Back to Journals » Journal of Inflammation Research » Volume 15
Apelin-13 Attenuates Lipopolysaccharide-Induced Inflammatory Responses and Acute Lung Injury by Regulating PFKFB3-Driven Glycolysis Induced by NOX4-Dependent ROS
Authors Yuan Y, Wang W, Zhang Y, Hong Q, Huang W, Li L, Xie Z, Chen Y, Li X, Meng Y
Received 11 November 2021
Accepted for publication 11 March 2022
Published 30 March 2022 Volume 2022:15 Pages 2121—2139
DOI https://doi.org/10.2147/JIR.S348850
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Yafei Yuan,1 Wei Wang,1 Yue Zhang,1 Qiaohui Hong,1 Wenhui Huang,1 Lijuan Li,1 Zhanzhan Xie,1 Yixin Chen,1 Xu Li,2,3 Ying Meng1
1Department of Respiratory and Critical Care Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 2Department of Emergency Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 3Ministry of Education, Key Laboratory of Hainan Trauma and Disaster Rescue, College of Emergency and Trauma, Hainan Medical University, Haikou, 571199, People’s Republic of China
Correspondence: Ying Meng, Department of Respiratory and Critical Care Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, People’s Republic of China, Tel/Fax +86 20-62787112, Email [email protected] Xu Li, Department of Emergency Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, People’s Republic of China, Email [email protected]
Purpose: Acute lung injury (ALI) is a life-threatening condition with limited therapeutic options. Macrophage inflammation plays a key role in the development of ALI. Abnormal glycolysis of macrophages contributes to the inflammatory response. However, the role of macrophage glycolysis in ALI still requires investigation. Apelin-13 has been shown to protect against ALI, whereas the underlying mechanisms remain unclear. In this study, we explored the effect of apelin-13 on lipopolysaccharide (LPS)-induced inflammation and ALI via regulation of glycolysis by modulating redox homeostasis in macrophages.
Methods: Serums from 34 patients with sepsis and 13 healthy volunteers were analyzed. In vivo, the protective effect of apelin-13 against LPS-induced ALI was evaluated using a mouse model of LPS-induced ALI. In vitro, mouse bone marrow macrophages (BMDMs) were pretreated with the antioxidant, NADPH oxidase (NOX) 4 (NOX4) small-interfering RNA (siRNA), the 6-phosphofructo-2 -kinase/fructose- 2,6-biphosphatase 3 (PFKFB3) siRNA, or the PFKFB3 overexpression plasmid before exposure to LPS.
Results: Serum apelin-13 levels were significantly elevated in patients with sepsis and sepsis-associated acute respiratory distress syndrome (ARDS) (P< 0.0001). In vivo, apelin-13 suppressed LPS-induced ALI and inflammatory cytokine production (P< 0.05). Furthermore, apelin-13 reduced hydrogen peroxide (H2O2) content, NOX4 protein levels, and glycolysis. In vitro, LPS stimulation elevated NOX4 protein levels and reactive oxygen species (ROS) production (P< 0.05). These changes resulted in the accumulation of glycolysis in BMDMs. Treatment with antioxidant or NOX4 siRNA inhibited LPS-induced glycolysis and inflammatory cytokine production (P< 0.05). Moreover, in vitro experiments revealed that PFKFB3 regulates the release of pro-inflammatory cytokines by modulating glycolysis. In contrast, the action of apelin-13 opposed the effects of LPS.
Conclusion: In conclusion, apelin-13 protects against LPS-induced inflammatory responses and ALI by regulating PFKFB3-driven glycolysis induced by NOX4-dependent ROS.
Keywords: acute lung injury, inflammation, mice, apelin-13, NADPH oxidase 4, glycolysis, PFKFB3
Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are common life-threatening lung diseases, primarily associated with acute and severe inflammation of the lungs.1 Many immune cells are involved in pulmonary inflammatory dysfunction.2,3 Among the various immune cells, macrophages play a crucial role in the host inflammatory response.2,4 Therefore, inhibition of the inflammatory response of macrophages may be a promising approach for treating ALI.
Lipopolysaccharide (LPS) is one of the most common microbial mediators in ALI.5 LPS is a major bioactive component of the cell wall of gram-negative bacteria and strongly stimulates the inflammatory response by recognizing the pattern recognition receptor toll-like receptor 4 (TLR4).6,7 LPS and other harmful stimuli have been associated with increased reactive oxygen species (ROS) production. Oxidative stress is defined as an imbalance between ROS production and the ability of cells to detoxify or scavenge these molecules. As a signaling molecule, ROS production plays a relevant role in the inflammatory response.8 NADPH oxidase (NOX) 4 (NOX4) is a non-phagocytic isoform of NOX and an essential source of cellular hydrogen peroxide (H2O2) and ROS.9 In recent years, the vital role of NOX4 in mediating macrophage function during inflammation has been emphasized. NOX4 knockdown protects against cecal ligation puncture-induced septic ALI10 and LPS-induced macrophage inflammation.11 However, the potential mechanism of NOX4 in the inflammatory phase of ALI remains to be explored.
Many studies have demonstrated that when exposed to different stimuli, the metabolic profile of activated macrophages can rapidly shift from oxidative phosphorylation to aerobic glycolysis (a phenomenon also known as the Warburg effect) to adopt an inflammatory state.12,13 As reported, enhanced glycolysis of macrophages leads to increased lactate production, which is thought to be associated with increased mortality in patients with sepsis.14 6-phosphofructo-2-kinase/ fructose- 2.6-biphosphatase 3 (PFKFB3) is one of the Critical drivers of glycolysis, which regulates glycolytic flux and produces fructose-2,6-bisphosphate (F2, 6BP), a potent allosteric activator of the glycolytic rate-limiting enzyme phosphofructokinase-1 (PFK1).15,16 Previous studies have shown that LPS stimulation promotes increased expression of PFKFB3 in macrophages, which contributes to increased production of interleukin (IL)-1β and IL-6.17 Various pathways regulating glycolysis have been identified, among which ROS represent one of the essential regulators.18,19 The appropriate ROS production within the macrophages to counteract pathogens or responses to cellular stress. The excessive ROS production influences the outcome of the inflammatory immune cell response, which in turn exacerbates abnormal metabolic events.18 Therefore, we hypothesized that LPS might activate ROS and subsequently activate glycolysis promoting the inflammatory phase of ALI.
The renin–angiotensin system (RAS) is involved in the development and pathogenesis of ALI.20–22 Apelin, an endogenous ligand for angiotensin type 1 receptor associated protein (APJ), is a group of small peptides.23 Previous studies have shown that the apelin /APJ system counteracts the effects of angiotensin converting enzyme (ACE)-angiotensin (Ang)-II-angiotensin II type 1 receptor (AT1R) axis and is a positive regulator of angiotensin converting enzyme-2 (ACE2) under many physiological and pathophysiological conditions.24–28 Recently, a study showed that rats with oleic acid-induced ARDS had higher lung tissue levels of apelin-13,29 and treatment with apelin-13 alleviated LPS-induced lung inflammation and injury.29–31 However, the exact molecular mechanism by which the apelin-13 protects against ALI is not fully understood. A study by Kong et al showed that apelin-13 enhanced AMPK phosphorylation to regulate mitochondrial function to improve the pulmonary endothelial barrier in ALI mice.30 Zhang et al reported that apelin-13 reduced IL-1β release, suppressed activation of NLRP3 inflammasome, and inhibited the NF-κB pathway in RAW264.7 cells.31 Nevertheless, the effect of apelin-13 on LPS-induced inflammation in primary macrophages remains unclear, and whether apelin-13 can be used to improve macrophage oxidative stress and thus glycolysis are still mostly unknown.
In this study, we explored the regulation of glycolysis through NOX4-dependent ROS in LPS-induced ALI and demonstrated that apelin- 13 attenuated macrophage inflammation and improved ALI by inhibiting the PFKFB3-driven glycolysis induced by NOX4-dependent ROS in vivo and in vitro.
Materials and Methods
Materials
LPS, Apelin-13, F13A (a selective APJ receptor antagonist), DPI (an NAPDH oxidase inhibitor, diphenylene iodonium), NAC (a superoxide inhibitor, N-Acetyl Cysteine) were purchased from Sigma-Aldrich (St. Louis, Missouri, USA). Mouse tumor necrosis factor (TNF)-α, IL-1β, IL-6 and Apelin-13 enzyme linked immunosorbent assay (ELISA) kits were purchased from Cusabio (Houston, TX, USA). The reactive oxygen species assay kit (DCF-DA) was from Applygen (Beijing, China). The H2O2 assay kit was from Beyotime (Shanghai, China). NOX4- small-interfering RNA (siRNA) and PFKFB3 siRNA were provided by GenePharma (Shang hai, China). Mouse PFKFB3 (NM_001177756) overexpressed plasmid was purchased from MiaoLing Plasmid Sharing Platform (Hubei, China). Other reagents are described below.
Animals
All experimental procedures involving mice were approved by the Committee on the Ethics of Animal Experiments of Southern Medical University and were performed in accordance with the National Institutes of Health guidelines. 8–10 weeks old male C57 mice (22–26g) were obtained from Southern Medical University Animal Center (Guangzhou, China) and housed in a standard environment with 12 h light, 12 h dark and free access to food and water.
Animal Treatments
A mouse model using LPS-induced ALI was used. Male C57 mice were randomly divided into three groups: the control group, the LPS group, and the LPS +Apelin group. Mice were treated once by intratracheal (i.t.) instillation of 5 mg/kg of LPS in saline (or with saline as a control). Mice in the LPS +Apelin group were injected with apelin (10 nmol/kg, intraperitoneal (i.p.)) 1 h after installation of LPS and 3 h after the initial dose of apelin-13. After 6 hours of LPS instillation, the mice were killed and the tissue of lung was obtained for scheduled tests.
H&E Staining
Paraffin-embedded mouse lung sections were stained by hematoxylin (DH0001, Leagene) and eosin (DH0055, Leagene). Images were captured by microscopy.
Immunohistochemical Staining
Paraffin-embedded mouse lung sections were incubated with primary antibodies against PFKFB3 (A5593, Bimake) at 4 C overnight. Then, the sections were incubated with an HRP-conjugated secondary antibody (GK500710, Gene Tech) for 30 min followed by DAB solution for seconds. Next, the nuclei were stained with hematoxylin and the sections were sealed with neutral gum. Images were captured by microscopy.
Immunofluorescence Staining
Tissue samples were sectioned, deparaffinized, and processed for staining. The tissue was incubated with primary antibodies overnight at 4°C. Then lung sections were stained with FITC and Cy3-conjugated secondary antibodies (A0562, A0521, beyotime) for an hour at room temperature, after which they were stained with DAPI (F6057, sigma). Images were captured with fluorescence microscopy (BX63, OLYMPUS). Primary antibodies here included anti-APJ (20341-1-AP, Proteintech), anti-F4/80 (MAB5580-SP, R&D Systems), anti-NOX4 (14347-1-AP, Proteintech), anti-PFKFB3 (A5593, Bimake).
Measurement of Lung Wet/Dry Weight Ratio
The lung wet/dry (W/D) weight ratio was calculated to assess the lung edema. Each lung was weighed before and after being dried in an oven at 80°C for at least 24 h until the weight was constant.
Analysis of BALF
The levels of TNF-α, IL-6, and IL-1β in the bronchoalveolar lavage fluid (BALF) samples were measured using ELISA according to the manufacturer’s instructions. Lactate in BALF was measured with Lactate Assay Kit according to the manufacturer’s instructions. The protein concentration in the BALF was assessed using the BCA protein assay kit (Bio-Rad Laboratories).
Cell Culture
Mouse macrophage cell line RAW 264.7 was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in DMEM (GIBCO) with 10% FBS (GIBCO) at 37 °C in a humidified atmosphere containing 5% CO2. Bone marrow-derived cell from C57BL/6 mice were cultured in DMEM media supplemented with 10% FBS and 1% penicillin/streptomycin and differentiated to bone marrow-derived macrophages (BMDMs) by recombinant murine granulocyte- macrophage colony stimulating factor (GM-CSF) (25 ng/mL; Miltenyi Biotech) for 7 days.
Measurement of Glucose Uptake, Lactate Production, LDH Activity and ATP Production
Glucose uptake assay kit was from Cayman Chemical Co (Ann Arbor, MI, USA). Lactate production and lactate dehydrogenase (LDH) enzymatic assay were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The ATP content assay kit was from Beyotime (Shanghai, China). All experiments were performed at least three times and the data were normalized by the cell numbers or protein content.
Western Blot Analysis
Relative protein expression levels were measured by Western blot as described previously.32 Antibodies used here were as follows: hexokinase (HK) 2 (22029-1-AP, Proteintech), pyruvate kinase M2 (PKM2) (15822-1-AP, Proteintech), PFKFB3 (A5593, Bimake), Lactate dehydrogenase A (LDHA) (21799-1-AP, Proteintech), NOX4 (14347-1-AP, Proteintech), β-actin (20536-1-AP, Proteintech), APJ (20341-1-AP, Proteintech) and secondary antibodies (92632210, 92632211, Licor). Protein bands were visualized by the Odyssey System from LI-COR Biosciences.
qRT-PCR
RNA was extracted by TRIzol (9109, Takara) and cDNA was synthesized using PrimeScript™ RT Master Mix (RR036A, Takara). Quantitative PCR was performed using TB Green™ Premix Ex Taq™ (RR420B, Takara) with Light Cycler® 480 (Roche). The primer sequences are as follows: TNF-α: Forward: GACTACGTGC- TCCTCACCCA, Reverse: TCTTGACGGCAGAGAGGAGG; IL-1β: Forward: TCGCAGCAGCACATCAACAAGAG, Reverse: AGGTCCACGGGAAAGACACAGG; IL-6: Forward: CTCCCAACAGACCTGTCTATAC, Reverse: CCATTGCACAACTCTTTTCTCA; APJ: Forward: CCAGTCTGAATGCGACTACG, Reverse: CTCCCG- GTAGGTATAAGTGGC; ACE: Forward: ACCAGAAGCCAGACAACAACTCAC, Reverse: CTTCCACGAACCTGTCAGCCTTG; AT1R: Forward: CTTGCTGCCTCGTCTACCACATG, Reverse: GTGCTCCTGAGAGGGTCCGAAG; ACE2: Forward: CTCTGGGAATGAGGACACGG, Reverse: CTTG- GGTTGGGCACTGCTTA; MAS: Forward: ATCAGTGTGGAGAGGTGCCTATCG, Reverse: ACGAATGCTGACTGGTGCTTGG; β-actin: Forward: GGCTGTATTCCCCTCCATCG, Reverse: CCAGTTGGTAACAATGCCATGT. The relative fold change was calculated by the comparative CT method.
Sepsis Patients and the Assessment of Patient Serum
Serums were collected from 34 sepsis patients (including nine sepsis-associated ARDS patients) and 13 healthy volunteers. Sepsis was described as a life-threatening organ dysfunction induced by a dysregulated host response to infection (sepsis 3.0);33 characterized as a ≥2 points increase in total Sequential Organ Failure Assessment score attributable to infection. The clinical definition of ARDS is based on clinical features and chest imaging according to the Berlin definition.34 The information about patients and healthy volunteers used as controls are listed in Table 1. Apelin-13 levels in the serum were determined using ELISA according to the manufacturer’s instructions. All participants provided informed consent to serum donation prior to participating in the study and all procedures were performed in accordance with the World Medical Association’s Declaration of Helsinki.
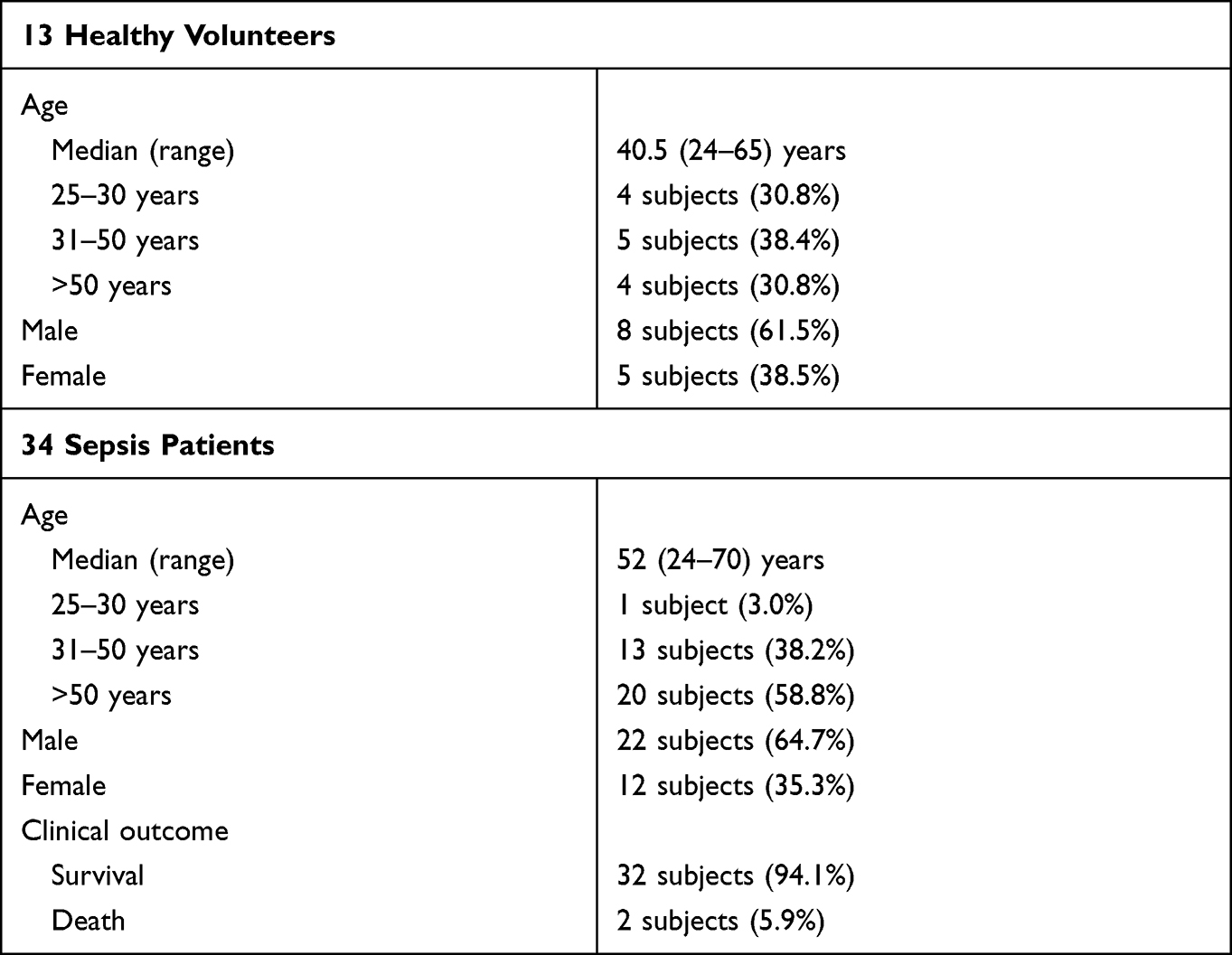 |
Table 1 Clinical Information for 13 Healthy Volunteers and 34 Sepsis Patients Enrolled in This Study |
Statistical Analysis
All the results are expressed as the mean ± SD. Data analysis was performed by SPSS 20.0 (SPSS Inc., Chicago, IL, USA). A two-tailed Student’s t-test was used for statistical analysis of two groups. ANOVA analyses were utilized when more than two groups were assessed. Statistical significance was defined as P < 0.05.
Results
Increased Serum Levels of Apelin-13 in Patients with Sepsis and Sepsis-Associated ARDS
A total of 47 participants with or without sepsis were studied. Serum samples were collected from 34 patients with sepsis (including nine patients with sepsis-associated ARDS) and 13 healthy volunteers were collected. Patients with sepsis and patients with sepsis-associated ARDS had significantly increased levels of apelin-13 compared with healthy controls (n=13) (Figure 1A, Table 1). Here we also showed that serum apelin-13 levels appear to be associated with the severity and lethality in patients with sepsis-associated ARDS, at least in some patients (Figure 1B), suggesting that the expression of apelin-13 may have a protective effect against sepsis-associated ARDS.
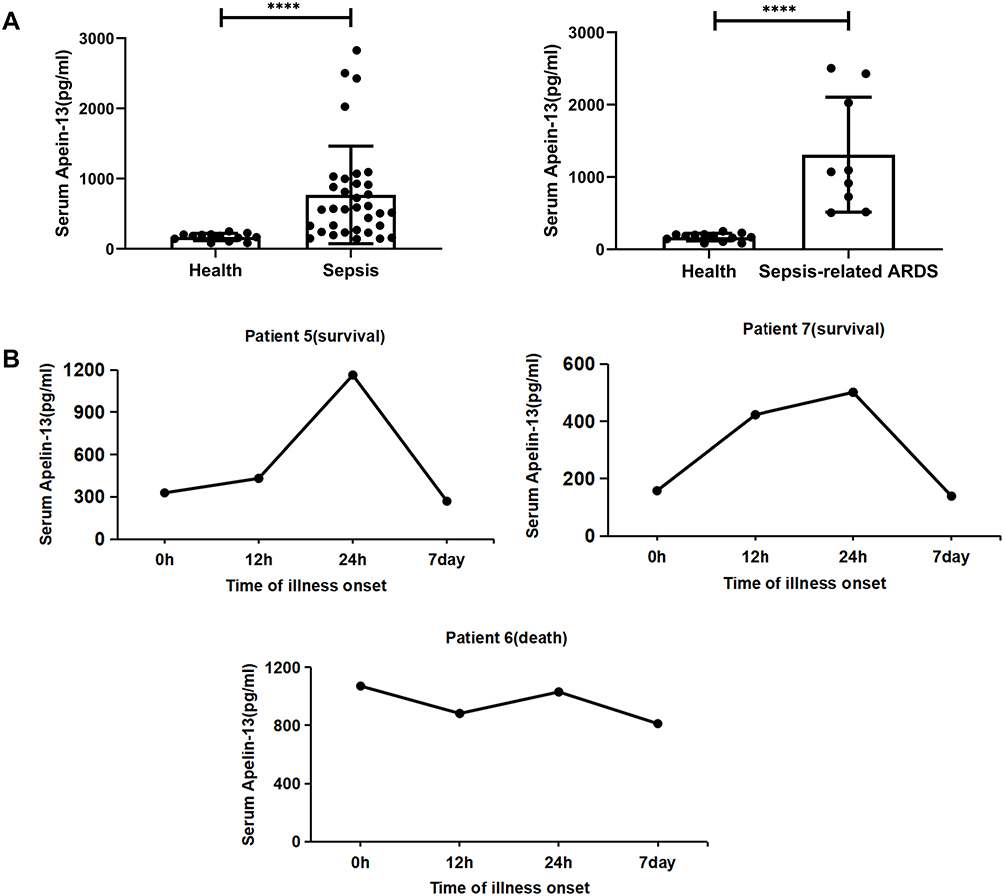 |
Figure 1 Serum apelin-13 levels increased in patients with sepsis and sepsis-associated ARDS. (A) Serum apelin-13 levels in healthy controls (n=13) and 34 patients with sepsis (n=34, including nine patients with sepsis-associated ARDS). Apelin-13 levels in serum were measured by ELISA. (B) Temporal changes of apelin-13 serum levels in three patients with sepsis-associated ARDS (patient 5, 6, and 7). Apelin-13 levels were measured by ELISA at the indicated time points after the visit for sepsis diagnosis. ****P<0.0001 versus healthy group. Abbreviations: ARDS, acute respiratory distress syndrome; ELISA, enzyme linked immunosorbent assay. |
Apelin-13 Attenuated Histological Damage and Lung Inflammation in Mice with LPS Induced Lung Injury
To demonstrate that increased apelin-13 expression after ALI may have a protective effect and explore its precise mechanism, we constructed an animal model of LPS-induced ALI (Figure 2A). Similar to our human data, serum apelin-13 levels were elevated in LPS-induced ALI mice (Figure 2B). H&E staining of lung tissues demonstrated that the main characteristics of ALI, including interstitial and alveolar infiltration of inflammatory cells and thickening of the alveolar septa, were evident in LPS-induced lungs, but were dramatically reduced after apelin-13 treatment (Figure 2C and D). Similarly, the total protein concentration in BALF and the lung W/D weight ratio were substantially increased in LPS group, which was attenuated by apelin-13 treatment (Figure 2E and F). Furthermore, the results of RT-qPCR and ELISA showed that, compared to the LPS group, apelin-13 significantly reduced the production of pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, in lung tissue and BALF (Figure 2G and H). These findings suggest that apelin-13 can effectively inhibit systemic inflammation in LPS-induced lung injury mice.
 |
Figure 2 Apelin-13 attenuated histological damage and lung inflammation in mice with LPS-induced lung injury. (A) The intervention dose regimen of apelin-13 in an experimental mouse model of acute lung injury. Mice were injected intraperitoneally with apelin-13 or vehicle 1 h after LPS instillation and 3 h after the initial dose of apelin-13. After 6 h of LPS instillation, the mice were killed and the tissue of lung was obtained for scheduled tests. (B) Expression of apelin-13 in the serum of the control group and the LPS treatment group. (C) Representative H&E-stained lung sections of different groups at 200x and 400 x original magnification. The alveolar walls were with intramural neutrophils (arrows). (D) Histopathological scores of different groups. (E) Total protein levels in BALF of each group. (F) The lung W/D weight ratio was assessed among experimental groups. (G) Levels of TNF-α, IL-1β, and IL-6 in lung tissues were detected by real-time PCR. (H) Levels of TNF-α, IL-1β, and IL-6 in BALF were detected by ELISA. Data were expressed as mean ± SD. *P<0.05 versus control group, #P<0.05 versus the LPS treatment group (n=6). Abbreviations: LPS, lipopolysaccharide; BALF, bronchoalveolar lavage fluid; W/D, wet/dry; ELISA, enzyme linked immunosorbent assay; TNF-α, tumor necrosis factor α; IL-1β, interleukin-1β; IL-6, interleukin-6. |
Apelin-13 Inhibited the Release of Pro-Inflammatory Cytokines in LPS-Treated Macrophages via the APJ Receptor
In in vivo studies, we found that apelin-13 significantly reduced lung inflammation. Macrophages, as one of the most important innate immune cells, play a crucial role in the highly inflammatory response to ALI. Therefore, we subsequently investigated the relationship between apelin-13 and macrophage inflammation. As showed in Figure 3A and B, compared with the control group, the expression of TNF-α, IL-6, and IL-1β were obviously increased in RAW264.7 cells after LPS stimulation. Treatment with apelin-13 at concentrations of 0.1 to 1 μM gradually decreased the levels of those pro-inflammatory cytokines. To determine whether this effect was also present in primary cells, we treated BMDMs with apelin-13 and observed the same results (Figure 3A and B). Stimulating BMDMs with LPS for the indicated periods of time (0, 3, 6 h), we also found that treating cells with apelin-13 before LPS stimulation inhibited the release of inflammatory cytokines (Figure 3C). These results suggest that apelin-13 reduces the secretions of TNF-α, IL-6, and IL-1β in LPS-activated macrophages.
 |
Figure 3 Apelin-13 inhibited the release of pro-inflammatory cytokines in LPS-treated macrophages through the APJ receptor. (A) RAW264.7 cells and BMDMs were treated with 1ug/mL LPS and the indicated concentrations of apelin-13 for 6 h. The mRNA levels of TNF-α, IL-1β, and IL-6 were measured by real-time PCR. (B) TNF-α, IL-1β, and IL-6 levels in culture supernatants were measured by ELISA. (C) BMDMs were treated with LPS (1ug/mL) or apelin-13 (1 umol/L) at different times. Pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 concentrations were measured in culture supernatants. (D) BMDMs were pre-incubated with apelin-13 (1umol/L) for 1 h before being exposing to 1ug/mL LPS for 6 h. APJ mRNA levels were measured by real-time PCR. (E) Protein levels of APJ were measured by Western blot analysis. (F) BMDMs were treated with apelin-13 (1 umol/L) or F13A (1 umol/L) for 1h before being stimulated with LPS (1 ug/mL) for 6 h. The mRNA levels of TNF-α, IL-1β, and IL-6 were determined by real-time PCR. (G) TNF-α, IL-1β, and IL-6 levels in culture supernatants were measured by ELISA. (H) Colocalization of APJ and F4/80 in lung sections was examined by immunofluorescence. Scale bar, 100 μm. Data were expressed as mean ± SD. *P<0.05 versus control group, #P<0.05 versus the LPS treatment group, &P< 0.05 vs the LPS + Apelin (n=3). Abbreviations: LPS, lipopolysaccharide; BMDMs, bone marrow-derived macrophages; ELISA, enzyme linked immunosorbent assay; TNF-α, tumor necrosis factor α; IL-1β, interleukin-1β; IL-6, interleukin-6. |
The G protein-coupled receptor APJ is one of the essential receptors for apelin-13. To determine whether APJ is involved in the inhibitory effect of apelin-13 on inflammatory cytokines release, we examined the expression of APJ in BMDMs cultured under different conditions. As shown in Figure 3D, the level of APJ increased in the LPS group compared to the control group. In addition, there appeared to be a positive autoregulation, and apelin-13 pretreatment enhanced LPS-induced mRNA and protein expression of APJ receptor (Figure 3D and E). RT-qPCR and ELISA results showed that LPS promotes the expression of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1β. Apelin-13 significantly inhibited these effects of LPS, and these inhibitory effects could be reversed by the APJ receptor antagonist F13A (Figure 3F and G). This result suggests that apelin-13 ameliorates LPS-induced inflammatory cytokines release mainly through the G protein-coupled receptor APJ. In vivo, immunofluorescence staining confirmed the colocalization of APJ and F4/80, a macrophage marker (Figure 3H). Therefore, apelin-13 inhibited the LPS-induced release of pro-inflammatory cytokines from macrophages through the APJ receptor.
Treatment of Apelin-13 Led to Down-Regulation of the ACE/AT1R Axis and Up-Regulation of ACE2 Expression in LPS-Treated Macrophages
Localized RAS is involved in the development of acute lung injury.20,21 Accumulating evidence shows that the apelin /APJ system interacts with the RAS.26,27 Therefore, we examined the expression of the RAS components in BMDMs cultured under different conditions. As shown in Figure 4A, LPS increased ACE and AT1R mRNA levels and decreased ACE2 mRNA levels. In contrast, apelin-13 decreased ACE and AT1R levels and increased ACE2 expression. Western blot analysis also showed the same trend (Figure 4B). MAS mRNA levels were reduced by LPS, while apelin-13 treatment had little effect on MAS expression levels (Figure 4A and B). These data indicate that apelin-13 can regulate RAS levels in BMDMs.
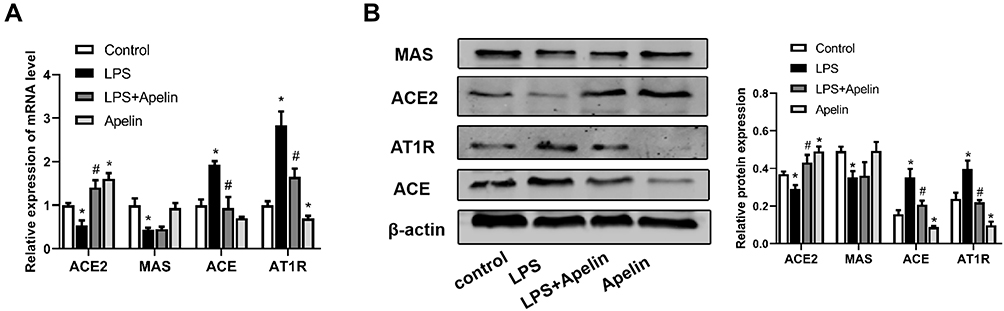 |
Figure 4 Treatment with apelin-13 led to down-regulation of the ACE/AT1R axis and up-regulation of ACE2 expression in LPS-treated macrophages. (A) BMDMs were treated with apelin-13 (1 umol/L) for 1 h before stimulation with LPS (1 ug/mL) for 6 h. The mRNA levels of ACE, AT1R, ACE2 and MAS were determined by real-time PCR. (B) Protein levels of ACE, AT1R, ACE2 and MAS were determined by Western blot analysis. Data were expressed as mean ± SD. *P<0.05 versus control group, #P<0.05 versus the LPS treatment group (n=3). Abbreviations: LPS, lipopolysaccharide; BMDMs, bone marrow-derived macrophages; ACE, angiotensin converting enzyme; AT1R, angiotensin II type 1 receptor; ACE2, angiotensin converting enzyme-2. |
Apelin-13 Suppressed Pro-Inflammatory Responses by Inhibiting Oxidative Stress in LPS-Treated Macrophages
As shown in Figure 5A and B, LPS significantly increased the production of TNF-α, IL-6, and IL-1β remarkably compared to the control group. Then we investigated the oxidation levels of BMDMs stimulated with LPS. As shown in Figure 5C, LPS enhanced the expression of NOX4 protein. Meanwhile, the concentration of H2O2 was increased after exposure to LPS (Figure 5G). Similarly, LPS stimulation resulted in noticeable increase in intercellular ROS production (Figure 5H and I). Apelin-13 obviously inhibited these increases, and these inhibitory effects could be reversed by the APJ receptor antagonist F13A. Meanwhile, pretreatment with NAPDH oxidase inhibitor DPI or ROS scavenger NAC inhibited the above effects of LPS. Analogously, the LPS-induced expression of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1β was suppressed by NOX4 siRNA (Figure 5D and E). The effect of NOX4 silencing was confirmed at the protein levels (Figure 5F). These data suggest that apelin-13 suppresses the LPS-induced expression of pro-inflammatory cytokines by inhibiting oxidative stress.
 |
Figure 5 Apelin-13 suppressed pro-inflammatory responses by inhibiting oxidative stress in LPS-treated macrophages. (A) BMDMs were treated with apelin-13 (1 umol/L), NAC (2 mmol/L), DPI (10 umol/L), or F13A (1 umol/L) for 1 h before stimulation with LPS (1 ug/mL) for 6 h. The mRNA levels of TNF-α, IL-1β, and IL-6 were determined by real-time PCR. (B) TNF-α, IL-1β, and IL-6 levels in culture supernatants were measured by ELISA. (C) Protein levels of NOX4 were measured by Western blot analysis. (D) BMDMs were transfected with NOX4 siRNA before stimulation with LPS (1 ug/mL) for 6 h. The mRNA levels of TNF-α, IL-1β, and IL-6 were determined by real-time PCR. (E) Concentrations of pro-inflammatory cytokines were detected in culture supernatants. (F) Protein levels of NOX4 were measured by Western blot analysis. (G) H2O2 concentrations in BMDMs were measured. (H and I) Intracellular ROS was detected by the probe DCF-DA. Data were expressed as mean ± SD. *P< 0.05 vs control group, #P< 0.05 vs the LPS group, &P< 0.05 vs the LPS + Apelin, $P< 0.05 vs NT siRNA group, ^P< 0.05 vs the LPS+NT siRNA (n=3). Abbreviations: LPS, lipopolysaccharide; BMDMs, bone marrow-derived macrophages; NAC, N-Acetyl Cysteine; DPI, diphenylene iodonium; TNF-α, tumor necrosis factor α; IL-1β, interleukin-1β; IL-6, interleukin-6; ELISA, enzyme linked immunosorbent assay; NOX4, NADPH oxidase (NOX) 4; siRNA, small-interfering RNA; H2O2, hydrogen peroxide; ROS, reactive oxygen species. |
Apelin-13 Attenuated the PFKFB3-Driven Glycolysis in LPS-Treated Macrophages
Recent studies have shown that increased aerobic glycolysis is essential for developing a pro-inflammatory phenotype in LPS-activated macrophages.35,36 We investigated whether apelin-13 inhibits inflammation by suppressing glycolysis in macrophages. BMDMs were stimulated with LPS for a specified duration (0, 3, or 6 h). We then evaluated glucose consumption, lactate production, LDH activity and ATP production in BMDMs. As shown in Figure 6A, LPS promoted significant increases in lactate levels, ATP production, glucose consumption and intracellular LDH activity in BMDMs, which are all indicators of enhanced glycolytic flux. Apelin-13 was shown to suppress LPS-induced glycolysis enhancement, and this effect was pronounced at 6h (Figure 6A). Meanwhile, extracellular acid ratio (ECAR) analysis showed that LPS treatment increased glycolysis and glycolytic activity in BMDMs, both of which were also reduced by apelin-13 (Figure 6B and C). Additionally, apelin-13 treatment increased Oxygen consumption rate (OCR) levels in BMDMs compared to the LPS group (Figure 6D and E). To elucidate the mechanism by which apelin-13 inhibits the enhanced glycolysis observed in LPS-stimulated BMDMs, we evaluated the expression of key glycolytic enzymes in these cells. Notably, PFKFB3 was significantly induced by LPS in BMDMs and the apelin-13 widely reduced its expression at the protein level (Figure 6F and G). Furthermore, the protein level of hypoxia-inducible factor 1-α (HIF-α) in LPS-treated BMDMs was markedly increased compared to the control group, while apelin-13 inhibited the aforementioned effects of LPS (Figure 6H).
 |
Figure 6 Apelin-13 attenuated PFKFB3-driven glycolysis in LPS-treated macrophages. (A) BMDMs were treated with LPS (1ug/mL) or apelin-13 (1 umol/L) at different times. Glucose consumption, lactate production, LDH activity, and ATP production were measured in different groups. (B) BMDMs were treated with apelin-13 (1 umol/L) for 1 h before stimulation with LPS (1 ug/mL) for 6 h. ECAR was measured using the Seahorse XF. (C) Glycolysis and glycolysis capacity were quantified and shown as histograms. (D) OCR was measured using the Seahorse XF. (E) Basal respiration and maximal respiration were quantified and shown as histograms. (F) Protein levels of HK2, PKM2, PFKFB3, and LDHA were analyzed by Western blot. (G) Quantification of HK2, PKM2, PFKFB3, and LDHA protein levels relative to β-actin is shown. (H) Protein level of HIF-α was analyzed by Western blot. (I) BMDMs were transfected with PFKFB3 siRNA before stimulation with LPS for 6 h. Protein level of PFKFB3 were measured by Western blot analysis. (J) The mRNA levels of TNF-α, IL-1β, and IL-6 were determined by real-time PCR. (K) TNF-α, IL-1β, and IL-6 levels in culture supernatants were measured by ELISA. (L) BMDMs were transfected with PFKFB3 plasmid. The protein level of PFKFB3 were measured by Western blot analysis. (M) The mRNA levels of TNF-α, IL-1β, and IL-6 in different groups were detected by real-time PCR. Data were expressed as mean ± SD. *P < 0.05 vs control group, #P< 0.05 vs the LPS group, &P< 0.05 vs the LPS + Apelin, $P< 0.05 vs NT siRNA group, ^P < 0.05 vs the LPS+NT siRNA (n=3). Abbreviations: LPS, lipopolysaccharide; BMDMs, bone marrow-derived macrophages; ECAR, Extracellular acid ratio; 2-DG, 2-deoxyglucose; OCR, Oxygen consumption rate; FCCP, Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone; Rot/AA, Rotenone & AntimycinA; LDHA, lactate dehydrogenase A; HK2, hexokinase 2; PKM2, pyruvate kinase M2; PFKFB3, 6-phosphofructo-2 -kinase/ fructose-2,6-biphosphatase 3; siRNA, small-interfering RNA; ELISA, enzyme linked immunosorbent assay; TNF-α, tumor necrosis factor α; IL-1β, interleukin-1β; IL-6, interleukin-6; HIF-α, Hypoxia-inducible factor 1-α. |
To further explore whether apelin-13 suppresses LPS-induced inflammatory responses by attenuating PFKFB3-driven glycolysis, we transfected BMDMs with PFKFB3 siRNA and PFKFB3 overexpression plasmid to test our hypothesis. Using PFKFB3 siRNA, we confirmed that pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) release was reduced in response to PFKFB3 siRNA (Figure 6J and K). The effect of PFKFB3 silencing was confirmed at protein levels (Figure 6I). As shown in Figure 6M, the reduction of pro-inflammatory cytokines expression in apelin-13 treated macrophages was reversed after PFKFB3 overexpression plasmid pretreatment. The effect of PFKFB3 overexpression was confirmed at the protein level (Figure 6L). Taken together, the above data demonstrate that apelin-13 can eliminate the PFKFB3-driven increase in glycolysis, which promotes the release of pro-inflammatory cytokines from BMDMs.
Apelin-13 Suppressed LPS-Induced Pro-Inflammatory Responses by Inhibiting the NOX4/ROS/PFKFB3-Driven Glycolysis
As shown in Figure 7A, apelin-13 inhibited LPS-induced enhancement of the glycolytic protein PFKFB3 and this inhibitory effect was reversed by the APJ receptor antagonist F13A. Pretreatment with NOX4 siRNA neutralized LPS-induced PFKFB3 enhancement (Figure 7B). This finding suggests that NOX4-derived ROS may be necessary for PFKFB3-driven glycolysis activation. Next, we used glucose consumption, lactate production, LDH activity, ATP production and ECAR to evaluate the glycolytic flux. Figure 7C–H showed that, after apelin-13 added to BMDMs stimulated by LPS, the glucose consumption, lactate production, LDH activity, ATP production and ECAR was significantly lower than those of stimulated by LPS alone, indicating that apelin-13 partially ameliorated LPS-induced glycolysis. Furthermore, apelin-13 treatment increased OCR levels in BMDMs compared to the LPS group (Figure 7I and J). However, the protective effect of apelin-13 was neutralized by F13A. Additionally, LPS-induced glycolysis was attenuated by NAC or DPI (Figure 7C–H), suggesting that apelin-13 suppressed LPS-induced inflammatory responses by regulating the NOX4/ROS/PFKFB3-driven glycolysis.
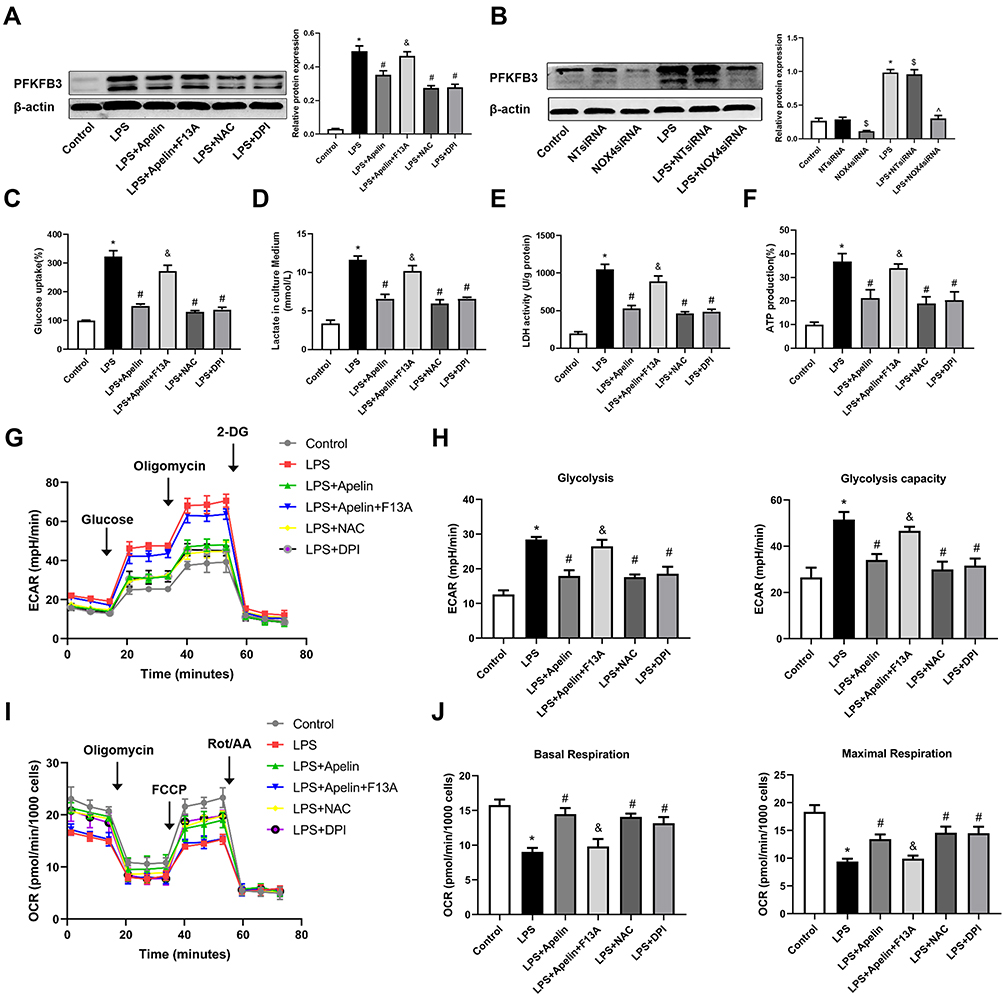 |
Figure 7 Apelin-13 suppressed LPS-induced pro-inflammatory responses by regulating the NOX4/ROS/PFKFB3-driven glycolysis. (A) BMDMs were treated with apelin-13 (1 umol/L), NAC (2 mmol/L) or DPI (10 umol/L) for 1 h before stimulation with LPS (1 ug/mL) for 6 h. BMDMs were pretreated with F13A (1 umol/L) for 1 h before stimulation with apelin-13. Protein levels of PFKFB3 were analyzed by Western blot. (B) BMDMs were transfected with NOX4 siRNA before stimulation with LPS for 6 h. PFKFB3 were analyzed by Western blot. (C–F) Glucose consumption, lactate production, LDH activity, and ATP production were measured. (G) ECAR was measured using the Seahorse XF. (H) Glycolysis and glycolysis capacity were quantified and displayed as histograms. (I) OCR was measured using the Seahorse XF. (J) Basal respiration and maximal respiration were quantified and shown as histograms. *P< 0.05 vs control group, #P< 0.05 vs the LPS group, &P< 0.05 vs the LPS + Apelin, $P< 0.05 vs NT siRNA group, ^P< 0.05 vs the LPS+NT siRNA (n=3). Abbreviations: LPS, lipopolysaccharide; BMDMs, bone marrow-derived macrophages; NAC, N-Acetyl Cysteine; DPI, diphenylene iodonium; ROS, reactive oxygen species; NOX4, NADPH oxidase (NOX) 4; siRNA, small-interfering RNA; PFKFB3, 6-phosphofructo-2 -kinase/ fructose-2,6-biphosphatase 3; LDH, lactate dehydrogenase; ECAR, Extracellular acid ratio; 2-DG, 2-deoxyglucose; FCCP, Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone; Rot/AA, Rotenone & AntimycinA; OCR, Oxygen consumption rate. |
Apelin-13 Alleviated Oxidation Levels and Reduced PFKFB3-Driven Glycolysis in LPS-Induced Lung Injury Mice
To confirm the regulation of oxidative stress and PFKFB3-driven glycolysis by apelin-13 in vivo, we evaluated oxidative levels, lactate levels, and PFKFB3 expression in lung tissue of mice. The results showed that the concentration of H2O2 (Figure 8A) and the NOX4 protein expression (Figure 8B) were increased in the LPS group compared to the control group. Apelin-13 treatment could suppress the aforementioned effects of LPS. Immunofluorescence staining showed that the expression of NOX4 were widely decreased in the LPS+Apelin13 group when compared to the LPS group (Figure 8C). As shown in Figure 8D, BALF lactic acid levels increased significantly in LPS-treated mice, whereas they were decreased considerably after intervention with apelin-13. In addition, western blotting and immunohistochemical staining showed that apelin-13 inhibited the LPS-induced upregulation of PFKFB3 in the lungs of mice (Figure 8E and F). Meanwhile, immunofluorescence staining confirmed the colocalization of PFKFB3 and F4/80, a macrophage marker (Figure 8G). Collectively, these results suggest that apelin-13 attenuates oxidative levels and reduces PFKFB3-driven glycolysis in mice with LPS-induced lung injury.
 |
Figure 8 Apelin-13 alleviated oxidation levels and reduced PFKFB3-driven glycolysis in mice with LPS-induced lung injury. (A) Mice were injected intraperitoneally with apelin-13 or vehicle after LPS instillation, and 6 h later, their lung were harvested. H2O2 concentrations in lung homogenates were tested. (B) NOX4 protein levels in the lungs were detected by Western blot. (C) Immunofluorescence images for NOX4 staining (red) in different groups of lung sections. Nuclei were stained with DAPI (blue). Scale bar, 100 μm. (D) Lactate concentrations were detected in different groups of BALF. (E) PFKFB3 protein levels in lung homogenates were assessed by Western blot. (F) The expression of PFKFB3 was detected by immunohistochemistry. (G) Immunofluorescence was performed to examine the colocalization of PFKFB3 (red) and F4/80 (green) in lung sections. Nuclei were stained with DAPI (blue). Scale bar, 100 μm. Data were expressed as mean ± SD. *P<0.05 versus control group, #P<0.05 versus the LPS treatment group (n=6). Abbreviations: LPS, lipopolysaccharide; H2O2, hydrogen peroxide; NOX4, NADPH oxidase (NOX) 4; PFKFB3, 6-phosphofructo-2 -kinase/ fructose-2,6-biphosphatase 3; BALF, bronchoalveolar lavage fluid. |
Discussion
In the present study, we demonstrated that apelin-13 alleviated the release of inflammatory cytokines from macrophages and improved ALI by regulating PFKFB3-driven glycolysis induced by NOX4-generated ROS (Figure 9). The principal findings included the following: (i) Serum apelin-13 levels were increased in patients with sepsis-associated ARDS; (ii) Apelin-13 upregulated ACE2 expression while downregulating the ACE/AT1R axis in LPS-treated BMDMs; (iii) Glycolysis is activated by LPS-induced NOX4-dependent ROS; (iiii) Apelin-13 attenuated macrophage inflammation and ameliorated ALI by regulating oxidative levels and PFKFB3-driven glycolysis in vivo and in vitro.
 |
Figure 9 Schematic diagram of the major signaling pathways involved in apelin-13 attenuates inflammatory responses and acute lung injury. NOX4-generated ROS and the consequent PFKFB3-driven glycolysis mediated macrophage inflammation and ALI. Apelin-13 attenuates macrophage inflammation and improves ALI by downregulating the NOX4/ROS/PFKFB3-driven glycolysis. Abbreviations: LPS, lipopolysaccharide; NOX4, NADPH oxidase (NOX) 4; ROS, reactive oxygen species; PFKFB3, 6-phosphofructo-2-kinase/fructose- 2,6-biphosphatase 3; TNF-α, tumor necrosis factor α; IL-1β, interleukin-1β; IL-6, interleukin-6. |
This study focused on apelin peptides, which are highly abundant in lung tissue.25,37 Previous studies have shown that the apelin /APJ system interacts with the RAS and that the apelin/APJ signaling pathway counteracts the ACE-Ang II-AT1R axis,24,26,38 and that apelin is a positive regulator of ACE2 in failing hearts.25 Furthermore, the RAS is involved in the development and pathogenesis of ALI.21,22,39 Activation of the RAS is involved in the process of respiratory syncytial virus-induced ALI and that was partially reversed by pharmacological inhibition of AT1R.39 Angiotensin-(1–7), produced by Ang II with ACE2, is protective in murine models of ventilator or acid aspiration-induced ALI.22 It has been reported that apelin-13 could protect against experimental ALI,29–31 and decrease IL-1β release in RAW 264.7 cells.31 However, the mechanisms by which apelin-13 protects against ALI are still not fully understood. Consistent with these findings, our results illustrated that apelin-13 could effectively protect mice from LPS-induced lung inflammation and injury. We hypothesized that the protective role of apelin-13 in inflammation and ALI could be associated with the decrease of ACE and AT1R levels and the increase of ACE2 levels. According to our data, LPS treatment increased the expression of the ACE/AT1R axis, both ACE and AT1R, and decreased the expression of ACE2 in BMDMs. Apelin-13 obviously constrained the aforementioned effects of LPS. These results suggested that apelin-13 increases ACE2 expression and decreases the activation of the ACE/AT1R axis induced by LPS in BMDMs.
Oxidative stress, an imbalance between excessive ROS production and the antioxidant capacity of cells, is a key factor that contributes to severe pulmonary inflammation in ALI/ARDS.8,40 The NOX enzyme family is a major source of ROS in macrophages.41,42 NOX4 is a non-phagocytic homolog of NOX, and NOX4 activation has been linked to pro-inflammatory responses induced by LPS.43,44 However, the underlying mechanisms of NOX4 in macrophages during the inflammatory phase of ALI remain unclear. Here, our results showed that oxidants, such as NOX4 and H2O2, were increased in LPS-induced ALI mice and in LPS-stimulated BMDMs, accompanied by the production of pro-inflammatory cytokines. Inhibition of NOX4-dependent ROS with DPI, NAC, or NOX4 siRNA abolished LPS-induced inflammatory effects, indicating that NOX4-driven ROS are important in LPS-induced inflammatory responses in macrophages. Although NOX4-generated ROS played a role in developing the macrophage inflammatory response, the exact molecular mechanism still needs to be further studied.
Severe metabolic abnormalities are another characteristic of sepsis-associated ALI, in which aerobic glycolysis is enhanced.45–47 Many studies have emphasized the importance of glycolytic metabolism in the regulation of effector function.48 Glycolytic metabolism has been identified as a vital driver of macrophage pro-inflammatory immune responses.49–51 Therefore, controlling the glycolysis of macrophages may be a potential strategy for treating sepsis-related ALI. Several previous studies have shown that PFKFB3 plays a central regulatory role in cellular metabolism.52,53 Here, we demonstrate that lactate production and PFKFB3 expression were remarkably increased together with inflammatory response after LPS treatment both in vivo and in vitro. LPS-induced release of pro-inflammatory cytokines could be ameliorated by PFKFB3 siRNA. Moreover, apelin-13 inhibited PFKFB3-driven glycolysis and ameliorated LPS-induced pro-inflammatory response. We proposed that the protective effect of apelin-13 against inflammation may be related to the reduction of PFKFB3-driven glycolysis. Pretreatment with the PFKFB3 overexpression plasmid was found to reverse the effect of apelin-13 on LPS-induced inflammatory cytokine release. Paradoxically, it has been reported that PFKFB3-driven macrophage glycolytic metabolism is a crucial component of innate antiviral defense.54 The discrepancy may be a result of different severity of lung injury and inflammation. Appropriate inflammatory responses are critical for pathogen elimination, but excessive inflammation may result in organ damage and increased mortality. Although PFKFB3-driven glycolysis is an important metabolic driver of macrophage function, excessive macrophage activation due to enhanced glycolytic activity can cause organ damage and even death.55,56 Recent studies have shown that deficiency of myeloid PFKFB3 protects mice from LPS-induced endotoxemia and hypoxia-induced pulmonary hypertension.57,58 Regulating excessive immune cell activation through metabolic reprogramming is seen as a promising approach to treating a variety of inflammatory diseases.55,59 Further studies are necessary to clarify the potential protect effect of apelin-13 in different severity and phase of septic ARDS.
ROS is one of the effector molecules that activate cellular aerobic glycolysis.60,61 Increasing evidence indicates that NOX4 plays a critical role in regulating glycolysis that depends on ROS production.62,63 Our findings have shown that LPS could enhance the NOX4-derived ROS in vivo and in vitro. Thus, we hypothesized that LPS-induced ROS might be involved in the process of glycolysis. Pretreatment with DPI, NAC, and NOX4 siRNA dramatically reduced the expression of PFKFB3, highlighting the essential role of NOX4-dependent ROS in the progress of glycolysis. In addition, antioxidants effectively attenuated lactate levels, ATP production, glucose consumption and intracellular LDH activity, which are indicators of enhanced glycolytic flux. Therefore, we presume that apelin-13 alleviates ALI and ameliorates macrophage inflammation by inhibiting NOX4-derived ROS and subsequent glycolytic activation.
The results in this study are consistent with our assumption. In vivo, apelin-13 treatment significantly attenuated LPS-induced oxidative stress and counteracted abnormal glycolysis, leading to the alleviation of ALI. In vitro, apelin-13 inhibited PFKFB3-driven glycolysis by suppressing NOX4-generated ROS. Furthermore, the inhibitory effects of apelin-13 could be weakened by F13A, the APJ receptor antagonist, suggesting that apelin-13 counteracts the effects of LPS through the APJ receptor. Although we demonstrated an essential role for NOX4-dependent ROS in LPS-induced glycolysis, the underlying signaling mechanism has not yet been clarified. HIF-1α is an important transcription factor in glucose metabolism that upregulates a series of genes involved in glucose transport and glycolysis.64,65 Current studies indicated that ROS could regulate glycolysis through HIF-1.60,66,67 Our findings show that the protein level of HIF-α was elevated in the LPS group, apelin-13 inhibited the aforementioned effect of LPS. This finding provides some critical information for our further study.
There are several limitations to this study. One limitation of this study is that we did not use LC-MS to detect glucose metabolites and circulating intermediates. Another limitation is that, as a potential therapeutic target for ALI, the effects of apelin-13 have not been estimated in patients. Future studies will focus on the therapeutic impact of apelin-13 in patients with ALI.
Conclusion
In conclusion, our study demonstrated that apelin-13 attenuates macrophage inflammation and improves ALI by regulating PFKFB3-driven glycolysis induced by NOX4-dependent ROS. Consequently, these results revealed a specific mechanism of apelin-13 attenuation of ALI, suggesting that apelin-13 may be a promising therapeutic agent for the prevention and treatment of ALI and macrophage inflammation.
Abbreviations
ALI, Acute lung injury; ARDS, Acute Respiratory Distress Syndrome; LPS, Lipopolysaccharide; BALF, Bronchoalveolar lavage fluid; W/D, Wet/dry; TNFα, Tumor necrosis factor α; IL-1β, Interleukin-1β; BMDMs, Bone marrow-derived macrophages; ACE, Angiotensin converting enzyme; AT1R, Angiotensin II type 1 receptor; ACE2, Angiotensin converting enzyme-2; NOX4, NADPH oxidase (NOX) 4; ROS, Reactive oxygen species; H2O2, Hydrogen peroxide; NAC, N-Acetyl Cysteine; DPI, Diphenylene iodonium; PFKFB3,6-phosphofructo-2 -kinase/ fructose- 2,6-biphosphatase 3; HIF-α, Hypoxia-inducible factor 1-α; 2-DG, 2-deoxyglucose; FCCP, Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone; Rot/AA, Rotenone & AntimycinA.
Ethics Approval and Consent to Participate
All experiments involving human subjects or animals were approved by the ethics committee of the Southern Medical University, Nanfang Hospital (Guangzhou, China) and the Experimental Animal Ethics Committee of Southern Medical University, respectively.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 81570064, 81870068, 82070063, 82170641) and the Key Laboratory of Emergency and Trauma (Hainan Medical University), Ministry of Education (grant. KLET-202102).
Disclosure
The authors report no conflicts of interest to declare.
References
1. Thompson BT, Chambers RC, Liu KD, Drazen JM. Acute respiratory distress syndrome. N Engl J Med. 2017;377(6):562–572. doi:10.1056/NEJMra1608077
2. Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol. 2020;11:1722. doi:10.3389/fimmu.2020.01722
3. Rahbarghazi R, Keyhanmanesh R, Aslani MR, et al. Bone marrow mesenchymal stem cells and condition media diminish inflammatory adhesion molecules of pulmonary endothelial cells in an ovalbumin-induced asthmatic rat model. Microvasc Res. 2019;121:63–70. doi:10.1016/j.cmvr.2018.10.005
4. Liu YC, Zou XB, Chai YF, et al. Macrophage polarization in inflammatory diseases. Int J Biol Sci. 2014;10(5):520–529. doi:10.7150/ijbs.8879
5. Opal SM. Endotoxins and other sepsis triggers. Contrib Nephrol. 2010;167:14–24. doi:10.1159/000315915
6. Yamamoto M, Akira S. Lipid A receptor TLR4-mediated signaling pathways. Adv Exp Med Biol. 2010;667:59–68. doi:10.1007/978-1-4419-1603-7_6
7. Gabarin RS, Manshu L, Zimmel PA, et al. Intracellular and extracellular lipopolysaccharide signaling in sepsis: avenues for novel therapeutic strategies. J Innate Immun. 2021;13(6):323–332. doi:10.1159/000515740
8. Liu W, Wu H, Chen L, et al. Park7 interacts with p47 (phox) to direct NADPH oxidase-dependent ROS production and protect against sepsis. Cell Res. 2015;25(6):691–706. doi:10.1038/cr.2015.63
9. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi:10.1152/physrev.00044.2005
10. Jiang J, Huang K, Xu S, et al. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol. 2020;36:101638. doi:10.1016/j.redox.2020.101638
11. Wang XL, Pan LL, Long F, et al. Endogenous hydrogen sulfide ameliorates NOX4 induced oxidative stress in LPS-stimulated macrophages and mice. Cell Physiol Biochem. 2018;47(2):458–474. doi:10.1159/000489980
12. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol. 2017;18(5):488–498. doi:10.1038/ni.3704
13. Russell DG, Huang L, VanderVen BC. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol. 2019;19(5):291–304. doi:10.1038/s41577-019-0124-9
14. Trzeciak S, Dellinger RP, Chansky ME, et al. Serum lactate as a predictor of mortality in patients with infection. Intensive Care Med. 2007;33(6):970–977. doi:10.1007/s00134-007-0563-9
15. Van Schaftingen E, Lederer B, Bartrons R, et al. A kinetic study of pyrophosphate: fructose-6-phosphate phosphotransferase from potato tubers. Application to a microassay of fructose 2,6-bisphosphate. Eur J Biochem. 1982;129(1):191–195. doi:10.1111/j.1432-1033.1982.tb07039.x
16. Klarer AC, O’Neal J, Imbert-Fernandez Y, et al. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2014;2(1):2. doi:10.1186/2049-3002-2-2
17. Finucane OM, Sugrue J, Rubio-Araiz A, et al. The NLRP3 inflammasome modulates glycolysis by increasing PFKFB3 in an IL-1β-dependent manner in macrophages. Sci Rep. 2019;9(1):4034. doi:10.1038/s41598-019-40619-1
18. Franchina DG, Dostert C, Brenner D. Reactive oxygen species: involvement in T cell signaling and metabolism. Trends Immunol. 2018;39(6):489–502. doi:10.1016/j.it.2018.01.005
19. Banerjee S, Ghosh S, Mandal A, et al. ROS-associated immune response and metabolism: a mechanistic approach with implication of various diseases. Arch Toxicol. 2020;94(7):2293–2317. doi:10.1007/s00204-020-02801-7
20. Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi:10.1038/nature03712
21. Deng W, Deng Y, Deng J, et al. Losartan attenuated lipopolysaccharide-induced lung injury by suppression of lectin-like oxidized low-density lipoprotein receptor-1. Int J Clin Exp Pathol. 2015;8(12):15670–15676.
22. Klein N, Gembardt F, Supé S, et al. Angiotensin-(1-7) protects from experimental acute lung injury. Crit Care Med. 2013;41(11):e334–43. doi:10.1097/CCM.0b013e31828a6688
23. Tatemoto K, Hosoya M, Habata Y, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251(2):471–476. doi:10.1006/bbrc.1998.9489
24. Pitkin SL, Maguire JJ, Bonner TI, et al. International union of basic and clinical pharmacology. LXXIV. Apelin receptor nomenclature, distribution, pharmacology, and function. Pharmacol Rev. 2010;62(3):331–342. doi:10.1124/pr.110.002949
25. Sato T, Suzuki T, Watanabe H, et al. Apelin is a positive regulator of ACE2 in failing hearts. J Clin Invest. 2013;123(12):5203–5211. doi:10.1172/JCI69608
26. Siddiquee K, Hampton J, McAnally D, et al. The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br J Pharmacol. 2013;168(5):1104–1117. doi:10.1111/j.1476-5381.2012.02192
27. Zhang J, Lin X, Xu J, et al. Apelin-13 reduces oxidative stress induced by uric acid via downregulation of renin-angiotensin system in adipose tissue. Toxicol Lett. 2019;305:51–57. doi:10.1016/j.toxlet.2019.01.014
28. Sabry MM, Ramadan NM, Al Dreny BA, et al. Protective effect of apelin preconditioning in a rat model of hepatic ischemia reperfusion injury; possible interaction between the apelin/APJ system, Ang II/AT1R system and eNOS. United Eur Gastroenterol J. 2019;7(5):689–698. doi:10.1177/2050640619826847
29. Fan XF, Xue F, Zhang YQ, et al. The apelin-APJ axis is an endogenous counterinjury mechanism in experimental acute lung injury. Chest. 2015;147(4):969–978. doi:10.1378/chest.14-1426
30. Kong X, Lin D, Liling L, et al. Apelin-13-mediated AMPK ameliorates endothelial barrier dysfunction in acute lung injury mice via improvement of mitochondrial function and autophagy. Int Immunopharmacol. 2021;101(PtB):108230. doi:10.1016/j.intimp.2021.108230
31. Zhang H, Chen S, Zeng M, et al. Apelin-13 administration protects against LPS-induced acute lung injury by inhibiting NF-κB pathway and NLRP3 inflammasome activation. Cell Physiol Biochem. 2018;49(5):1918–1932. doi:10.1159/000493653
32. Meng Y, Pan M, Zheng B, et al. Autophagy attenuates angiotensin ii-induced pulmonary fibrosis by inhibiting redox imbalance-mediated NOD-like receptor family pyrin domain containing 3 inflammasome activation. Antioxid Redox Signal. 2019;30(4):520–541. doi:10.1089/ars.2017.7261
33. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for Sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
34. Ranieri VM, Rubenfeld GD, Rubenfeld GD, et al.; ARDS Definition Task Force. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307(23):2526–2533. doi:10.1001/jama.2012.5669
35. Cui H, Banerjee S, Guo S, et al. IFN regulatory factor 2 inhibits expression of glycolytic genes and lipopolysaccharide-induced proinflammatory responses in macrophages. J Immunol. 2018;200(9):3218–3230. doi:10.4049/jimmunol.1701571
36. Virga F, Cappellesso F, Stijlemans B, et al. Macrophage miR-210 induction and metabolic reprogramming in response to pathogen interaction boost life-threatening inflammation. Sci Adv. 2021;7(19):eabf0466. doi:10.1126/sciadv.abf0466
37. Hosoya M, Kawamata Y, Fukusumi S, et al. Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin. J Biol Chem. 2000;275(28):21061–21067. doi:10.1074/jbc.M908417199
38. Kalea AZ, Batlle D. Apelin and ACE2 in cardiovascular disease. Curr Opin Investig Drugs. 2010;11(3):273–282.
39. Gu H, Xie Z, Li T, et al. Angiotensin-converting enzyme 2 inhibits lung injury induced by respiratory syncytial virus. Sci Rep. 2016;6:19840. doi:10.1038/srep19840
40. Kellner M, Noonepalle S, Lu Q, et al. ROS signaling in the pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS). Adv Exp Med Biol. 2017;967:105–137. doi:10.1007/978-3-319-63245-2_8
41. Herb M, Schramm M. Functions of ROS in macrophages and antimicrobial immunity. Antioxidants. 2021;10(2):313. doi:10.3390/antiox10020313
42. Lambeth JD, Neish AS. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu Rev Pathol. 2014;9:119–145. doi:10.1146/annurev-pathol-012513-104651
43. Park HS, Jung HY, Park EY, et al. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol. 2004;173(6):3589–3593. doi:10.4049/jimmunol.173.6.3589
44. Hwangbo H, Ji SY, Kim MY, et al. Anti-Inflammatory effect of auranofin on palmitic acid and LPS-induced inflammatory response by modulating TLR4 and NOX4-mediated NF-κB signaling pathway in RAW264.7 macrophages. Int J Mol Sci. 2021;22(11):5920. doi:10.3390/ijms22115920
45. Cheng SC, Scicluna BP, Arts RJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. 2016;17(4):406–413. doi:10.1038/ni.3398
46. Gong Y, Lan H, Yu Z, et al. Blockage of glycolysis by targeting PFKFB3 alleviates sepsis-related acute lung injury via suppressing inflammation and apoptosis of alveolar epithelial cells. Biochem Biophys Res Commun. 2017;491(2):522–529. doi:10.1016/j.bbrc.2017.05.173
47. Bar-Or D, Carrick M, Tanner A, et al. Overcoming the Warburg effect: is it the key to survival in sepsis? J Crit Care. 2018;43:197–201. doi:10.1016/j.jcrc.2017.09.012
48. Voss K, Hong HS, Bader JE, et al. A guide to interrogating immunometabolism. Nat Rev Immunol. 2021;21(10):637–652. doi:10.1038/s41577-021-00529-8
49. Viola A, Munari F, Sánchez-Rodríguez R, et al. The metabolic signature of macrophage responses. Front Immunol. 2019;10:1462. doi:10.3389/fimmu.2019.01462
50. Ip WKE, Hoshi N, Shouval DS, et al. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356(6337):513–519. doi:10.1126/science.Aal3535
51. Palsson-McDermott EM, Curtis AM, Goel G, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab. 2015;21(1):65–80. doi:10.1016/j.cmet.2014.12.005
52. Cao Y, Zhang X, Wang L, et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc Natl Acad Sci USA. 2019;116(27):13394–13403. doi:10.1073/pnas.1821401116
53. Fu-Long L, Jin-Ping L, Ruo-Xuan B, et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat Commun. 2018;9(1):508. doi:10.1038/s41467-018-02950-5
54. Jiang H, Shi H, Sun M, et al. PFKFB3-driven macrophage glycolytic metabolism is a crucial component of innate antiviral defense. J Immunol. 2016;197(7):2880–2890. doi:10.4049/jimmunol.1600474
55. Troha K, Ayres JS. Metabolic adaptations to infections at the organismal level. Trends Immunol. 2020;41(2):113–125. doi:10.1016/j.it.2019.12.001
56. Zheng Z, He M, Zhang X, et al. Enhanced glycolytic metabolism contributes to cardiac dysfunction in polymicrobial sepsis. J Infect Dis. 2017;215(9):1396–1406. doi:10.1093/infdis/jix138
57. Jiean X, Wang L, Yang Q, et al. Deficiency of myeloid Pfkfb3 protects mice from lung edema and cardiac dysfunction in LPS-induced endotoxemia. Front Cardiovasc Med. 2021;8:745810. doi:10.3389/fcvm.2021.745810
58. Wang L, Zhang X, Cao Y, et al. Mice with a specific deficiency of Pfkfb3 in myeloid cells are protected from hypoxia-induced pulmonary hypertension. Br J Pharmacol. 2021;178(5):1055–1072. doi:10.1111/bph.15339
59. Van Wyngene L, Vandewalle J, Libert C, et al. Reprogramming of basic metabolic pathways in microbial sepsis: therapeutic targets at last? EMBO Mol Med. 2018;10(8):e8712. doi:10.15252/emmm.201708712
60. Shi DY, Xie FZ, Zhai C, et al. The role of cellular oxidative stress in regulating glycolysis energy metabolism in hepatoma cells. Mol Cancer. 2009;8:32. doi:10.1186/1476-4598-8-32
61. Pinheiro CH, Silveira LR, Nachbar RT, et al. Regulation of glycolysis and expression of glucose metabolism-related genes by reactive oxygen species in contracting skeletal muscle cells. Free Radic Biol Med. 2010;48(7):953–960. doi:10.1016/j.freeradbiomed.2010.01.016
62. Gupta P, Jagavelu K, Mishra DP. Inhibition of NADPH oxidase-4 potentiates 2-deoxy-D-glucose-induced suppression of glycolysis, migration, and invasion in glioblastoma cells: role of the Akt/HIF1α/HK-2 signaling axis. Antioxid Redox Signal. 2015;23(8):665–681. doi:10.1089/ars.2014.5973
63. Zeng C, Wu Q, Wang J, et al. NOX4 supports glycolysis and promotes glutamine metabolism in non-small cell lung cancer cells. Free Radic Biol Med. 2016;101:236–248. doi:10.1016/j.freeradbiomed.2016.10.500
64. Loor G, Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 2008;15(4):686–690. doi:10.1038/cdd.2008.13
65. Miikkulainen P, Högel H, Rantanen K, et al. HIF prolyl hydroxylase PHD3 regulates translational machinery and glucose metabolism in clear cell renal cell carcinoma. Cancer Metab. 2017;5:5. doi:10.1186/s40170-017-0167-y
66. Fruehauf JP, Meyskens FL. Reactive oxygen species: a breath of life or death?. Clin Cancer Res. 2007;13(3):789–794. doi:10.1158/1078-0432.CCR-06-2082
67. Schumacker PT. SIRT3 controls cancer metabolic reprogramming by regulating ROS and HIF. Cancer Cell. 2011;19(3):299–300. doi:10.1016/j.ccr.2011.03.001
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.