Back to Journals » International Journal of Nanomedicine » Volume 13
Apically targeted oral micelles exhibit highly efficient intestinal uptake and oral absorption
Authors Wang JL, Wang LF, Li Y, Wang XH, Tu PF
Received 13 August 2018
Accepted for publication 9 October 2018
Published 26 November 2018 Volume 2018:13 Pages 7997—8012
DOI https://doi.org/10.2147/IJN.S183796
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Jinling Wang, Lifang Wang, Ying Li, Xiaohui Wang, Pengfei Tu
School of Chinese Materia Medica, Modern Research Center for Traditional Chinese Medicine, Beijing University of Chinese Medicine, Beijing, 100029, China
Introduction: Polymeric micelles (PMs) hold promise for improving solubility and oral absorption of poorly soluble drugs. Unfortunately, the oral absorption of PMs is also limited by intestinal epithelium. To improve the oral delivery efficiency of micelles, transporter-mediated micelles could enhance the transport efficiency across the epithelial barrier, and they have attracted more attention.
Methods: Peptide transporter 1 (PepT1)-mediated micelles (Val-PMs/Phe-PMs) were designed by grafting valine (or phenylalanine) onto the surface of curcumin (Cur)-loaded-D-α-tocopheryl polyethylene glycol 1000 succinate micelles (TP-PMs). The oral absorption mechanism and oral bioavailability were further investigated in vitro and in vivo.
Results: The cellular study showed that Val-PMs/Phe-PMs had a high PepT1 affinity, resulting in a higher drug uptake and transcellular transport than TP-PMs. In rats, Val-PMs/Phe-PMs exhibited higher intestinal accumulation in the apical side of the intestinal epithelium than TP-PMs, promoting drug diffusion across epithelial barrier. The oral bioavailability of Cur was significantly improved by Val-PMs/Phe-PMs, which was about 10.50- and 3.40-fold greater than that of Cur-Sol and TP-PMs, respectively.
Conclusion: PepT-1-mediated micelles, using PepT1 as a target on intestinal epithelium, have unique functions with intestine and prove promising for oral delivery of poorly water-soluble drugs.
Keywords: PepT1, micelles, epithelial barrier, curcumin, oral delivery
Introduction
Oral administration is commonly used for drug delivery because oral drug delivery is more safe and convenient and offers high patient compliance. However, effective oral drug delivery holds great challenges due to physicochemical properties of the drug and biologic barrier in gastrointestinal (GI) tract, especially the intestinal epithelium. Therefore, effective strategies for improving oral absorption are urgently needed. Nanocarriers, such as liposomes,1 nanoparticles,2,3 nanocrystals,4 and micelles,5 have unique physicochemical properties to overcome intestinal epithelial barrier and have attracted more attention in improving the efficiency of oral drug delivery.6
Nowadays, polymeric micelles (PMs) are promising vehicles to increase the oral absorption of hydrophobic drugs. PMs could self-assemble in aqueous medium and encapsulate hydrophobic drug in the inner core. PMs exhibited numerous advantages in oral delivery, such as the nanosize, the improved solubility, stability and permeability of insoluble drug in GI tract, and the involvement of the lymphatic transport. However, limited absorption and transport of PMs by the intestinal epithelium is still the main great challenge, and the enhanced oral absorption of PMs is also necessary and urgent.7–10 Recently, membrane transporter-targeted nanocarriers have been put forward and have attracted more attention, which could selectively identify and target certain transporters, such as bile acid transporter,11,12 and thereby result in more transcellular delivery of nanocarriers.13
Peptide transporter 1 (PepT1) is an oligopeptide transporter and abundantly expressed in the apical (AP) membrane of the intestinal epithelium.14,15 Due to high capacity and broad substrate specificity, PepT1 could efficiently identify and transport amino acid and oligopeptide (di/tripeptides), including L-valine, L-phenylalanine, and L-Val-L-Val.16,17 It is determined that targeting PepT1 could have a high affinity for epithelium and improve intestinal permeability and oral absorption of the drug due to the amino group.18 Recently, PepT1 was widely applied as an attractive target in oral prodrug, but it is seldom chosen as a target to enhance the oral absorption of micelles. However, functional micelles targeting PepT1 might be more effective in enhancing the oral absorption of the micelles by modifying micellar surface with a free amino group (such as L-valine or L-phenylalanine), which could have a high affinity for the intestinal epithelial transporter and improve the intestinal uptake and transport of the micelles. Therefore, PepT-1-targeting micelles would be very helpful in increasing the oral absorption of micelles.
D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) is an amphiphilic polymer formed by esterification of vitamin E succinate with polyethylene glycol (PEG).19,20 TPGS has been approved as a safe and effective solublizer and stabilizer for water-insoluble and unstable drug.21 TPGS is also a P-glycoprotein (P-gp) inhibitor and widely used as an absorption enhancer for improving the oral absorption of insoluble drug.22–24 Our previous research showed that D-α-tocopheryl polyethylene glycol 1000 succinate micelle (TP-PMs) had significantly increased the oral bioavailability of curcumin (Cur); however, the improved transport efficiency of Cur across epithelium by TP-PMs is still limited.25 Thereafter, to achieve a better transport efficiency and oral absorption of TP-PMs, PepT1-mediated TP-PMs by grafting free amino group onto TP-PMs surface have been proposed, which could specifically recognize PepT1 on epithelium, enhance the affinity of TP-PMs to epithelium, facilitate the transport across intestine, and further increase the oral absorption of TP-PMs.
This study focused on the design of transporter-mediated micelles to overcome the intestinal epithelial barriers and further enhance the oral bioavailability of micelles. In this study, PepT1 transporter-mediated micelles were developed to further facilitate the oral absorption of micelles and to investigate whether PepT1 targeting could improve micelles transport efficiency across the intestinal epithelium and oral absorption in vivo. Briefly, valine (or phenylalanine) was conjugated with TPGS (Val-TPGS or Phe-TPGS), leaving the free amino group for PepT1 recognition and used as a nanocarrier. Cur has anti-inflammatory, antioxidant, and antineoplastic activities with low solubility and poor stability and is used as a model drug. Then, in in vitro Caco-2 cells and in vivo rat model, the transport efficiency and oral absorption of PepT-1-mediated micelles (Val-PMs or Phe-PMs) were investigated, with the Cur solution (Cur-Sol) and non-PepT1-targeting micelles (TP-PMs) as control. Finally, there was a clear understanding on the effects of PepT1 on improving transport across epithelium and oral absorption of micelles. To our knowledge, the literature seldom reported grafting of the PepT1-targeting group on the surface of micelles, so it is novel to involve the target of PepT1 into oral micelles and investigate the mechanism of transport and oral absorption of PepT1-targeting micelles.
Materials and methods
Materials
Cur, TPGS, leptin, and glycylsarcosine (Gly-Sar) were all purchased from Sigma-Aldrich Co. (St Louis, MO, USA). N-benyloxycarbonyl-valine (Cbz-Val-OH) and N-benyloxycarbonyl-phenylalanine (Cbz-Phe-OH) were gained from GL Biochem Ltd. (Shanghai, China). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 4-dimethylaminopyridine (DMAP) were obtained from Zhenjiang Pukang Pharm Co. Ltd (Zhejiang, China). Penicillin-streptomycin solution and Hank’s buffered salt solution (HBSS) were obtained from Hyclone (Logan, USA). MTT and trypsin were also purchased from Sigma-Aldrich Co. All solvents used in the investigation were of analytical reagent grade.
Synthesis of Val-TPGS copolymer and Phe-TPGS copolymer
Val-TPGS and Phe-TPGS copolymer were synthesized similarly in two steps. Cbz-Val-OH (5 mmol) or Cbz-Phe-OH (5 mmol) was initially activated with EDC (8 mmol) and DMAP (5 mmol) under ice bath for 1 hour. Then, TPGS (5 mmol) was added to the mixture solution of Cbz-Val-OH or Cbz-Phe-OH and reacted at 30°C under nitrogen for another 12 hours. The product was purified by silica column chromatography. Finally, the Cbz group in the product was removed by Pd/C (10%) for 4 hours under H2 at 30°C. The final product of Val-TPGS or Phe-TPGS was obtained as a faint yellow waxy solid after drying in a vacuum drier.
The chemical structures of Val-TPGS and Phe-TPGS copolymer were characterized by 1H-nuclear magnetic resonance (1H-NMR) spectrometry (500 Hz; Varian, Palo Alto, CA, USA).
The critical micelle concentrations (CMCs) of Val-TPGS and Phe-TPGS copolymer were determined using pyrene as the fluorescence probe. Briefly, aliquots of 3×10−6 mol/L pyrene solution in acetone were transferred into a series of 10 mL volumetric flasks and the acetone was evaporated under nitrogen gas streams. Val-TPGS, Phe-TPGS, or TPGS solution was added to the volumetric flasks at different copolymer concentrations ranging from 2×10−4 to 1 mg/mL. The mixed solution of polymer and pyrene was ultrasound scanned for 4 hours and equilibrated overnight in dark at room temperature. The samples were measured using a fluorescence spectrophotometer (F-4500; Hitachi Ltd., Tokyo, Japan) with the emission wavelength at 338 nm and the excitation fluorescence at 375 and 386 nm. The CMC was measured as the cross-point when extrapolating the intensity ratio I375/I386 at low and high concentration tangent.
Preparation and characterization of Cur nanomicelles
Preparation of Val-PMs and Phe-PMs
Film hydration technique was applied to prepare Val-PMs and Phe-PMs. Briefly, Cur and Val-TPGS (or Phe-TPGS) were codissolved in dichloromethane under mild stirring and evaporated to form a thin film by rotary vacuum evaporation. Subsequently, the film was hydrated in 4 mL PBS (pH 7.4) and stirred for 2 hours at 37°C. Cur-loaded micelles were obtained after centrifugation (12,000 rpm, 10 minutes, and 4°C) to remove the unloaded drug. TP-PMs were used as a negative control and prepared in a similar way as described above. The empty micelles were prepared with the same procedure without Cur. Coumarin-6 (C6)-loaded micelles (C6-Val, C6-Phe, or C6-TP) were prepared by the same method, except for C6 instead of Cur.
Size, zeta potential, and particle morphology
The particle size and zeta potential of micelles were determined by dynamic light scattering using a Zetasizer (Nano ZS90; Malvern Instruments, Malvern, UK). Measurements were repeated in triplicate. The morphology was observed by transmission electron microscopy (TEM; Tecnai 20 200 kV, FEI). Samples were negatively stained with 2% phosphotungstic acid before observation by TEM.
Drug encapsulation efficiency (EE) and drug loading
EE estimated the amount of Cur encapsulated in micelles to the amount of the drugs added. Meanwhile, the drug loading efficiency (DL) was calculated as the amount of the drug encapsulated in micelles to the weight of lyophilized micelles. The EE and DL of Cur-PMs were studied in our previous report.
Stability of Cur-PMs
For the storage stability study, TP-PMs, Val-PMs, and Phe-PMs were stored at 37°C and 4°C for over 15 days. Particle size, polydispersity, and drug loading content were measured to evaluate the stability of micelles.
Physical status of Cur in micelles
The physical state of Cur in Val-PMs was characterized by differential scanning calorimetry (DSC) analysis of Cur powder, a physical mixture of Cur and polymer, lyophilized blank Phe-TPGS micelles, and lyophilized Phe-PMs using a TA-60 WS Thermal Analyzer (Shimadzu, Japan) over the temperature range of 30°C–200°C at 10°C/min.
In vitro drug release
The release profiles of Cur from TP-PMs, Val-PMs, and Phe-PMs were conducted in simulated intestinal fluid (SIF, pH 1.2) or simulated gastric fluid (SGF, pH 6.8) medium containing 0.5% (w/v) Tween 80 by a modified dialysis method. One milliliter of PMs was sealed in dialysis bags (molecular weight cut off =14,000) and incubated in 50 mL of preheated release medium at 37°C with gentle shaking (100 rpm) in the dark. At predetermined time intervals, 1 mL of medium was withdrawn and supplemented immediately with the same volume of preheated fresh release medium. The released content of Cur was analyzed by HPLC as previously described.25 Experiments were carried out in triplicate.
Caco-2 cell culture
Human colon cancer Caco-2 cells were purchased from National Platform of Experimental Cell Resources for Sci-Tech (Beijing, China) and cultured in a culture flask containing DMEM supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) nonessential amino acids, 100 U/mL penicillin, and 100 g/mL streptomycin, with a constant incubated temperature of 37°C in 5% CO2. After the proliferation, Caco-2 cells were digested with 0.25% trypsin.
For the in vitro cellular uptake model, Caco-2 cells were seeded in a 24-well sterile plate at a density of 1×105 cells/well and cultured for 15 days. For normal cells, the culture medium was exchanged every 2 days for the first week and thereafter every day. To build high-PepT1-expression Caco-2 cell (leptin-treated [LP] cells), the medium was exchanged every 2 days for the first week and then everyday with medium containing leptin during the 15-day incubation for cellular uptake.
To establish in vitro transcellular model, Caco-2 cells were seeded on inserts with polycarbonate membrane (Transwell, 12-well, 0.4 μm pore size; Corning Incorporated, Corning, NY, USA). Four hundred microliters DMEM was added to the AP side and 600 μL was added to basolateral (BL) side. The medium in both AP and BL sides was changed every 2 days for the first week and everyday thereafter during 21 days incubation. Transepithelial electrical resistance (TEER) values were evaluated to detect the integrity of cell monolayer with a Millicell® electrical resistance system (Millipore, Bedford, MA, USA). After 21 days incubation, the inserts with TEER value above 300 Ω·cm2, suggesting the formation of integrated cell monolayers and complete tight junction, were selected for further transport studies.
Quantitative real-time PCR (qPCR)
qPCR was performed to verify the PepT1-targeting efficiency of the micelles. Initially, normal Caco-2 cells and LP cells were cultured as described above and then incubated with TP-PMs, Val-PMs, and Phe-PMs for another 2 hours, with HBSS as a control. Meanwhile, the LP cells were incubated with Glysar along with Val-PMs (or Phe-PMs) for 2 hours. Then, the cells were washed with cold PBS twice. The total mRNA from cells was extracted with EZNA total RNA kit (Omega, Bio-tek, Inc, Norcross, GA, USA) according to the manufacturer’s instructions. Total RNA was reverse transcribed and analyzed by qPCR as described previously.27 β-actin was used as the control, and the data for each sample were expressed relative to the expression level of β-actin. The target genes with PepT1 and β-actin mRNA primers are as shown in Table 1.
 | Table 1 Target genes with PepT1 and β-actin mRNA primers |
Cellular uptake of PMs
The cellular uptake measurement was investigated quantitatively by ultra performance liquid chromatography-tandem mass spectrometer (UPLC-MS/MS) and qualitatively by confocal laser scanning microscopy (CLSM). In the quantitative determination by UPLC-MS/MS, Caco-2 cells were grown in 24-well plates and cultured as described above. Caco-2 cells (including normal cells and LP cells) were incubated with Cur-Sol, TP-PMs, Val-PMs, and Phe-PMs (20 μg/mL) for 30, 60, and 90 minutes. Additionally, to evaluate the concentration effects on cells, Cur-Sol and Cur-loaded micelles were incubated with 10 and 20 μg/mL of Cur for 1 hour on normal and LP Caco-2 cells. Competitive inhibition with Glysar was measured among Val-PMs, Phe-PMs, Val-PMs+Glysar, and Phe-PMs+Glysar (20 μg/mL) after 1 hour incubation with LP cells. At the established time points, the medium was removed and cellular monolayer was washed with 4°C PBS thrice. Then, the final cell lysis solutions were achieved after treating with 0.1% Triton X-100 for 1 hour. Finally, the drug content and protein amount were detected with UPLC-MS/MS and BCA protein assay kit.
CLSM was used to qualitatively evaluate the imaging and internalization of cells after uptake of micelles. In terms of CLSM, Caco-2 cells (including normal cells and LP cells) were seeded in laser confocal culture dish (15 mm) and then were incubated with C6-Sol and C6-loaded micelles (including TP-PMs, Val-PMs, Phe-PMs, the mixture of Val-PMs and Glysar [Val-PMs + Glysar], and the mixture of Phe-PMs and Glysar [Phe-PMs + Glysar]) for 1 hour. After rinsing with 4°C PBS, cells were fixed with 4% paraformaldehyde for 10 minutes. To evaluate the function of actin in the internalization of PMs, Caco-2 cells were further stained with rhodamine-phalloidin (Sigma-Aldrich Co.) to mark F-actin. Then, after labeling the nuclei with DAPI, the treated Caco-2 cells were imaged by CLSM (Olympus Corporation, Tokyo, Japan).
Endocytosis pathways of Cur-PMs
The endocytosis pathway of Val-PMs and Phe-PMs was investigated with specific endocytosis inhibitors and detected by UPLC-MS/MS. Initially, cell monolayers were preincubated with chlorpromazine (20 μg/mL), quercetin (40 μg/mL), indomethacin (100 μg/mL), β-cyclodextrin (β-CD, 2 mg/mL), and amiloride (40 μg/mL) for 1 hour at 37°C, respectively. Then, the cells were incubated with Val-PMs and Phe-PMs (20 μg/mL of Cur) in the presence of corresponding endocytosis inhibitors for further 1 hour at 37°C. Meanwhile, to investigate the energy-mediated endocytosis, the cells were precultured for 1 hour at 4°C and then incubated with Val-PMs and Phe-PMs at 4°C for another 1 hour. After coincubation, Caco-2 cells were treated and measured as described above.
Transcellular investigation of micelles across Caco-2 cell monolayer
For transcellular studies, 400 μL test solutions of Cur-Sol and Cur-PMs (30 μg/mL Cur) were added to AP side and 600 μL of fresh HBSS was added to BL side. At a determined time point, 200 μL medium was aspirated from BL side and the same volume fresh HBSS was replenished. The obtained samples were detected by UPLC-MS/MS. Briefly, 50 μL of sample was mixed with 50 μL glimepiride (internal standard [IS]) and 100 μL acetonitrile, vortexed for 3 minutes, and centrifugated for 10 minutes. The concentration of Cur in the supernatant was determined and the apparent permeability (Papp) coefficients for Cur were calculated according to the following equation:
|
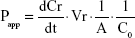
where dCr/dt is the permeation rate of drug, Vr the volume of the receiver compartment, A the surface area of the inserts, and C0 is the initial concentration of Cur at AP side.
Transcytosis mechanism of PMs in Caco-2 cells
The transcytosis mechanism of Phe-PMs across Caco-2 cells was identified through specific inhibitors. Briefly, cellular monolayer was preincubated for 1 hour with 400 μL of different concentrations of endocytosis inhibitors, including β-CD, chlorpromazine, quercetin, indomethacin, and amiloride, respectively. Then, the mixtures of Phe-PMs and the corresponding endocytosis inhibitors were incubated on Caco-2 cell monolayer for another 2 hours at 37°C. Then, the drug content in the BL side was quantified by UPLC-MS/MS and the Papp values were evaluated as above.
Biodistribution of PMs in intestinal tract
Male Sprague-Dawley rats (weighing 220±20 g) were provided by Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All animal experiments were performed in the Animal Center of Beijing University of Chinese Medicine under the guidelines for the Use and Care of Animals approved by the Beijing University of Chinese medicine Committee of Ethics. The in vivo studies were approved by the Animal Care Ethical Committee of Beijing University of Chinese medicine. Rats were fasted overnight with free access to water prior to the experiments.
To study the biodistribution of PMs in intestine after oral administration, Cur-Sol, TP-PMs, Val-PMs, and Phe-PMs were administrated to the fasted rats at 50 mg/kg of Cur by oral gavage. At 0.5 hour, the rats were anesthetized, the abdomen opened, and the intestinal segments of duodenum, jejunum, and ileum were removed. Every selected intestinal segment was everted, rinsed, accurately weighted, and homogenized with 1 mL acetonitrile on a TissueLyser II (QIAGEN, Germany). Then, 50 μL intestinal tissue homogenate and 50 μL glimepiride (IS) were vortexed with 1 mL ethyl acetate and centrifuged at 12,000 ×g for 10 minutes. Then, the supernatant was dried, redissolved, and analyzed by UPLC-MS/MS method as described below. The Cur concentration in each intestinal segment was normalized by the weight of selected tissue.
To visualize the intestinal absorption of PMs, C6-labeled micelles were utilized to visualize the absorption in mice by CLSM. Briefly, C6-Sol, C6-TP, C6-Val, and C6-Phe were administered at 5 mg/kg of C6 by oral gavage. After 0.5 hour, the rats were anesthetized, and the intestinal tissues of duodenum, jejunum, and ileum were excised, carefully everted, frozen in cryoembedding media (OCT) at −20°C and sectioned at 20 μm (Leica CM1950; Leica Biosystems, Germany). Afterward, the sections were fixed with 4% buffer paraformaldehyde for 10 minutes, stained with rhodamine-phalloidin (Sigma-Aldrich Co.) and DAPI, and imaged under the CLSM.
Lymphatic transport
The potential lymphatic transport pathway of TP-PMs, Val-PMs, and Phe-PMs was investigated in cycloheximide (CHM)-treated rat model according to the previous report.28 Briefly, Sprague-Dawley rats were fasted for 12 hours and then intraperitoneally injected with CHM. The rats treated with the same volume of saline were used as control. After 1 hour, the rats were administered with TP-PMs, Val-PMs, and Phe-PMs (50 mg/kg) by oral gavage. Then, blood was collected into heparinized tubes by puncture of the retro-orbital sinus at 0.083, 0.167, 0.25, 0.33, 0.5, 0.75, 1, 1.5, and 2 hours after oral administration. The heparinized blood samples were centrifuged at 4,000 ×g for 15 minutes and stored at −80°C until analysis.
Pharmacokinetic studies in rats
Sprague-Dawley rats weighing 220±20 g were randomly divided into four groups (n=6). Cur-Sol, TP-PMs, Val-PMs, and Phe-PMs were orally given to fasted rats at a dose of 50 mg/kg of Cur. Blood samples (0.4 mL) were sampled at certain points into heparinized tubes after oral gavage. The plasma samples were obtained by centrifugation at 4,000 ×g for 15 minutes and stored at −20°C until analysis by UPLC-MS/MS.
The concentration of Cur in plasma was determined by UPLC-MS/MS after ethyl acetate extraction with glimepiride as IS. Fifty microliters of glimepiride (IS) and 50 μL plasma were mixed with 1 mL ethyl acetate, vortexed for 3 minutes, and centrifuged at 12,000 ×g for 10 minutes. The upper organic layers were evaporated, reconstituted in 100 μL mobile phase, and centrifuged for another 10 minutes at 12,000 ×g. An aliquot of 5 μL was injected into the UPLC-MS/MS for analysis. Chromatographic separation was performed on an ACQUITY UPLC system (Waters Corp., Milford, MA, USA) and an ACQUITY UPLC™ BEH C18 column (100 mm × 2.1 mm, 1.7 μm; Waters Corp.) by gradient elution of 0.1% formic acid and acetonitrile at a flow rate of 0.4 mL/min. Mass spectrometer determination was operated on TQS triple-quadrupole tandem mass spectrometer (Waters Corp.) with a negative ESI mode. The ion transitions chosen for multiple reactions monitoring were m/z 367.11→176 for Cur and m/z 492.13→367.05 for IS, respectively. In addition, the pharmacokinetic parameters were calculated by noncompartmental model.
Statistical analysis
Results were expressed as mean ± SD. A Student’s t-test or one-way ANOVA was applied to test the significance in the experiments. Statistical differences were significant at P<0.05 and very significant at P<0.01.
Results and discussion
Synthesis and characterization of Val-TPGS copolymer and Phe-TPGS copolymer
The copolymer of Val-TPGS or Phe-TPGS was initially synthesized by the esterification reaction of the hydroxyl in TPGS and the carboxyl group in Cbz-Val-OH (or Cbz-Phe-OH) and then removed the Cbz with Pd/C to obtain the free amino group for PepT1 targeting. The synthetic scheme is presented in Figure S1. The chemical structure of the final product was confirmed by 1H-NMR in CDCl3 (shown in Figure S2). As shown in Figure S2A and B, the intense peaks at 7.4 ppm (the phenyl ring in Cbz), 5.0 ppm (methylene protons in Cbz), and 5.4 ppm (N-acyl group) were attributed to the characteristic peaks for Cbz-Phe-OH and Cbz-Val-OH, and the intense peaks at 3.5 ppm (methylene protons of the PEG) and under 3.0 ppm (protons of vitamin E succinate) were the characteristic peaks for TPGS, indicating that Cbz-Val-OH or Cbz-Phe-OH was successfully attached onto TPGS. After removing the Cbz group, the characteristic peaks of the phenyl ring (7.4 ppm) and methylene protons (5.0 ppm) of Cbz in Cbz-Val-OH or Cbz-Phe-OH disappeared, but the intense peak at 5.3 ppm for amino group (NH2) appeared (Figure S2C and D), confirming that valine or phenylalanine was successfully conjugated to TPGS.
The CMCs of Phe-TPGS and Val-TPGS were determined as 9.55 and 8.51 μg/mL, all lower than that of TPGS (15.07 μg/mL; Figure S3). It indicated that Phe-TPGS and Val-TPGS displayed better self-assembling ability and better stability than TPGS upon the dilution of biologic fluids such as GI tract and blood circulation.
Characterization of Cur-PMs
The Val-PMs and Phe-PMs were prepared by film-dispersion method, with TP-PMs as negative control. The physicochemical characteristics of Cur-loaded micelles were determined and summarized in Table 2.
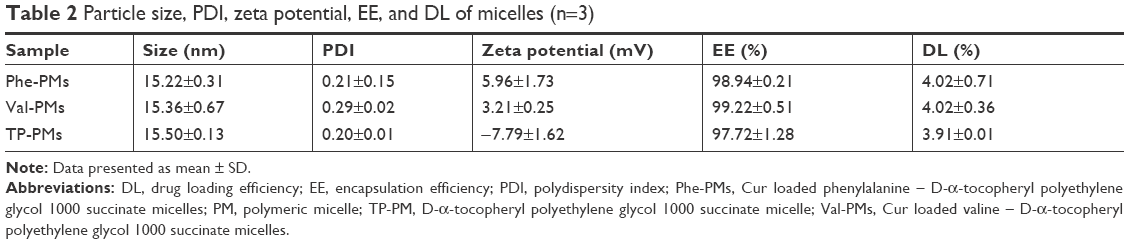 | Table 2 Particle size, PDI, zeta potential, EE, and DL of micelles (n=3) |
TP-PMs, Val-PMs, and Phe-PMs displayed a narrow size distribution (polydispersity index below 0.30) and similar average size of 15 nm (Figures 1A and S4A; Table 2). TEM images visualized the narrow size distribution and spherical morphology of TP-PMs, Val-PMs, and Phe-PMs (Figures 1B and S4B). The negative charge of TP-PMs (−7.79±1.62 mV) was mainly contributed by the carboxylic acid groups of TPGS. However, due to the modification of amino acid and amine groups on the TPGS surface, the zeta potential of Phe-PMs (5.96±1.73 mV) and Val-PMs (3.21±0.25 mV) increased and became positive. It indicated that Val-PMs or Phe-PMs had the electropositive PEG surface, which might be liable to interact with the cell and enhance the cellular uptake of the micelles. Due to the strong interaction between Cur and hydrophobic core of the micelles, the EE of Cur in three micelles all reached up to 97.7% and the drug loading was about 4.0%.
The physical state of Cur in Phe-PMs was evaluated by DSC. As shown in Figure 1C, the melting peak of Cur was clear in Cur powder and the physical mixture of Cur and nanocarrier, but completely disappeared in Phe-PMs. The result indicated that Cur was in an amorphous or molecular state in micelles.
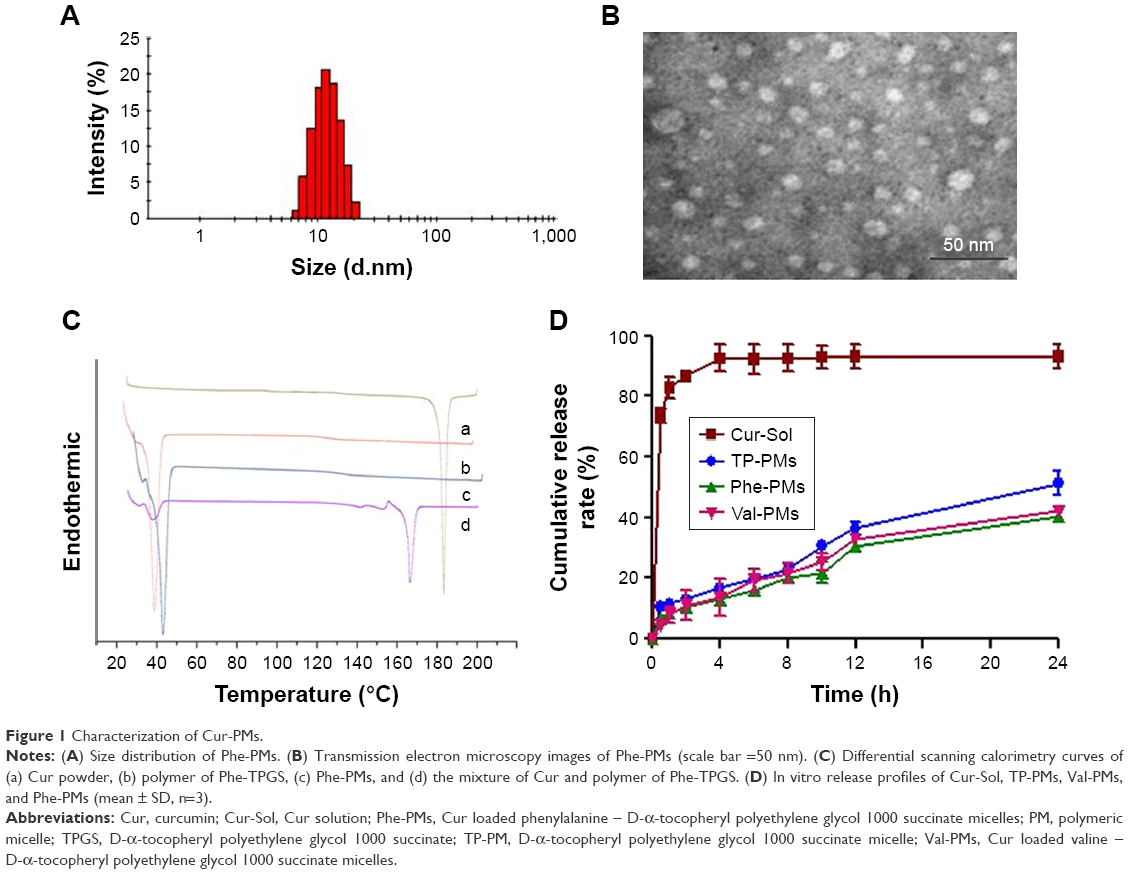 | Figure 1 Characterization of Cur-PMs. |
For the storage stability study, the micelles were stored at 4°C and room temperature, and the particle size and the content of Cur-PMs were monitored over 15 days (Figure S5). The particle size of TP-PMs, Val-PMs, and Phe-PMs showed no significant change at 4°C over 15 days and at room temperature over 10 days. However, the particle size of TP-PMs increased at day 10 at room temperature, which was due to the Ostwald ripening phenomenon, but no visible precipitation was observed. The encapsulation rate of TP-PMs, Val-PMs, and Phe-PMs was as high as 90% at 4°C and room temperature over 15 days, indicating that the Cur-loaded micelles had good storage stability.
The drug release profiles of Val-PMs, Phe-PMs, and TP-PMs were evaluated in both SGF and SIF in vitro. As shown in Figure 1D, the cumulative release of Cur-Sol was 70% at 2 hours in SGF and more than 90% within another 2 hours in SIF, indicating that Cur exhibited relatively fast release and the dialysis bag had no detention on drug release. In contrast, the drug release from micelles was significantly slower than Cur-Sol. Less than 15% of Cur was released from three micelles in SGF within 2 hours, which was useful for reducing the drug leakage in stomach during oral delivery and thereby improved the stomach stability. Meanwhile, <55% release of Cur from TP-PMs, Val-PMs, and Phe-PMs was observed in SIF at 24 hours, suggesting that micelles not only improved the stability of Cur but also exhibited a sustained-release behavior. However, the cumulative release of Cur from Val-PMs and Phe-PMs was slower than that from TP-PMs, especially after 12 hours (P<0.05), indicating that the release stability of Val-PMs and Phe-PMs was stronger in comparison with TP-PMs due to the relatively lower CMC values of Val-TPGS and Phe-TPGS. In conclusion, TP-PMs, Val-PMs, and TP-PMs all exhibited a sustained-release profile and good release stability.
qPCR for PepT1 targeting
The intestinal epithelium is the main barrier restricting the oral delivery of low-permeability drugs or nanoparticles.29 PepT1 is highly expressed on the intestinal epithelium, so targeting PepT1 could overcome the epithelium barrier and increase the cellular uptake and transport of drug or nanoparticles across intestinal epithelium. Caco-2 has been recognized as an excellent cell model for mimicking the intestinal epithelium. As previous results, the expression of PepT1 could be increased in LP Caco-2 cells.30,31 To verify the higher expression level of PepT1 in LP Caco-2 cells than in normal cells (Normal) and to study the PepT1-targeting efficiency of Val-PMs and Phe-PMs, qPCR analysis was applied. As shown in Figure 2, a markedly higher expression of PepT1 was found in LP cells than in normal Caco-2 cells (P<0.01), proving that PepT1 was highly expressed on Caco-2 cells after leptin coincubation. With regard to the PepT1-targeting efficiency, TP-PMs had the similar PepT1 level in LP cells and normal cells, demonstrating that TP-PMs and TPGS nanocarrier did not have PepT1-targeting ability. Excitingly, PepT1 expression level in LP cells was significantly decreased after coincubation with Val-PMs or Phe-PMs in comparison with control group and TP-PMs (P<0.01), suggesting that valine and phenylalanine decorated on the surface of micelles could efficiently combine with PepT1 and then Val-PMs and Phe-PMs possessed PepT1-targeting ability. Glysar, the substrate of PepT1, decreased the expression of PepT1 after coincubation with Val-PMs or Phe-PMs in LP cells compared with sole Val-PMs or Phe-PMs (P<0.01), once again proving that Val-PMs and Phe-PMs have strong affinity for PepT1.
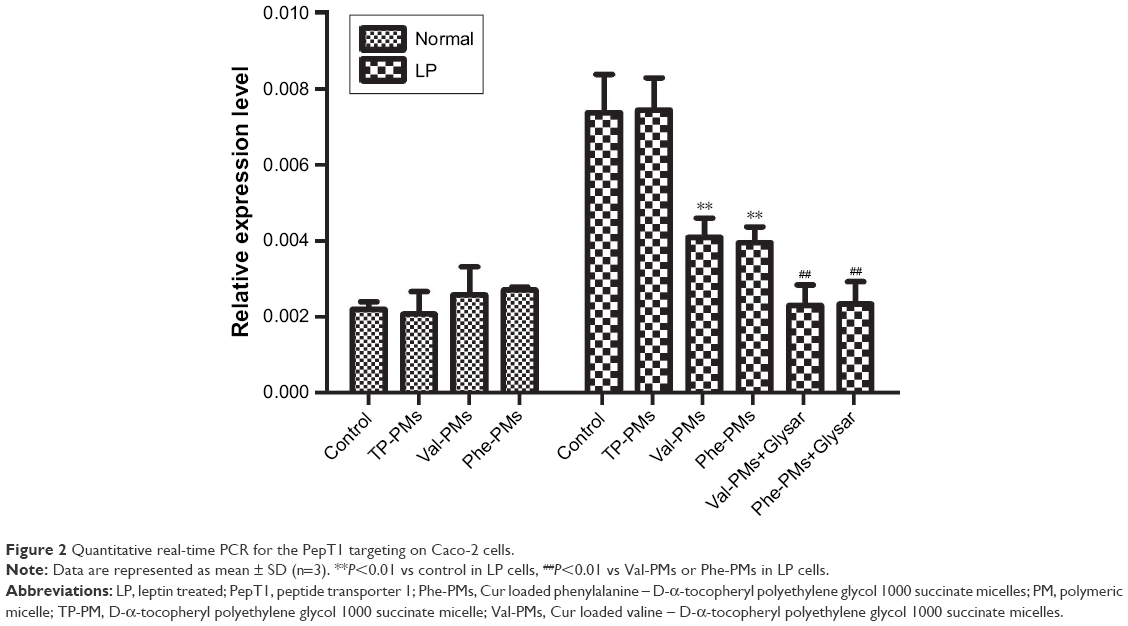 | Figure 2 Quantitative real-time PCR for the PepT1 targeting on Caco-2 cells. |
Cellular uptake
PepT1-targeting efficiency of micelles
Figures 3 and 4 displayed the cellular uptake of Cur-PMs by immunofluorescence analysis and UPLC-MS/MS analysis in both normal and LP Caco-2 cells. Apparently, the intracellular amount of Cur-Sol and Cur-PMs was significantly enhanced with the extension of incubation time and the increase of Cur concentration, implying a time- and concentration-dependent endocytosis (Figure 3). In normal Caco-2 cells, the cellular uptake of TP-PMs, Val-PMs, and Phe-PMs were similar and were all about 2.5-fold higher than that of Cur-Sol at the same concentration and same time point. Meanwhile, the cellular uptake of TP-PMs displayed no significant differences between normal and LP cells. However, the cellular uptake of Val-PMs and Phe-PMs in LP cells were similar (P>0.05), but were significantly higher than that of TP-PMs in LP and those in normal cells (P<0.01). As a result, the improved cellular uptake of Val-PMs and Phe-PMs is mainly due to the selective identification and target PepT-1.
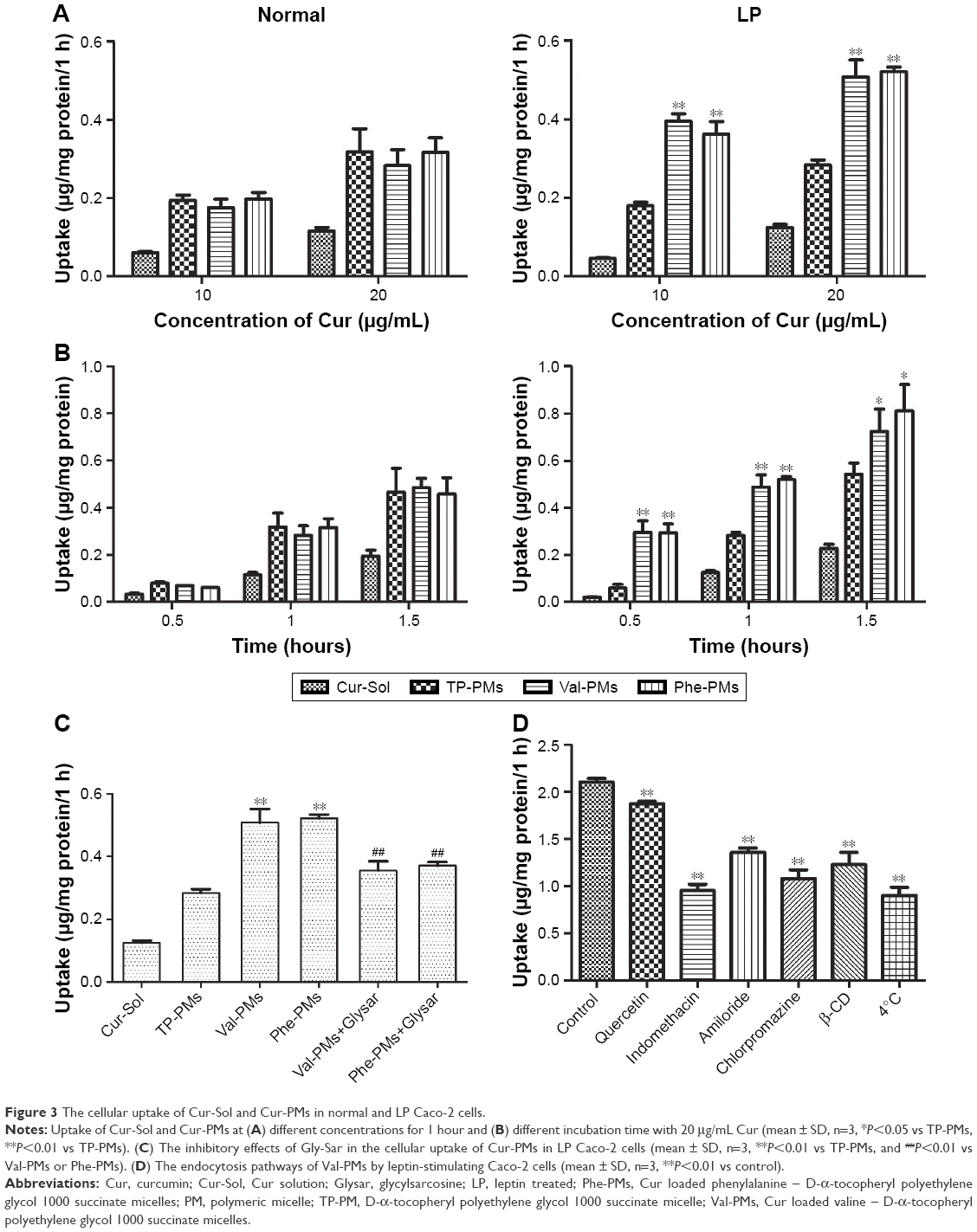 | Figure 3 The cellular uptake of Cur-Sol and Cur-PMs in normal and LP Caco-2 cells. |
To further confirm the affinity of micelles to PepT1 and subcellular distribution of micelles, C6 was used as a fluorescence probe, and CLSM images of cellular uptake of C6-PMs are shown in Figure 4. The green fluorescence intensity from C6-PMs was visibly stronger than that from C6-Sol in both normal and LP cells, suggesting that a higher amount of C6 was taken up by cells when encapsulated into micelles. In normal Caco-2 cell model, similar fluorescence signals were observed in C6-Val, C6-Phe, and C6-TP groups. However, LP cells treated with C6-Val and C6-Phe exhibited stronger fluorescence in comparison with that treated with C6-TP, once again confirming that C6-Val and C6-Phe have PepT1-targeting action and could efficiently enhance the internalization of drug in a PepT1-mediated way.
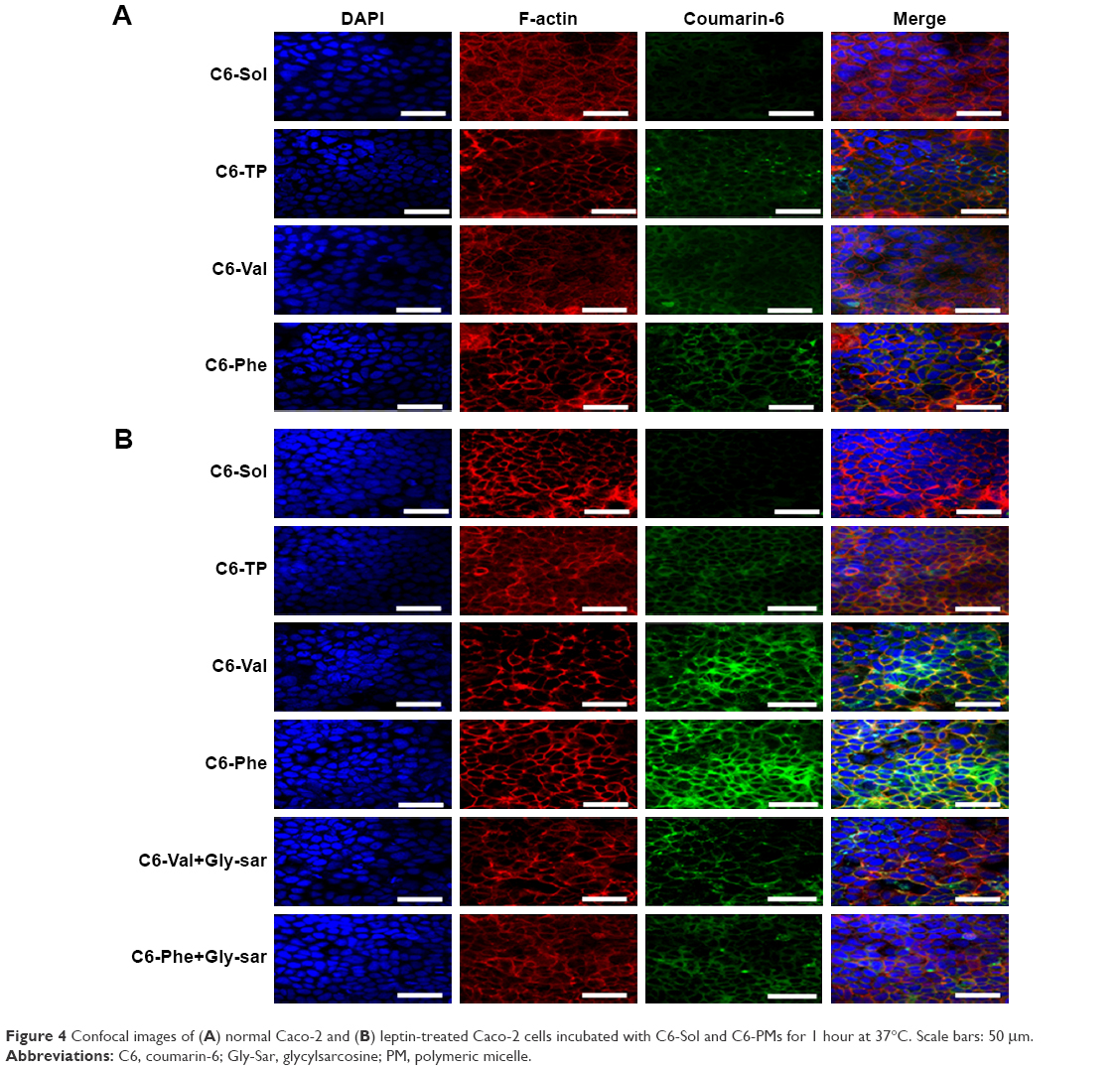 | Figure 4 Confocal images of (A) normal Caco-2 and (B) leptin-treated Caco-2 cells incubated with C6-Sol and C6-PMs for 1 hour at 37°C. Scale bars: 50 μm. |
Involvement of actins in the endocytosis
The subcellular colocalization of C6-PMs was investigated by immunofluorescence analysis. As shown in Figure 4, the green fluorescence spots of C6-PMs were mainly colocalized with the red fluorescence spots of F-actin at the cell junctions or on the surface of cell membranes, but were hardly observed in cell nucleus (blue fluorescence spots). Meanwhile, C6-PMs triggered cell disorder and fuzziness of F-actin. Actin, as an important linker among multiple organelles and endosome vesicles, played important roles in the internalization and endocytosis at the intestinal epithelial cell.7,32 Actins could also reflect the fluidity of cellular membranes. As a result, F-actin participated in the internalization of C6-Val, C6-Phe, and C6-TP by Caco-2 cells, and could regulate the intracellular location of micelles and the fluidity of cellular membrane.
Competitive inhibition assay
To further assess the interaction of Val-PMs and Phe-PMs with PepT1, the inhibitory effect of Glysar on the uptake of Val-PMs and Phe-PMs was assayed qualitatively and quantitatively. Glysar, a competing inhibitor for PepT1,33 significantly decreased the cellular uptake of Val-PMs and Phe-PMs to LP cells (Figure 3C). Furthermore, the green fluorescence intensity of C6-Val and C6-Phe in LP cells was significantly depressed after coincubation with Glysar (Figure 4B). In conclusion, these results once again revealed that the PepT1 was involved in the targeting and internalization of Val-PMs and Phe-PMs in Caco-2 cells.
Endocytosis mechanism
Specific endocytosis inhibitors were utilized to identify the endocytosis mechanism of Val-PMs and Phe-PMs by Caco-2 cells (Figures 3D and S6). The cellular uptake of both Val-PMs and Phe-PMs was reduced by approximately 57% at 4°C (P<0.01) and reached to half of the cellular uptake at 37°C, verifying the involvement of the active and energy-dependent endocytic mechanism in Val-PMs and Phe-PMs. β-CD,34 an inhibitor of lipid raft/caveolae-dependent endocytosis by depleting of cholesterol, caused a significant decrease of the intracellular uptake of both PepT1-targeting micelles. The addition of indomethacin,35 an inhibitor of cyclooxygenase, significantly reduced Cur uptake by nearly 54% for Val-PMs and 45% for Phe-PMs, further suggesting the involvement of caveolae-mediated endocytosis. In the presence of chlorpromazine,36 an inhibitor of clathrin-mediated endocytosis, and amiloride,37 an inhibitor of macropinocytosis, the intracellular uptake of both PMs was significantly reduced (P<0.05), demonstrating that clathrin-mediated endocytosis and macropinocytosis were involved in the internalization of Val-PMs and Phe-PMs. However, quercetin,26 an inhibitor of caveolae- and clathrin-independent endocytosis, significantly decreased the cellular uptake of Val-PMs, but not of Phe-PMs. Therefore, caveolae-mediated, clathrin-mediated endocytosis and macropinocytosis were involved in the cellular uptake of both Val-PMs and Phe-PM, and only the pathway of caveolae- and clathrin-independent endocytosis existed in Val-PMs but not in Phe-PMs. Meanwhile, TP-PMs were internalized through caveolae-mediated, clathrin-mediated and energy-mediated endocytosis, but not through macropinocytosis and caveolae- and clathrin-independent endocytosis (Figure S6). In summary, the surface modification amino on micelles may cause different endocytic pathway of micelles in Caco-2 cells, because the micelles with different surface modification may bind or adsorb different cell-surface membrane proteins, influencing the endocytosis or transport of micelles.
Transport of Cur-PMs through Caco-2 monolayer
Efficient transcytosis of drug across the intestinal epithelia, permeability from AP to BL, is essential for the improvement of oral drug delivery. Therefore, the Papp values of Cur-Sol, TP-PMs, Val-PMs, and Phe-PMs across Caco-2 cell monolayer were evaluated (Figure 5A). All Cur-PMs were more permeable than Cur-Sol. The Papp for TP-PMs, Val-PMs, and Phe-PMs were 14.37-, 18.5-, and 17.4-fold higher than that for Cur-Sol, respectively, suggesting that Cur encapsulated in micelles could significantly improve the BL exocytosis of Cur and increase the drug transport across Caco-2 cell monolayer. Furthermore, the Papp values of Val-PMs and Phe-PMs were 1.29- and 1.21-fold greater than TP-PMs, respectively (P<0.05). Therefore, PepT-1-mediated transcytosis may be responsible for improving the transmembrane transport efficiency of micelles.
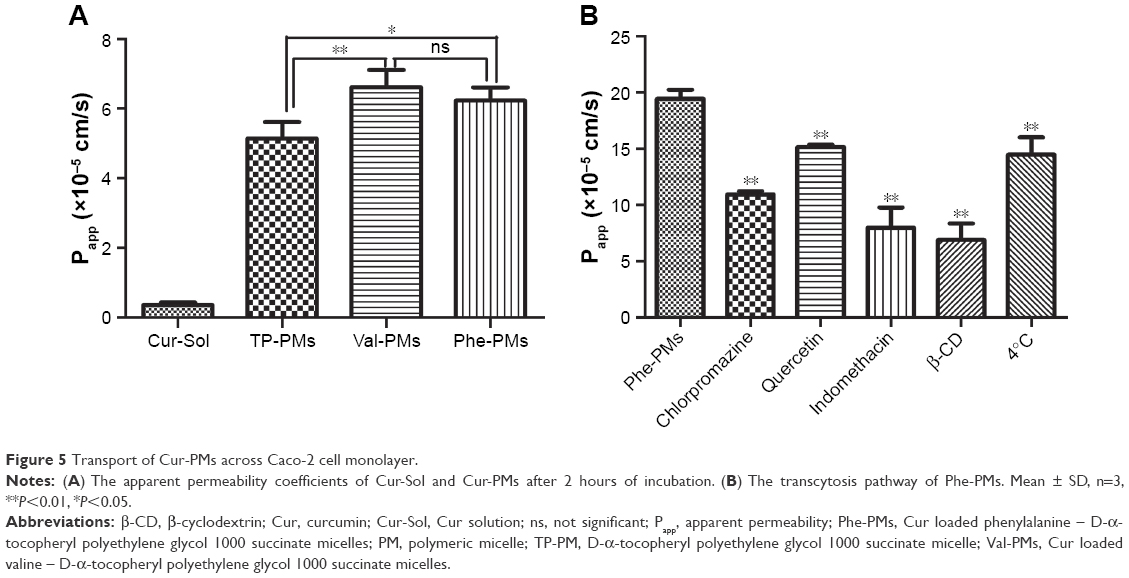 | Figure 5 Transport of Cur-PMs across Caco-2 cell monolayer. |
Transport mechanism across Caco-2 cells
Paracellular transport and transcellular transport were two major transport pathways of drugs or preparations.38 Paracellular transport refers to the transport across tight junctions between epithelial cells. TEER was measured to evaluate the integrity of cell monolayer and the state of cell junction. During the transcytosis, TEER values did not have significant changes, which indicated that the cell junctions were hardly opened and the paracellular route was not involved in the transport pathway of PMs.
Furthermore, transcellular transport refers to transport across the epithelial cells by passing through from the AP membrane to BL membrane. As shown in Figure 5B, the Papp value of Phe-PMs was significantly decreased at 4°C compared to that at 37°C, indicating the existence of energy-dependent and active transcytosis of Phe-PMs through Caco-2 cell monolayer. Furthermore, the transcellular transport mechanism of Phe-PMs across the Caco-2 monolayer was also evaluated with the inhibitors. The permeability of Phe-PMs was significantly reduced by the addition of β-CD, indomethacin, chlorpromazine, and amiloride, showing that multiple pathways (including caveolae- and clathrin-mediated transcytosis, macropinocytosis, and caveolae- and clathrin-independent transcytosis) were involved in the transcellular transport pathway of Phe-PMs and would be responsible for the improved transcellular transport across the Caco-2 cells.
Biodistribution of PMs in the intestine
The intestinal absorption of Cur was significantly varied with the intestinal segments (Figure 6). The absorption of Cur-Sol and Cur-PMs was highest in duodenum and then gradually decreased in jejunum and ileum. Obviously, Cur-loaded micelles significantly improved the intestinal absorption of Cur in different intestinal segments. The concentration of Cur from TP-PMs obviously increased 8.30- and 10.99-fold in duodenum and jejunum in comparison with Cur-Sol (P<0.01), but no significant differences in ileum, indicating that the affinity and adhesion with the intestinal epithelial cells were significantly improved by TP-PMs due to the nanosize, PEGylated surface modification, and P-gp inhibition. The absorption of Val-PMs and Phe-PMs in different intestinal segments exhibited no statistical difference (P>0.05), but was obviously higher than that of Cur-Sol and TP-PMs. The absorption of Val-PMs and Phe-PMs in duodenum, jejunum, and ileum was approximately increased by 1.44-, 2.55-, and 3.32-fold compared to that of TP-PMs, respectively. These results demonstrated that Val-PMs and Phe-PMs exhibited higher intestinal permeability due to the high PepT1 targeting with intestinal epithelium, so PepT1-targeting nanocarrier and nanoparticles should be a good vehicle to enhance the intestinal targeting and then increase the intestinal permeability of drugs.
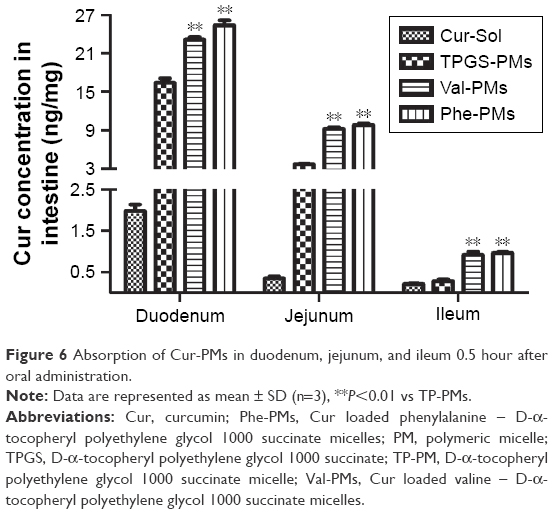 | Figure 6 Absorption of Cur-PMs in duodenum, jejunum, and ileum 0.5 hour after oral administration. |
Furthermore, intestinal distribution and penetration of C6-labeled PMs in duodenum, jejunum, and ileum were also visualized by CLSM. As shown in Figure 7, C6-TP, C6-Val, and C6-Phe exhibited stronger green fluorescence signals compared to C6-Sol in every intestinal segment. The green fluorescence of C6-PMs was stronger in the duodenum, followed by the jejunum and ileum after oral administration. Meanwhile, the green fluorescence of C6-PMs was observed not only on the brush border of intestinal villi but also in the inner of the villi. The results once again proved that micelles were able to slip through the mucus layer, adhered to the surface of the intestinal epithelium, and then significantly enhanced drug absorption by enterocytes. Excitingly, C6-Val and C6-Phe exhibited stronger green fluorescence in duodenum and jejunum compared to C6-TP, which was in accordance with the quantitative results and also verified the higher transport efficiency of Val-PMs and Phe-PMs. Above all, PepT1-targeting micelles (Val-PMs and Phe-PMs) could significantly enhance the bioadhesion and penetration of micelles to the intestinal mucosa and then obviously improve the intestinal absorption of micelles in vivo.
 | Figure 7 The confocal laser scanning microscopy images of (A) duodenum, (B) jejunum, and (C) ileum at 0.5 hour after oral administration of C6-Sol and C6-PMs. Scale bars: 100 μm. |
Lymphatic transport in vivo
The potential lymphatic transport pathway of Cur-PMs was investigated with lymphatic transport inhibitor (CHM).28,39 The plasma concentration–time curves of Cur-PMs in saline-treated rats and CHM-treated rats are shown in Figure 8A, and the major pharmacokinetic parameters are summarized in Table S1. The plasma concentrations of Val-PMs, Phe-PMs, and TP-PMs in CHM-treated rats were significantly decreased in comparison with those in normal rats. When the rats were treated with CHM, intestinal lymphatic transport was blocked and the plasma concentration of Cur in Cur-PMs was significantly reduced within 2 hours. The Cmax of Cur in TP-PMs, Val-PMs, and Phe-PMs had reduced almost 32%, 43%, and 36%, and the AUC(0–2 h) of them had significantly decreased 30%, 60%, and 52% in CMH-treated mice. Therefore, lymphatic transport pathway was involved in the oral absorption of Cur-PMs. Furthermore, lymphatic targeting transport was facilitated to increase the uptake of drug into lymphocytes, bypass the liver, and then get directly into the systemic circulation.40 As a consequence, intestinal lymphatic transport could be helpful for the improvement of oral absorption of micelles.
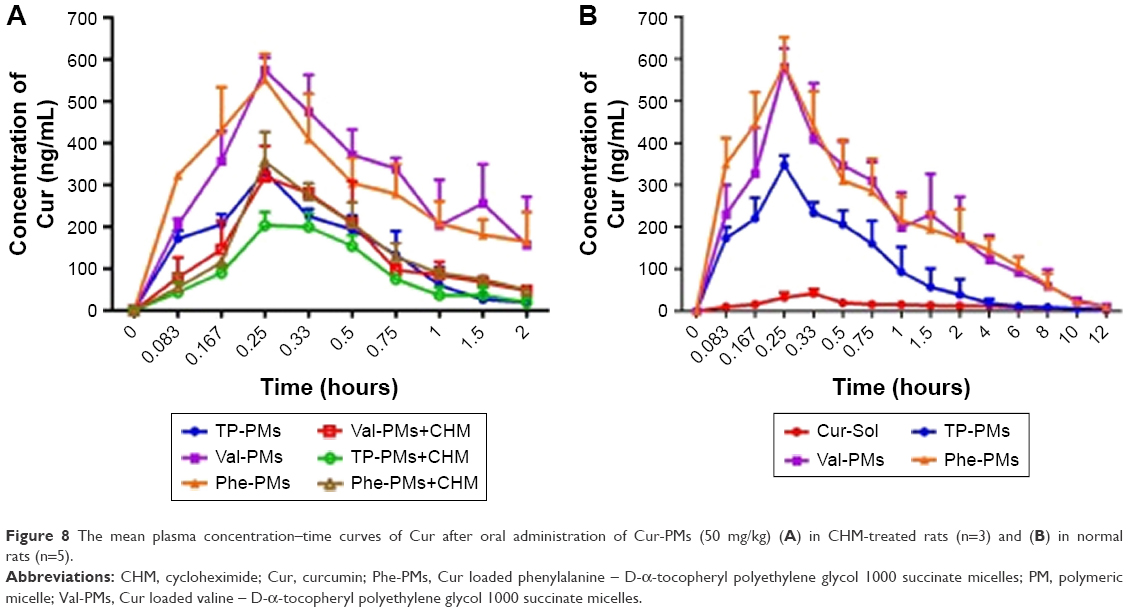 | Figure 8 The mean plasma concentration–time curves of Cur after oral administration of Cur-PMs (50 mg/kg) (A) in CHM-treated rats (n=3) and (B) in normal rats (n=5). |
Pharmacokinetics in vivo
Pharmacokinetics was studied in rats to evaluate the in vivo behavior and the absorption efficiency of Cur-PMs after administration. The plasma concentration–time profiles of Cur had significant differences among Cur-Sol, TP-PMs, Val-PMs, and Phe-PMs (Figure 8B). The major pharmacokinetic parameters are summarized in Table S2. Cur-loaded micelles showed significant increase in plasma concentration of Cur. The Cmax, AUC(0–12 h), and t1/2 of TP-PMs was 8.27-, 3.17-, and 2.24-fold higher than those of Cur-Sol, respectively. It once again demonstrated that TP-PMs could significantly enhance the oral absorption and prolong the blood circulation time of Cur as a result of the increase of drug solubility and intestinal membrane adhesion in vivo as previously reported. As for Val-PMs and Phe-PMs, the Cmax of Val-PMs (580.40±39.34 μg/L) and Phe-PMs (588.60±55.08 μg/L) were approximately 1.67- and 1.70-fold higher than that of TP-PMs (all P<0.01), the AUC(0–12 h) were probably 3.41- and 3.57-fold greater than that of TP-PMs (all P<0.01), and the t1/2 were approximately 1.79- and 1.40-fold longer than that of TP-PMs (P<0.05). It demonstrated that Val-PMs and Phe-PMs had stronger capability of improving the systemic circulation concentration of drug in vivo. According to the in vivo and in vitro results mentioned above, due to the high PepT1-targeting ability, Val-PMs and Phe-PMs could obviously enhance the adhesion of micelles to intestinal mucosa, improve the transmembrane transport efficiency of TP-PMs across the intestinal epithelium, and ultimately significantly enhance the oral absorption of Cur and even TP-PMs. Lymphatic transport, bypassing the first-pass hepatic metabolism, was also an important absorption route for the enhancement of oral absorption of Val-PMs and Phe-PMs (Figure 9). Accordingly, PepT-1-mediated targeting micelles could be a promising vehicle for the oral delivery of drugs.
 | Figure 9 Schematic diagram of potential transport mechanisms of PepT1-mediated micelles. |
Conclusion
PepT1 is overexpressed in intestinal epithelium and is an important uptake transporter in oral administration. Our results confirmed the effects of PepT1-mediated micelles on uptake and transport in cells and even oral absorption in rats. In vitro and in vivo results demonstrated that Val-PMs and Phe-PMs improved the transport efficiency across the intestinal epithelium, which were adhered to the PepT1 on epithelia surfaces, entered into the villi and transcytosis across epithelium, finally leading to a significant increase of oral absorption of drugs. In summary, PepT1-mediated micelles, by modifying the micelles’ surface with amino or dipeptide, should be a promising vehicle to enhance the transport across the intestinal epithelial barrier and increase oral absorption. Modifying PepT1 target to micelles may provide some information for the rational design of nanocarriers for oral delivery of poorly soluble drugs.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (grant number 81503262), Outstanding Person Foundation of Beijing University of Chinese Medicine (BUCM-2019-JCRC003), and Young Talents Project of China Association of Traditional Chinese medicine(CACM-2018-QNRC2-C04).
Disclosure
The authors report no conflicts of interest in this work.
References
Zhao M, Lee SH, Song JG, Kim HY, Han HK. Enhanced oral absorption of sorafenib via the layer-by-layer deposition of a pH-sensitive polymer and glycol chitosan on the liposome. Int J Pharm. 2018;544(1):14–20. | ||
Kang SH, Revuri V, Lee SJ, et al. Oral siRNA delivery to treat colorectal liver metastases. ACS Nano. 2017;11(10):10417–10429. | ||
Ren T, Wang Q, Xu Y, et al. Enhanced oral absorption and anticancer efficacy of cabazitaxel by overcoming intestinal mucus and epithelium barriers using surface polyethylene oxide (PEO) decorated positively charged polymer-lipid hybrid nanoparticles. J Control Release. 2018;269:423–438. | ||
Zhang H, Hu H, Zhang H, et al. Effects of PEGylated paclitaxel nanocrystals on breast cancer and its lung metastasis. Nanoscale. 2015;7(24):10790–10800. | ||
Hou J, Sun E, Zhang ZH, et al. Improved oral absorption and anti-lung cancer activity of paclitaxel-loaded mixed micelles. Drug Deliv. 2017;24(1):261–269. | ||
Fan W, Xia D, Zhu Q, Hu L, Gan Y. Intracellular transport of nanocarriers across the intestinal epithelium. Drug Discov Today. 2016;21(5):856–863. | ||
He B, Lin P, Jia Z, et al. The transport mechanisms of polymer nanoparticles in Caco-2 epithelial cells. Biomaterials. 2013;34(25):6082–6098. | ||
Shan W, Zhu X, Tao W, et al. Enhanced oral delivery of protein drugs using zwitterion-functionalized nanoparticles to overcome both the diffusion and absorption barriers. ACS Appl Mater Interfaces. 2016;8(38):25444–25453. | ||
Shalaby KS, Soliman ME, Casettari L, et al. Determination of factors controlling the particle size and entrapment efficiency of noscapine in PEG/PLA nanoparticles using artificial neural networks. Int J Nanomedicine. 2014;9:4953–4964. | ||
Tay CY, Cai P, Setyawati MI, et al. Nanoparticles strengthen intracellular tension and retard cellular migration. Nano Lett. 2014;14(1):83–88. | ||
Mahmud F, Jeon OC, Al-Hilal TA, et al. Absorption mechanism of a physical complex of monomeric insulin and deoxycholyl-l-lysyl-methylester in the small intestine. Mol Pharm. 2015;12(6):1911–1920. | ||
Mahmud F, Jeon OC, Alam F, et al. Oral pemetrexed facilitates low-dose metronomic therapy and enhances antitumor efficacy in lung cancer. J Control Release. 2018;284:160–170. | ||
Li L, Di X, Zhang S, et al. Large amino acid transporter 1 mediated glutamate modified docetaxel-loaded liposomes for glioma targeting. Colloids Surf B Biointerfaces. 2016;141:260–267. | ||
Spanier B, Rohm F. Proton coupled oligopeptide transporter 1 (PepT1) function, regulation, and influence on the intestinal homeostasis. Compr Physiol. 2018;8(2):843–869. | ||
Dranse HJ, Waise TMZ, Hamr SC, et al. Physiological and therapeutic regulation of glucose homeostasis by upper small intestinal PepT1-mediated protein sensing. Nat Commun. 2018;9(1):1118. | ||
Fang L, Wang M, Gou S, Liu X, Zhang H, Cao F. Combination of amino acid/dipeptide with nitric oxide donating oleanolic acid derivatives as PepT1 targeting antitumor prodrugs. J Med Chem. 2014;57(3):1116–1120. | ||
Tao W, Zhao D, Sun M, et al. Enzymatic activation of double-targeted 5′-O-L-valyl-decitabine prodrug by biphenyl hydrolase-like protein and its molecular design basis. Drug Deliv Transl Res. 2017;7(2):304–311. | ||
Gourdon B, Chemin C, Moreau A, et al. Functionalized PLA-PEG nanoparticles targeting intestinal transporter PepT1 for oral delivery of acyclovir. Int J Pharm. 2017;529(1–2):357–370. | ||
Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. | ||
Liang D, Wang AT, Yang ZZ, Liu YJ, Qi XR. Enhance cancer cell recognition and overcome drug resistance using hyaluronic acid and α-tocopheryl succinate based multifunctional nanoparticles. Mol Pharm. 2015;12(6):2189–2202. | ||
Yang C, Wu T, Qi Y, Zhang Z. Recent advances in the application of vitamin E TPGS for drug delivery. Theranostics. 2018;8(2):464–485. | ||
Cheng B, Pan H, Liu D, et al. Functionalization of nanodiamond with vitamin E TPGS to facilitate oral absorption of curcumin. Int J Pharm. 2018;540(1–2):162–170. | ||
Pham AC, Gavin P, Libinaki R, Ramirez G, Boyd BJ. A new lipid excipient, phosphorylated tocopherol mixture, TPM enhances the solubilisation and oral bioavailability of poorly water soluble CoQ10 in a lipid formulation. J Control Release. 2017;268:400–406. | ||
Beig A, Fine-Shamir N, Porat D, Lindley D, Miller JM, Dahan A. Concomitant solubility-permeability increase: vitamin E TPGS vs. amorphous solid dispersion as oral delivery systems for etoposide. Eur J Pharm Biopharm. 2017;121:97–103. | ||
Wang J, Ma W, Tu P. The mechanism of self-assembled mixed micelles in improving curcumin oral absorption: in vitro and in vivo. Colloids Surf B Biointerfaces. 2015;133:108–119. | ||
Wang J, Sun J, Chen Q, et al. Star-shape copolymer of lysine-linked di-tocopherol polyethylene glycol 2000 succinate for doxorubicin delivery with reversal of multidrug resistance. Biomaterials. 2012;33(28):6877–6888. | ||
Wang J, Ma W, Guo Q, et al. The effect of dual-functional hyaluronic acid-vitamin E succinate micelles on targeting delivery of doxorubicin. Int J Nanomedicine. 2016;11:5851–5870. | ||
Zhang Z, Ma L, Jiang S, et al. A self-assembled nanocarrier loading teniposide improves the oral delivery and drug concentration in tumor. J Control Release. 2013;166(1):30–37. | ||
Lundquist P, Artursson P. Oral absorption of peptides and nanoparticles across the human intestine: opportunities, limitations and studies in human tissues. Adv Drug Deliv Rev. 2016;106(Pt B):256–276. | ||
Tao W, Zhao D, Sun M, et al. Intestinal absorption and activation of decitabine amino acid ester prodrugs mediated by peptide transporter PEPT1 and enterocyte enzymes. Int J Pharm. 2018;541(1–2):64–71. | ||
Zhang Y, Sun J, Sun Y, Wang Y, He Z. Prodrug design targeting intestinal PepT1 for improved oral absorption: design and performance. Curr Drug Metab. 2013;14(6):675–687. | ||
Apodaca G. Endocytic traffic in polarized epithelial cells: role of the actin and microtubule cytoskeleton. Traffic. 2001;2(3):149–159. | ||
Hu Y, Xie Y, Wang Y, Chen X, Smith DE. Development and characterization of a novel mouse line humanized for the intestinal peptide transporter PEPT1. Mol Pharm. 2014;11(10):3737–3746. | ||
Kanatani I, Ikai T, Okazaki A, et al. Efficient gene transfer by pullulan-spermine occurs through both clathrin- and raft/caveolae-dependent mechanisms. J Control Release. 2006;116(1):75–82. | ||
Sato K, Nagai J, Mitsui N, Ryoko Yumoto, Takano M. Effects of endocytosis inhibitors on internalization of human IgG by Caco-2 human intestinal epithelial cells. Life Sci. 2009;85(23–26):800–807. | ||
Bewersdorff T, Vonnemann J, Kanik A, Haag R, Haase A. The influence of surface charge on serum protein interaction and cellular uptake: studies with dendritic polyglycerols and dendritic polyglycerol-coated gold nanoparticles. Int J Nanomedicine. 2017;12:2001–2019. | ||
Hasan M, Nishimoto A, Ohgita T, et al. Faint electric treatment-induced rapid and efficient delivery of extraneous hydrophilic molecules into the cytoplasm. J Control Release. 2016;228:20–25. | ||
Mukaizawa F, Taniguchi K, Miyake M, et al. Novel oral absorption system containing polyamines and bile salts enhances drug transport via both transcellular and paracellular pathways across Caco-2 cell monolayers. Int J Pharm. 2009;367(1–2):103–108. | ||
Bhalekar MR, Upadhaya PG, Madgulkar AR, Kshirsagar SJ, Dube A, Bartakke US. In-vivo bioavailability and lymphatic uptake evaluation of lipid nanoparticulates of darunavir. Drug Deliv. 2016;23(7):2581–2586. | ||
Yáñez JA, Wang SW, Knemeyer IW, Wirth MA, Alton KB. Intestinal lymphatic transport for drug delivery. Adv Drug Deliv Rev. 2011;63(10–11):923–942. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.