Back to Journals » Biologics: Targets and Therapy » Volume 13
Antitumor and antibacterial properties of virally encoded cationic sequences
Authors Colle JH ![]() , Périchon B, Garcia A
, Périchon B, Garcia A ![]()
Received 12 January 2019
Accepted for publication 22 March 2019
Published 25 June 2019 Volume 2019:13 Pages 117—126
DOI https://doi.org/10.2147/BTT.S201287
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Doris Benbrook
Jean-Hervé Colle,1,* Bruno Périchon,2,* Alphonse Garcia1,3
1Laboratoire E3 des Phosphatases-Unité RMN, Institut Pasteur, Paris, France; 2Unité de Biologie des Bactéries pathogènes à Gram-positif, Institut Pasteur, Paris, France; 3Département de Biologie Structurale et Chimie et pôle Dde-Design de la Biologie, Institut Pasteur, Paris, France
*These authors contributed equally to this work
Objective: The objective of this study was to test our Viral Quinta Columna Strategy (VQCS), a new biological hypothesis predicting that specific multifunctional virally encoded cationic domains may have the capacity to penetrate human cells and interact with PP2A proteins to deregulate important human intracellular pathways, and may display LL37 cathelicidin-like antagonistic effects against multiple pathogens such as bacteria or viruses.
Methods: We comparatively analyzed the host defense properties of adenodiaphorins and of some specific cationic sequences encoded by different viruses using two distinct biological models: U87G, a well-characterized cell tumor model; and a group B Streptococcus agalactiae NEM316 ΔdltA, highly sensitive to LL37 cathelicidin.
Results: We found that the adenovirus type 2 E4orf4 is a cell-permeable protein containing a new E4orf464–95 protein transduction domain, named large adenodiaphorin or LadD64–95. Interestingly, the host defense LL37 peptide is the unique cathelicidin in humans. In this context, we also demonstrated that similarly to LL37 LadD64–95, several virally encoded cationic sequences including the C-terminus HIV-1 89.6 Vpr77–92, shorter adenodiaphorins AdD67–84/AdD/69–84/AdD69–83, as well as HIV-2 Tat67–90 and JC polyomavirus small t115–134, displayed similar toxicity against Gram-positive S. agalactiae NEM316 ΔdltA strain. Finally, LadD64–95, adenodiaphorin AdD67–84, AdD69–84, and LL37 and LL17–32 cathelicidin peptides also inhibited the survival of human U87G glioblastoma cells.
Conclusion: In this study, we demonstrated that specific cationic sequences encoded by four different viruses displayed antibacterial activities against S. agalactiae NEM316 ΔdltA strain. In addition, HIV-1 Vpr71–92 and adenovirus 2 E4orf464–95, two cationic penetrating sequences that bind PP2A, inhibited the survival of U87G glioblastoma cells. These results illustrate the host defense properties of virally encoded sequences and could represent an initial step for future complete validation of the VQCS hypothesis.
Keywords: cationic sequences, PP2A, cancer, viruses, bacteria
Introduction
Protein transduction domains (PTDs) and derived cell-penetrating peptides (cpps) are small peptide sequences derived from the few proteins that are naturally able to penetrate cells.1–3 Furthermore, cpps usually contain short sequences with a positive charge resulting from several lysine and arginine residues, and are able both to deliver themselves and to deliver large micromolecules.4 In addition, some cationic anti-microbial peptides (CAMPs), that have some similar physicochemical properties to cpps, can also have cell-penetrating properties, suggesting that they could be highly efficient therapeutic tools.5,6 CAMPs, such as the unique anti-microbial human cathelicidin LL37 peptide, are naturally produced by the innate immune system and mediate a widespread anti-microbial activity against Gram-positive and Gram-negative bacteria such as Staphylococcus aureus and Escherichia coli, and also against fungi and enveloped viruses.7,8 Conversely, Gram-positive bacteria have evolved the ability to increase their positive surface charge through D-alanylation of teichoic acid, thus gaining resistance to CAMPs.9 In addition, both LL37 and its C-terminal fragment LL17–32 (also known as FK16) exhibit cytotoxicity against distinct tumor cells.7,10
The PP2A family of serine/threonine protein phosphatases, a major target for cationic sequences, is critically involved in the regulation of numerous intracellular pathways.11 In addition, various viruses encode specific proteins that interact with PP2A holoenzymes to specifically deregulate the intracellular pathways of their hosts.12 In this regard, two distinct small viral cationic proteins, HIV-1 Vpr and adenovirus type 2 (ad2) E4orf4, interact with a trimeric PP2A holoenzyme ABC, named PP2A1, to specifically induce p53-independent cell death.13,14
The C-terminus sequence of HIV-1 Vpr is a multifunctional domain with cell-penetrating, PP2A-mediated cell death and bactericidal anti-E. coli effects.13,15 Similarly to HIV-1 Vpr77–92, the ad2 E4orf464–95 (LadD64–95) sequence contains residues required for PP2A binding, nuclear localization, and cell death.14,16
The Viral Quinta Columna Strategy (VQCS) is a new biological hypothesis that is based on combinatorial physical and biological properties, including cell penetration, PP2A interaction, and LL37-like host defense effects, that could be mediated by specific virally encoded cationic domains.17 Consistent with this hypothesis, in this study we found that E4orf4 large adenodiaphorin (LadD64–95) penetrating sequence, and cathelicidin active LL37 and LL17–32 peptides inhibit survival of human U87G glioblastoma cells. In addition, similarly to LL37, some virally encoded cationic domains, such as HIV-1 Vpr77–92, LadD64–95, AdD67–84, AdD69–84, AdD69–83, HIV-2 Tat67–90, and polyoma JC small t115–134 antigen, displayed similar toxicities against Streptococcus agalactiae NEM316 ΔdltA strain, a powerful Gram-positive bacterial model.18
Materials and methods
Cells
We used human glioblastoma U87G (kindly provided by Pr Marie Dutreix, Curie Institute, Orsay, France) and dermal human primary fibroblasts (DHF; Tebu-bio:
Peptides
Chemical solid-phase peptide synthesis of 15 NH2-biotinylated peptides (listed in Table 1) was commercially realized by the French Proteogenix company at >95% purity (for profile see website:
 | Table 1 List of peptides |
Kits and reagents
We used streptavidin horseradish peroxidase (HRP) conjugate (Euromedex, Strasbourg, France), 3,3ʹ-diaminobenzidine tetrahydrochloride (DAB) of the DAB Substrate Kit for Peroxidase (Vector Laboratories, Les Ulis, France), cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail Protease Inhibitor Cocktail Tablets (Roche, Meylan, France), and O-phenylenediamine dihydrochloride (OPD) tablets (Sigma Chemical Co, St Louis, MO, USA).
Quantification of peptide internalization
As previously described,11 before incubation, the peptides were pre-incubated for 1 hour with streptavidin–HRP conjugate in a 4/1 ratio. Cells at 80% confluence were incubated with different concentrations of peptide in 24-well plates. After 6 hours, cells were washed twice in PBS, trypsinized (Trypsin EDTA; Invitrogen, Les Ulis, France), harvested in 1 mL of PBS, and counted. Cells were lysed in 300 μL of 0.1 M Tris (pH 7.4) and 0.5% Nonidet P-40 buffer for 10 minutes on ice. A total of 50 μL of cell lysate was mixed with 50 μL of OPD buffer (51.4 mM Na2HPO4 and 24.3 mM citric acid), then mixed with 100 μL of OPD solution (one OPD tablet; Sigma) in 100 mL of OPD buffer to which 40 μL of 30% hydrogen peroxide was added just before use. After 10–20 minutes, the reaction was stopped by adding 100 μL of 1 M HCl, and optical density (OD) was read at 490 nm. The assays were performed in triplicate. We used Gen5 detection software (BioTek, Colmar, France) for data capture and export into Excel, and Microsoft Excel software 2016 for macOS for statistical analyses in histograms with error bars indicating the SD.
Cytotoxicity assays
As previously described,11 3,000 cells were incubated for 24 hours with different concentrations of pharmacological agents. Cell cytotoxicity was analyzed by a colorimetric assay using MTT for adherent cells, as described by the manufacturer (Sigma).
The assays were performed in triplicate. We used Gen5 detection software (BioTek) for data capture and export into Excel. Histograms with error bars indicating the SD were created using Microsoft Excel software.
Bacterial strain and antibacterial susceptibility test
The S. agalactiae mutant NEM 316 ΔdltA strain, which is characterized by a complete absence of D-alanine due to the insertional inactivation of dltA, has been described previously.18
The minimum inhibitory concentrations (MICs) of each peptide were tested in Todd–Hewitt broth (THB) buffered with 50 mM HEPES in 96-well Costar polypropylene microplates (Costar, Cambridge, MA, USA) by a dilution method. Bacteria (106 CFU) were added in triplicate to wells containing increasing concentrations of the anti-microbial peptides. Plates were incubated for 24 hours at 37°C and then read (OD 600 nm) using a microplate reader (Synergy 2; BioTek) for bacterial growth. The MIC90 was considered to be the peptide concentration that inhibited 90% of growth.
Western blot analyses
As previously described,11 exponentially growing cells (105 cells) were seeded overnight in 24-well culture cell plates, in a sub-confluent monolayer, prior to pharmacological treatments. For preparation of the extract, cells were rinsed in cold PBS, scraped, pelletized, and lysed in RIPA buffer 89900 supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (78442; Thermo Scientific, Les Ulis, France), according to the manufacturer's instructions, and finally sonicated for 2 minutes at 50% pulse.
The protein concentration in each sample extract was quantified using a Bio-Rad Protein Assay (Bio-Rad Laboratories, Les Ulis, France). Lanes were loaded with the material corresponding to 20–40 μg of cell protein extract. The following primary antibodies were used: anti-phospho-AKT (Ser 473) (D9E) and anti-AKT (pan) (C67E7) (Cell Signaling Technology, Saint-Cyr-l'École, France), and anti-HP1γ (2MOD-1G6) (Euromedex). Goat peroxidase-labeled anti-rabbit IgG or horse peroxidase-labeled anti-mouse IgG antibodies (Vector Laboratories) were used as secondary antibodies. Immunoreactivities were visualized using Pierce ECL Western Blotting Substrate and the myECL Imager (Thermo Scientific) and ImageJ 1.45s software (National Institutes of Health, USA,
Results
LadD, a PTD encoded by the human ad2 E4orf4 protein, inhibits the phosphatidylinositol-3 (PI3)-kinase-dependent survival pathway of human U87G glioblastoma cells
The ad2 E4orf466–74 residues contain an arginine/lysine-rich motif (RAKRRDRRR) located inside the multifunctional E4orf464–95 domain that is involved in nuclear localization,16 and is partially homologous to the HIV-1 TAT47–57 cell-penetrating sequence (Tat cpp) (Table 2). This observation suggests that ad2 E4orf4 may be a new cell-penetrating protein. Consistent with this hypothesis, the use of chemical synthesis of peptides containing biotinylated ad2 E4orf4 sequences (detailed sequences shown in Table 2) indicated that the full-length wild-type E4orf41–114 polypeptide, the E4orf464–95 containing the E4orf466–74 cationic stretch, named large adenodiaphorin (LadD or LadD64–95), and the shorter E4orf464–78 sequence, named adenodiaphorin (adD or AdD64–78), can deliver streptavidin–HRP into U87G glioblastoma cells with similar cargo efficiencies to the HIV-1 Tat peptide (Figure 1, upper panel). Mutations either resulting from the deletion of the E4orf464–95 domain (Eorf4Δ64–95) or involving (R→A) substitution within the LadD64–95 sequence (LadDmut) or within the shorter adenodiaphorin adD64–78 (adDmut) sequence suppressed this cargo effect.
 | Table 2 Sequence of N-terminus biotinylated peptides used in this study |
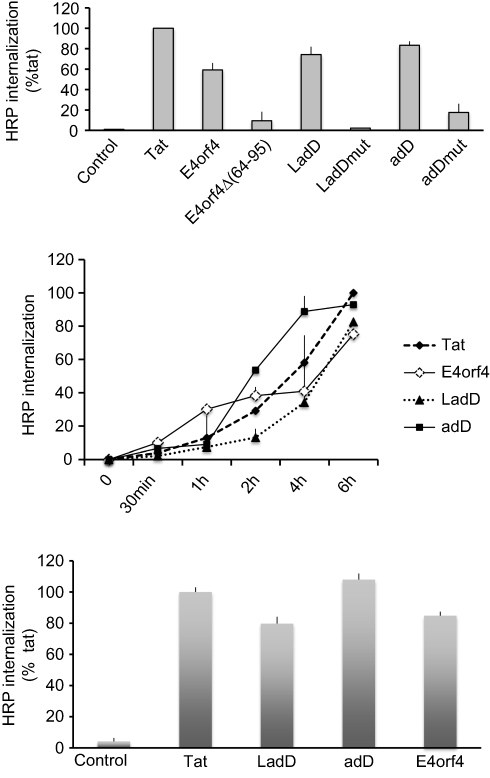 | Figure 1 Effects of human adenovirus type 2 E4orf4 peptides on intracellular delivery of streptavidin–peroxidase in human U87G glioblastoma cells and in human DHF cells. Streptavidin–peroxidase coupled with 125 nM of biotinylated peptides was incubated at 37°C for 6 hours (upper panel) or 0–6 hours (middle panel) with U87G, and for 6 hours (lower panel) with DHF cells. Internalized complexes were visualized by a colorimetric test, OPD, as described previously.11,13 HRP internalization of E4orf4 peptides is expressed as % of Tat-mediated HRP peptide (incubated for 6 hours) used as positive control. SD is shown for n=3. For negative control (Control), no peptide, no HRP, HRP alone, or cargo-inactive DPT-sh1 peptide (VKKKIKREIKI) was used, giving similar results. 6.88±0.96 ng and 4.29±0.82 ng of HRP, respectively, were internalized by 105 U87G (upper and middle panels) and by 105 DHF (lower panel) cells following 6-hour incubation with 125 ng of biotinylated-Tat peptide complexed with streptavidin–peroxidase.Abbreviations: adD, adenodiaphorin; DHF, dermal human fibroblast; HRP, horseradish peroxidase; LadD, large adenodiaphorin; OPD, O-phenylenediamine dihydrochloride. |
In addition, both LadD and adD adenodiaphorin sequences displayed Tat-like kinetic cargo delivery properties (Figure 1, middle panel). Finally, as illustrated in Figure 1 (lower panel), E4orf4 as well as LadD and adD adenodiaphorin sequences also displayed similar Tat cargo delivery in DHF cells.
Furthermore, the cytotoxicity of LadD64–95 and adD64–78 penetrating peptides was investigated in U87G cells by the MTT assay. As shown in Figure 2 (upper panel), treatment of U87G cells with increasing amounts of LadD64–95 for 24 hours resulted in a dose-dependent reduction in cell viability of the U87G glioblastoma cells. In contrast, no significant toxicity was detected in the presence of adD peptide. In addition, and in contrast to adD64–78, LadD64–95 inhibited AKT phosphorylation in U87G glioblastoma cells (Figure 2, lower panel). The two short penetrating sequences, AdD64–78 and Tat47–57 cpps, containing homologous nuclear localization signals,16,19 respectively, RAKRRDRRR in adenovirus E4orf4 adD and RKKRRQRRR in HIV-1 Tat (single-letter amino acid code; basic residues are highlighted in bold type), displayed similar biological properties, such as cell penetration (Figure 1) without toxicity (Figure 2). In contrast, the larger adenodiaphorin LadD64–95, containing the previously described PP2A binding sequence required for cell toxicity,14 inhibited survival of U87G cells. Finally, as shown in Figure 1 (upper panel), we confirmed that the suppression of cationic properties resulting from the substitution of R with A in LadD (LadDmut) and adD (adDmut) also ablated cell penetration. Since we have previously demonstrated that both Akt basal constitutive activity and U87G survival are downregulated by specific PI3K/Akt pharmacological inhibitors,20 these results indicate that LadD64–95, but not adD64–78, inhibited the constitutively active PI3K/Akt pathway required for the survival of U87G cells. In addition, although wild-type LadD and adD adenodiaphorin sequences have similar penetrating properties in U87G and in non-transformed DHFs (Figure 1, upper and lower panels), no toxicity was detected in DHF cells (not shown) where, in contrast to U87G, the PI3K/Akt survival pathway is not constitutively activated.20
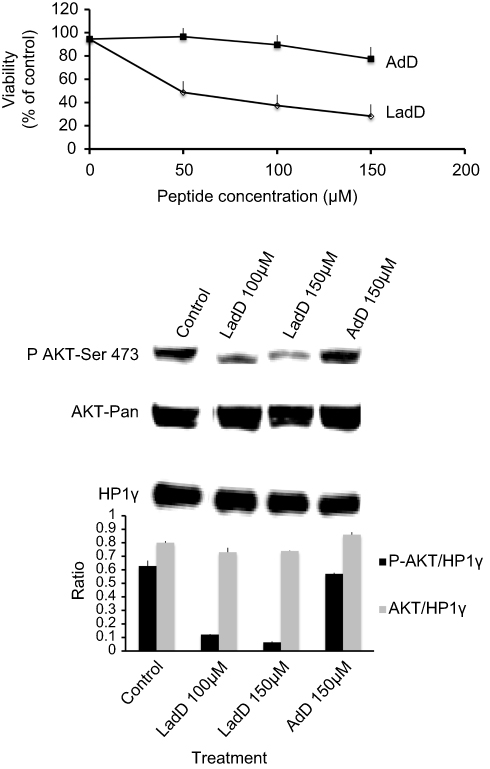 | Figure 2 LadD inhibits a constitutively active PI3K/Akt survival pathway in U87G cells. Upper panel: cells were treated for 24 hours with LadD and adD peptides (0–150 μM), and cell viability was assessed by the MTT test (n=3). The lower panel shows a Western blot probed with the antibody specifically recognizing phosphorylated Akt-pSer473 (D9E rabbit mAb from cell signaling). The same blot was reprobed with the antibody specifically recognizing total Akt (C67E rabbit mAb from cell signaling) and with the antibody specifically recognizing HP1γ (2MOD-1G6 mouse mAb from Euromedex) that was used as loading control. Cells were untreated (control) or treated for 5 hours at 37°C with 100 μM or 150 μM of LadD, or with 150 μM of AdD peptides. The Western blot was quantified with ImageJ software, and AKT/HP1γ and pAKT/HP1γ normalized ratios, corresponding to the quotient of AKT or pAKT versus HP1γ expression, are illustrated in a histogram shown in the lower panel (n=3). Abbreviations: adD, adenodiaphorin; DHF, dermal human fibroblast; LadD, large adenodiaphorin; mAb, monoclonal antibody. |
Comparative analyses of the effects of human cathelicidin LL37 and several virally encoded peptides on growth of D-alanylated mutants of human pathogen group B S. agalactiae and on the survival of human U87G glioblastoma cells
In addition to HIV-1 Tat cpp, the arginine/lysine-rich motif (RAKRRDRRRR) localized in LadD64–95 is also partially homologous to virally encoded arginine/lysine-rich motifs deduced from HIV-1 Vpr, HIV-2 Tat, and JC polyomavirus small t proteins, suggesting that the derived viral sequences may display common properties. In this regard, as mentioned in the Introduction, these virally encoded cationic sequences have similar physical characteristics to the anti-microbial LL37 molecule, suggesting that these sequences may behave as cathelicidin-like host defense molecules.17 Therefore, to test this hypothesis, we comparatively investigated the anti-microbial activities of these viral peptides with the two human LL-37 and LL17–32 molecules by monitoring the bacterial growth of S. agalactiae NEM 316 ΔdltA strain, a Gram-positive bacterial model that is highly sensitive to human LL37 cathelicidin.18
As shown in Table 3 (column 4), the MIC90 values of LL37, HIV-1 Vpr, adenodiaphorins, and JC polyomavirus small t115–134 lie in a similar range (12.5–25 μM). The C-terminal LL17–32 fragment is more efficient than the full-length LL37 (MIC90 12.5 μM and 6.25 μM, respectively). In addition, and surprisingly, HIV-2 Tat67–90 is slightly more active than LL17–32 (MIC90 3.1 μM and 6.25 μM, respectively). As expected, no antibacterial activity was observed with Tat47–57 cpp, used as a negative control.
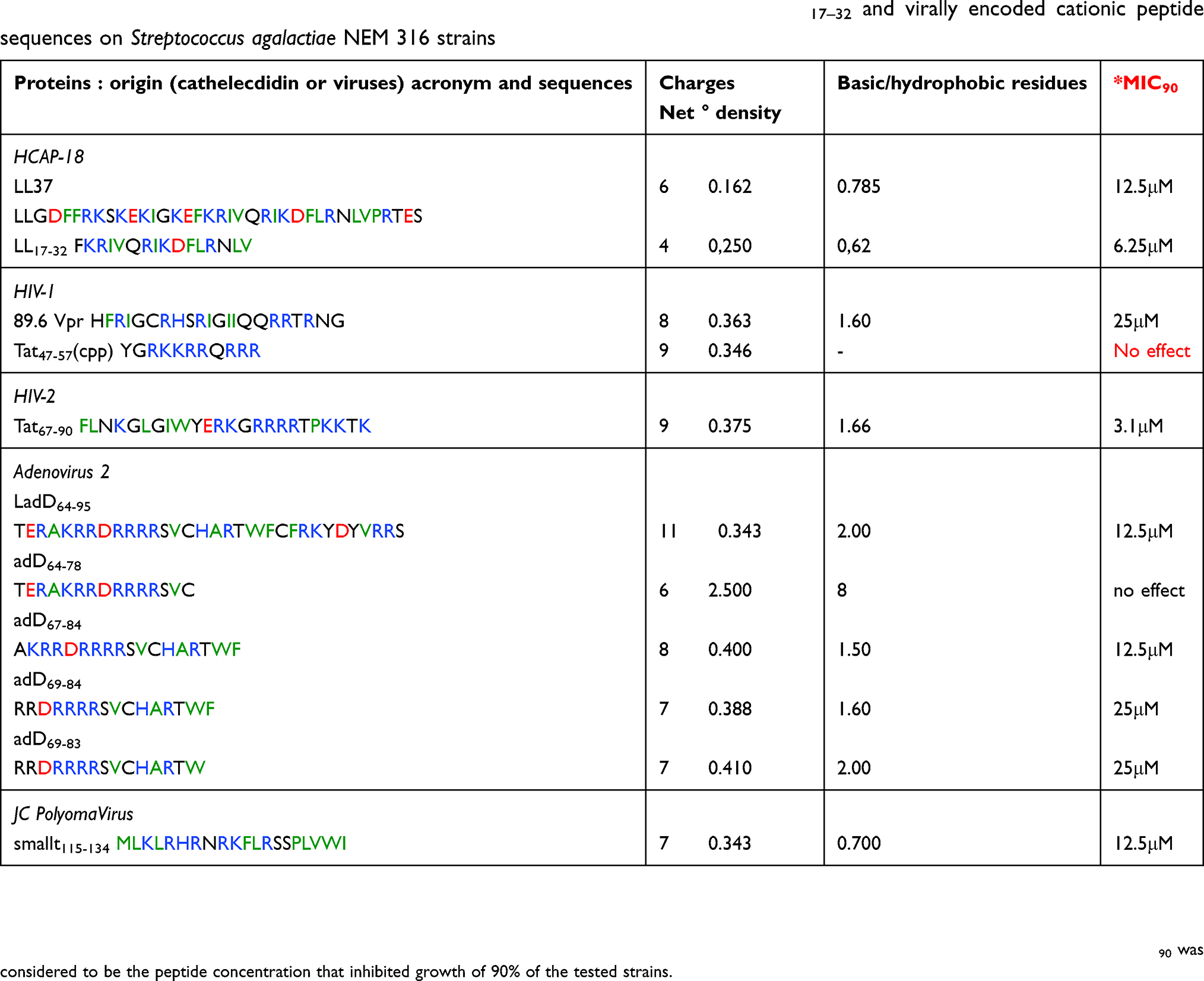 | Table 3 Physical characteristics and antibacterial effects of cathelicidins LL37 and LL17–32 and virally encoded cationic peptide sequences on Streptococcus agalactiae NEM 316 strains |
We also comparatively investigated the effect of human cathelicidin LL37 and virally encoded cationic peptides on survival of human U87G glioblastoma cells. We performed the MTT assay to monitor cytotoxicity of full-length LL-37 and C-terminal LL17–32, also named FK16,21 peptides in the U87G glioblastoma cell line. As shown in Figure 3 (upper panel), consistent with the previously described toxic effect in colon cancer cells,21 both LL37 and LL17–32 peptides provoke a similar and important reduction in the viability of U87G cells in a dose-dependent manner. Furthermore, as illustrated in Figure 3 (lower panel), we observed a dose-dependent reduction in U87G cell viability with adD67–84 and adD69–84, but not adD69–83 (F84 deleted mutant). In addition, no significant toxicity was observed with HIV-2 Tat67–90 and JC polyomavirus small t115–134 treatments.
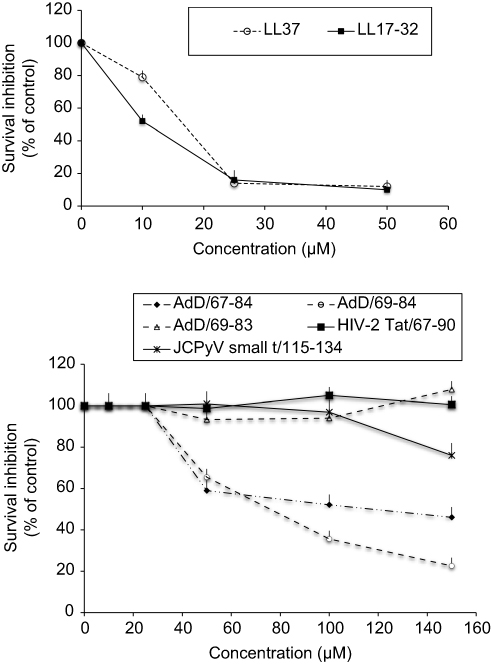 | Figure 3 Effect of cathelicidin LL-37/LL17–32, adenodiaphorin67–84/69–84/69–83/deletion mutants, HIV-2 Tat67–90, and JC polyomavirus small t115–134 sequences, on viability of U87G glioblastoma cells. U87G cells were treated for 24 hours with the LL37 and LL17–32 cathelicidin peptides (0–50 μM) (upper panel) or with virally encoded LadD deletion mutants (AdD67–84, AdD69–84, and AdD69–83), HIV-2 Tat67–90, and JC polyomavirus small t115–134 cationic peptides (0–150 μM) (lower panel). Cell viability was assessed by the MTT test (n=3). Abbreviations: adD, adenodiaphorin; LadD, large adenodiaphorin. |
Discussion
The VQCS model predicts that multifunctional specific virally encoded cationic sequences may have the capacity to penetrate cells and to deregulate important human intracellular signaling pathways, such as PP2A-mediated pathways, but may also display LL37 cathelocidin-like antagonistic effects against multiple pathogens such as bacteria or viruses.17 In this study, following the identification of ad2 E4orf4 cationic penetrating sequences, named adenodiaphorins, we tested the VQCS hypothesis by comparatively analyzing host defense properties of adenodiaphorins and some specific cationic sequences encoded by different viruses using two distinct biological models: U87G, a well-characterized cell tumor model; and a group B S. agalactiae NEM316 ΔdltA highly sensitive to LL37 cathelicidin.
Host defense properties of adenovirus E4orf4 adenodiaphorin penetrating sequences
Adenoviruses are non-enveloped double-stranded DNA viruses that can infect human tissues to provoke mild gastrointestinal and respiratory symptoms, and are often associated with pediatric patients.22 The common species C adenoviruses (serotypes Ad1, Ad2, Ad5, and Ad6) infect more than 80% of the human population early in life and they also form latent infections in human lymphocytes.23 In this study, we found that the full-length Ad2 E4orf41–114 sequence is a new cell-penetrating polypeptide and we demonstrated that the E4orf464–95/LadD64–95 sequence, previously characterized as a multifunctional PP2A-binding domain and as a PP2A-dependent death sequence, is also a new PTD. In addition, we analyzed the potential host defense properties of the anti-microbial LL-37 and LL17–32 cathelicidin peptides with specific virally encoded cationic peptides, including E4orf4 adenodiaphorin sequences, against both glioblastoma cells and S. agalactiae NEM 316 ΔdltA strain.
Using U87G glioblastoma cells, we demonstrated that LL-37, the only cathelicidin found in humans, and its shorter active fragment LL17–32, are potent inhibitors of U87G glioblastoma cell survival. It has been reported that LL37 can mediate a dual role in tumorigenesis. First, as a positive factor, LL37 can promote the growth of ovarian,24 lung,25 and breast cancers.26 Second, LL37 can promote tumor suppression in gastric cancer,27 acute myeloid leukemia,28 and lymphocytic leukemia.29 Since U87G is a highly radio-resistant glioblastoma cell line, our results suggest that LL17–32 may be beneficial in the treatment of glioblastomas. Previous work established that PP2A1 inactivates Akt and PP2A1 inhibition activates tumor survival pathways associated with cancer progression.30 Furthermore, we have previously reported that two PP2A activators, the immunosuppressant FTY72020 and the peptide-mimetic DPT-Cog,31 downregulated a constitutively active PI3K–Akt tumor survival pathway controlled by PP2A in radio-resistant U87G glioblastoma cells. The ad2 E4orf4 protein interacts with the regulatory Bα-subunit of PP2A1 to specifically induce p53-independent death of human cancer cells.14,32 In addition, the E4orf464–95 domain (here renamed LadD64–95) is involved in cell penetration, PP2A and Src binding, nuclear localization, and cell death mediated by the viral E4orf4 protein.14,16,32–34 In this regard, we found in this study that LadD inhibits the PI3K-dependent pathway required for survival of U87G cells. We also identified the shortest adenodiaphorin, adD69–84, that inhibits U87G survival. The F84 deletion in adD69–83, critically involved in PP2A1 binding,14 stops the inhibition of U87G cell survival mediated by the adD69–84 RRDRRRRSVCHARTWF sequence. In contrast to LL37 and LadD69–84, no toxicity was detected in the presence of HIV-2 Tat67–90 and JC polyomavirus small t115–134 cationic peptides.
Using S. agalactiae NEM 316 ΔdltA strain, in agreement with previous work,18 we found that LL17–32 displayed better antibacterial activity (MIC=6.25 μM) than LL37 (MIC=12.5 μM). These data clearly suggest that similarly to LL37, some virally encoded adenodiaphorin sequences could act as endogenous host defense peptides. In addition, we identify adD69–84 as the shortest dual antitumor and antibacterial active adenodiaphorin. Our data are also consistent with a regulatory model based on two distinct host defense mechanisms mediated by adenodiaphorins. First, similarly to full-length E4orf4,14,32 adenodiaphorins could kill tumor cells by interacting with the PP2A1 target. Second, similarly to many anti-microbial cationic peptides, adenodiaphorins could kill their bacterial targets after interaction with anionic components of the bacterial membrane.35 In agreement with this hypothetical model, in adD69–83 the deletion of residue F84, required for both E4orf4-mediated PP2A binding and tumor cell death,14 prevents U87G cell death induced by AdD69–84 but retains the antibacterial effect. adD69–84 and adD69–83 have the same net charge of +7, suggesting that, similarly to LL37 and other CAMPs, adenodiaphorins could kill their bacterial targets by disrupting membrane integrity.35 Together, these results suggest that, consistent with HIV-1 Vpr's biological effects, PP2A intracellular interaction in human cells and LL37-like membrane disruption in bacteria may represent a general property shared by some virally encoded sequences, including HIV-1 Vpr and adenodiaphorin molecules.
Antibacterial properties of HIV-1 Vpr, HIV-2 Tat67–90, and JC polyomavirus small t115-13-90 sequences
We have previously established that the cell-penetrating C-terminal domain of HIV-1 Vpr 89.6 isolate can interact with the structural A subunit of PP2A1 to induce cell death.13 In addition, antibacterial effects of C-terminal HIV-1 Vpr against E. coli have been previously reported.15 Here, we found an anti-Gram-positive bacterial effect of the HIV-1 Vpr C-terminal sequence (Vpr71–92) against S. agalactiae NEM 316 ΔdltA strain. In addition, consistent with our VQCS hypothesis, we found an antibacterial effect of HIV-2 Tat67–90 and JC polyomavirus small t115-134 cationic sequences against the same strain.
Conclusion
In this study, we found that ad2 E4orf4 is a cell-permeable protein containing a new E4orf464–95 PTD. We also demonstrated that, similarly to the unique human cathelicidin LL37 host defense peptide, LadD64–95 and several virally encoded cationic sequences, including the C-terminus HIV-1 89.6 Vpr77–92, shorter adenodiaphorins AdD67–84/AdD69–84/AdD69–83, and HIV-2 Tat67–90 and JC polyomavirus small t 115-134, displayed similar toxicity against Gram-positive S. agalactiae NEM316 ΔdltA strain. Finally, LadD64–95, AdD67–84, AdD69–84, and cathelicidin LL37 and LL17–32 peptides, also inhibit the survival of human U87G glioblastoma cells. HIV-1 Vpr peptides were previously identified in serum and in the cerebrospinal fluid of HIV-1-infected individuals.36,37 In addition, given that E4orf4 protein can be detected late in the infectious cycle,34 E4orf4 sequences may, similarly to HIV-1 Vpr, be liberated after the lysing of infected cells and circulate in biological fluids. Together, in agreement with the potential infective effects predicted by the VQCS hypothesis, our results suggest that the presence of virally encoded cationic peptides, such as adenodiaphorins and HIV-1 Vpr peptides, which could circulate in biological fluids, may define a new paradigm for a potential virally mediated innate immunity. In addition, it is noteworthy that anti-biofilm effects and wound-healing properties of LL-37 have already been shown, suggesting that LL37, or its derivatives, could be used to develop new therapeutic strategies against biofilm-mediated infections to treat polymicrobially infected wounds through topical application.38,39 Since the results in Table 3 clearly indicate that viral peptides work in the same LL37 concentration range, in accordance with the VQCS hypothesis,17 we can postulate that some peptides containing virally encoded sequences may behave as LL37 derivatives and may be used against infected wounds.
Our results represent the first experimental data consistent with the VQCS hypothesis. Furthermore, in conjunction with future work involving other viruses, microbes, and parasites, the mimicry of host defense peptides of viral origin may represent a promising approach to design new therapeutic molecules with anti-infective and antitumor effects, as previously suggested with cellular host defense sequences.40
Acknowledgments
The present study was supported by Institut Pasteur. The authors thank Patrick Trieu Cuot for providing S. agalactiae ΔdltA strain.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi:10.1016/0092-8674(88)90263-2
2. Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450.
3. Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi:10.1126/science.285.5433.1569
4. Pooga M, Langel U. Synthesis of cell-penetrating peptides for cargo delivery. Methods Mol Biol. 2005;298:77–89.
5. Rodriguez Plaza JG, Morales-Nava R, Diener C, et al. Cell penetrating peptides and cationic antibacterial peptides: two sides of the same coins. J Biol Chem. 2014;289(21):14448–144457. doi:10.1074/jbc.M113.515023
6. Ruchi O, Arpita Y. The remarkable cationic peptides: a boon to pharmaceutical sciences? J Pharm Pharm Sci. 2018;21(1):60–72. doi:10.18433/jpps29455
7. Murakami M, Ohtake T, Dorschner RA, Schittek B, Garbe C, Gall RL. Cathelicidin anti-microbial peptide expression in sweat, an innate defense system for the skin. J Invest Dermatol. 2002;119:1090–1095. doi:10.1046/j.1523-1747.2002.19507.x
8. Kim JE, Kim BJ, Jeong MS, et al. Expression and modulation of LL-37 in normal human keratinocytes, HaCaT cells, and inflammatory skin diseases. J Korean Med Sci. 2005;20:649–654. doi:10.3346/jkms.2005.20.4.649
9. Weidenmaier C, Peschel A. Teichoic acids and related cell-wall glycopolymers in gram-positive physiology and host interactions. Nat Rev Microbiol. 2008;6:276–287. doi:10.1038/nrmicro1861
10. Wu WK, Wang G, Coffelt SB, et al. Emerging roles of the host defense peptide LL-37 in human cancer and its potential therapeutic applications. Int J Cancer. 2010;127:1741–1747. doi:10.1002/ijc.25499
11. Guergnon J, Dessauge F, Dominguez V, et al. Use of penetrating peptides interacting with PP1/PP2A proteins as a basis for a new drug phosphatase technology. Mol Pharmacol. 2006;69:1115–1124. doi:10.1124/mol.105.019364
12. Guergnon J, Godet AN, Galioot A, et al. PP2A targeting by viral proteins: a widespread biological strategy from DNA/RNA tumor viruses to HIV-1. Bba. 2011;1812:1498–1507.
13. Godet AN, Guergnon J, Croset A, et al. PP2A1 binding, cell transducing and apoptotic properties of Vpr77-92 a new functional domain of HIV-1 Vpr proteins. PLoS One. 2010;5:e13760. doi:10.1371/journal.pone.0013760
14. Marcellus RC, Chan H, Paquette D, Thirlwell S, Boivin D, Branton PE. Induction of p53-independent apoptosis by the adenovirus E4orf4 protein requires binding to the balpha subunit of protein phosphatase 2A. J Virol. 2000;74:7869–7877. doi:10.1128/JVI.74.17.7869-7877.2000
15. Faill P, Castelli LA, Azad AA, Macreadie IG. Bactericidal properties of HIV-1 Vpr C terminal sequences. Protein Pept Let. 1997;4:383–390.
16. Miron M, Gallouzi IE, Lavoie JN, Branton PE. Nuclear localization of the adenovirus E4orf4 protein is mediated through an arginine-rich motif and correlates with cell death. Oncogene. 2004;23:7458–7468. doi:10.1038/sj.onc.1207444
17. Garcia A. The viral quinta columna strategy: a new biological hypothesis to study infections in humans. Med Hypotheses. 2018;113:9–12. doi:10.1016/j.mehy.2018.02.007
18. Saar-Dover R, Bitler A, Nezer R, et al. D-alanylation of lipoteichoic acids confers resistance to cationic peptides in group B streptococcus by increasing the cell wall density. PLoS Pathog. 2012;8:e1002891. doi:10.1371/journal.ppat.1002891
19. Ruben S, Perkins A, Purcell R, et al. Structural and functional characterization of human immunodeficiency virus tat protein. J Virol. 1989;63:1–8.
20. Colle JH, Falanga PB, David-Watine B, Dutreix M, Garcia A. FTY720 overcomes resistance of human U87G glioma cells expressing irradiation-induced SA-β-beta-gal biomarker. Curr Top Pharmacol. 2015;19:13–19.
21. Ren SX, Shen J, Cheng ASL, et al. FK-16 Derived from the anticancer peptide LL-37 Induces caspase-independent apoptosis and autophagic cell death in colon cancer cells. PLoS One. 2013;8:e63641. doi:10.1371/journal.pone.0063641
22. Ghebremedhin B. Human adenovirus: viral pathogen with increasing importance. Eur J Microbiol Immunol. 2014;4:26–33. doi:10.1556/EuJMI.4.2014.1.2
23. Garnett CT, Erdman D, Xu W, Gooding LR. Prevalence and quantitation of species C adenovirus DNA in human mucosal lymphocytes. J Virol. 2002;76:10608–10616. doi:10.1128/JVI.76.21.10608-10616.2002
24. Coffelt SB, Waterman RS, Florez L, et al. Ovarian cancers overexpress the antimicrobial protein hCAP-18 and its derivative LL-37 increases ovarian cancer cell proliferation and invasion. Int J Cancer. 2008;122:1030–1039. doi:10.1002/ijc.23186
25. von Haussen J, Koczulla R, Shaykhiev R, et al. The host defence peptide LL-37/hCAP-18 is a growth factor for lung cancer cells. Lung Cancer. 2008;59:12–23. doi:10.1016/j.lungcan.2007.07.014
26. Heilborn JD, Nilsson MF, Jimenez CI, et al. Antimicrobial protein hCAP18/LL-37 is highly expressed in breast cancer and is a putative growth factor for epithelial cells. Int J Cancer. 2005;114:713–719. doi:10.1002/ijc.20795
27. Wu WK, Sung JJ, To KF, et al. The host defense peptide LL-37 activates the tumor-suppressing bone morphogenetic protein signaling via inhibition of proteasome in gastric cancer cells. J Cell Physiol. 2010;223:178–186.
28. An LL, Ma XT, Yang YH, Lin YM, Song YH, Wu KF. Marked reduction of LL-37/hCAP-18, an antimicrobial peptide, in patients with acute myeloid leukemia. Int J Hematol. 2005;81:45–47. doi:10.1532/IJH97.A10407
29. Yang YH, Zheng GG, Li G, Zhang B, Song YH, Wu KF. Expression of LL-37/hCAP-18 gene in human leukemia cells. Leuk Res. 2003;27:947–950. doi:10.1016/S0145-2126(03)00020-1
30. Resjö S, Göransson O, Härndahl L, Zolnierowicz S, Manganiello V, Degerman E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal. 2002;14:231–238. doi:10.1016/S0898-6568(01)00238-8
31. Colle JH, Garcia A. The new APOE analog DPT-Cog inhibits PI3k/Akt-dependant survival of human radio-resistant U87G glioblastoma cells. Curr Top Pharmacol. 2016;20:33–37.
32. Shtrichman R, Sharf R, Barr H, Dobner T, Kleinberger T. Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc Natl Acad Sci USA. 1999;96:10080–10085. doi:10.1073/pnas.96.18.10080
33. Branton PE, Roopchand DE. The role of adenovirus E4orf4 protein in viral replication and cell killing. Oncogene. 2001;20:7855–7865. doi:10.1038/sj.onc.1204862
34. Champagne C, Landry MC, Gingras MC, Lavoie JN. Activation of adenovirus type 2 early region 4 ORF4 cytoplasmic death function by direct binding to Src kinase domain. J Biol Chem. 2004;279:25905–25915. doi:10.1074/jbc.M400933200
35. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi:10.1038/415389a
36. Levy DN, Refaeli Y, MacGregor RR, Weiner DB. Serum Vpr regulates productive infection and latency of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1994;91:10873–10877. doi:10.1073/pnas.91.23.10873
37. Hoshino S, Su B, Konishi M, et al. Vpr in plasma of HIV type 1-positive patients is correlated with the HIV type 1 RNA titers. AIDS Res Hum Retroviruses. 2007;2:391–397. doi:10.1089/aid.2006.0124
38. Carretero M, Escamez MJ, Garcia M, et al. In vitro and in vivo wound healing-promoting activities of human cathelicidin LL-37. J Invest Dermatol. 2008;128:223–236. doi:10.1038/sj.jid.5701043
39. Duplantier AJ, van Hoek ML. The human cathelicidin antimicrobial peptide LL-37 as a potential treatment for polymicrobial infected wounds. Front Immunol. 2013;4:143. doi:10.3389/fimmu.2013.00143
40. Fjell CD, Hiss JA, Hancock REW, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov. 2012;11:37–51. doi:10.1038/nrd3591
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.