Back to Journals » The Application of Clinical Genetics » Volume 11
Aortic calcification in Gaucher disease: a case report
Authors Alsahli S ![]() , Bubshait DK, Rahbeeni ZA
, Bubshait DK, Rahbeeni ZA ![]() , Alfadhel M
, Alfadhel M ![]()
Received 21 July 2018
Accepted for publication 21 September 2018
Published 17 October 2018 Volume 2018:11 Pages 107—110
DOI https://doi.org/10.2147/TACG.S180995
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Saud Alsahli,1,2 Dalal K Bubshait,3 Zuhair A Rahbeeni,4 Majid Alfadhel1,2
1Medical Genomic Research Department, King Abdullah International Medical Research Center, King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia; 2Division of Genetics, Department of Pediatrics, King Abdulaziz Medical City, Ministry of National Guard-Health Affairs (MNGHA), Riyadh, Saudi Arabia; 3Department of Pediatrics, College of Medicine, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia; 4Department of Medical Genetics, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
Abstract: Gaucher disease is the most common sphingolipid storage disease and is present in all ethnic groups. Its symptoms span all systems including the cardiovascular system. The health care provider should be vigilant regarding this potentially fatal complication. Gaucher disease type IIIC has been linked to causing oculomotor apraxia and cardiac calcification. We report a Saudi girl who developed valvular and aortic calcification in late childhood and died as a result of her cardiovascular complications. This report further strengthens the association and reminds the clinicians that patients with D409H mutation need echocardiographic evaluation annually.
Keywords: Gaucher disease, aortic calcification, D409H
Introduction
Gaucher disease (GD) is the prototypical and the most known among storage diseases, and one of the earliest lysosomal diseases to be described. It is called after Dr Gaucher, a French dermatologist who described a 32-year-old lady with splenic enlargement in 1882. It was not until 1962 when the biochemical abnormalities in GD were unpuzzled. GD is caused by deficient activity of lysosomal β-glucocerebrosidase (encoded by GBA gene), an enzyme that converts glucocerebroside to ceramide and glucose.1 GD is multisystem autosomal recessive disease, and it has been classified into three known clinical phenotypes based on the age of onset, progression, and neurological involvement. GD type I (OMIM #230800) is the nonneuronopathic type, and it is characterized by the presence of bone disease, hepatosplenomegaly, anemia, thrombocytopenia, and, to a lesser extent, pulmonary and renal involvement.2 Neurological symptoms are present in type II and III. GD type II (OMIM #230900) is the acute neuronopathic form, and it is characterized by hepatosplenomegaly and extensive central nervous system damage in infancy. Type III GD (OMIM #231000) has a later onset and less severe course. Patterson et al3 divided type III into type IIIA, which is characterized by myoclonus and dementia, and type IIIB, characterized by early onset of isolated horizontal supranuclear gaze palsy and aggressive systemic disease. Type IIIC (OMIM #231005) has been proposed recently and was discovered in patients harboring the homozygous missense variant (D409H) in GBA gene. They present with cardiac valvular calcifications and neurological manifestations.4 It is believed that other genetic and environmental factors play a role in the final encountered phenotype.5 GD shows poor genotype–phenotype correlation, and it has been found that the phenotype differs even between monozygotic twins.6,7
Case report
The patient was an 11-year-old Saudi girl who was referred to our center from a local hospital at age of 4 years when she was discovered to have hepatosplenomegaly and pancytopenia. She was born at full term to a healthy Saudi mother following an uneventful pregnancy. Her perinatal history as well as developmental history were unremarkable. Family history was significant for consanguinity and a history of an older brother deceased at age of 12 years with GD type III based on molecular and enzymatic testing with a similar clinical picture of his sister. On physical examination, her weight was in the 10th percentile and height was in the 3rd percentile. Her heart rate and blood pressure were within normal range. The liver and spleen were palpable 5 and 7 cm below the costal margin, respectively. Cardiac examination was normal at the time of presentation. Neurological examination was normal apart from oculomotor apraxia. Examinations of other systems were unremarkable. Complete blood count showed pancytopenia as follows: white blood cells: 4×109/L (5–15), hemoglobin: 11.7 g/dL (11.5–13.5), platelets: 106×109/L (140–350). Echocardiography was normal. β-glucocerebrosidase activity was low (0.02 U/g protein). Abdominal ultrasound showed mild enlargement of the liver with a measurement of 13.5 cm and diffuse increased echogenicity. Also, there was moderate splenomegaly, with a splenic measurement of 14.2 cm. She was subsequently diagnosed with GD, which was confirmed by genetic testing that showed a homozygous mutation in the GBA gene (NM_000157.3(GBA):c.1342G>C: p.(Asp448His)). This variant is known as D409H, and we will use the old nomenclature for consistency. The family refused enzyme replacement therapy due to social reasons. Years later, she presented at 11 years of age with a history of syncope after exertion and dyspnea. On examination, her weight and height were below the 3rd percentile. She continued to have hepatosplenomegaly and pancytopenia and has normal neurological status. Complete blood count showed the following values: white blood cells: 9.6×109/L (5–15), hemoglobin: 9.7 g/dL (11.5–15.5), platelets: 109×109/L (140–350). Abdominal ultrasound showed mild hepatomegaly, with a measurement of 15.6 cm, and moderate splenomegaly, with a measurement of 17 cm. There was no focal lesion and normal parenchyma. A repeated echocardiography revealed thickened mitral valve leaflets with severe mitral regurgitation, thickened aortic valve leaflets, fixed right aortic cusp with mild regurgitation, severely dilated left atrium, depressed biventricular systolic function, right ventricular outflow tract obstruction, and peak end-diastolic gradient of 18 mmHg. Cardiac computed tomography scan (Figure 1) showed diffuse thickening of the aortic root extending along the ascending aorta and aortic arch with calcifications. There was minimal fluid surrounding the aorta, thickening of the aortic valve leaflets and mitral valve, and marked enlargement of the left atrium. She was started on antifailure medications without significant improvement, as observed on repeated echocardiography. She was scheduled for surgery, but unfortunately, she deteriorated rapidly and died before the surgery.
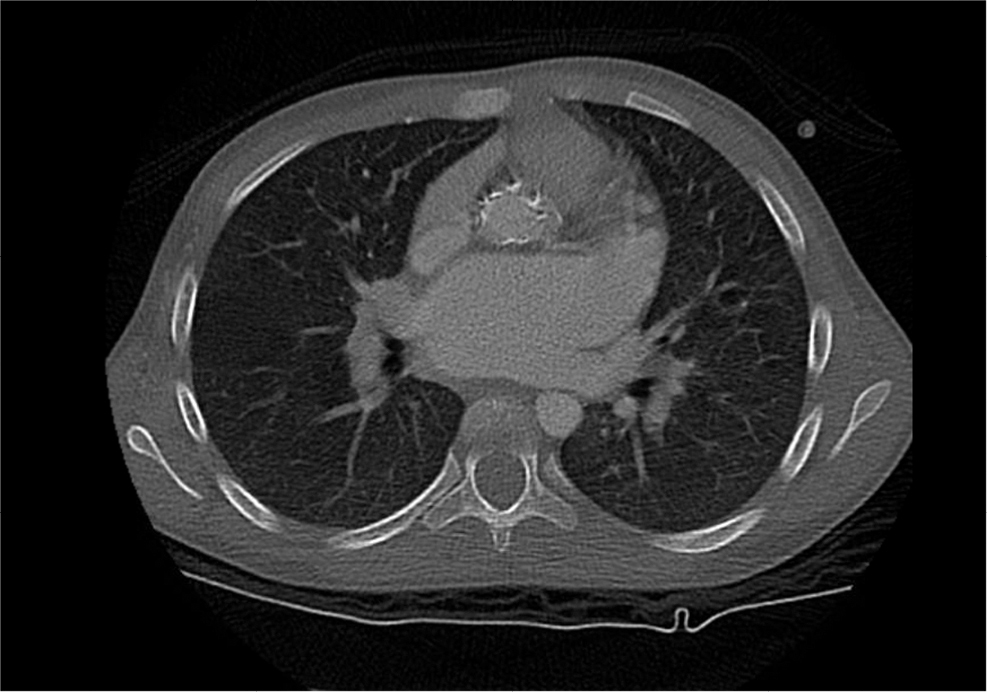 | Figure 1 Cardiac CT scan showing calcification of the aorta. Abbreviation: CT, computed tomography. |
Discussion
We report a case of a Saudi girl who was homozygous for the D409H mutation and developed valvular and aortic calcifications. Aortic and valve calcification in GD has been previously reported in multiple ethnic groups. All the patients reported had the variant D409H, which is known to cause GD type III. This variant has been reported in most ethnic groups with most reports from the Middle East. Casta et al8 was the first to report aortic calcification in GD. He described a 15-year-old boy with diffuse corneal deposits and calcification of ascending aorta and aortic and mitral valves.8 Uyama et al9 reported three Japanese siblings with supranuclear gaze palsies, corneal opacities, communicating hydrocephalus, deformed toes, deafness, calcified aortic valve, and mitral stenosis. Those siblings were homozygous for the D409H mutation.10 Abrahamov et al11 described 12 Arab patients with oculomotor apraxia, calcifications of the aortic or mitral valves, and corneal opacities. All of them were homozygous for the D409H mutation.11 Chabás et al12 has previously shown that three siblings with GD harboring the variant D409H had calcification of the ascending aorta and of the aortic and mitral valves. They also had neurological findings in the form of tonic–clonic seizures in one patient and ophthalmoplegia and saccadic eye movements in two patients.12 Bohlega et al13 reported four siblings with abnormal eye movements and calcification of the ascending aorta, aortic and mitral valves, and aortic root. George et al14 reported a Palestinian boy with a severe mitral and aortic valves thickening and calcification as well as thickening and calcification of the ascending aorta, transverse arch, and isthmus.
Cindik et al15 reported a 14-year-old Turkish girl with hydrocephalus, biatrial and left ventricular dilatation, severe aortic and mitral valve insufficiency, and aortic and mitral valve stenosis and thickening. Talluto and Silverman16 reported a 13-year-old Mexican–American girl with severe aortic valve stenosis and generalized calcification involving the aortic and coronary arteries and aortic and mitral valves. Altunbas et al17 reported widespread calcification in the ascending aorta, left main coronary artery, and carotid arteries in a Turkish patient.17 Saleh et al19 and Abdelwahab et al18 described three Egyptian patients with aortic calcification with severe stenosis and left ventricular hypertrophy. One patient expired due to sudden cardiac death.19 Kör et al20 reported a 15-year-old Turkish boy with aortic and mitral valves thickening with fibrosis, aortic and mitral valve regurgitation, aortic valve stenosis, and widespread calcification involving the ascending and transverse aorta, left main coronary artery, and the left carotid and subclavian artery. The main distinguishing feature of D409H mutation is that it leads to calcification of the aorta and mitral and aortic valves, as illustrated above. How the disease results in aortic and valvular calcification is poorly understood. Since the disease was acquired in almost all cases, it has been proposed that it is caused by valvulitis triggered by deposition of Gaucher cells or by the release of cytokines.13,21 This association between D409H mutation and cardiac calcification necessitates comprehensive cardiac evaluation in patients with GD, particularly those homozygous for the D409H mutation. Some of the reported patients received enzyme replacement therapy that did not seem to stop the calcification, which further supports the need for cardiac surveillance in those patients. We herein recommend annual echocardiogram for patients harboring the mutation D409H as a measure to detect the disease earlier.
Informed consent
The patient’s guardian has provided written informed consent for publication of the case details.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis. 2017;2(1–2):1–71. | ||
Brady RO, Barton NW, Grabowski GA. The role of neurogenetics in Gaucher disease. Arch Neurol. 1993;50(11):1212–1224. | ||
Patterson MC, Horowitz M, Abel RB, et al. Isolated horizontal supranuclear gaze palsy as a marker of severe systemic involvement in Gaucher’s disease. Neurology. 1993;43(10):1993–1997. | ||
Mireles SA, Seybold J, Williams G. Undiagnosed type IIIc Gaucher disease in a child with aortic and mitral valve calcification: perioperative complications after cardiac surgery. J Cardiothorac Vasc Anesth. 2010;24(3):471–474. | ||
Koprivica V, Stone DL, Park JK, et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet. 2000;66(6):1777–1786. | ||
Lachmann RH, Grant IR, Halsall D, Cox TM. Twin pairs showing discordance of phenotype in adult Gaucher’s disease. QJM. 2004;97(4):199–204. | ||
Elstein D, Gellman A, Altarescu G, et al. Disease severity in sibling pairs with type 1 Gaucher disease. J Inherit Metab Dis. 2010;33(1):79–83. | ||
Casta A, Hayden K, Wolf WJ. Calcification of the ascending aorta and aortic and mitral valves in Gaucher’s disease. Am J Cardiol. 1984;54(10):1390–1391. | ||
Uyama E, Takahashi K, Owada M, et al. Hydrocephalus, corneal opacities, deafness, valvular heart disease, deformed toes and leptomeningeal fibrous thickening in adult siblings: a new syndrome associated with beta-glucocerebrosidase deficiency and a mosaic population of storage cells. Acta Neurol Scand. 1992;86(4):407–420. | ||
Uyama E, Uchino M, Ida H, Eto Y, Owada M. D409H/D409H genotype in Gaucher-like disease. J Med Genet. 1997;34(2):175. | ||
Abrahamov A, Elstein D, Gross-Tsur V, et al. Gaucher’s disease variant characterised by progressive calcification of heart valves and unique genotype. Lancet. 1995;346(8981):1000–1003. | ||
Chabás A, Cormand B, Grinberg D, et al. Unusual expression of Gaucher’s disease: cardiovascular calcifications in three sibs homozygous for the D409H mutation. J Med Genet. 1995;32(9):740–742. | ||
Bohlega S, Kambouris M, Shahid M, Al Homsi M, Al Sous W. Gaucher disease with oculomotor apraxia and cardiovascular calcification (Gaucher type IIIC). Neurology. 2000;54(1):261–263. | ||
George R, Mcmahon J, Lytle B, Clark B, Lichtin A. Severe valvular and aortic arch calcification in a patient with Gaucher’s disease homozygous for the D409H mutation. Clin Genet. 2001;59(5):360–363. | ||
Cindik N, Ozcay F, Süren D, et al. Gaucher disease with communicating hydrocephalus and cardiac involvement. Clin Cardiol. 2010;33(1):E26–E30. | ||
Talluto CJ, Silverman NH. Aortic and mitral valve stenosis with regurgitation: not due to rheumatic heart disease. Echocardiography. 2011;28(2):E24–E27. | ||
Altunbas G, Ercan S, Inanç IH, Ozer O, Kervancıoğlu S, Davutoğlu V. Extensive vascular and valvular involvement in Gaucher disease. Asian Cardiovasc Thorac Ann. 2015;23(4):446–448. | ||
Abdelwahab M, Blankenship D, Schiffmann R. Long-term follow-up and sudden unexpected death in Gaucher disease type 3 in Egypt. Neurol Genet. 2016;2(2):e55. | ||
Saleh Y, Almaghraby A, Hammad B, Mokhtar A, Abdel-Hay MA. Gaucher disease causing sudden cardiac death. Egypt Heart J. 2016;68(3):201–204. | ||
Kör Y, Keskin M, Başpınar O. Severe cardiac involvement in Gaucher type IIIC: a case report and review of the literature. Cardiol Young. 2017;27(7):1426–1429. | ||
Veinot JP, Elstein D, Hanania D, Abrahamov A, Srivatsa S, Zimran A. Gaucher’s disease with valve calcification: possible role of Gaucher cells, bone matrix proteins and integrins. Can J Cardiol. 1999;15(2):211–216. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.