Back to Journals » Drug Design, Development and Therapy » Volume 20
Apigenin Upregulates Regulatory B Cells, Suppresses Class Switch Recombination, and Restores Immune Homeostasis to Treat Autoimmune Arthritis and Sepsis
Authors Chen H ![]() , Huang Y, He Y, Zha M, Liu X, Lin T
, Huang Y, He Y, Zha M, Liu X, Lin T ![]() , Li L, He Z, Zheng SG, Shao Y
, Li L, He Z, Zheng SG, Shao Y ![]() , Luo A, Sun J
, Luo A, Sun J ![]()
Received 3 March 2026
Accepted for publication 7 May 2026
Published 25 May 2026 Volume 2026:20 603143
DOI https://doi.org/10.2147/DDDT.S603143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Hong Chen,1,* Yilian Huang,2,3,* Yufu He,1,4,5,* Menglei Zha,1 Xin Liu,1 Tianqiong Lin,2,6 Lei Li,1 Ziyi He,7 Song Guo Zheng,1,8,9 Yiming Shao,4,5 Aoxiang Luo,2 Jianbo Sun1
1Dongguan Key Laboratory of Chronic Inflammatory Diseases, The First Dongguan Affiliated Hospital, School of Pharmacy, Guangdong Medical University, Dongguan, Guangdong, 523710, People’s Republic of China; 2School of Nursing, Guangdong Pharmaceutical University, Guangzhou, Guangdong, 510310, People’s Republic of China; 3Liwan District Maternal and Child Health Hospital, Guangzhou, Guangdong, 510000, People’s Republic of China; 4Dongguan Key Laboratory of Sepsis Translational Medicine, The First Dongguan Affiliated Hospital, The Second School of Clinical Medicine, Guangdong Medical University, Dongguan, Guangdong, 523710, People’s Republic of China; 5The Intensive Care Unit, The First Dongguan Affiliated Hospital, The Second School of Clinical Medicine, Guangdong Medical University, Dongguan, Guangdong, 523710, People’s Republic of China; 6The Third Affiliated Hospital, Guangzhou University of Traditional Chinese Medicine, Guangzhou, Guangdong, 510378, People’s Republic of China; 7Dongguan Central Blood Station, Dongguan, Guangdong, 523930, People’s Republic of China; 8Department of Immunology, School of Cell and Gene Therapy, Songjiang Institute and Songjiang Hospital Affiliated to the Shanghai Jiao Tong University School of Medicine, Shanghai, 201600, People’s Republic of China; 9State Key Laboratory of Innovative Immunotherapy, Shanghai Jiao Tong University, Shanghai, 201600, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jianbo Sun, Dongguan Key Laboratory of Chronic Inflammatory Diseases, the First Dongguan Affiliated Hospital, School of Pharmacy, Guangdong Medical University, Dongguan, Guangdong, 523710, People’s Republic of China, Tel +86 076989190089, Fax +86 07690600, Email [email protected] Aoxiang Luo, School of Nursing, Guangdong Pharmaceutical University, Guangzhou, Guangdong, 510310, People’s Republic of China, Tel +86 02034055935, Email [email protected]
Background: Restoring immune homeostasis is crucial for inflammation resolution, in which B cells play dual regulatory roles. A promising therapeutic strategy involves simultaneously suppressing proinflammatory B-cell activity while enhancing regulatory B cells (Bregs). Natural anti-inflammatory compounds, such as the flavonoid apigenin (API), may achieve this dual immunomodulation, yet the precise mechanisms of API on B cells remain incompletely understood.
Purpose: This study aimed to elucidate how API modulates B cells to exert immunoregulatory and protective effects in inflammatory conditions.
Design: In vitro B-cell functional and molecular assays were integrated with two mechanistically distinct in vivo models—collagen-induced arthritis (CIA) in DBA/1 mice and cecal ligation and puncture (CLP)–induced sepsis in C57BL/6 mice—to evaluate API’s dual immunomodulatory effects and therapeutic potential.
Methods: B cells from mouse splenocytes (n=5 per group) or human PBMCs (n=5 healthy donors) were assessed for differentiation, cytokine production, class switch recombination (CSR), and signaling pathways. p38 MAPK involvement was examined using pathway-specific inhibitors. In vivo, disease severity, survival, immune phenotypes, cytokines, and histopathology were evaluated.
Results: API increased IL-10⁺ Breg frequency by ~2-fold (P < 0.001) and reduced Aicda expression by ~60% (P < 0.001), while suppressing plasma cell differentiation via Prdm1 downregulation. These effects were p38 MAPK-dependent. In CIA mice, API reduced arthritis scores by ~45% (P < 0.01); in CLP-induced sepsis, 7-day survival improved from 0% to 70% (P < 0.05), with attenuated lung injury and pro-inflammatory cytokine levels. Collectively, API promotes immune homeostasis restoration in both chronic autoimmune and acute inflammatory settings.
Conclusion: API restores immune homeostasis in both chronic autoimmune and acute inflammatory settings by coordinating Breg differentiation and suppressing pathogenic B-cell responses, highlighting B-cell-centric immune rebalancing as a promising therapeutic strategy for inflammatory diseases.
Plain Language Summary: Excessive inflammation is a defining feature of both chronic autoimmune diseases and acute systemic inflammatory conditions. Restoring immune balance is critical for resolving inflammation—a process in which B cells play dual roles by both promoting and suppressing immune responses. A rational therapeutic strategy, therefore, is to simultaneously curb proinflammatory B cell activity while enhancing the function of immunosuppressive regulatory B cells (Bregs). Natural anti-inflammatory compounds derived from herbal medicine offer a promising source of agents capable of achieving this dual immunomodulation. Apigenin (API), a naturally occurring flavonoid, is known for its broad anti-inflammatory effects. However, whether and how it exerts dual regulatory actions on B cells remains unclear.
In this study, we show that API promotes the differentiation of regulatory B cells (Bregs) via activation of the p38 MAPK signaling pathway, suppresses antibody class-switch recombination (a process that generates pathogenic antibodies) through downregulation of the gene Aicda, and inhibits plasma cell differentiation by reducing Prdm1 and IL-6 expression. These coordinated actions underlie its therapeutic efficacy in two distinct models of immune dysregulation. In a mouse model of collagen-induced arthritis—which mimics human rheumatoid arthritis—API alleviated joint inflammation and bone erosion. In a cecal ligation and puncture-induced sepsis model—which replicates severe systemic infection—API improved survival and attenuated acute lung injury. Together, these findings identify B-cell–centered immune rebalancing using natural anti-inflammatory compounds as a promising therapeutic approach for both chronic autoimmune and acute inflammatory conditions. The diagram illustrates the immune status in inflammatory diseases and the therapeutic effects of apigenin (API). On the left, the inflammatory state in diseases like rheumatoid arthritis and sepsis lung injury is depicted, where elevated pro-inflammatory cytokines (TNF-α, IL-6, IL-1β) and increased pro-inflammatory cells (macrophages, dendritic cells, T cells) lead to immune imbalance, with reduced regulatory B cells (Breg). On the right, apigenin (API) treatment restores immune balance in these diseases: it activates p38 MAPK signaling to expand Breg cells and increase IL-10 production, while suppressing antibody diversification, plasma cell differentiation, and pro-inflammatory responses. This reduces inflammation and promotes tissue homeostasis, as shown by the normalization of joint and lung conditions. The diagram includes labels for immune cells, antibodies, API, and cytokines, with arrows indicating increases or decreases in activity.Diagram comparing immune imbalance in inflammatory diseases (rheumatoid arthritis, sepsis lung injury) with apigenin (API)-mediated restoration of immune balance and anti-inflammatory effects.
Keywords: apigenin, regulatory B cells, class switch recombination, inflammation
Introduction
Excessive inflammatory responses represent a common hallmark of chronic autoimmune disorders and acute systemic inflammatory conditions.1–3 Restoring immune homeostasis is crucial for timely resolution of inflammation, a process in which B cells play a dual regulatory role—both promoting and suppressing immune responses.4 On the one hand, B cells drive immunity through antigen presentation, antibody production, secretion of proinflammatory cytokines, and expression of co-stimulatory molecules.5–7 On the other hand, they contribute to immune tolerance by generating regulatory B cells (Bregs), which produce anti-inflammatory cytokines such as IL-10, IL-35, and TGF-β1,8,9 facilitate regulatory T cell (Treg) induction, and promote autoantigen clearance.10
In autoimmune diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus, and multiple sclerosis, proinflammatory B cells are frequently hyperactivated, resulting in elevated antibody and proinflammatory cytokine production, while Bregs are often reduced in both frequency and function.11,12 A similar imbalance is observed in acute inflammatory settings such as sepsis, further underscoring the broad relevance of B-cell dysregulation.13,14 Thus, a promising therapeutic strategy lies in simultaneously dampening proinflammatory B-cell activity and enhancing the differentiation and function of Bregs. Natural anti-inflammatory compounds derived from herbal medicine may offer a valuable source of agents capable of achieving this dual immunomodulatory goal.
Apigenin (API) is a natural flavonoid compound widely present in various fruits and vegetables, and has multiple biological effects such as antioxidation, anti-inflammation, anti-tumor and anti-diabetic.15,16 It can inhibit the production of pro-inflammatory factors, and regulate pyroptosis of immune cells, to regulate the immune homeostasis.17–20 In RA therapy, API can reduce the expression of IL-6, IL-1β and TNF-α in synovial cells of the joint and exert anti-inflammatory effects by regulating the function of macrophages.21–23 Furthermore, in models of pulmonary inflammation and asthma, API alleviates airway inflammation and hyper responsiveness by modulating immune cell activation and cytokine production.17,24 Although these studies have revealed the potential of API in anti-inflammation and immune regulation, the regulatory mechanisms of API on Bregs and CSR remain unclear.
Specifically, no prior study has demonstrated that API can coordinately enhance Breg differentiation while suppressing class-switch recombination (CSR) and plasma cell generation, particularly across distinct disease models of chronic autoimmunity and acute inflammation. We hypothesized that API might be the anti-inflammatory herbal compound capable of achieving the dual immunomodulatory goal. Herein, we investigated whether API is capable of suppressing proinflammatory B-cell activity and enhancing the differentiation and function of Bregs to alleviate RA and sepsis.
Materials and Methods
Animals
All animal procedures were approved by the relevant Institutional Animal Care and Use Committees and conducted in accordance with the ARRIVE guidelines and AVMA euthanasia recommendations. All mice were maintained on a 12-h light/dark cycle at 25 ± 2°C and acclimatized for at least 3 days. Surgical anesthesia was induced and maintained with sodium pentobarbital (50 mg/kg, i.p). At the end of experiments or upon reaching predetermined humane endpoints, mice were anesthetized by isoflurane overdose (5% in oxygen) and peripheral blood was subsequently collected for flow cytometric analysis prior to cervical dislocation for euthanasia. All efforts were made to minimize animal suffering.
Twenty-eghint 8-week-old male DBA/1 mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. For the collagen-induced arthritis (CIA) experiment, mice were randomly divided into four groups (n=7 per group) and housed under specific pathogen-free (SPF) conditions at Sun Yat-sen University. All animal procedures were approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University (Approval No. SYSU-IACUC-2020-000414).
8-week-old male C57BL/6 mice (20–22 g) were obtained from Guangdong Zhiyuan Biomedicine Technology Co., Ltd. For the cecal ligation and puncture (CLP)-induced sepsis experiment, mice were randomly assigned to four groups (n=10 per group for evaluation of survival rate, or n=6 per group for the organ injury study) and maintained under SPF conditions at Guangdong Medical University. The animal protocol was approved by the Institutional Animal Care and Use Committee of Guangdong Medical University (Approval No. GDMU-2023-000003).
In addition, to evaluate the effect of API on Breg generation under physiological conditions, a separate group of 8-week-old male C57BL/6J mice was randomly divided into two groups (n=5 per group): a control group fed a normal diet and an API-treated group fed a diet supplemented with 2 g/kg API for 6 consecutive days.
Isolation of Peripheral Blood Mononuclear Cells (PBMCs)
This study was approved by the Medical Ethics Committee of the First Dongguan Affiliated Hospital of Guangdong Medical University (Approval No. YS202310007) and was conducted in accordance with the principles of the Declaration of Helsinki. Peripheral blood samples were obtained from five healthy donors (three males and two females, aged 20–40 years) after written informed consent was obtained from all participants. PBMC isolation and subsequent experiments were performed using cells from each individual donor as independent biological replicates (n=5). Lymphocytes were isolated from PBMCs following previously reported protocols.25
Collagen-Induced Arthritis Model
Collagen-induced arthritis was established in DBA/1 mice using a single immunization protocol. Mice were randomly divided into four groups (n=7 per group): wild-type control (WT), CIA model (CIA), CIA with API prevention (CIA-Prevention, API diet from day 0), and CIA with API therapy (CIA-Therapy, API diet from day 21). On day 0, under anesthesia, mice received a subcutaneous injection at the tail base with 100 µL of an emulsion containing immune-grade bovine type II collagen (1 mg/mL; Chondrex, 20022) and Mycobacterium tuberculosis (2 mg/mL; Chondrex, 7001). Control group mice were injected with an equal volume of PBS.26 Starting from day 21, when clinical symptoms (joint redness and swelling) became apparent, mice in the API treatment groups received a diet supplemented with 2 g/kg apigenin and were maintained on this diet until the experimental endpoint. Mice were monitored for disease development. Body weight was measured every two days. Paw thickness and clinical arthritis scores were recorded twice weekly. On day 42, all mice were euthanized and tissue samples were collected for subsequent analyses.
Cecal Ligation and Puncture (CLP) Model
Cecal ligation and puncture (CLP) was used to induce polymicrobial sepsis in C57BL/6 mice. Two separate experiments were performed to assess (1) survival and (2) organ injury. For both experiments, mice were randomly divided into four groups: Sham, CLP model, CLP + API (20 mg/kg), and CLP + API (60 mg/kg). API (purity >98%), suspended in saline containing 0.5% sodium carboxymethyl cellulose, was administered by daily oral gavage for 7 consecutive days before surgery. Control and CLP model mice received the vehicle only. One hour after the last API administration, mice were anesthetized with isoflurane, and CLP was performed through a midline abdominal incision. The cecum was exposed, ligated at 50% of its length, and punctured twice with a 21-gauge needle before being returned to the abdominal cavity. Sham-operated mice underwent the same surgical procedure without ligation and puncture. All mice received 1 mL of sterile saline subcutaneously for fluid resuscitation post-operation. For the survival study, mice (n=10 per group) were monitored daily for 7 days, and survival was recorded. All surviving mice were euthanized at the end of the 7-day observation period. For the organ injury study, a separate cohort of mice (n=6 per group) was euthanized at 24 h post-CLP, and lung tissues and blood samples were collected for histopathological analysis, Evans blue quantification, wet-to-dry weight ratio measurement, and cytokine ELISA.27
H&E Staining and Histopathological Score
Tissues (limb joints and lungs) were fixed in 4% paraformaldehyde at room temperature for 48 hours. Joint samples underwent subsequent decalcification in 10% EDTA. All tissues were then dehydrated, embedded in paraffin, and sectioned for hematoxylin and eosin (H&E) staining.
Histopathological assessments were performed by two blinded examiners. Joint inflammation was scored based on comprehensive evaluation of H&E-stained sections. The detailed scoring criteria are shown in Tables S1 and S2. Lung injury was scored by assessing four parameters: inflammatory cell infiltration, alveolar epithelial cell necrosis, hemorrhage, and interstitial edema. Each parameter was graded from 0 to 3, yielding a total possible score of 0–12 per sample. The detailed scoring criteria are shown in Table S3.
Micro-CT Analysis of Bone and Joint in Mice
Bone architecture in mouse paws was analyzed using high-resolution micro-CT (Viva CT 40, Scanco, Switzerland). Specimens were scanned with the following parameters: 55 kV voltage, 145 µA current, 200 ms integration time, and a voxel size of 17.5 µm. Acquired projection images were reconstructed and processed using manufacturer-provided software, including noise reduction with a Gaussian filter.
For quantitative analysis, three-dimensional (3D) reconstructions were generated. A standardized grayscale threshold was applied to segment bone from soft tissue. Bone morphometry was assessed in the second, third, and fourth metatarsals by defining a consistent volume of interest (VOI) extending 1 mm proximally and distally from the center of each metatarsophalangeal joint. The calculated parameters included bone volume (BV), bone volume fraction (BV/TV), trabecular thickness (TB.TH), and trabecular number (Tb.N).
Immune Cell Preparation, Staining and Flow Cytometry Analysis
Following euthanasia, samples including blood, mesenteric lymph nodes (MLN), and spleens were collected. Single-cell suspensions were prepared. Blood was processed with red blood cell (RBC) lysis buffer (YuanYe, R20172-100 mL). MLNs and spleens were mechanically dissociated through a 40 µm cell strainer, and residual RBCs were lysed when necessary.
For intracellular cytokine detection, cells were resuspended at 2×106 cells/mL in complete RPMI-1640 medium and stimulated for 5 h at 37°C with a cocktail containing LPS (10 µg/mL), PMA (50 ng/mL), ionomycin (500 ng/mL), and monensin (2 µM).
Post-stimulation, cells were stained with LIVE/DEAD™ Fixable Near-IR dye, followed by surface staining with antibodies against CD19. Cells were then fixed, permeabilized (Cytofix/Cytoperm Plus Kit, BD Biosciences), and stained intracellularly with antibodies against IL-10 and IL-6. Stained cells were fixed in 1.5% formaldehyde.28,29
Samples were acquired on a CytoFLEX (Beckman Coulter) or LSRFortessa™ (BD Biosciences) flow cytometer. Data were analyzed using FlowJo software (v10, BD Biosciences).
Western Blot Analysis
Proteins were extracted from cell lysates using RIPA buffer (Beyotime, P0013B) supplemented with phosphatase and protease inhibitors. Lysates were centrifuged at 12,000 rpm for 15 min at 4°C, and supernatants were collected for quantification using a BCA Protein Assay Kit (Beyotime, P0010).
Protein samples were denatured, separated by 10% SDS-PAGE, and transferred onto PVDF membranes (Millipore, ISEQ00010) using a wet transfer system.30 Membranes were blocked with 5% skim milk in TBST for 30 min at room temperature and subsequently incubated overnight at 4°C with the following primary antibodies: ERK (1:8000, Proteintech, 11257-1-AP-100UL), p-ERK (1:1000, Proteintech, 28733-1-AP-100UL), JNK (1:5000, Proteintech, 24164-1-AP-100UL), p-JNK (1:1000, Proteintech, 80024-1-RR-100UL), p38 (1:5000, Proteintech, 14064-1-AP-100UL), and p-p38 (1:1000, CST, 4511S).
After washing with TBST, membranes were incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. Protein bands were visualized using an ECL substrate and imaged with a chemiluminescence imaging system.
RT-qPCR
Total RNA was extracted from mouse B cells using TRNzol Universal Reagent. Subsequently, RNA was reverse-transcribed into complementary DNA (cDNA) using the HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, China, R312-01/02). Quantitative PCR was performed using the Hieff® Fast Cell Direct SYBR Green RT-qPCR Kit (Yeasen, China, 11172ES) according to the manufacturer’s instructions. Primer sequences used for amplification are listed in Tables 1 (mouse) and 2 (human). All reactions were performed in triplicate. Gene expression levels were quantified using the 2−ΔΔCt method, normalized to the housekeeping genes Gapdh (mouse) or β-ACTIN (human).
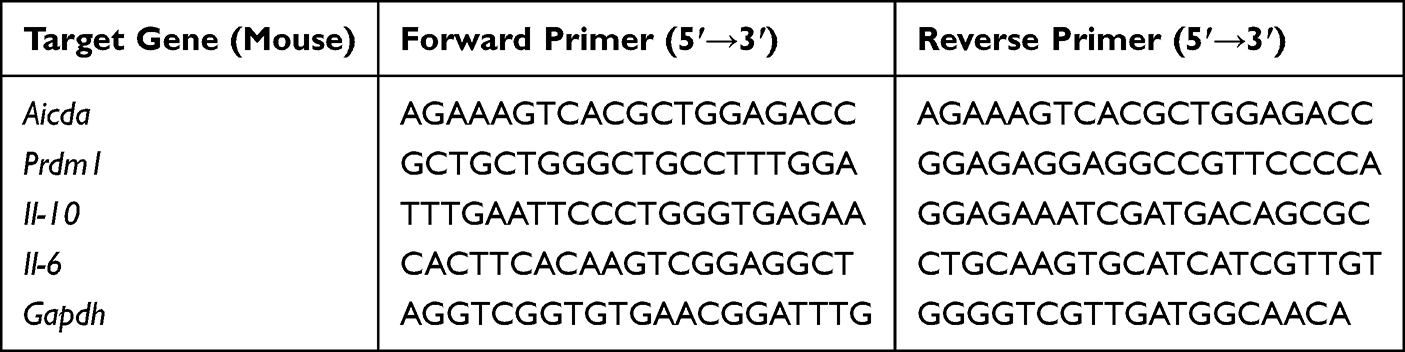 |
Table 1 Mouse Primer Sequences |
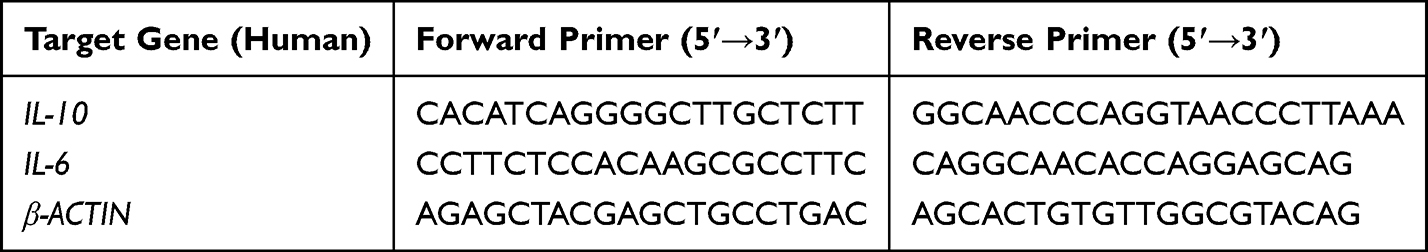 |
Table 2 Human Primer Sequences |
Enzyme-Linked Immunosorbent Assay
The concentrations of cytokines in the collected supernatants were quantified using a commercial ELISA kit (Elabscience, China, E-EL-M0046-96T) according to the manufacturer’s instructions. Absorbance was measured at 450 nm using a microplate reader (PerkinElmer 2104). Data are expressed as nanograms of cytokine per milligram of total protein (ng/mg protein) for each sample.
RNA-Seq and Data Analysis
RNA-Seq libraries were prepared from poly(A)-enriched mRNA and sequenced on an MGI 2000 platform (BGI, China). Clean reads were aligned to the mouse reference genome (mm10) using STAR (v1.5.1). Gene expression quantification and differential expression analysis were performed with HTSeq-count (v0.11.3) and DESeq2 (v1.28.1), respectively. Differentially expressed genes (DEGs) were defined by a false discovery rate (FDR) < 0.05 and an absolute fold change > 2. Subsequent data processing and visualization were performed in RStudio or TBtools.29 The RNA-seq data generated in this study have been deposited in the Genome Sequence Archive (GSA) of the National Genomics Data Center (CNCB) under accession number CRA038922. The data can be accessed via the following link: https://ngdc.cncb.ac.cn/gsa/s/vwo6XXcW.
Statistical Analysis
All experimental data are presented as mean ± standard deviation (Mean ± SD) and were statistically analyzed by GraphPad Prism 8.0 software. Flow cytometry data were processed and analyzed by FlowJo V10 software. The comparison between the two groups of quantitative data was analyzed by Student ‘s t test. The differences among multiple groups were analyzed by one-way ANOVA. A p-value of 0.05 was considered statistically significant. To ensure comparability, the data from different batches were standardized prior to pooling for statistical analysis and the results were presented as “Relative level”. This standardization process involved converting the absolute values of each group within a batch to a ratio relative to the mean value of the control group in that same batch. All in vitro experiments (including Western blotting, RT-qPCR, ELISA, etc) were performed using cells isolated from at least five independent biological samples (individual mice or human donors) as biological replicates. For in vivo studies, each experimental group consisted of 5–7 mice per cohort, representing independent biological replicates. For each biological sample, technical replicates (eg, triplicate or quadruplicate wells) were averaged in each RT-qPCR or ELISA experiment.
Results
Apigenin Promotes the Generation of Breg in vitro
API has anti-inflammatory effects, but its influence on Bregs remains unknown. To detect whether API affects the differentiation of Bregs, Bregs extracted from mouse spleens were treated with LPS and API for 48 hours. Subsequently, cells were collected after stimulation with PMA/ionomycin/monensin (PIM) for the last 5 hours for detection. Flow cytometry analysis revealed that API treatment significantly increased the proportion of Bregs (defined as IL-10⁺ B cells) and concurrently decreased the frequency of IL-6⁺ B cells (Figure 1A and B). At the concentrations tested, API did not affect overall cell viability, confirming that its immunomodulatory effects were not attributable to cytotoxicity (Figure S1). Consistent with these phenotypic changes, API upregulated IL-10 mRNA and downregulated IL-6 mRNA transcript levels compared to the blank control (Figure 1C). This regulatory pattern was further confirmed at the protein level, as ELISA measurements of culture supernatants showed elevated IL-10 and reduced IL-6 secretion following API treatment (Figure 1D). Taken together, these results demonstrate that API promotes the generation of functional Bregs while suppressing pro-inflammatory cytokine production.
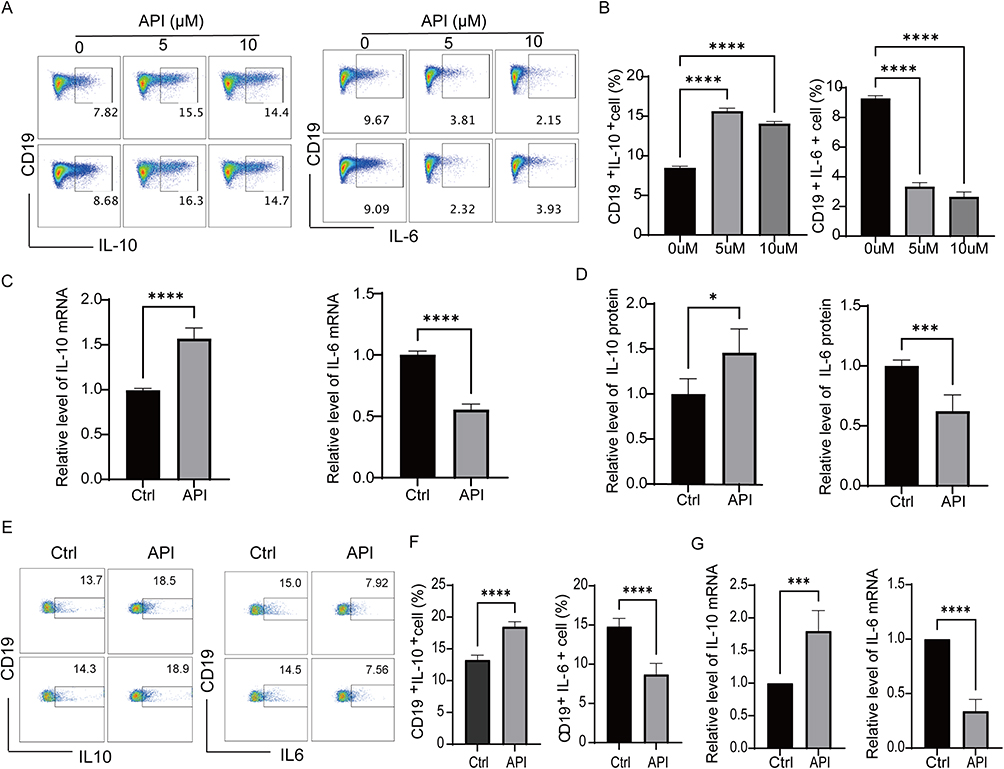 |
Figure 1 Apigenin promotes Breg generation in vitro. (A) Representative flow cytometry plots showing cytokine expression in splenic B cells isolated from C57BL/6J mice. Cells were stimulated with LPS (10 μg/mL) for 48 h in the absence or presence of the indicated concentrations of API (5 or 10 μM), and then treated with L+PIM (PMA, ionomycin, and monensin) for the last 5 h to block cytokine secretion. (B) Quantification of the percentage of IL-10⁺ and IL-6⁺ B cells under the same treatment conditions as in (A). (C) RT-qPCR analysis of IL-10 and IL-6 mRNA expression in purified mouse B cells cultured with or without 5 μM API for 48 h. (D) ELISA determination of IL-10 and IL-6 protein levels in the culture supernatants of the same cells described in (C). (E and F) Representative flow cytometry plots and statistical analysis of human PBMCs stimulated with LPS (10 μg/mL) in the absence or presence of 5 μM API for 48 h, and treated with L+PIM for the last 5 h. (G) RT-qPCR analysis of IL10 and IL-6 mRNA expression in human PBMCs under the same conditions as in (E). Data are presented as mean ± SD from at least five independent biological replicates (each replicate used cells from a separate mouse or individual human donor). *p < 0.05, ***p < 0.001, ****p < 0.0001, compared with the Ctrl or 0 µM group. |
A similar regulatory effect was observed in human peripheral blood mononuclear cells (PBMCs). When stimulated with LPS, API treatment enhanced the generation of human Bregs and reduced IL-6 production (Figure 1E and F). Consistent with the flow cytometry results, API also increased IL-10 mRNA expression and decreased IL-6 mRNA expression in these cells (Figure 1G). Collectively, these findings suggest that API helps maintain immune homeostasis by modulating Breg biogenesis and cytokine secretion, thereby inhibiting inflammatory responses and counteracting immune overactivation.31 This observation is consistent with recent evidence highlighting the immunomodulatory potential of flavonoids. Flavonoids have been recognized as key modulators of immune cells, including the regulation of regulatory B cell differentiation and cytokine production in inflammatory conditions.32
Apigenin Promotes the Generation of Breg in vivo
Next, we evaluated the ability of API to enhance Breg generation in vivo. To comprehensively assess its immunomodulatory potential across different physiological contexts, we performed studies in both healthy C57BL/6J mice and a murine model of collagen-induced arthritis (CIA) in DBA/1J mice (Figure 2A–D). Healthy mice were administered API via a diet containing 2 g/kg for 6 days. Subsequent flow cytometric analysis of their PBMCs showed that this treatment significantly increased the frequency of Bregs within the B-cell compartment relative to the untreated control (Figure 2B and C). A consistent effect was observed in the CIA model. In this autoimmune setting, API administration significantly increased the proportion of splenic Bregs and markedly reduced the production of the pro-inflammatory cytokine IL-6 (Figure 2E and F). Taken together, these in vivo results demonstrate that API effectively promotes the differentiation of regulatory B cells while suppressing key inflammatory mediators across both physiological and pathological models. These findings provide a compelling mechanistic basis for the therapeutic potential of API in mitigating autoimmune pathology.
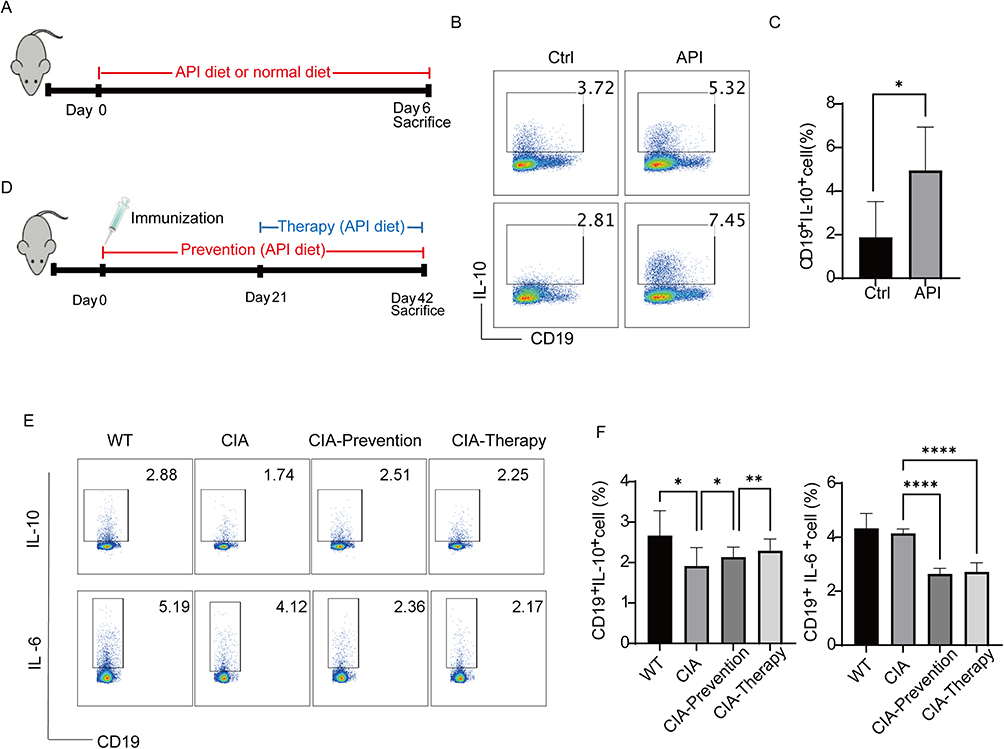 |
Figure 2 Apigenin promotes Breg generation in vivo. (A) Schematic of the experimental procedure. C57BL/6J mice were randomly divided into two groups: Control (normal diet) and API-treated (diet supplemented with 2 g/kg apigenin) for 6 days. Peripheral blood was collected for in vitro analysis. (B) Representative flow cytometry plots of IL-10⁺ cells among CD19⁺ B cells from the spleens of the mice described in (A). Splenocytes were cultured with LPS (10 μg/mL) for 48 h and stimulated with L+PIM for the last 5 h. (C) Quantification of splenic Breg (IL-10⁺CD19⁺) frequency. (D) Schematic of API treatment in the collagen-induced arthritis (CIA) model. DBA/1J mice were immunized with type II collagen on day 0. API was administered via diet (2 g/kg) either from day 0 (prevention) or day 21 (therapy) until day 42. (E) Representative flow cytometry plots of IL-10⁺ cells among CD19⁺ B cells from the spleens of CIA mice. Splenocytes were cultured as in (B). (F) Quantification of splenic Breg frequency (left) and IL-6⁺ B cell frequency (right) from CIA mice. Data in (C and F) are presented as mean ± SD. *p < 0.05, **p < 0.01 and ****p < 0.0001 compared to Ctrl or CIA group with the same stimuli. |
Apigenin Inhibits Antibody Class Switch Recombination in vitro
An in vitro class switch recombination (CSR) model was established to evaluate the effect of API on antibody class switching in B cells. Switching rates to IgG1 (induced by LPS+IL-4 for 72 h) and IgG3 (induced by LPS for 96 h) were quantified by flow cytometry. API treatment significantly reduced the frequency of both IgG1+ B cells and IgG3+ B cells without markedly affecting cell viability, suggesting that API may inhibit antibody diversification by interfering with the CSR process in B cells, as illustrated in Figure 3A.
Given that Aicda is a key enzyme for class switch recombination and Prdm1 is a core transcription factor regulating cell differentiation and antibody secretion.33 The downregulation of Aicda indicates that API directly inhibits the core molecular machinery driving CSR. CSR mediates the diversification of antibody effector functions, and its dysregulation promotes the production of class-switched, high-affinity pathogenic autoantibodies. Concurrent downregulation of Prdm1 suggests that API also impedes the commitment to plasma cell fate, thereby limiting the pool of long-lived antibody-secreting cells capable of sustaining autoreactive humoral responses. RT-qPCR analysis revealed that API treatment for 72 or 96 h significantly downregulated the mRNA levels of both Aicda and Prdm1 (Figure 3B and C). Collectively, these results demonstrate that API attenuates antibody class switching and plasma cell generation, thereby restricting the pro-inflammatory potential of B cells.
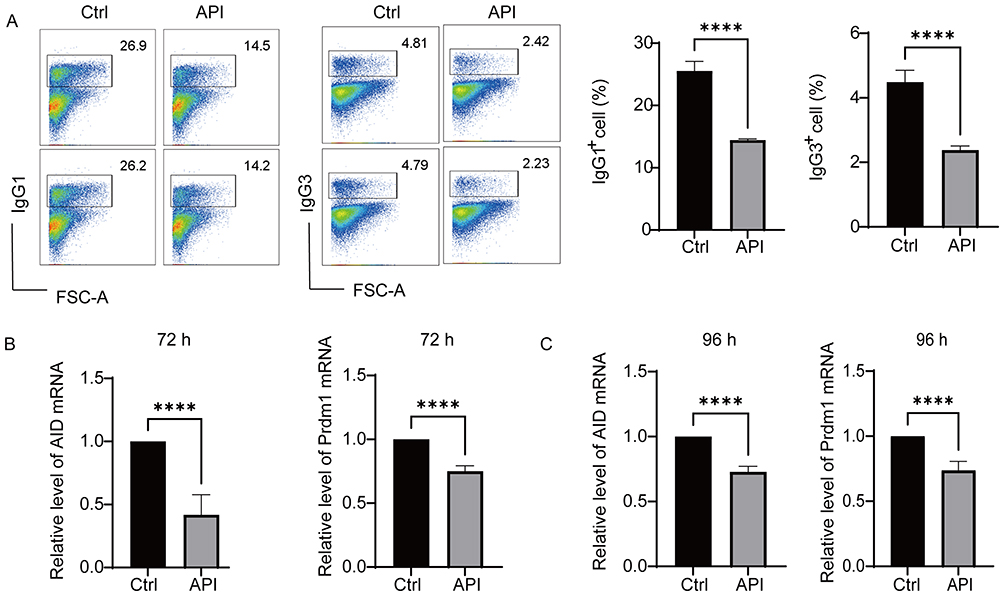 |
Figure 3 Apigenin inhibits antibody class switch recombination in vitro. (A) Purified mouse splenic B cells were stimulated with LPS (10 μg/mL) plus IL-4 (20 ng/mL) for 72 h in the presence or absence of 5 μM API to assess IgG1 switching, or with LPS alone (10 μg/mL) for 96 h to assess IgG3 switching. Cell viability and the percentages of IgG1⁺ and IgG3⁺ B cells were determined by flow cytometry. (B and C) RT-qPCR analysis of Aicda (AID) and Prdm1 mRNA levels in B cells cultured with LPS + IL-4 for 72 h (B) or with LPS for 96 h (C). Data represent five independent experiments and are shown as mean ± SD. ****p < 0.0001, compared with the Ctrl group. |
Apigenin Activates the p38 MAPK Pathway in B Cells
To elucidate the mechanism by which API regulates Breg differentiation, we performed RNA-seq analysis on purified B cells stimulated with LPS for 72 h with or without API. KEGG pathway analysis of the differentially expressed genes (DEGs) revealed significant enrichment of the MAPK signaling pathway across all DEGs, as well as in both up-regulated and down-regulated gene subsets (Figure 4A–Figure 4C).
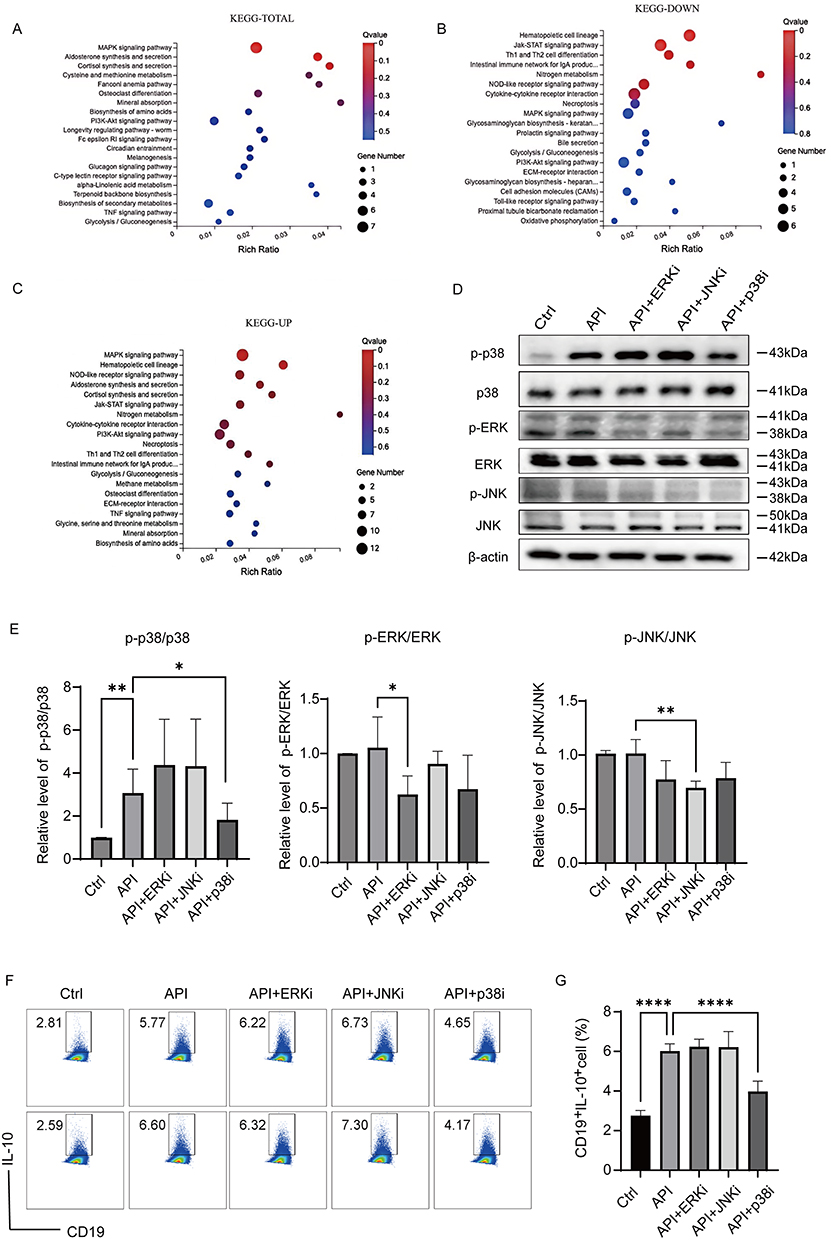 |
Figure 4 Apigenin upregulates Bregs via activation of the p38 MAPK pathway. Purified mouse splenic B cells were treated with LPS (10 μg/mL) and 5 μM API for 48 h, and then stimulated with L+PIM for the last 5 h before RNA-seq or protein analysis. (A–C) KEGG pathway enrichment analysis of all differentially expressed genes (DEGs) (A), down-regulated DEGs (B), and up-regulated DEGs (C). (D and E) Immunoblot analysis of phosphorylated and total p38, ERK, and JNK. Band intensities were semi-quantified using ImageJ, and the p-protein/total protein ratios are shown in (E). (F and G) Effect of MAPK inhibitors on API-induced Breg generation. B cells were cultured for 48 h with LPS and API in the presence or absence of the indicated inhibitors: ERK inhibitor (ERKi, 5 μM), p38 inhibitor (p38i, 5 μM), or JNK inhibitor (JNKi, 1 μM). Breg frequency was measured by flow cytometry. Data are mean ± SD from five independent biological replicates. *p < 0.05, **p < 0.01, ****p < 0.0001, compared with the API group. |
We then validated whether API directly activates MAPK signaling. Western blot analysis showed that API treatment significantly enhanced phosphorylation of p38, but not of ERK or JNK, in LPS-stimulated B cells (Figure 4D and Figure 4E). These results are consistent with previous studies showing that pharmacological or genetic disruption of p38 MAPK signaling impairs Breg differentiation and suppressive function, underscoring the pathway’s indispensable role in Breg biology.34
We next used specific kinase inhibitors to assess the functional relevance of these pathways in API-induced Breg generation. Inhibition of p38 MAPK markedly suppressed API-induced Breg differentiation, returning Breg frequencies to levels comparable to the untreated control (Figure 4F and Figure 4G). In contrast, ERK and JNK inhibitors had no significant effect. Consistent with this, the p38 inhibitor selectively attenuated API-induced p38 phosphorylation, whereas ERK and JNK inhibitors did not inhibit p38 activation. The efficacy of each inhibitor was confirmed by reduced phosphorylation of its respective target kinase.
Together, these data demonstrate that API selectively activates the p38 MAPK signaling pathway to promote the generation of IL-10–producing regulatory B cells.
Apigenin Inhibits Synovial Inflammation and Bone Loss in CIA Mice
Based on the CIA model, this study systematically evaluated the preventive and therapeutic effects of API intervention. Body weight changes, clinical arthritis scores, and joint histopathology were monitored. The results showed that from day 21 post-immunization, mice in the CIA group began to lose weight and developed acute symptoms, including joint redness and swelling. In contrast, body weight remained stable in both the API prevention and treatment groups, which also exhibited a significant delay in arthritis onset and significantly lower clinical scores compared to the CIA group (Figures 5A–C and S2). Histopathological examination of ankle joints revealed marked differences among the experimental groups (Figure 5D and F). Wild-type (WT) mice displayed normal synovial architecture with a thin synovial lining, intact cartilage surfaces, and no evidence of inflammatory cell infiltration. In contrast, CIA mice exhibited severe synovial hyperplasia, extensive infiltration of inflammatory cells, prominent cartilage erosion, and focal areas of bone destruction. Both the CIA-Prevention group (API administered from day 0) and the CIA-Therapy group (API administered from day 21) showed significant attenuation of these pathological features. Specifically, API-treated mice presented with reduced synovial thickening, diminished inflammatory infiltrates, and notably better preservation of cartilage and bone structures compared with untreated CIA controls. Semiquantitative scoring confirmed that API treatment significantly lowered both inflammation scores (Figure 5G) and bone destruction scores (Figure 5E) relative to the CIA group. These findings align with recent studies demonstrating that various natural compounds can achieve similar protective outcomes in rheumatoid arthritis, including the suppression of synovial hyperplasia, angiogenesis, and osteoclastogenesis.35
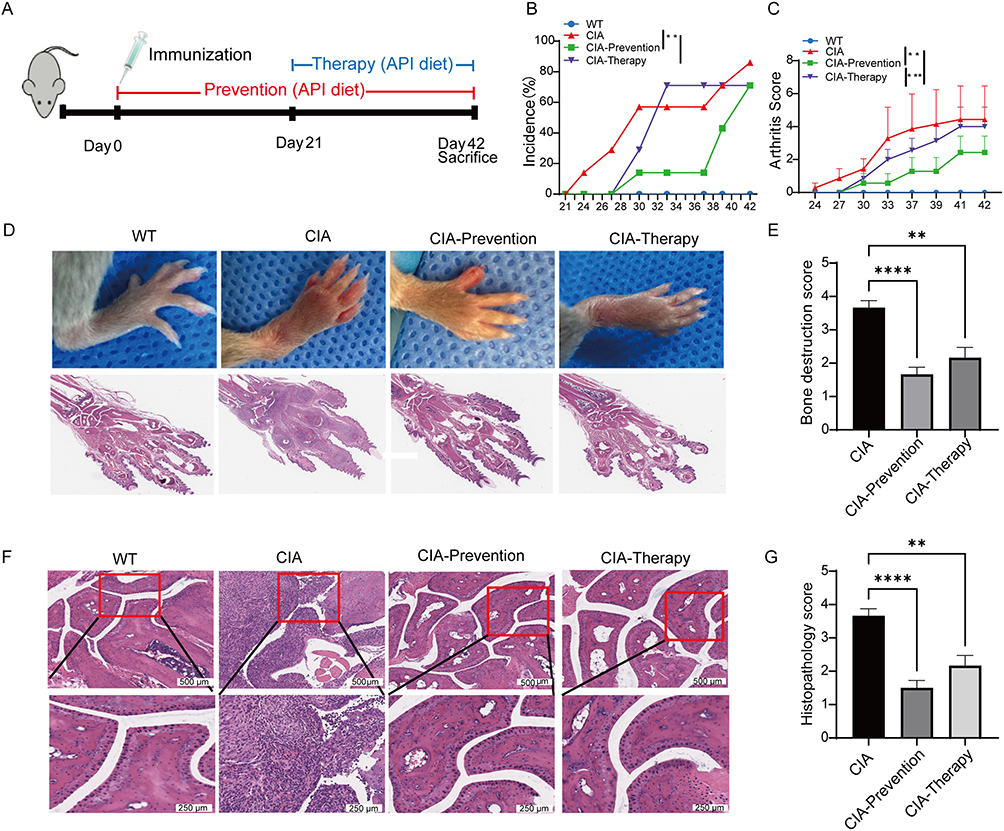 |
Figure 5 Apigenin ameliorates synovial inflammation in CIA mice. (A) Experimental design: DBA/1J mice were immunized on day 0 and received API diet (2 g/kg) from day 0 (prevention) or day 21 (therapy). (B) Incidence of visible paw arthritis. (C) Clinical arthritis scores over time. (D) Representative photographs and H&E-stained joint sections from each group. (E) Bone destruction score based on histological analysis of ankle joints. (F) Representative H&E-stained ankle joint sections. Upper panels: low-magnification (scale bar=500 μm). Lower panels: high-magnification of the regions outlined by red boxes in the corresponding upper panels (scale bar=250 μm). (G) Inflammation score of ankle joint sections. Data are shown as mean ± SD from at least six mice per group. **p < 0.01, ***p < 0.001, ****p < 0.0001, compared with the CIA group. |
To further assess the protective effect of API on bone architecture, micro-CT was used for 3D imaging and bone histomorphometric analysis of mouse ankle joints. 3D reconstructions revealed severe bone destruction in the ankle joints and metatarsals of the CIA group, characterized by rough and blurred articular surfaces. In contrast, the API prevention and treatment groups maintained clear, intact articular surfaces and preserved bone structure (Figure 6A–C). Quantitative analysis showed that bone volume (BV), bone volume fraction (BV/TV), and trabecular thickness (Tb.Th) were significantly lower in the CIA group than in the API-treated groups (Figure 6D–F). Together, these data demonstrate that API intervention effectively mitigates arthritis-induced bone destruction, highlighting its distinct bone-protective properties.
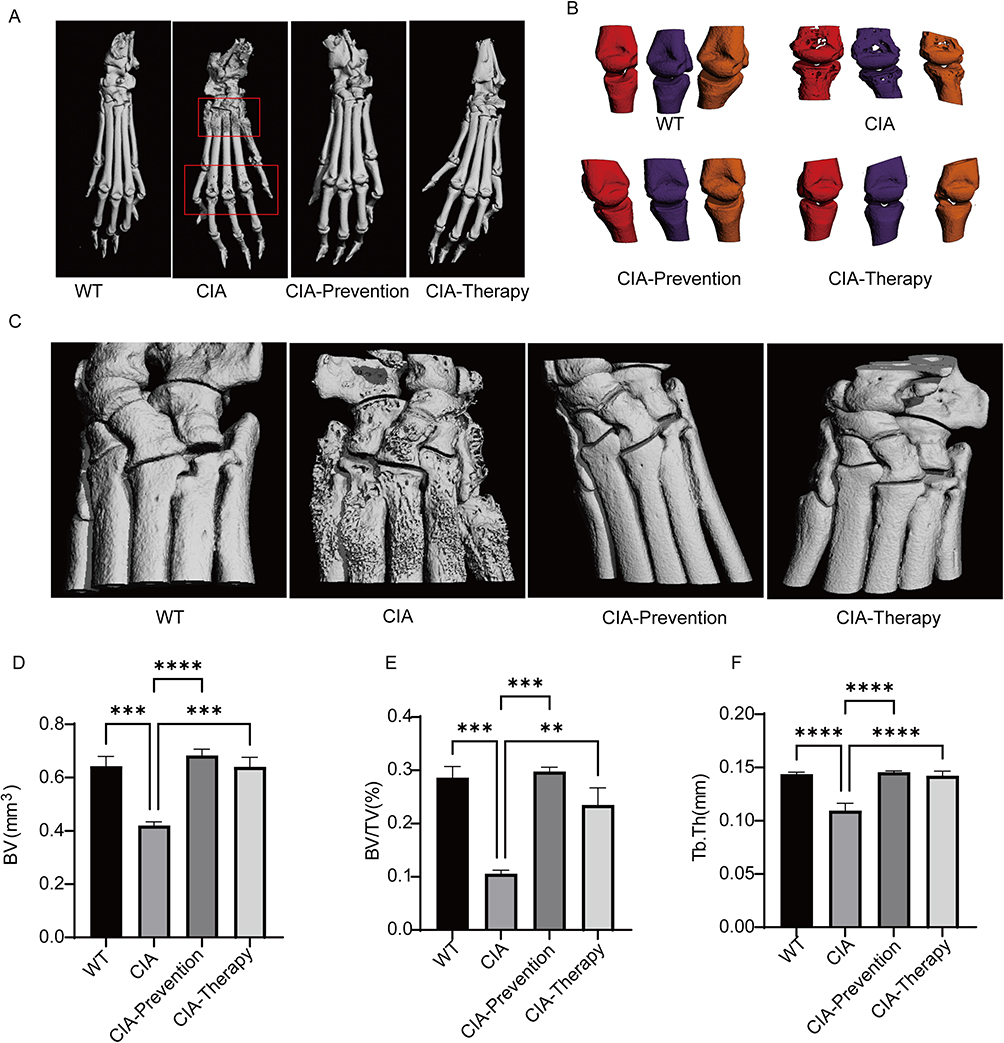 |
Figure 6 Effect of Apigenin on bone destruction in CIA mice. (A) Three-dimensional micro-CT reconstructions of the whole paw. (B) Enlarged 3D images of the second to fourth metatarsals. (C) Micro-CT 3D images of ankle joints. (D) Bone volume (BV). (E) Bone volume/total volume ratio (BV/TV). (F) Trabecular thickness (Tb.Th). Data are presented as mean ± SD. **p < 0.01, ***p < 0.001, ****p < 0.0001, compared with the CIA group. |
Apigenin Ameliorates Survival and Lung Injury in Septic Mice
Based on the CLP-induced sepsis model, this study systematically evaluated the protective effect of API. Because of the high mortality rate in sepsis, the 7-day survival rate of mice was first examined. Results showed that the survival rate was 0% in the CLP model group, while API intervention exhibited a dose-dependent protective effect: the survival rates were 40% in the low-dose group (20 mg/kg) and 70% in the high-dose group (60 mg/kg) (Figure 7A), indicating that API significantly counteracts lethal sepsis outcomes.
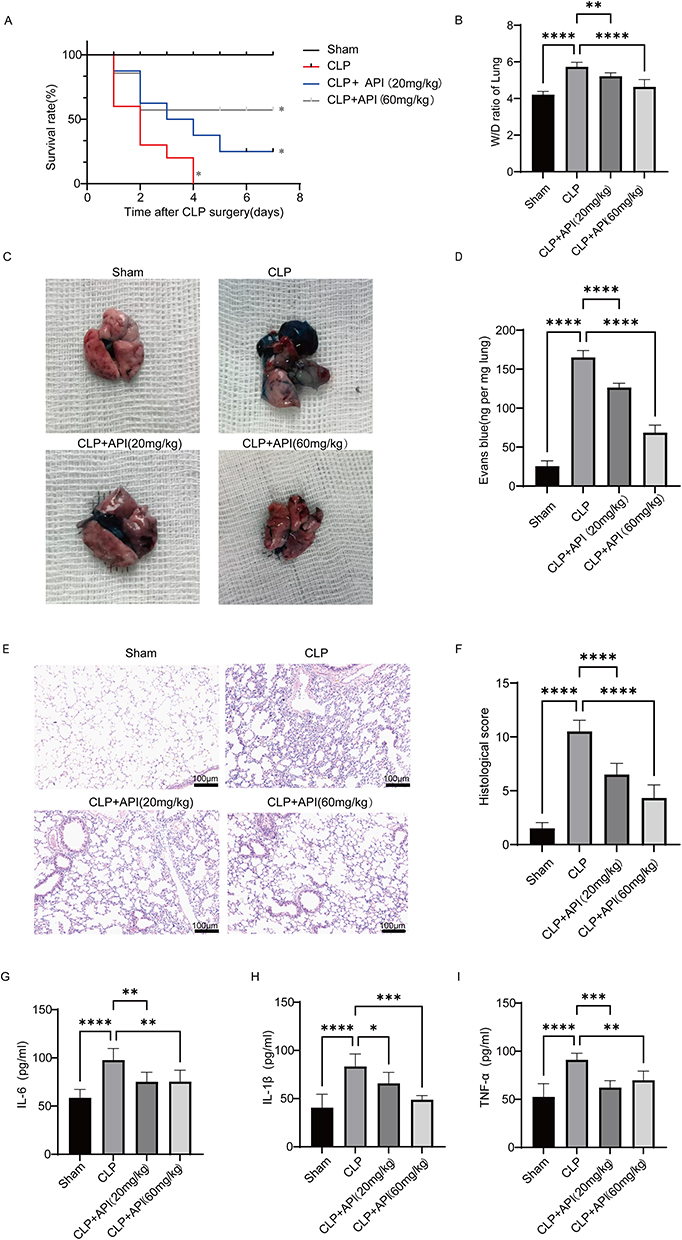 |
Figure 7 Apigenin ameliorates survival and lung injury in septic mice. (A) Seven-day survival curves after cecal ligation and puncture (CLP). (B) Lung wet-to-dry (W/D) weight ratio at 24 h post-CLP, an indicator of pulmonary edema. (C) Representative macroscopic images of Evans Blue (EB)-stained lungs. Blue staining denotes dye extravasation and increased vascular permeability. (D) Quantitative measurement of EB content in lung tissue. (E) Representative H&E-stained lung sections (original magnification, ×100; scale bar=100 μm). (F) Semi-quantitative histopathological injury scores. (G–I) Plasma concentrations of IL-6 (G), IL-1β (H), and TNF-α (I) at 24 h post-CLP measured by ELISA. Data are shown as mean ± SD from at least six mice per group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, compared with the CLP group. |
Given the improved survival, we further assessed whether API alleviates critical organ injury by evaluating its impact on acute lung injury. Results showed that compared with the model group, API (especially at 60 mg/kg) significantly reduced the lung wet/dry weight ratio (Figure 7B) and Evans blue accumulation in lung tissue (Figure 7C and D), demonstrating that API effectively mitigated pulmonary edema and decreased vascular permeability. Histopathological examination of lung tissue (Figure 7E and F) revealed marked differences among groups. Lung sections from sham-operated mice displayed normal alveolar architecture with thin alveolar septa and no evidence of inflammatory infiltration or hemorrhage. In contrast, CLP mice exhibited severe lung injury, characterized by thickened alveolar septa, extensive inflammatory cell infiltration, intra-alveolar hemorrhage, and interstitial edema. API treatment, particularly at the high dose (60 mg/kg), significantly attenuated these pathological changes, as evidenced by reduced septal thickening, diminished inflammatory infiltrates, and less pronounced hemorrhage and edema. In support of these observations, a recent study by Geng et al reported that apigenin significantly improves sepsis-induced lung inflammation and injury by promoting macrophage polarization toward the anti‑inflammatory M2 phenotype in a TLR7‑dependent manner, independently corroborating the protective effect of apigenin against septic acute lung injury.36
Because API was observed to alleviate lung parenchymal injury, to explore the underlying mechanism, the systemic inflammatory response was examined. ELISA results showed that high-dose API (60 mg/kg) significantly suppressed the elevation of pro-inflammatory cytokines IL-6, IL-1β, and TNF-α in plasma (Figure 7G–I), suggesting that its protective effect may be associated with the suppression of systemic hyperinflammation.
Discussion
Restoration of immune homeostasis, rather than indiscriminate immunosuppression, has emerged as a central paradigm in the treatment of inflammatory and autoimmune diseases.37,38 In this study, we identify apigenin (API), a naturally occurring flavonoid, as a potent immunoregulatory agent that reprograms B-cell fate decisions toward a regulatory phenotype while concomitantly restraining pathogenic antibody diversification.15,39 Our findings reveal a unified cellular and molecular mechanism through which API exerts therapeutic effects across both chronic autoimmunity and acute systemic inflammation.
A principal finding of this work is that API robustly promotes the differentiation of IL-10-producing regulatory B cells (Bregs) via selective activation of the p38 MAPK signaling pathway. While p38 MAPK is classically associated with inflammatory responses, accumulating evidence suggests that its function is highly context- and cell type–dependent.40,41 Our data extend this concept by demonstrating that p38 activation in B cells preferentially supports the acquisition of an immunoregulatory program, characterized by increased IL-10 production and suppression of pro-inflammatory cytokines such as IL-6. Pharmacological inhibition of p38 abrogated API-induced Breg expansion, underscoring the essential role of this pathway in mediating API’s immunomodulatory effects. The importance of the MAPK signaling pathway in rheumatoid arthritis pathogenesis is further supported by recent studies demonstrating that apigenin derivatives inhibit RA fibroblast-like synoviocyte activation via this pathway, and that genetic association studies have identified shared risk genes between autoimmune thyroid disease and RA that converge on immune signaling networks.42,43
In parallel with Breg induction, API markedly suppressed class switch recombination (CSR) and plasma cell differentiation, as evidenced by reduced expression of Aicda and Prdm1 and decreased generation of IgG1⁺ and IgG3⁺ B cells.44,45 These findings suggest that API enforces a coordinated B-cell regulatory state that limits antibody diversification and effector differentiation while enhancing immunosuppressive capacity.46 Such dual regulation is particularly relevant in autoimmune settings, where aberrant antibody production and dysregulated B–T cell interactions drive chronic inflammation and tissue destruction.47,48 Notably, our findings align with and extend recent work showing that natural compounds can attenuate RA progression through multiple mechanisms, including the inhibition of synovial hyperplasia, angiogenesis, and osteoclastogenesis in CIA models.49,50
The functional relevance of this B-cell–centered mechanism was substantiated in two distinct disease models. In collagen-induced arthritis, API attenuated synovial inflammation, preserved bone architecture, and reduced clinical severity, consistent with the restoration of Breg-mediated immune tolerance. In the cecal ligation and puncture model of sepsis, API significantly improved survival and mitigated acute lung injury, accompanied by suppression of systemic pro-inflammatory cytokines.36 Despite the divergent pathophysiology of RA and sepsis, both conditions share a common feature of immune dysregulation, and our data suggest that reinforcement of the Breg network represents a convergent therapeutic strategy.
Several limitations of the present study should be acknowledged. Although we establish p38 MAPK as a critical mediator of API-induced Breg differentiation, the upstream molecular targets through which API engages B-cell signaling remain undefined. Whether API directly interacts with specific B-cell surface receptors (eg, G protein-coupled receptors or Toll-like receptors) to initiate downstream signaling cascades requires further exploration.51,52 In addition, while our human PBMC data support the translational relevance of these findings, validation in patient-derived samples from individuals with RA or sepsis will be essential to confirm clinical applicability.
Finally, the poor solubility and bioavailability of API remain major obstacles to clinical translation.53 Advances in nanotechnology-based drug delivery systems and structure-guided optimization offer promising avenues to overcome these pharmacokinetic limitations.54–57 By integrating immunological mechanism, disease relevance, and translational perspective, our study positions apigenin as a prototype of mechanism-based immunoregulatory therapy aimed at rebuilding immune homeostasis rather than merely suppressing inflammation.
These findings support a paradigm in which B-cell–intrinsic immune reprogramming represents a viable therapeutic strategy for both chronic autoimmune and acute inflammatory diseases.
Conclusion
This study demonstrates that API promotes regulatory B cell differentiation via activation of the p38 MAPK pathway, suppresses antibody class-switch recombination by downregulating Aicda, and inhibits plasma cell generation through reduced expression of Prdm1 and IL-6. These mechanisms underpin its therapeutic efficacy in two distinct models of immune dysregulation. In collagen-induced arthritis, API alleviated joint inflammation and bone erosion, while in cecal ligation and puncture-induced sepsis, it improved survival and attenuated lung injury. Collectively, these findings position API as a promising immunomodulatory agent capable of restoring immune homeostasis in both chronic autoimmune and acute inflammatory settings.
Abbreviations
Aidca, Activation-Induced Cytidine Deaminase; API, Apigenin; Breg(s), Regulatory B Cell(s); CIA, Collagen-Induced Arthritis; CLP, Cecal Ligation and Puncture; CSR, Class Switch Recombination; ERK, Extracellular Signal-Regulated Kinase; H&E, Hematoxylin and Eosin; IL, Interleukin; JNK, c-Jun N-terminal Kinase; MAPK, Mitogen-Activated Protein Kinase; PBMC(s), Peripheral Blood Mononuclear Cell(s); PFA, Paraformaldehyde; Prdm1, PR Domain Zinc Finger Protein 1; p38, p38 Mitogen-Activated Protein Kinase; RA, Rheumatoid Arthritis; SA-ALI, Sepsis-Associated Acute Lung Injury; TGF-β, Transforming Growth Factor Beta.
Data Sharing Statement
The RNA-seq data generated in this study have been deposited in the Genome Sequence Archive (GSA) of the National Genomics Data Center (CNCB) under accession number CRA038922. The data can be accessed via the following link: https://ngdc.cncb.ac.cn/gsa/s/vwo6XXcW.
Ethics Approval and Informed Consent
Experiments with human PBMCs were approved by the Ethics Committee of The First Dongguan Affiliated Hospital of Guangdong Medical University (Approval No. YS202310007) and were conducted in accordance with the principles of the Declaration of Helsinki. Peripheral blood samples were obtained from five healthy donors (three males and two females, aged 20–40 years) after written informed consent was obtained from all participants.
All animal procedures were approved by the relevant Institutional Animal Care and Use Committees and conducted in strict accordance with the ARRIVE guidelines and the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals. The CIA experiment was approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University (Approval No. SYSU-IACUC-2020-000414), and the CLP experiment was approved by the Institutional Animal Care and Use Committee of Guangdong Medical University (Approval No. GDMU-2023-000003).
Consent for Publication
All authors have read and approved the final version of this manuscript. This work is original, has not been published previously, and is not currently under consideration elsewhere.
Acknowledgments
The authors sincerely thank all the volunteers who participated in this study. We gratefully acknowledge the clinical staff of The First Dongguan Affiliated Hospital of Guangdong Medical University for their professional assistance in the collection and processing of samples. We thank our laboratory technician for skilled technical support in establishing animal models and performing flow cytometry assays. Finally, we extend our thanks to all colleagues who facilitated and supported this research.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work is supported by National Nature Science Foundation of China (Grant No. 82370969) to J.S.; Guangdong Basic and Applied Basic Research Foundation (Grant No. 2024A151540164, and 2022A1515010678) to J.S.; Dongguan Science and Technology of Social Development Program (Grant No. 20221800905652) to J.S.; Guangdong Basic and Applied Basic Research Foundation (Grant No. 2022A1515140101) to L.L.; Clinical plus Basic Innovation Project of Guangdong Medical University (Grant No. 4SG24012G and GDMULCJC2025115) to J.S.; Talent Development Foundation of The First Dongguan Affiliated Hospital of Guangdong Medical University (Grant No. GCC2022003) to J.S.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Leuti A, Fazio D, Fava M, Piccoli A, Oddi S, Maccarrone M. Bioactive lipids, inflammation and chronic diseases. Adv Drug Deliv Rev. 2020;159:133–19. doi:10.1016/j.addr.2020.06.028
2. Luo H. Global burden and cross-country inequalities in six major immune-mediated inflammatory diseases from 1990 to 2021: a systemic analysis of the Global Burden of Disease Study 2021. Autoimmun Rev. 2024;23(10):103639. doi:10.1016/j.autrev.2024.103639
3. Shi HW, Yang BC, Ren YQ, Xue Y. Applications of antioxidant nanoparticles in immune-mediated inflammatory diseases. Antioxidants. 2025;14(9):1128. doi:10.3390/antiox14091128
4. Maerz JK, Trostel C, Lange A, et al. Bacterial immunogenicity is critical for the induction of regulatory B cells in suppressing inflammatory immune responses. Front Immunol. 2020;10:3093. doi:10.3389/fimmu.2019.03093
5. Deacy AM, Gan SKE, Derrick JP. Superantigen recognition and interactions: functions, mechanisms and applications. Front Immunol. 2021;12:731845. doi:10.3389/fimmu.2021.731845
6. Krapić M, Kavazović I, Mikašinović S, et al. NK cell-derived IFNγ mobilizes free fatty acids from adipose tissue to promote early B cell activation during viral infection. Nat Metab. 2025;7(5):985–1003. doi:10.1038/s42255-025-01273-2
7. Nadeem A, Ahmad SF, Al-Harbi NO, et al. Imbalance in pro-inflammatory and anti-inflammatory cytokines milieu in B cells of children with autism. Mol Immunol. 2022;141:297–304. doi:10.1016/j.molimm.2021.12.009
8. Catalán D, Mansilla MA, Ferrier A, et al. Immunosuppressive mechanisms of regulatory B cells. Front Immunol. 2021;12:611795. doi:10.3389/fimmu.2021.611795
9. Yang Q, Zhang Z, Chen Z, et al. Flot2 deficiency facilitates B cell-mediated inflammatory responses and endotoxic shock. Immunology. 2023;170(4):567–578. doi:10.1111/imm.13692
10. Aoun M, Coelho A, Krämer A, et al. Antigen-presenting autoreactive B cells activate regulatory T cells and suppress autoimmune arthritis in mice. J Exp Med. 2023;220(11):e20230101. doi:10.1084/jem.20230101
11. Xie G, Chen X, Gao Y, et al. Age-associated B cells in autoimmune diseases: pathogenesis and clinical implications. Clin Rev Allergy Immunol. 2025;68(1):18. doi:10.1007/s12016-025-09021-w
12. Su QY, Jiang ZQ, Song XY, Zhang SX. Regulatory B cells in autoimmune diseases: insights and therapeutic potential. J Autoimmun. 2024;149:103326. doi:10.1016/j.jaut.2024.103326
13. Dong X, Tu H, Qin S, Bai X, Yang F, Li Z. Insights into the roles of B cells in patients with sepsis. J Immunol Res. 2023;2023:7408967. doi:10.1155/2023/7408967
14. Wang C, Xu H, Gao R, et al. CD19⁺CD24ʰiCD38ʰi regulatory B cells deficiency revealed severity and poor prognosis in patients with sepsis. BMC Immunol. 2022;23(1):28. doi:10.1186/s12865-022-00528-x
15. Taha M, Eldemerdash OM, Elshaffei IM, Yousef EM, Soliman AS, Senousy MA. Apigenin attenuates hippocampal microglial activation and restores cognitive function in methotrexate-treated rats: targeting the miR-15a/ROCK-1/ERK1/2 pathway. Mol Neurobiol. 2023;60(7):3770–3787. doi:10.1007/s12035-023-03299-7
16. Sun D, Cao C, Sun F, et al. Development of apigenin monoclonal antibodies and an immunoassay for the quantification of apigenin in celery. Food Chem. 2025;497:146989. doi:10.1016/j.foodchem.2025.146989
17. Feng X, Weng D, Zhou F, et al. Activation of PPARγ by a natural flavonoid modulator, apigenin ameliorates obesity-related inflammation via regulation of macrophage polarization. EBioMedicine. 2016;9:61–76. doi:10.1016/j.ebiom.2016.06.017
18. Gongpan P, Xu T, Zhang Y, et al. Apigenin alleviates inflammation as a natural IRAK4 inhibitor. Phytomedicine. 2025;139:156519. doi:10.1016/j.phymed.2025.156519
19. Liu T, Gao H, Zhang Y, et al. Apigenin ameliorates hyperuricemia and renal injury through regulation of uric acid metabolism and JAK2/STAT3 signaling pathway. Pharmaceuticals. 2022;15(11):1442. doi:10.3390/ph15111442
20. Miyoshi M, Liu S. Collagen-induced arthritis models. Methods Mol Biol. 2024;2766:3–7. doi:10.1007/978-1-0716-3682-4_1
21. Zhou XW, Wang J, Tan WF. Apigenin suppresses innate immune responses and ameliorates lipopolysaccharide-induced inflammation via inhibition of STING/IRF3 pathway. Am J Chin Med. 2024;52(2):471–492. doi:10.1142/S0192415X24500204
22. Kuru Bektaşoğlu P, Demir D, Koyuncuoğlu T, et al. Possible anti-inflammatory, antioxidant, and neuroprotective effects of apigenin in the setting of mild traumatic brain injury: an investigation. Immunopharmacol Immunotoxicol. 2023;45(2):185–196. doi:10.1080/08923973.2022.2130076
23. Monu AP, Saquib M, Biswas S, Biswas S. Targeting TNF-α-induced expression of TTR and RAGE in rheumatoid arthritis: apigenin’s mediated therapeutic approach. Cytokine. 2024;179:156616. doi:10.1016/j.cyto.2024.156616
24. Yu H, Huang X, Zhu HH, et al. Apigenin ameliorates non-eosinophilic inflammation, dysregulated immune homeostasis and mitochondria-mediated airway epithelial cell apoptosis in chronic obese asthma via the ROS-ASK1-MAPK pathway. Phytomedicine. 2023;111:154646. doi:10.1016/j.phymed.2023.154646
25. Lipsit SWL, Wilkinson J, Scruten E, et al. Kinome profiling of peripheral blood mononuclear cells collected prior to vaccination reveals biomarkers and potential mechanisms of vaccine unresponsiveness in pigs. Sci Rep. 2020;10(1):11546. doi:10.1038/s41598-020-68039-6
26. Zhou N, Zou F, Cheng X, et al. Porphyromonas gingivalis induces periodontitis, causes immune imbalance, and promotes rheumatoid arthritis. J Leukoc Biol. 2021;110(3):461–473. doi:10.1002/JLB.3MA0121-045R
27. Steven S, Helmstaedter J, Pawelke F, et al. P717 Glucagon-like peptide 1 (GLP-1) improves endothelial dysfunction and vascular inflammation in polymicrobial sepsis induced by cecal ligation and puncture (CLP). Eur Heart J. 2019;40(Suppl_1):
28. Arantes AQ, Leal CT, Silva CA, et al. Decreased activity of NK cells in myeloproliferative neoplasms. Blood. 2015;126(23):1637. doi:10.1182/blood.v126.23.1637.1637
29. Zou F, Qiu Y, Huang Y, et al. Effects of short-chain fatty acids in inhibiting HDAC and activating p38 MAPK are critical for promoting B10 cell generation and function. Cell Death Dis. 2021;12(6):582. doi:10.1038/s41419-021-03880-9
30. Leija R, Duong J, Arevalo J, et al. The final pathway of carbohydrate oxidation: revisiting the mLOC. Physiology. 2023;38(S1). doi:10.1152/physiol.2023.38.s1.5731313
31. Attiq A, Jalil J, Husain K, Mohamad HF, Ahmad A. Luteolin and apigenin derived glycosides from Alphonsea elliptica abrogate LPS-induced inflammatory responses in human plasma. J Ethnopharmacol. 2021;275:114120. doi:10.1016/j.jep.2021.114120
32. Liu Y, Jiao A. Flavonoids as immunoregulators: molecular mechanisms in regulating immune cells and their therapeutic applications in inflammatory diseases. Front Immunol. 2025;16:1703672. doi:10.3389/fimmu.2025.1703672
33. Moroney JB, Chupp DP, Xu Z, Zan H, Casali P. Epigenetics of the antibody and autoantibody response. Curr Opin Immunol. 2020;67:75–86. doi:10.1016/j.coi.2020.09.004
34. Barrio L, Román-García S, Díaz-Mora E, et al. B cell development and T-dependent antibody response are regulated by p38γ and p38δ. Front Cell Dev Biol. 2020;8:189. doi:10.3389/fcell.2020.00189
35. Jiao Y, Wang Z, Diao W, et al. Increased alleviation of bone destruction in individuals with rheumatoid arthritis via the coinhibition of the METTL3 and YTHDF1 axis by the combination of triptolide and medicarpin. Engineering. 2025;48:277–291. doi:10.1016/j.eng.2025.03.014
36. Geng J, Zheng Z, Li L, et al. Apigenin attenuated sepsis induced acute lung injury via polarizing macrophage towards M2 by blocking miR-146a→TLR7 interaction. Int Immunopharmacol. 2025;152:114446. doi:10.1016/j.intimp.2025.114446
37. Ho P, Cahir-McFarland E, Fontenot JD, et al. Harnessing regulatory T cells to establish immune tolerance. Sci Transl Med. 2024;16(738):eadm8859. doi:10.1126/scitranslmed.adm8859
38. Dixit N, Fanton C, Langowski JL, et al. NKTR-358: a novel regulatory T-cell stimulator that selectively stimulates expansion and suppressive function of regulatory T cells for the treatment of autoimmune and inflammatory diseases. J Transl Autoimmun. 2021;4:100103. doi:10.1016/j.jtauto.2021.100103
39. Ahmedy OA, Abdelghany TM, El-Shamarka MEA, Khattab MA, El-Tanbouly DM. Apigenin attenuates LPS-induced neurotoxicity and cognitive impairment in mice via promoting mitochondrial fusion/mitophagy: role of SIRT3/PINK1/Parkin pathway. Psychopharmacology. 2022;239(12):3903–3917. doi:10.1007/s00213-022-06262-x
40. Maeyer RPHD, van de MRC, Louie R, et al. Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat Immunol. 2020;21(6):615–625. doi:10.1038/s41590-020-0646-0
41. Zheng Y, Cheng D, Wang X, et al. Magnolol attenuates hyperlipidemia-induced endothelial disorder by alleviating oxidative stress, inflammation, and mitochondrial dysfunction via p38 MAPK-FoxO1 axis. Free Radic Biol Med. 2026;245:361–373. doi:10.1016/j.freeradbiomed.2026.01.011
42. Cao D, Fan Q, Li Z, et al. Transcriptomic profiling revealed the role of apigenin-4’-O-α-L-rhamnoside in inhibiting the activation of rheumatoid arthritis fibroblast-like synoviocytes via MAPK signaling pathway. Phytomedicine. 2022;102:154201. doi:10.1016/j.phymed.2022.154201
43. Peng Z, Huang W, Tang M, et al. Investigating the shared genetic architecture between hypothyroidism and rheumatoid arthritis. Front Immunol. 2024;14:1286491. doi:10.3389/fimmu.2023.1286491
44. Ochiai K, Shima H, Tamahara T, et al. Accelerated plasma-cell differentiation in Bach2-deficient mouse B cells is caused by altered IRF4 functions. EMBO J. 2024;43(10):1947–1964. doi:10.1038/s44318-024-00077-6
45. Pati S, Mukherjee S, Dutta S, et al. Tumor-associated CD19⁺CD39− B regulatory cells deregulate class-switch recombination to suppress antibody responses. Cancer Immunol Res. 2023;11(3):364–380. doi:10.1158/2326-6066.CIR-21-1073
46. Wu F, Gao J, Kang J, et al. B cells in rheumatoid arthritis: pathogenic mechanisms and treatment prospects. Front Immunol. 2021;12:750753. doi:10.3389/fimmu.2021.750753
47. Jang S, Kwon EJ, Lee JJ. Rheumatoid arthritis: pathogenic roles of diverse immune cells. Int J Mol Sci. 2022;23(2):905. doi:10.3390/ijms23020905
48. Rosser EC, Mauri C. The emerging field of regulatory B cell immunometabolism. Cell Metab. 2021;33(6):1088–1097. doi:10.1016/j.cmet.2021.05.008
49. Pan Y, Liu X, Wang S, et al. Coniferyl aldehyde in ginger-eucommiae cortex enhances osteoarthritis treatment by modulating ALDOA and H3K23la histone lactylation. Phytomedicine. 2026;152:157852. doi:10.1016/j.phymed.2026.157852
50. Zheng H, Deng Y, Li B, et al. Xuetongsu attenuates bone destruction in rheumatoid arthritis by suppressing RANKL/RANK/NFATc1 pathway to inhibit osteoclastogenesis and bone resorption. Bone Jt Res. 2025;14(11):1016–1032. doi:10.1302/2046-3758.1411.BJR-2025-0259.R1
51. Chen B, Guan X, Gunning WT, et al. Negative modulation of B cell activation by melanocortin 1 receptor signaling protects against membranous nephropathy. J Am Soc Nephrol. 2023;34(3):467–481. doi:10.1681/ASN.2022050605
52. Schön MP, Schön M. Imiquimod: mode of action. Br J Dermatol. 2007;157(Suppl 2):8–13. doi:10.1111/j.1365-2133.2007.08265.x
53. Li B, Hu Y, Wu T, et al. Apigenin-oxymatrine binary co-amorphous mixture: enhanced solubility, bioavailability, and anti-inflammatory effect. Food Chem. 2022;373(Pt B):131485. doi:10.1016/j.foodchem.2021.131485
54. Teixeira S, Carvalho MA, Castanheira EMS. Functionalized liposome and albumin-based systems as carriers for poorly water-soluble anticancer drugs: an updated review. Biomedicines. 2022;10(2):486. doi:10.3390/biomedicines10020486
55. Wu Y, Wan S, Yang S, et al. Macrophage cell membrane-based nanoparticles: a new promising biomimetic platform for targeted delivery and treatment. J Nanobiotechnol. 2022;20(1):542. doi:10.1186/s12951-022-01746-6
56. Zeng J, Zhang Y, Gao Y, et al. Biomimetic ginsenoside Rb1 and probucol co-assembled nanoparticles for targeted atherosclerosis therapy via inhibition of oxidative stress, inflammation, and lipid deposition. ACS Nano. 2025;19(25):22968–22987. doi:10.1021/acsnano.5c02492
57. Zhong D, Xie W, Liao Z, et al. YTHDF1 transcriptionally activated by TCF4 suppresses osteoblast ferroptosis in titanium nanoparticle-induced osteolysis by accelerating GPX4 and SLC7A11 translation. J Nanobiotechnol. 2025;23(1):783. doi:10.1186/s12951-025-03861-6
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.