDox was prepared in water and phosphate-buffered saline and stored for 12 weeks in various conditions (-20°C, 4°C, 20°C, and 37°C in dark, and 4°C and 20°C under ambient light). During storage, a very low amount of prematurely released drug molecules were detected (maximum of 7.5% for ApoDox prepared in PBS and 4.4% for ApoDox prepared in water). Fourier-transform infrared spectra revealed no significant differences in any of the samples after storage. Most of the ApoDox prepared in phosphate-buffered saline and ApoDox prepared in water and stored at -20°C formed very large aggregates (up to 487% of original size). Only ApoDox prepared in water and stored at 4°C showed no significant increase in size or shape. Although this storage caused slower internalization to LNCaP prostate cancer cells, ApoDox (2.5 µM of Dox) still retained its ability to inhibit completely the growth of 1.5×104 LNCaP cells after 72 hours. ApoDox stored at 20°C and 37°C in water was not able to deliver Dox inside the nucleus, and thus did not inhibit the growth of the LNCaP cells. Overall, our study demonstrates that ApoDox has very good stability over the course of 12 weeks when stored properly (at 4°C), and is thus suitable for use as a nanocarrier in the specific delivery of anticancer drugs to patients.
Keywords: anticancer therapy, doxorubicin-loaded apoferritin, encapsulation, long-term stability, protein nanocarriers
Introduction
Doxorubicin (Dox), an anthracycline antibiotic discovered in 1969,1 is a potent chemotherapeutic drug used in the treatment of various solid and hematological malignancies. These include the most prevalent ones, such as breast,2 lung,3 or bladder4 cancer, but also less common ones, such as soft-tissue sarcoma5 or Hodgkin’s lymphoma.6 There are several mechanisms through which Dox achieves its high efficacy: intercalation into DNA,7 inhibition of topoisomerase II,8 and free-radical formation.9 These lead to apoptosis, necrosis, autophagy, or senescence. Which of these mechanisms will be the cause of cell death or cell-growth arrest is highly influenced by the individual patient, cell/cancer type, concentration of Dox, and the duration of treatment.10 Due to its high efficacy, Dox has been included in the biannual World Health Organization’s Model List of Essential Medicines since it was first published in 1977.11
The drawback of Dox usage, as well as any other conventional antitumor drugs, is its nonselectivity for cancer cells, resulting in high toxicity for nonmalignant cells. Dox in clinical practice severely affects the heart (causing cumulative dose-limiting cardiotoxicity), liver, and hematopoiesis.12 The consequences of this toxicity can be apparent immediately after administration of the drug, but they can also take many years to manifest.13 Dox causes the formation of reactive oxygen species, and iron oxidation induces a release of cytochrome C from mitochondria, leading to apoptosis.14 This is most prominent in the heart, where these mechanisms lead to cardiac hypertrophy, but they are also prominent in cells of brain and liver. Dox interference with mitochondrial complexes I and IV, which leads to superoxide increase and vitamin E and antioxidant decrease, has been shown to cause damage to glomeruli in rabbits and mice.15
Various approaches to mitigate these side effects have been taken. One approach is the coadministration of Dox with a cardioprotective agent.16 The most studied and clinically used cardioprotective agent is dexrazoxane. This reduces the reactive oxygen species formed by Dox activity, and serves as a chelator of iron. By combining these effects, it reduces the risk of heart failure by almost 80%.17 However, previous research has shown that dexrazoxane administration may increase the incidence of secondary malignancies.18
Another approach in mitigating the adverse effects is the encapsulation of Dox inside a suitable nanocarrier,19 allowing for targeted delivery to diseased tissue while avoiding healthy cells. These nanosized carriers can target diseased tissue and cells via passive targeting (due to their size)20 or active targeting (due to specific moieties on their surface).21 Various materials have been tested for targeted Dox delivery, including inorganic,22 polymeric,23 lipid-based,24 or proteins.25 The first US Food and Drug Administration-approved nanoformulation was liposome-encapsulated Dox (Doxil®/Caelyx® or Myocet®). This nanoformulation was proven to increase the therapeutic index of Dox by significant lowering its toxicity for normal cells.26
The major drawback of liposomal Dox is its short shelf life. While separated, Dox and liposomes can be stored for up to 18 months. The shelf life of liposome-encapsulated Dox, however, is only 5 days when stored at 2°C–8°C.27 Moreover, bare liposomes are often recognized by the mononuclear phagocyte system and removed from the organism, lowering their efficacy. Therefore, stealth coating with polyethylene glycol (PEGylation) is often used.28 However, PEGylated nanocarriers cause other adverse effects, such as swelling of palms and feet (palmar–plantar erythrodysesthesia).26
Due to these reasons, the potential use of alternative nanocarriers naturally present in the human body is being investigated. Nanocarriers prepared using ubiquitous proteins or protein cages appear to be a suitable alternative.25 We have previously employed an ubiquitous protein, apoferritin (Apo), to be a potential carrier for Dox, with a simple-to-use encapsulation protocol (creating ApoDox).29 Also, heavy-chain ferritin,30 as well as other ferritins, have been used for delivery of anticancer drugs,31 small nutrients,32 and imaging molecules.33 Subsequently, we modified the Apo surface with small molecules,34 as well as antibodies for active targeting to cells of a chosen cancer type. We proved that surface-modified ApoDox retained the high toxicity of Dox for targeted cancer cells while sparing 50% of nonmalignant cells.21
In this work, we evaluated the long-term stability of ApoDox. For 12 weeks, we continuously monitored changes in undesirable premature Dox release during storage, its optical properties, and the size and ζ-potential of the Apo nanocarrier, as well as its toxicity for prostate cell lines.
Materials and methods
Chemicals
All chemicals of American Chemical Society purity were obtained from Sigma-Aldrich (St Louis, MO, USA), unless otherwise stated. The pH was measured using an InoLab (Xylem, Rye Brook, NY, USA).
Encapsulation of Dox into Apo and its storage
9,600 μL of 1 mg·mL−1 DOX was added to 960 μL of 50 mg·mL−1 horse-spleen Apo and 4,800 μL of water, and 120 μL of 1 M hydrochloric acid was added to decrease the pH of the solution and disintegrate the Apo. The solution was mixed for 15 minutes, then 120 μL of 1 M sodium hydroxide was added to increase the pH and encapsulate the Dox inside Apo (creating ApoDox). The solution was mixed for 15 minutes and divided into two equal parts. The parts were diafiltered three times with water or phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 1.8 mM KH2PO4, and 4.3 mM Na2HPO4, pH 7.4), respectively, using an Amicon® Ultra (0.5 mL 3K; Merck Millipore, Billerica, MA, USA) at 6,000 g for 15 minutes and filled to 24,000 μL with the same solvent used for diafiltration. The samples were divided into 300 μL aliquots to avoid repeated freeze–thaw cycles and stored for 12 weeks at −20°C, 4°C, 20°C, and 37°C, and two of the samples stored at 4°C and 20°C were also stored under ambient light for evaluation of light influence on the stability of the sample.
Characterization of nanocarrier changes during storage
Every week of storage, aliquots from all storage conditions were collected, and prematurely released drug molecules were removed by diafiltration with the respective solvent using the Amicon Ultra at 6,000 g for 15 minutes. The amount of released drug was determined by measurement of free Dox fluorescence compared with fluorescence of the whole sample. Fluorescence measurement was performed using an Infinite 200 Pro (Tecan, Männedorf, Switzerland) with excitation wavelength of 480 nm and emission wavelengths of 515–815 nm. The encapsulated drug was evaluated by absorbance-scan measurement using the Infinite 200 Pro with wavelengths in the range of 230–850 nm.
Visualization of nanocarriers prior to removal of released drug molecules was performed using transmission electron microscopy (TEM) with negative staining technique. For this purpose, an organotungsten compound, Nano-W (Nanoprobes, Yaphank, NY, USA) was utilized. Then, 4 μL of samples was deposited onto 400-mesh copper grids coated with a continuous carbon layer. Dried grids were imaged by TEM (Tecnai F20; FEI, Hillsboro, OR, USA) at 80,000× magnification.
The average size of nanocarriers was determined by quasielastic dynamic light scattering with a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). Prior to removal of released drug, nanocarriers were diluted 100× with distilled water, placed into polystyrene latex cells, and measured at a detector angle of 173°, wavelength of 633 nm, and temperature of 25°C, with refractive index of dispersive phase 1.45 and 1.333 for dispersive environment. For each measurement, Zen0040 disposable cuvettes (Brand GmbH, Wertheim, Germany) were used, containing 50 μL of sample. Equilibration time was 120 seconds. Measurements were performed in hexaplicate.
The surface ζ-potential of the nanocarrier diluted 20× was measured using the Zetasizer Nano ZS. For each measurement, disposable cells (DTS1070) were employed. The number of runs varied between 20 and 40, and calculations considered the diminution of particle concentration based on the Smoluchowski model, with an F(ka) of 1.5 and an equilibration time of 120 seconds. Measurements were performed in triplicate.
Fourier-transform infrared (FT-IR) spectra were collected using a Nicolet iS10 with diamond attenuated total reflection (ATR) attachment (Thermo Fisher Scientific, Waltham, MA, USA). The sample solution was supplied dropwise (5 μL) on the diamond crystal of the ATR cell, and thin film was measured after spontaneous evaporation of the solvent. IR spectra were recorded from 650 to 4,000 cm−1 at a resolution of 2 cm−1. Each spectrum was acquired by adding together 64 interferograms. Spectra were acquired at 22°C for each sample. Omnic software was used for IR-spectra recording, and JDXView version 0.2 software was used for further spectra evaluation.
Influence of nanocarrier on prostate cancer cell line
The human prostate adenocarcinoma-derived cell line LNCaP, purchased from Public Health England (London, UK), was maintained in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum. Cells were incubated in 5% CO2 in air at 37°C. Cells (105) in 1 mL medium were seeded in each of six wells in 12-well culture plates and cultivated for 21 hours. After cultivation, the medium was discarded and replaced with 90 μL of fresh medium containing 34 μM Dox/ApoDox, followed by incubation for 2 hours. Live cells were washed with 200 μL of phosphate buffered saline (PBS) and stained with Hoechst 33342 and CellRox Green stains (Thermo Fisher Scientific) in dilutions of 1:2,000 and 1:500, respectively, and the cells were incubated for 30 minutes. Then cells were washed with 200 μL PBS and studied under inverted fluorescence microscopy (IX 71S8F-3; Olympus, Tokyo, Japan). Cell morphology was recorded under ambient light, the 4′,6-diamidin-2-fenylindol (DAPI) filter was used (excitation 360–370 nm, emission 420–460 nm, dichroic mirror 400 nm) for visualization of nuclei, redox stress was monitored using the fluorescein isothiocyanate filter (excitation 460–495 nm, emission 510–550 nm, dichroic mirror 505 nm), and Dox fluorescence was monitored using the Texas Red filter (excitation 545–580 nm, emission 610 nm, dichroic mirror 600 nm) at 200× magnification. Photographs were recorded, merged, and processed using Stream Basic software.
An xCelligence real-time cell analysis dual purpose instrument (Hoffman-La Roche Ltd, Basel, Switzerland) was used to evaluate the in vitro cytotoxicity of ApoDox after 12 weeks of storage at various conditions through real-time label-free monitoring of cell impedance. The background impedance signal was measured using 100 μL cell-culture media with ApoDox. Then, 1.5×104 LNCaP cells were seeded in each well of 16-well plates (E-plates; Hoffman-La Roche), followed by incubation at 37°C in atmosphere with 5% CO2. Final concentrations of ApoDox were 2.5 μM in 200 μL total volume in each well. Proliferation, spreading, and cell-attachment kinetics were monitored every 15 minutes for the first 26 hours, then every hour for 72 hours. Experiments were performed in two independent repetitions.
Descriptive statistics
Results are expressed as mean ± standard deviation, unless noted otherwise. Differences between groups were analyzed using Student’s paired t-test and analysis of variance. Unless noted otherwise, the threshold for significance was P<0.05. Statistica 12 software (Statistica, Tulsa, OK, USA) was employed for analyses.
Results and discussion
Experimental design
Ferritins are proteins that serve as one of the detoxifying factors, preventing the formation of reactive oxygen species by transforming ferrous ions into their insoluble ferric form.35 Subsequently, they reversibly store these ferric ions and can transfer them to the site of action.36 Ferritins are found in most organisms and bacteria, including humans,37 and there is very high (85%–90%) interspecies homology of ferritin sequences from the same tissue with low (40%–50%) intraspecies homology of ferritins from different tissues.38
Without the iron content, ferritins self-assemble into hollow rhombic dodecahedral cage Apo, with outer diameter of 12–13 nm and inner diameter of 7–8 nm. The 450–475 kDa shell39 is composed of 24 heavy and light subunits. Horse-spleen Apo (450 kDa), the best-studied Apo,38 can reversibly dissociate and associate based on the surrounding pH.40 It has been shown that while disassembled, Apo can be mixed with drug molecules, and these are encapsulated within the Apo cavity once reassembled.29,34,41
This easy-to-use encapsulation protocol was employed in this work. Figure 1 shows the design of the whole experiment. Horse-spleen Apo was mixed with Dox at a 1:155 molar ratio. The pH was lowered from 6.7 to 2.7 to disassemble the Apo structure, and the mixture was shaken for 15 minutes to create a homogeneous solution. After mixing, the pH was returned back to neutral (7.2), and the mixture was incubated for a further 15 minutes to enable the reassembly of the Apo structure and encapsulation of Dox molecules within (creating ApoDox).
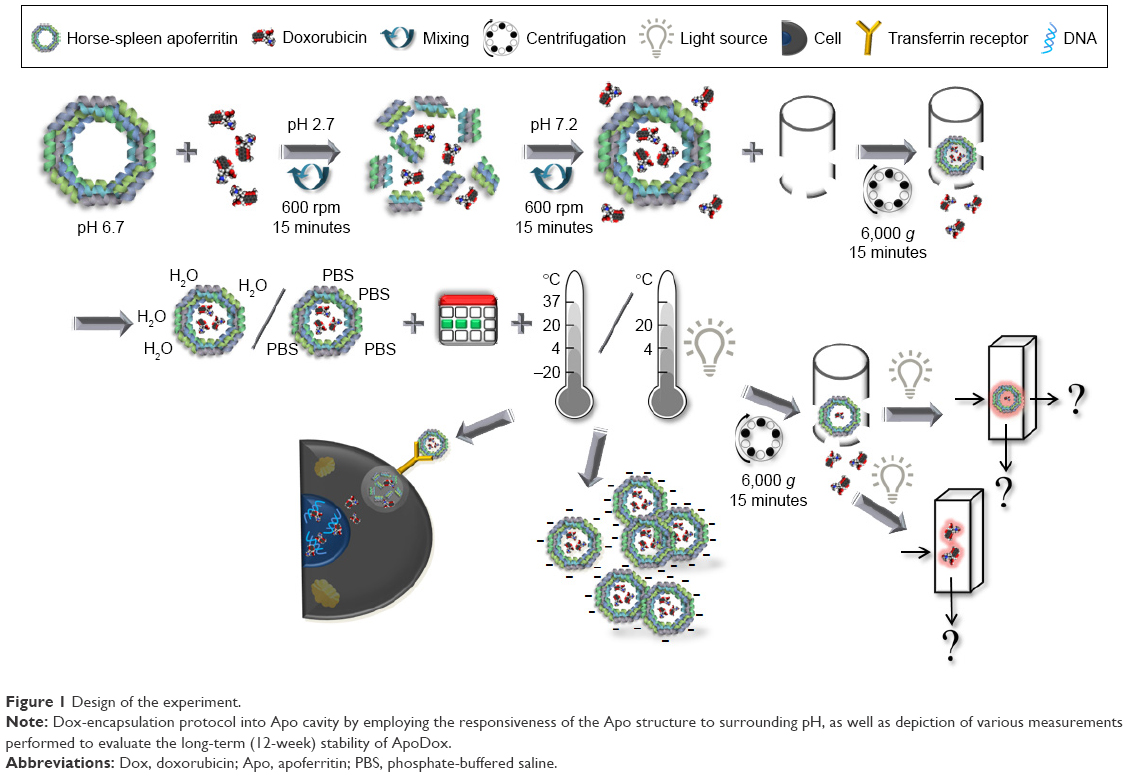 | Figure 1 Design of the experiment. |
The aim of this study was to evaluate the long-term stability of ApoDox in various conditions. For this purpose, free Dox molecules were removed from ApoDox by diafiltration and filled to 1 mg·mL−1 of Apo with water (final pH 5.6) or PBS (final pH 7.0), which was used as a model of the physiological environment. Prepared samples were aliquoted to avoid repeated freeze–thaw cycles and excess protein loss caused by repeated diafiltration. These aliquots were stored for 12 weeks in the dark and at various temperatures: −20°C, 4°C, 20°C, and 37°C. Since free Dox molecules are degradable by light,42 in addition to storage in dark, aliquots stored at 4°C and 20°C were also under direct ambient light to evaluate the influence of illumination during storage on the stability of ApoDox.
Each week of storage, aliquots were collected from the various storage conditions and subjected to multiple analyses. Prematurely released Dox was removed by diafiltration, and the amount of both encapsulated and released Dox was measured using its unique optical properties. Changes in size and surface ζ-potential of nanocarriers were also evaluated each week, as well as their ability to enter cells and release the Dox cargo within them for therapeutic activity.
Optical properties of encapsulated and prematurely released Dox
Dox structure is mainly formed by anisole, quinone, and hydroquinone, along with several residues, such as the amine group, ketone groups, and hydroxyl groups.43 The resulting conjugated system of bonds in Dox tetracene structure with partially disrupted aromaticity causes typical Dox absorbance, with a maximum at approximately 480 nm, as well as its fluorescence, with emission maximum at approximately 600 nm.44
Changes in optical properties of an encapsulated drug, both absorbance and fluorescence, can be caused by different amounts of prematurely released drug, changes in the molecular structure of the drug, or even the structure of the nanocarrier. Absorbance (Figure 2A and C) and fluorescence (Figure 2B and D) of the encapsulated drug in nanocarriers prepared in water (Figure 2A and B) and PBS (Figure 2C and D) were collected every week. The observed changes were rapid, and did not significantly change on a week-to-week basis. The results are thus presented as average change throughout the 12-week period.
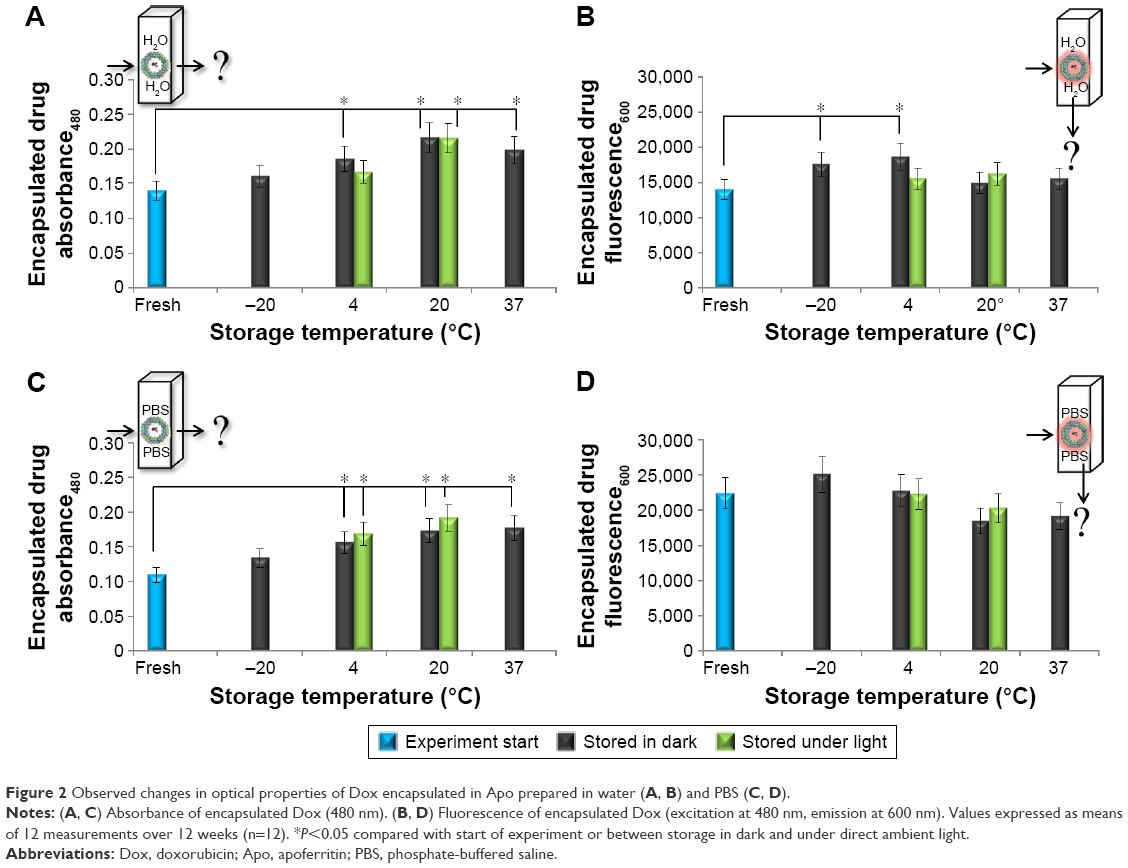 | Figure 2 Observed changes in optical properties of Dox encapsulated in Apo prepared in water (A, B) and PBS (C, D). |
Significant (P<0.05) increases in encapsulated drug absorbance were observed in samples stored at most storage temperatures and solutions. No significant changes were observed between samples stored in dark and under ambient light. At the start of the experiment, absorbance of the encapsulated drug was 0.14 AU for ApoDox prepared in water and 0.11 AU for ApoDox prepared in PBS. The highest absorbance change for nanocarriers prepared in water compared with the state at the start of the experiment was observed for ApoDox stored at 20°C (55% increase, both in dark and under ambient light) and 37°C (42% increase). ApoDox stored at 4°C showed a 33% increase in encapsulated drug absorbance when stored in dark and a nonsignificant 19% increase when stored under ambient light. ApoDox stored at −20°C showed 15% (insignificant) increase in encapsulated drug absorbance.
Absorbance changes for ApoDox prepared in PBS followed the same trend as those prepared in water when compared with freshly prepared ApoDox. The highest absorbance change was again observed for ApoDox stored at 20°C (58% increase for storage in dark and 75% increase for storage under light) and 37°C (62% increase). ApoDox stored at 4°C also showed high change with 42% increase when stored in dark and 54% increase when stored under direct ambient light. ApoDox stored at −20°C showed an insignificant 22% increase in encapsulated drug absorbance. These changes in drug absorbance could account for decreased amounts of prematurely released drug or some structural changes.
Almost no significant (P<0.05) changes were observed in encapsulated drug fluorescence. At the start of the experiment, the fluorescence of the encapsulated drug was 13,936 AU for ApoDox prepared in water and 22,333 AU for ApoDox prepared in PBS. The only significant increases (compared with freshly prepared samples) for ApoDox prepared in water were observed for ApoDox stored at 4°C (34% for storage in dark and nonsignificant 11% for storage under light) and −20°C (25%). ApoDox stored at 37°C showed a nonsignificant 11% increase in encapsulated drug fluorescence, and ApoDox stored at 20°C showed nonsignificant changes, with 9% increase for storage in dark and 16% increase for storage under ambient light. The increased fluorescence could also account for decreased amounts of prematurely released drug or could indicate some structural changes in the nanocarrier.
Encapsulated drug fluorescence for nanocarriers prepared in PBS did not significantly increase in any of the tested temperatures, contrary to the results from ApoDox prepared in water. The highest fluorescence increase for nanocarriers prepared in PBS was observed for ApoDox stored at −20°C (12%). ApoDox stored at 20°C and 37°C showed a slight decrease in fluorescence (18% for storage in dark and at 20°C and 15% for storage at 37°C). These results did not correspond to the results from encapsulated drug absorbance.
To help explain observed changes, the amount of prematurely released drug from nanocarriers stored under different conditions was evaluated each week. For this evaluation, only Dox fluorescence was employed, since the concentration of the released drug was too low for absorbance detection. The observed changes did not significantly change on a week-to-week basis. The results are thus presented as an average percentage of released drug molecules throughout the 12 week period. Figure 3 shows the percentage of released drug from nanocarriers prepared in water (Figure 3A) and PBS (Figure 3B). Premature release of cargo molecules, whether in patient organism or during storage, is one of the most undesirable properties of a nanocarrier, since it can lead to increased toxicity for healthy cells. Lower premature drug release decreases the nonspecific interactions of the drug molecules with these healthy cells.45
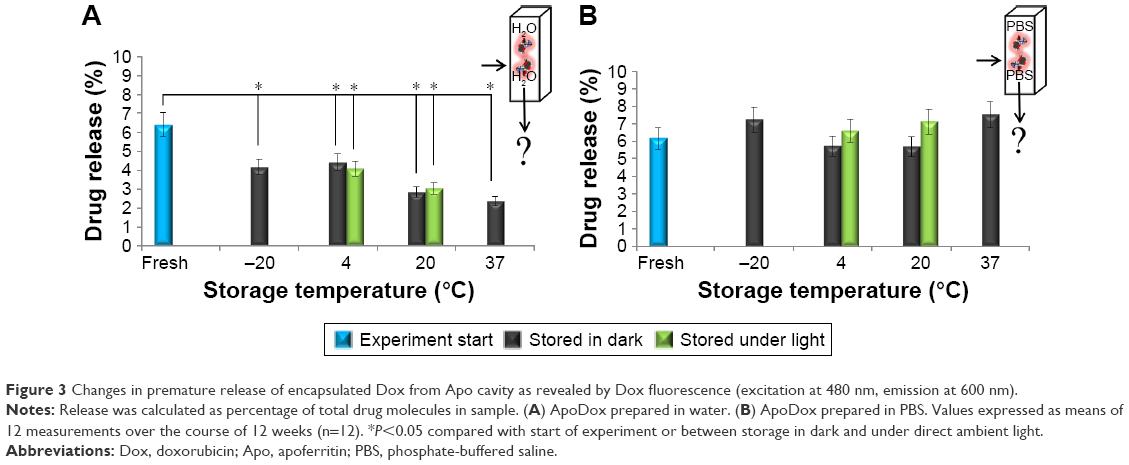 | Figure 3 Changes in premature release of encapsulated Dox from Apo cavity as revealed by Dox fluorescence (excitation at 480 nm, emission at 600 nm). |
All samples prepared in water and stored at all storage temperatures showed significant (P<0.05) decreases in the amount of prematurely released drug compared to the freshly prepared sample, which is highly favorable. No significant influence of light was detected at the start of the experiment, the percentage of unencapsulated drug was 6.4% for ApoDox prepared in water. The lowest undesired drug release after storage was observed for ApoDox stored at 37°C (2.4%) and 20°C (2.8% for samples stored in dark and 3% for samples stored under ambient light). ApoDox stored at −20°C showed 4.2% release, and ApoDox stored at 4°C showed 4.4% release. The decrease in the prematurely released drug correlated with the observed higher absorbance and fluorescence values of encapsulated drug for samples stored in water (Figure 2).
On the contrary, none of the samples prepared in PBS showed any significant (P<0.05) changes in the amount of prematurely released drug, which is in contrast to the results from encapsulated drug absorbance and correlates with encapsulated drug fluorescence for samples stored in PBS (Figure 2B and D). At the start of the experiment, the percentage of unencapsulated drug was 6.1% for ApoDox prepared in water. The only samples that showed slight decrease in the amount of prematurely released drug were ApoDox stored at 4°C and 20°C, both in dark (5.7%). The highest increase in the amount of prematurely released drug was observed for ApoDox stored at 37°C (7.5%). ApoDox stored at −20°C showed 7.2% release, ApoDox stored under ambient light at 20°C showed 7.1% release, and ApoDox stored under ambient light at 4°C showed 6.6% release. Overall, our premature-release results indicate that the encapsulation of Dox in Apo is stable, with very low premature release over the course of 12 weeks.
Structural changes in ApoDox
Size, shape, and surface charge are among the most important parameters influencing the in vivo biodistribution of nanoparticles.46 With nanocarriers of suitable size (20–100 nm), it is possible to utilize fully the enhanced permeability and retention (EPR) effect47 while avoiding extravasation from normal blood vessels (for particles below 10 nm)48 and removal from body through renal clearance (for particles below 5 nm) or elimination by the reticuloendothelial system (RES, for particles larger than 100 nm).49 The EPR effect is caused by 1) tumor angiogenesis, where the newly formed blood vessels are underdeveloped and contain large pores, making them leaky, thus increasing accumulation of nanoparticles in tumor tissue; and 2) lack of lymphatic vessels in tumors, thus increasing retention of nanoparticles in the tissue.50 Moreover, there are various modes of entry to cells with which nanoparticles can engage, ie, clathrin-or caveolae-mediated endocytosis, phagocytosis, or macropinocytosis. The size of these nanoparticles has a great impact on the mode of utilized cellular internalization, and thus which microenvironment the nanoparticles will face upon internalization.46
Nanoparticle shape can also greatly influence both internalization in cells and removal by the RES. Macrophages rapidly internalize exogenous spherical nanoparticles, shortening their circulation time, and preventing their accumulation in the site of action. In the case of elliptical nanoparticles, the same internalization is observed only if the contact with nanoparticles was along their major axis, whereas contact along the minor axis meant prolonged circulation time and thus higher chance of accumulation in tumors.46 Cubic shape favorably influences internalization to target cells, where HeLa cells have been shown to be able to internalize 3 μm cubic nanoparticles, even though they are technically unable to perform phagocytosis.51
Surface charge influences the internalization of nanoparticles in cells, but its neutralization (as well as increased hydrophilicity) can also slow the opsonization process and removal by the RES.46 The higher the charge of nanocarriers is (both cationic and anionic), the easier they are opsonized and removed from the circulation.52 Also, positively charged nanocarriers induce opsonization more than negatively charged nanocarriers. In contrast, negatively charged nanocarriers are unable to enter cells via micropinocytosis, and have to be internalized using other forms of endocytosis.53
One of the reasons Apo was chosen in this experiment was its ubiquitous presence in nature and natural property to self-assemble into uniform icosahedral nanocages that are highly stable and do not form aggregates in the physiological environment.38 However, Dox was found to form aggregates containing up to 40 molecules at 0.5 mg·mL−1. This aggregation is highly dependent on Dox concentration (aggregates of more Dox molecules with higher concentration) and its charge (protonated Dox creates fewer aggregates than neutral Dox).54 The isoelectric point of Dox has been found to be 8.8.55
Changes in shape and size of ApoDox nanocarriers were observed using TEM, and weekly changes in surface ζ-potential were investigated using dynamic light scattering. Figure 4 shows the average changes in shape and ζ-potential for nanocarriers stored in water (Figure 4A) and PBS (Figure 4B).
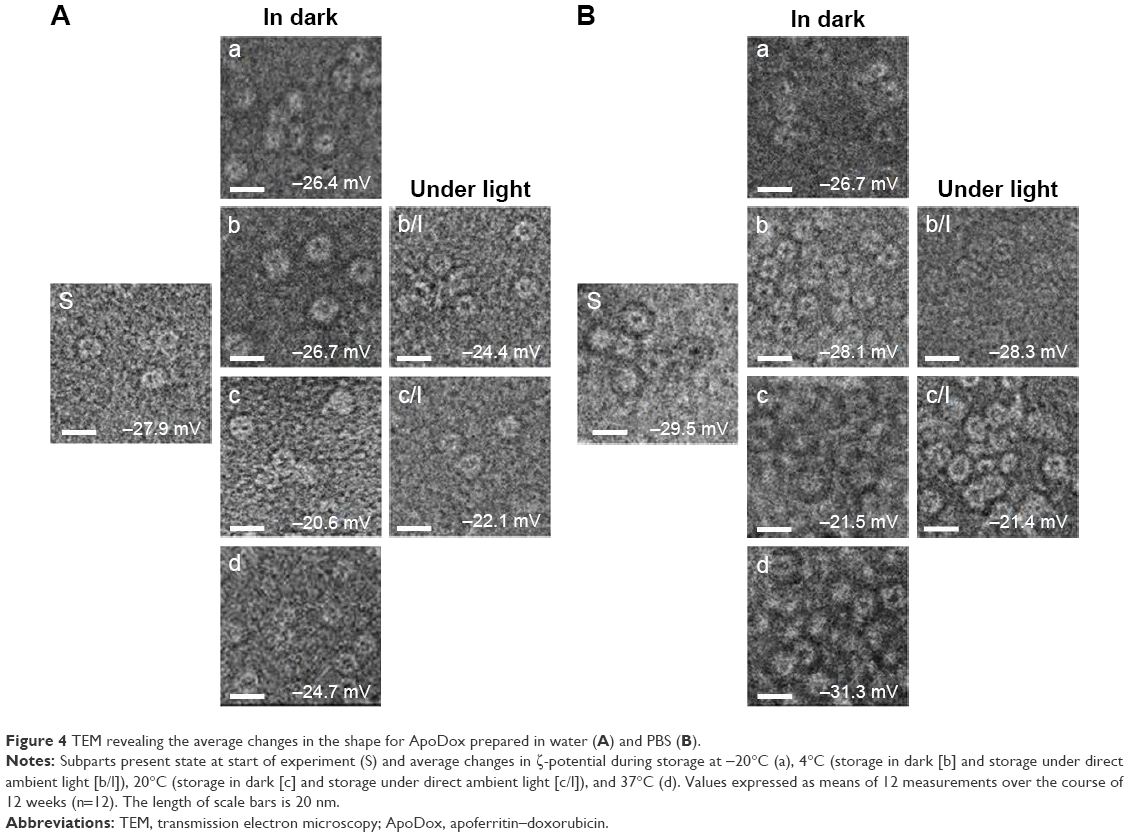 | Figure 4 TEM revealing the average changes in the shape for ApoDox prepared in water (A) and PBS (B). |
At the start of the experiment, ApoDox stored in water (Figure 4A, pH 5.6) showed individual Apo nanocarriers with the cavity filled with Dox. Samples stored for 12 weeks at 4°C and in dark (pH 5.7) still showed individual nanocarriers with no change in shape or size, while samples stored at 4°C under light (pH 5.4) showed aggregation of multiple nanocarriers, changing both the size and shape of the nanocarrier, while other nanocarriers remained individual. Most of the ApoDox nanocarriers stored at −20°C (pH 6) formed large multinanocarrier aggregates, changing shape from regular, icosahedral particles to irregular particles. This was probably caused by the higher amount of neutral Dox molecules. ApoDox stored at 20°C (both in dark [pH 5.5] and under light [pH 5.4]), as well as ApoDox stored at 37°C (pH 5.6), also showed formation of aggregates, while some nanocarriers remained individual. These aggregates were probably caused by Dox instability at increased temperatures.56 The ζ-potential of samples stored at most storage conditions remained similar to that measured in freshly prepared samples, with the exception of samples stored at 20°C, where negative ζ-potential was lowered from −27.9 mV to −20.6 mV (for storage in dark) and −22.1 mV (for storage under light), showing lower stability.
ApoDox stored in PBS (Figure 4B, pH 7.0) formed aggregates even at the start of the experiment, which could again have been caused by a presence of neutral Dox on the surface of nanocarriers, connecting multiple nanocarriers together. These aggregates remained similar to those found at the start of the experiment in the case of storage at −20°C (pH 6.9). It can be seen that storage at other temperatures (pH 7) caused formation of even larger aggregates of many nanocarriers with a very low number of individual nanocarriers. Such formation of large aggregates explains the increased absorbance of encapsulated drug observed in the previous part of the experiment. The ζ-potential of samples stored under most storage conditions remained similar to that measured in freshly prepared samples, with the exception of samples stored at 20°C, where the negative ζ-potential was lowered from −29.5 mV to −21.5 mV (for storage in dark) and −21.4 mV (for storage under light), showing lower stability of the nanocarrier.
We further investigated changes in nanocarrier size. The observed changes were again rapid, and did not significantly change on a week-to-week basis. The results are thus presented as average change throughout the 12-week period (Figure 5A and C). Also, ATR FT-IR spectra of the nanocarriers were collected (Figure 5B and D).
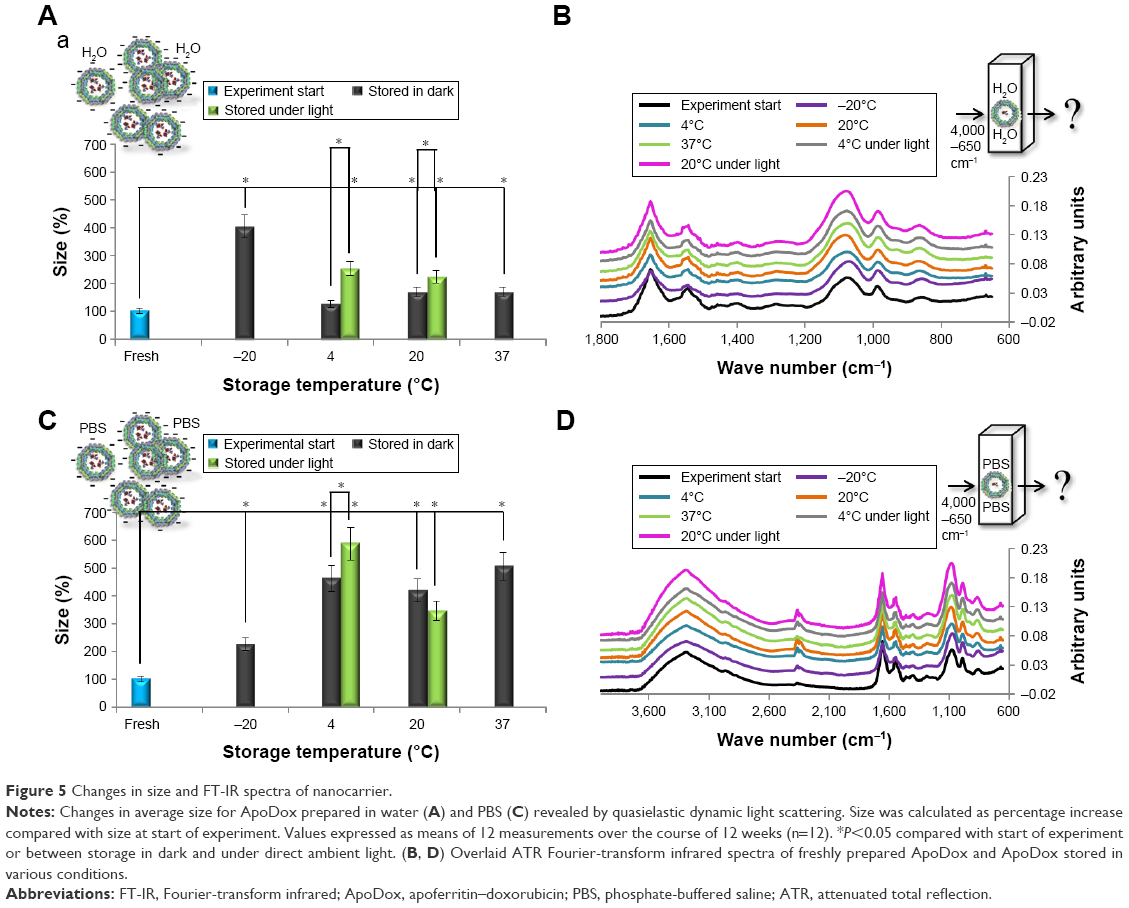 | Figure 5 Changes in size and FT-IR spectra of nanocarrier. |
There was a significant (P<0.05) increase in size of ApoDox nanocarriers in samples stored at most storage temperatures and solutions, as well as between most samples stored in dark and under ambient light. The highest change in average size for ApoDox prepared in water (Figure 5A) was observed in ApoDox stored at −20°C (304% increase in size), corresponding to the changes observed using TEM. ApoDox stored at 4°C and in dark showed no significant increase in size, while samples stored under ambient light showed 152% increase in size. ApoDox stored at 20°C showed 67% increase in size while stored in dark and 123% increase in size while stored under ambient light. ApoDox stored at 37°C showed 68% increase in size. ATR FT-IR spectra showed no changes in any of the ApoDox samples stored at various conditions (Figure 5B).
Significant (P<0.05) increase in size was observed in all samples prepared in PBS (Figure 5C). The highest increase was observed for ApoDox stored at 4°C (362% for storage in dark and 487% for storage under ambient light). ApoDox stored at −20°C showed 126% increase in size, while ApoDox stored at 20°C showed 318% (stored in dark) and 244% (stored under light) increase in size. ApoDox stored at 37°C showed 405% increase in size. ATR FT-IR spectra showed no changes in any of the ApoDox samples stored in various conditions (Figure 5D). The large differences observed between samples stored in dark and under ambient light could have been caused by photodegradation of Dox molecules.42 These molecules were shown to be encapsulated not only within the Apo cavity (up to 28 Dox molecules per Apo molecule)41 but also attached by Dox–Apo π–π interactions on the external surface of Apo.43 The degraded Dox products could have been one of the reasons for the formation of the observed aggregates.
In vitro assessment of differently stored ApoDox influence on cancer cells
Probably the most important property of a nanocarrier is its ability to internalize into target cells and the ability of its cargo to reach the organelle where it can effectively inhibit cell growth.53 In our previous work, we proved that the encapsulation of Dox inside Apo has similar influence on cancer cells as free Dox, whereas normal, noncancer cells are significantly protected from Dox influence.21 Therefore, in the present experiment we tested the internalization of ApoDox stored at various conditions using only the androgen-dependent metastatic prostate cancer cell line LNCaP.
LNCaP cells were treated with Dox, either free or encapsulated in freshly prepared ApoDox, or ApoDox stored at various conditions. After 2 hours of treatment, nuclei were stained with Hoechst 33342 to help distinguish whether the encapsulated Dox was able to get to its site of action properly. Moreover, since one of the main mechanisms of Dox inhibition is the formation of free oxygen radicals,14 cell redox stress was investigated by staining with CellRox Green stains. Figure 6 shows the morphology, stained nuclei, and redox stress of these cells for ApoDox prepared in water.
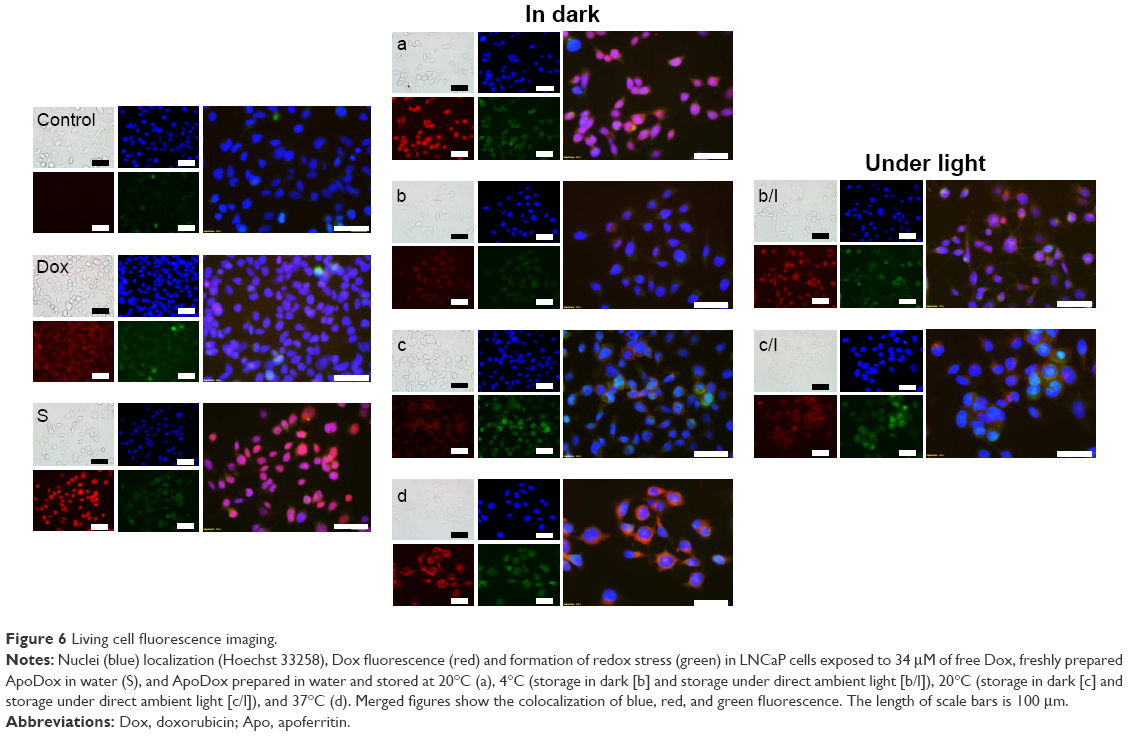 | Figure 6 Living cell fluorescence imaging. |
Ambient-light microscopy revealed that the treated cells were more round as they started to detach from the surface of the well and also at formation of first apoptotic bodies, both of which are the first sign of onset of apoptosis. The untreated cells (Figure 6 [Control]) showed blue nucleus fluorescence with no red (Dox) fluorescence and very low redox stress, almost always in the cytoplasm. Cells treated with free Dox (Figure 6 [Dox]) showed red Dox fluorescence and higher oxidative stress, both colocalized in nuclei. Cells treated with freshly prepared ApoDox (Figure 6 [S]) showed much higher intensity of Dox fluorescence than cells treated with free Dox, which indicated that ApoDox internalized in the cells more quickly than free Dox, probably through the overexpressed transferrin receptors on the surface of LNCaP cells.57 These cells also showed medium redox stress, mainly in the cytoplasm. ApoDox stored at −20°C (Figure 6 [a]) showed very similar results as those obtained with freshly prepared ApoDox: high Dox fluorescence in nuclei and medium redox stress in cytoplasm. ApoDox stored at 4°C and in dark (Figure 6 [b]) showed some fluorescence in both nuclei and cytoplasm, indicating slower internalization, as well as medium redox stress in both nuclei and cytoplasm. ApoDox stored at 4°C under ambient light (Figure 6 [b/l]) showed higher Dox fluorescence and redox stress, but mostly in cytoplasm, although there was also observable Dox fluorescence in nuclei. ApoDox stored at 20°C and in dark (Figure 6 [c]) showed higher Dox fluorescence than ApoDox stored at 4°C; however, this was localized exclusively in cytoplasm, proving that no or negligible Dox was able to get inside the nucleus. This could have been caused by different internalization mechanisms of large aggregates or slower release of drug molecules from these aggregates in endosomes; however, the precise mechanism needs to be further elucidated. These cells also showed high levels of cytoplasmic redox stress. ApoDox stored at 20°C under ambient light (Figure 6 [c/l]) showed similar results, with slightly higher Dox fluorescence and redox stress localized only in cytoplasm. ApoDox stored at 37°C (Figure 6 [d]) showed very high Dox fluorescence, but again almost always localized in the cytoplasm (only a very small percentage was observed in nuclei).
Figure 7 shows the morphology, stained nuclei, and redox stress of these cells for ApoDox prepared in PBS. Ambient-light microscopy again revealed the starting apoptosis in treated cells, which became more rounded as they started to detach from the surface of the well and apoptotic bodies became visible. Untreated cells (Figure 7 [Control]) showed only blue nucleus fluorescence with negligible red (Dox) fluorescence and very low redox stress, almost always in the cytoplasm. Cells treated with free Dox (Figure 7 [Dox]) showed red Dox fluorescence only in nuclei. Also, higher oxidative stress was observed in the nucleus. Cells treated with freshly prepared ApoDox (Figure 7 [S]) showed much higher intensity of Dox fluorescence than cells treated with free Dox, which was probably again caused by faster internalization of ApoDox due to overexpressed transferrin receptors. However, this Dox fluorescence was mostly localized in the cytoplasm with smaller percentages in nuclei. Also, redox stress was observed mainly in the cytoplasm. ApoDox stored at −20°C (Figure 7 [a]) showed Dox fluorescence in both cytoplasm and nuclei, where we also observed redox stress. ApoDox stored at 4°C and in dark (Figure 7 [b]) showed only very low Dox fluorescence, as well as redox stress in cytoplasm. ApoDox stored at 4°C under ambient light (Figure 7 [b/l]) showed very high Dox fluorescence in cytoplasm, as well as redox stress, with only low percentage of Dox and redox-stress fluorescence in nuclei. ApoDox stored at 20°C and in dark (Figure 7 [c]) showed medium Dox fluorescence, localized only in the cytoplasm. However, redox stress was observed in both nuclei and cytoplasm. ApoDox stored at 20°C under ambient light (Figure 7 [c/l]) again showed very high Dox fluorescence and redox stress in cytoplasm, with a small percentage in nuclei. ApoDox stored at 37°C (Figure 7 [d]) showed Dox fluorescence and redox stress mainly in cytoplasm (only a very small percentage was observed in nuclei).
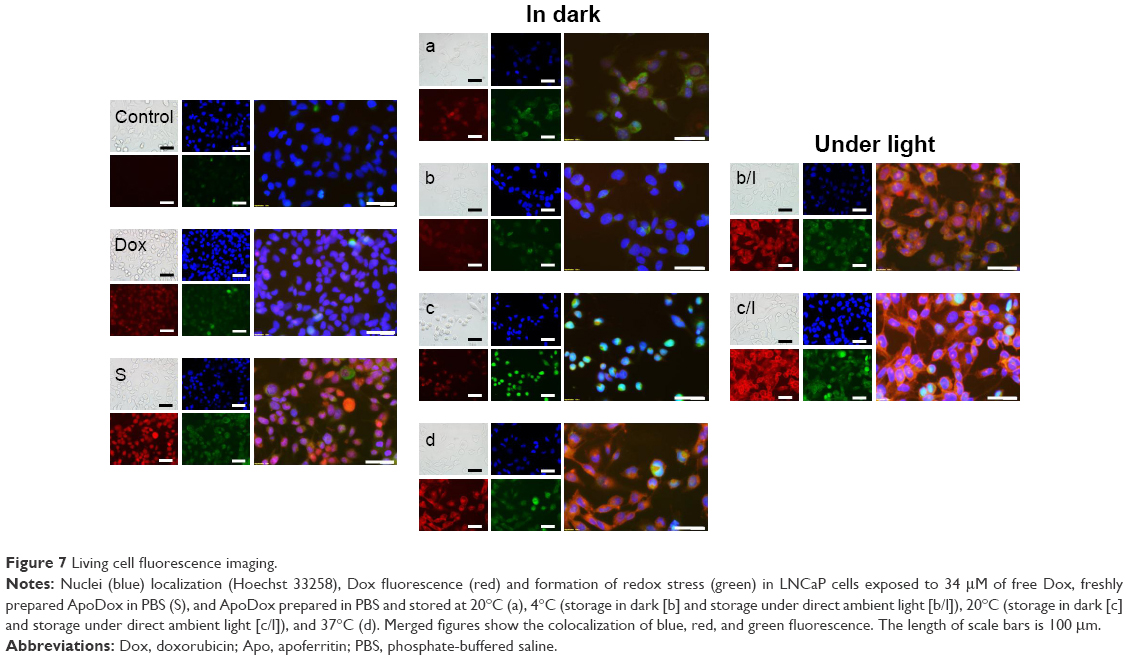 | Figure 7 Living cell fluorescence imaging. |
Next, real-time label-free monitoring of cell impedance was used to evaluate the 72-hour in vitro toxicity of ApoDox stored at various conditions for LNCaP cells. Since the experiment revealed that ApoDox prepared in PBS formed very large aggregates and was mostly unable to deliver the Dox cargo into nuclei, only ApoDox prepared in water was used for the cytotoxicity assay.
Control cells still proliferating can be seen in Figure 8 as a dark blue line. All cells treated with free Dox (Figure 8 [red]) were dead after 55 hours of treatment. After 72 hours of treatment, freshly prepared ApoDox in water (Figure 8 [black]) showed inhibition of 95% of cells. ApoDox stored at 20°C (Figure 8 [orange]) and 37°C (Figure 8 [light green]) were mostly unable to retain their ability to kill cancer cells, showing only 23% and 9% of dead cells, respectively. Storage under light showed similar results between temperatures, with 76% of dead cells for storage at 4°C (Figure 8 [gray]) and 58% of dead cells for storage at 20°C (Figure 8 [pink]), respectively.
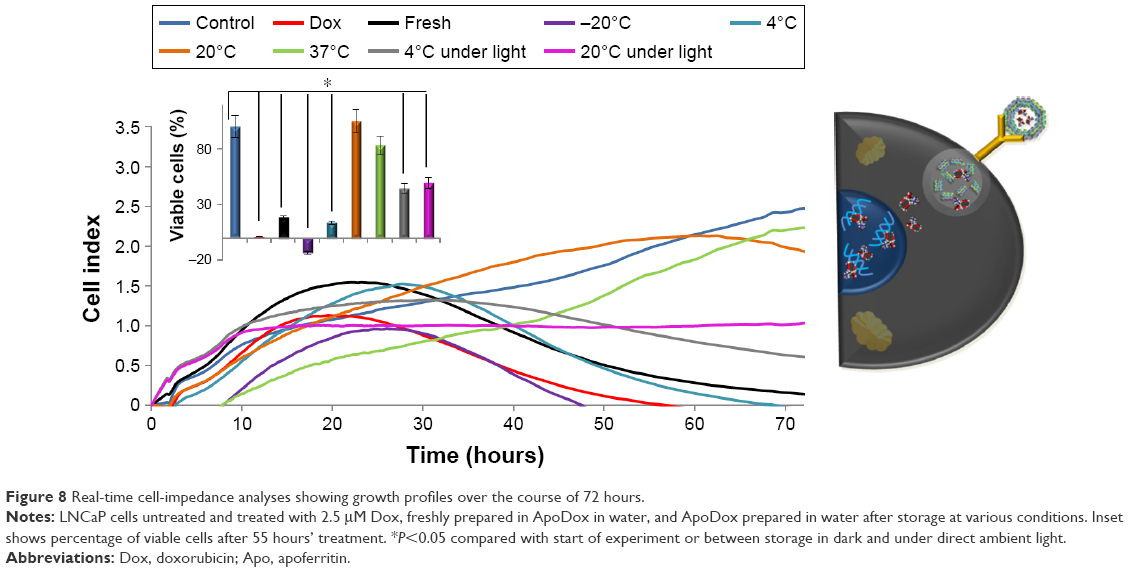 | Figure 8 Real-time cell-impedance analyses showing growth profiles over the course of 72 hours. |
The highest cytotoxicity was obtained using ApoDox stored at −20°C (Figure 8 [purple]). It was able to kill all cells within 46 hours of treatment, even faster than in the case of free Dox, corresponding to its fast internalization and induction of redox stress. However, this storage temperature is not suitable for use in patients, since ApoDox stored at −20°C creates very large aggregates that would be removed rapidly from the body by the RES.49 All cells had died after 68 hours of treatment with ApoDox stored at 4°C and in dark (Figure 8 [cyan]), which was slightly better than in the case of freshly prepared ApoDox. Moreover, storage at this temperature showed the highest stability with regard to size, shape, and surface charge. Storage at 4°C in water was thus evaluated as the most stable condition.
Conclusion
In conclusion, Apo is a natural nanocarrier that has a suitable cargo-release mechanism for delivery to cancer cells. In this study, we performed a comprehensive investigation of the long-term stability of the ApoDox nanocarrier stored at various conditions. We prepared the ApoDox nanocarrier in two different solvents (water and PBS), and stored it for 12 weeks in various conditions (−20°C, 4°C, 20°C, and 37°C in dark and 4°C and 20°C under ambient light). We tested the optical properties of the encapsulated cargo; the amount of prematurely released drug molecules; size, shape, ζ-potential, and the FT-IR spectra of the whole nanocarrier; and the ability to internalize into cancer cells and deliver the drug to nuclei. Many of the tested storage conditions caused formation of large aggregates of multiple nanocarriers, which would be unsuitable for use in patients. The optimal storage conditions seem to be 4°C in dark and in water, where the ApoDox showed very good stability over the course of 12 weeks. The obtained data are very helpful for future use of Apo as a nanocarrier for anticancer therapy. Long-term stability of a suitable nanocarrier can help overcome the higher cost of nanocarrier-based treatment compared with conventional anticancer drugs. The ApoDox nanocarrier will be tested further in in vivo experiments.
Acknowledgments
The authors gratefully acknowledge financial support from the Grant Agency of the Czech Republic (GA CR 17-12816S), IGA MENDELU IP_28/2016, and CEITEC 2020 (LQ1601). They also express thanks to Michal Horak for perfect technical assistance. The authors acknowledge the CF CEITEC – Core Facility Cryo-Electron Microscopy and Tomography, supported by the CIISB research infrastructure (LM2015043 funded by MEYS CR), for their support with obtaining the TEM micrographs.
Disclosure
The authors report no conflicts of interest in this work.
References
Dimarco A, Gaetani M, Scarpinato B. Adriamycin (NSC-123, 127): a new antibiotic with antitumor activity. Cancer Chemother Rep. 1969;53(1):33–37. | ||
Pilco-Ferreto N, Calaf GM. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int J Oncol. 2016;49(2):753–762. | ||
Lv LX, An XM, Li HY, Ma LX. Effect of miR-155 knockdown on the reversal of doxorubicin resistance in human lung cancer A549/dox cells. Oncol Lett. 2016;11(2):1161–1166. | ||
Shang C, Guo Y, Zhang H, Xue YX. Long noncoding RNA HOTAIR is a prognostic biomarker and inhibits chemosensitivity to doxorubicin in bladder transitional cell carcinoma. Cancer Chemother Pharmacol. 2016;77(3):507–513. | ||
Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1B and randomised phase 2 trial. Lancet. 2016;388(10043):488–497. | ||
Bürkle C, Borchmann P. Diagnostik und Therapie des Hodgkin-Lymphoms [Diagnosis and treatment of Hodgkin lymphoma]. Onkologe. 2016;22(8):603–616. German. | ||
Agudelo D, Bourassa P, Bérubé G, Tajmir-Riahi HA. Review on the binding of anticancer drug doxorubicin with DNA and tRNA: structural models and antitumor activity. J Photochem Photobiol B Biol. 2016;158:274–279. | ||
Kumar A, Ehrenshaft M, Tokar EJ, Mason RP, Sinha BK. Nitric oxide inhibits topoisomerase II activity and induces resistance to topoisomerase II-poisons in human tumor cells. Biochim Biophys Acta. 2016;1860(7):1519–1527. | ||
Agustini FD, Arozal W, Louisa M, et al. Cardioprotection mechanism of mangiferin on doxorubicin-induced rats: focus on intracellular calcium regulation. Pharm Biol. 2016;54(7):1289–1297. | ||
Meredith AM, Dass CR. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism. J Pharm Pharmacol. 2016;68(6):729–741. | ||
World Health Organization. WHO model list of essential medicines. 2015. Available from: http://www.who.int/medicines/publications/essentialmedicines/en. Accessed September 20, 2016. | ||
Li XY, Wu MY, Pan LM, Shi JL. Tumor vascular-targeted co-delivery of anti-angiogenesis and chemotherapeutic agents by mesoporous silica nanoparticle-based drug delivery system for synergetic therapy of tumor. Int J Nanomedicine. 2016;11:93–105. | ||
Mercurio V, Pirozzi F, Lazzarini E, et al. Models of heart failure based on the cardiotoxicity of anticancer drugs. J Card Fail. 2016;22(6):449–458. | ||
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–170. | ||
Xia CC, Yadav AK, Zhang K, et al. Synchrotron radiation (SR) diffraction enhanced imaging (DEI) of chronic glomerulonephritis (CGN) mode. J Xray Sci Technol. 2016;24(1):145–159. | ||
Fanous I, Dillon P. Cancer treatment-related cardiac toxicity: prevention, assessment and management. Med Oncol. 2016;33(8):84. | ||
Schuler MK, Gerdes S, West A, et al. Efficacy and safety of dexrazoxane (DRZ) in sarcoma patients receiving high cumulative doses of anthracycline therapy: a retrospective study including 32 patients. BMC Cancer. 2016;16:619. | ||
Shaikh F, Dupuis LL, Alexander S, Gupta A, Mertens L, Nathan PC. Cardioprotection and second malignant neoplasms associated with dexrazoxane in children receiving anthracycline chemotherapy: a systematic review and meta-analysis. J Natl Cancer Inst. 2016;108(4):djv357. | ||
Luhmann T, Meinel L. Nanotransporters for drug delivery. Curr Opin Biotechnol. 2016;39:35–40. | ||
Maeda H, Tsukigawa K, Fang J. A retrospective 30 years after discovery of the enhanced permeability and retention effect of solid tumors: next-generation chemotherapeutics and photodynamic therapy problems, solutions, and prospects. Microcirculation. 2016;23(3):173–182. | ||
Dostalova S, Cerna T, Hynek D, et al. Site-directed conjugation of antibodies to apoferritin nanocarrier for targeted drug delivery to prostate cancer cells. ACS Appl Mater Interfaces. 2016;8(23):14430–14441. | ||
Tian DY, Wang WY, Li SP, Li XD, Sha ZL. A novel platform designed by Au core/inorganic shell structure conjugated onto MTX/LDH for chemo-photothermal therapy. Int J Pharm. 2016;505(1–2):96–106. | ||
Jena SK, Sangamwar AT. Polymeric micelles of amphiphilic graft copolymer of α-tocopherol succinate-g-carboxymethyl chitosan for tamoxifen delivery: synthesis, characterization and in vivo pharmacokinetic study. Carbohydr Polym. 2016;151:1162–1174. | ||
Lindqvist A, Friden M, Hammarlund-Udenaes M. Pharmacokinetic considerations of nanodelivery to the brain: using modeling and simulations to predict the outcome of liposomal formulations. Eur J Pharm Sci. 2016;92:173–182. | ||
Agudelo D, Berube G, Tajmir-Riahi HA. An overview on the delivery of antitumor drug doxorubicin by carrier proteins. Int J Biol Macromol. 2016;88:354–360. | ||
Alphandery E, Grand-Dewyse P, Lefevre R, Mandawala C, Durand-Dubief M. Cancer therapy using nanoformulated substances: scientific, regulatory and financial aspects. Expert Rev Anticancer Ther. 2015;15(10):1233–1255. | ||
Kim HS, Wainer IW. Simultaneous analysis of liposomal doxorubicin and doxorubicin using capillary electrophoresis and laser induced fluorescence. J Pharm Biomed Anal. 2010;52(3):372–376. | ||
Suk JS, Xu QG, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(Pt A):28–51. | ||
Tmejova K, Hynek D, Kopel P, et al. Electrochemical behaviour of doxorubicin encapsulated in apoferritin. Int J Electrochem Sci. 2013;8(12):12658–12671. | ||
Liang MM, Fan KL, Zhou M, et al. H-ferritin-nanocaged doxorubicin nanoparticles specifically target and kill tumors with a single-dose injection. Proc Natl Acad Sci U S A. 2014;111(41):14900–14905. | ||
Cutrin JC, Crich SG, Burghelea D, Dastru W, Aime S. Curcumin/Gd loaded apoferritin: a novel “theranostic” agent to prevent hepatocellular damage in toxic induced acute hepatitis. Mol Pharm. 2013;10(5):2079–2085. | ||
Zang J, Chen H, Guanghua Z, Wang F, Ren F. Ferritin cage for encapsulation and delivery of bioactive nutrients: from structure, property to applications. Crit Rev Food Sci Nutr. Epub 2016 Mar 16. | ||
Crich SG, Cadenazzi M, Lanzardo S, et al. Targeting ferritin receptors for the selective delivery of imaging and therapeutic agents to breast cancer cells. Nanoscale. 2015;7(15):6527–6533. | ||
Blazkova I, Nguyen HV, Dostalova S, et al. Apoferritin modified magnetic particles as doxorubicin carriers for anticancer drug delivery. Int J Mol Sci. 2013;14(7):13391–13402. | ||
Bulvik BE, Berenshtein E, Meyron-Holtz EG, Konijn AM, Chevion M. Cardiac protection by preconditioning is generated via an iron-signal created by proteasomal degradation of iron proteins. PLoS One. 2012;7(11):e48947. | ||
Asano T, Komatsu M, Yamaguchi-Iwai Y, Ishikawa F, Mizushima N, Iwai K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol. 2011;31(10):2040–2052. | ||
Smith JL. The physiological role of ferritin-like compounds in bacteria. Crit Rev Microbiol. 2004;30(3):173–185. | ||
Gallois B, dEstaintot BL, Michaux MA, et al. X-ray structure of recombinant horse L-chain apoferritin at 2.0 angstrom resolution: implications for stability and function. J Biol Inorg Chem. 1997;2(3):360–367. | ||
Haussler W. Structure and dynamics in apoferritin solutions with paracrystalline order. Chem Phys. 2003;292(2–3):425–434. | ||
Kim M, Rho Y, Jin KS, et al. pH-dependent structures of ferritin and apoferritin in solution: disassembly and reassembly. Biomacromolecules. 2011;12(5):1629–1640. | ||
Kilic MA, Ozlu E, Calis S. A novel protein-based anticancer drug encapsulating nanosphere: apoferritin-doxorubicin complex. J Biomed Nanotechnol. 2012;8(3):508–514. | ||
Wood MJ, Irwin WJ, Scott DK. Photodegradation of doxorubicin, daunorubicin and epirubicin measured by high-performance liquid-chromatography. J Clin Pharm Ther. 1990;15(4):291–300. | ||
Konecna R, Nguyen HV, Stanisavljevic M, et al. Doxorubicin encapsulation investigated by capillary electrophoresis with laser-induced fluorescence detection. Chromatographia. 2014;77(21–22):1469–1476. | ||
Dai XW, Yue ZL, Eccleston ME, Swartling J, Slater NK, Kaminski CF. Fluorescence intensity and lifetime imaging of free and micellar-encapsulated doxorubicin in living cells. Nanomed Nanotechnol Biol Med. 2008;4(1):49–56. | ||
Yang K, Luo HQ, Zeng M, Jiang YY, Li JM, Fu XL. Intracellular pH-triggered, targeted drug delivery to cancer cells by multifunctional envelope-type mesoporous silica nanocontainers. ACS Appl Mater Interfaces. 2015;7(31):17399–17407. | ||
Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. | ||
Svenson S. Theranostics: are we there yet? Mol Pharm. 2013;10(3):848–856. | ||
Appelbe OK, Zhang Q, Pelizzari CA, Weichselbaum RR, Kron SJ. Image-guided radiotherapy targets macromolecules through altering the tumor microenvironment. Mol Pharm. 2016;13(10):3457–3467. | ||
Liu T, Chao Y, Gao M, et al. Ultra-small MoS2 nanodots with rapid body clearance for photothermal cancer therapy. Nano Res. 2016;9(10):3003–3017. | ||
Svenson S. Clinical translation of nanomedicines. Curr Opin Solid State Mater Sci. 2012;16(6):287–294. | ||
Gratton SE, Ropp PA, Pohlhaus PD, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105(33):11613–11618. | ||
Zamboni WC, Torchilin V, Patri AK, et al. Best practices in cancer nanotechnology: perspective from NCI nanotechnology alliance. Clin Cancer Res. 2012;18(12):3229–3241. | ||
Stylianopoulos T, Jain RK. Design considerations for nanotherapeutics in oncology. Nanomed Nanotechnol Biol Med. 2015;11(8):1893–1907. | ||
Fülöp Z, Gref R, Loftsson T. A permeation method for detection of self-aggregation of doxorubicin in aqueous environment. Int J Pharm. 2013;454(1):559–561. | ||
Righetti PG, Menozzi M, Gianazza E, Valentini L. Protolytic equilibria of doxorubicin as determined by isoelectric-focusing and electrophoretic titration curves. FEBS Lett. 1979;101(1):51–55. | ||
Beijnen JH, Wiese G, Underberg WJ. Aspects of the chemical-stability of doxorubicin and seven other anthracyclines in acidic solution. Pharm Weekbl Sci. 1985;7(3):109–116. | ||
Liu AY. Differential expression of cell surface molecules in prostate cancer cells. Cancer Res. 2000;60(13):3429–3434. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.