")
Back to Journals » International Journal of Nanomedicine » Volume 14
Antitumor activity of the bioreductive prodrug 3-(2-nitrophenyl) propionic acid-paclitaxel nanoparticles (NPPA-PTX NPs) on MDA-MB-231 cells: in vitro and in vivo
Authors Duan X, Yao X, Zhang S, Xu M, Hao Y, Li Z, Zheng X, Liu M, Li Z, Li H, Wang J, Feng Z, Zhang X
Received 5 September 2018
Accepted for publication 27 November 2018
Published 24 December 2018 Volume 2019:14 Pages 195—204
DOI https://doi.org/10.2147/IJN.S186556
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Xiao-Chuan Duan,1,2,* Xin Yao,1,2,* Shuang Zhang,1,2 Mei-Qi Xu,1,2 Yan-Li Hao,1,2 Zhan-Tao Li,1,2 Xiu-Chai Zheng,2 Man Liu,1,2 Zhuo-Yue Li,1,2 Hui Li,1 Jing-Ru Wang,1,2 Zhen-Han Feng,1 Xuan Zhang1,2
1Beijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery Systems, School of Pharmaceutical Sciences, Peking University, Beijing 100191, People’s Republic of China; 2Department of Pharmaceutics, School of Pharmaceutical Sciences, Peking University, Beijing 100191, People’s Republic of China
*These authors contributed equally to this work
Background: 3-(2-Nitrophenyl) propionic acid-paclitaxel (NPPA-PTX) is a paclitaxel (PTX) bioreductive prodrug synthesized by our lab. We hypothesize that NPPA-PTX can self-assemble to form nanoparticles (NPs).
Materials and methods: In the present research, the theoretical partition coefficient (XlogP) and Hansen solubility parameters of NPPA-PTX were calculated. NPPA-PTX nanoparticles prepared by NPPA-PTX and DSPE-PEG (NPPA-PTX:DSPE-PEG =1:0.1, w/w) (NPPA-PTX@PEG NPs) were prepared and characterized. The cellular uptake, in vitro antitumor activity, in vivo targeting effect, tumor distribution, in vivo antitumor activity, and safety of NPPA-PTX@PEG NPs were investigated.
Results: Our results indicate that NPPA-PTX can self-assemble to form NPPA-PTX@PEG NPs. Both the cellular uptake and safety of NPPA-PTX@PEG NPs were higher than those of Taxol. NPPA-PTX@PEG NPs could target tumor tissues by a passive targeting effect. In tumor tissues, NPPA-PTX@PEG NPs could completely transform into active PTX. The in vivo antitumor activity of NPPA-PTX@PEG NPs was confirmed in MDA-MB-231 tumor-bearing nude mice.
Conclusion: The bioreductive prodrug NPPA-PTX could self-assemble to form NPs. The safety and antitumor activity of NPPA-PTX@PEG were confirmed in our in vitro and in vivo experiments. The NPPA-PTX@PEG NPs developed in this study could offer a new way of preparing bioreductive prodrug, self-assembled NPs suitable for antitumor therapy.
Keywords: bioreductive prodrug, 3-(2-nitrophenyl) propionic acid-paclitaxel, nanoparticles, antitumor activity, in vitro, in vivo
Introduction
Hypoxia is a common feature of solid tumors.1 One of the therapeutic strategies for targeting tumor hypoxia is to use bioreductive prodrugs, which can be activated by enzymatic reduction in hypoxic tumor tissues.2 Bioreactive prodrugs can respond to the hypoxic microenvironment of tumors by producing the parent drug to implement antitumor activity. Until now, five types of bioreactive prodrugs have been shown to be capable of selectively targeting hypoxic cells. These bioreactive prodrugs include nitro (hetero)-cyclic compounds, aromatic N-oxides, aliphatic N-oxides, quinone, and transition metal complexes.3
Many nanomaterials, such as organic, inorganic, lipid, protein, or polymeric compounds, are being used as carrier materials for constructing antitumor nanomedicine systems loaded into antitumor agents.4,5 Currently, some prodrug nanomedicine systems have been reported.6–9 However, only limited research studies involved bioreactive prodrug nanomedicine systems. Recently, an angiogenesis vessel-targeting nanoparticle (AVT-NP) consisting of a photosensitizer, angiogenic vessel-targeting peptide, and bioreductive prodrug (tirapazamine [TPZ]) was developed for a chemo-photo synergistic cancer therapy.10 In this AVT-NP, the bioreductive prodrug TPZ was loaded in the hydrophobic core of a TPC-GX1 assembling micelle. We speculate that many bioreductive prodrug nanomedicine systems could be designed and developed in future research.
For antitumor nanomedicine systems, the ratio of antitumor agent to carrier material is relatively low (below 1:10 or less), leading to a low antitumor agent occupancy rate. To overcome this shortcoming, small molecule modified anticancer drug conjugates (SMMDCs), which have a higher drug occupancy, have been synthesized. Interestingly, these can self-assemble into NPs in water, and their antitumor activity has been confirmed in vitro and in vivo;11 eg, conjugated linoleic acid-paclitaxel conjugate (CLA-PTX) can self-assemble into NPs.12 These SMMDC NPs, with higher drug loading and an aqueous solvent with organic solvent-free characteristics, can be prepared by a simple precipitation method.11 The theoretical partition coefficient (XlogP) and Hansen solubility parameters of the SMMDCs might be the key factors for forming self-assembled NPs.13
3-(2-Nitrophenyl) propionic acid-paclitaxel (NPPA-PTX) is a paclitaxel (PTX) bioreductive prodrug synthesized by our lab.14 The antitumor activity of NPPA-PTX was confirmed by our in vitro and in vivo experiments.
In the present research, we hypothesize that NPPA-PTX could self-assemble to form NPs. The XlogP and Hansen solubility parameters of NPPA-PTX were first calculated. Then, the NPPA-PTX@PEG NPs was prepared and characterized. The cellular uptake and in vitro antitumor activity of NPPA-PTX@PEG NPs were evaluated in MDA-MB-231 cells. In addition, the in vivo targeting effect, tumor distribution, and antitumor activity of NPPA-PTX@PEG NPs were investigated in MDA-MB-231 tumor-bearing nude mice. The safety of NPPA-PTX@PEG NPs was evaluated in healthy Institute of Cancer Research (ICR) mice by maximum tolerated dose (MTD) experiments.
Materials and methods
Materials
NPPA was purchased from Beijing OUHE Tech. Co., Ltd. (Beijing, China). PTX was obtained from Mei-Lian Co., Ltd. (Chongqing, China). NPPA-PTX was synthesized from NPPA and PTX according to our previous report.14 Cremophor EL (CrEL) was obtained from BASF (Ludwigshafen, Germany). PTX injection (Taxol) was obtained from a local hospital in Beijing (Bristol-Myers Squibb, New York, NY, USA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine [methoxy(polyethylene glycol)2000] (DSPE-PEG) was obtained from NOF Corporation (Tokyo, Japan). Sulforhodamine B (SRB) was purchased from Sigma-Aldrich Co, (St Louis, MO, USA). Cell culture media L-15, penicillin, streptomycin, FBS, and L-glutamine were all obtained from Thermo Fisher Scientific (Waltham, MA, USA). All other chemicals were of analytical or HPLC grade.
The MDA-MB-231 cell lines and human breast cancer cells (estrogen receptor negative) were obtained from the Chinese Academy of Sciences Cell Bank (Beijing, China) and cultivated in L-15 medium, which was supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin. The cultures were maintained at 37°C and 95% relative humidity.
Animals
Female BALB/c nude mice and healthy ICR mice (all supplied by the Experimental Animal Center of Peking University, Beijing, China, weighing 18–20 g) were acclimatized for 7 days prior to the experiment and allowed free access to a standard diet and water with the maintained condition of 25°C temperature and 50% relative humidity. All care and handling of animals were performed with the approval of the Experimental Animal Center of Peking University Health Science Center. Additionally, this study was performed following the National Institutes of Health guidelines for the use of experimental animals.
For preparing the MDA-MB-231 tumor-bearing nude mice model, the female BALB/c nude mice were injected subcutaneously with 0.2 mL MDA-MB-231 cell suspension (5×106 cells) with 20% basement membrane matrix into the right armpit.15
XlogP and Hansen solubility parameters of NPPA-PTX
The XlogP and Hansen solubility parameters (dispersion [δd], polar [δp], hydrogen bonding [δh], and total solubility parameter [δt]) of NPPA-PTX were calculated according to our previous report.13
Preparation of NPPA-PTX@PEG NPs
NPPA-PTX@PEG NPs was prepared by precipitation as shown in our previous report.12 Briefly, both NPPA-PTX and DSPE-PEG (NPPA-PTX:DSPE-PEG =1:0.1, w/w) were dissolved in DMSO. One milliliter of this DMSO solution (1 mg/mL) was added dropwise to 20 mL of distilled water with continuous and gentle stirring (300–500 rpm) at room temperature. Under these conditions, the NPPA-PTX NPs (NPPA-PTX@PEG NPs) occurred spontaneously. Then, the resulting NP dispersion was dialyzed and ultrafiltrated. The concentration of NPPA-PTX in the obtained NPPA-PTX@PEG NPs was ~7.2 mg/mL (if necessary, adjusted with distilled water).
Characterization of NPPA-PTX@PEG NPs
Particle size and distribution
The particle size, particle size distribution, and zeta potential of NPPA-PTX@PEG NPs were all assayed by dynamic light scattering measurements on a Nano ZS instrument (Malvern Instruments, Malvern, UK).
Transmission electron microscopy (TEM)
TEM was performed using a JEM 1400 microscope (JEOL, Tokyo, Japan) operating at 140 kV using a permeable carbon-coated copper grid to examine the morphology of NPPA-PTX@PEG NPs.
X-ray diffraction
The X-ray diffraction patterns of the samples were measured using a D/MAX 2000 rotating anode X-ray diffractometer (Rigaku Corporation, Tokyo, Japan) equipped with a Cu-Kα X-ray source (λ=1.541 nm, 40 kV/100 mA). All X-ray diffraction data were collected over the 2θ range from 3° to 40° at a step size of 0.02° increments, for quantitative analysis. Profile fits of the data were performed using Origin 8.5.1 software.
In vitro cellular uptake
MDA-MB-231 cells (3×105 cells/well) were seeded in 6-well plates and incubated for 24 hours at 37°C. Then, the cells were treated with free NPPA-PTX solution or NPPA-PTX@PEG NPs (10 μM) for 2, 4, or 6 hours at 4°C or 37°C. The cellular uptake efficiency was determined as in our previous report.16 In brief, after incubation, the cells were washed with PBS and lysed with SDS. After that, the lysate was taken for the determination of the protein concentration using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific) in accordance with the manufacturer’s instructions. Then, the remaining lysate was extracted with acetonitrile and centrifuged. The resulting supernatant was dried using nitrogen. Finally, the dry samples were redissolved in the mobile phase (acetonitrile:distilled water =6:4) for the HPLC analysis. Three wells were measured for each sample. The cellular uptake of free NPPA-PTX solution or NPPA-PTX@PEG NPs was calculated using the following formula:
|
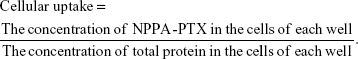
In vitro cytotoxicity
The in vitro cytotoxicity of NPPA-PTX@PEG NPs was measured using SRB.17 MDA-MB-231 cells (1×104 cells/well) were seeded in 96-well plates, incubated for 24 hours, and then treated with NPPA-PTX@PEG NPs at 37°C for 48 hours. The cell viability was determined using SRB, which allowed quantification of the living cells by measuring absorbance at 540 nm by a 96-well plate reader (model 680; Bio-Rad Laboratories Inc., Hercules, CA, USA). The half-maximal inhibitory concentration (IC50) was calculated according to the dose–effect curves using GraphPad Prism 6/SPSS software.
In vivo imaging in mice
The in vivo imaging of the NPPA-PTX@PEG NPs was evaluated in MDA-MB-231 tumor-bearing nude mice. When the tumor volume reached ~300 mm3, these mice were given a tail vein injection of 5% glucose injection or Alexa Fluor 680-labeled NPPA-PTX@PEG NPs. In vivo fluorescence imaging was performed using an IVIS Spectrum in vivo imaging system (PerkinElmer Inc., Waltham, MA, USA) at different times (2, 4, 6, 8, 10, and 24 hours). The mice were sacrificed at 24 hours, and the heart, liver, spleen, lung, kidneys, and tumor were collected immediately. The fluorescence intensities in the different tissues were photographically recorded.18,19
Distribution of tumor tissues
When the tumor volume reached ~400 mm3, the mice were randomly divided into two treatment groups (nine mice per group). Then, each group was given a tail vein injection of Taxol (15 mg/kg PTX) or NPPA-PTX@PEG NPs (18.10 mg/kg, equimolar to 15 mg/kg PTX). The mice were executed at 1, 4, and 8 hours (each group containing three mice). The tumor tissues were weighed (1 g) and homogenized with 3 mL PBS, followed by extraction and HPLC analysis.
In vivo antitumor activity
The in vivo antitumor activity of NPPA-PTX@PEG NPs was evaluated in MDA-MB-231 tumor-bearing nude mice. When the tumor volume reached ~100–200 mm3, the mice were randomly divided into four groups. Then, each group was given a tail vein injection of 5% glucose as the control, Taxol (10 mg/kg PTX, q3d), NPPA-PTX@PEG NPs (12 mg/kg, equimolar to 10 mg/kg PTX, q3d), or NPPA-PTX@PEG NPs (36 mg/kg, equimolar to 30 mg/kg PTX, single-dose). The tumor volume was measured every 2 days using the calculation based on the equation (a×b2)/2, where a and b are the length and width of the tumor, respectively. After 21 days, all of the mice were sacrificed, and the tumor tissues were removed and weighed.
MTD
The MTD of NPPA-PTX@PEG NPs was determined using a dose escalation method in healthy ICR mice (50% male and 50% female). The animals (n=10, per dose group) received intravenous injections of NPPA-PTX@PEG NPs (81, 121, and 181 mg/kg, equimolar to 70, 100, and 150 mg/kg PTX, respectively), Taxol (20, 25, and 30 mg/kg), or 5% glucose as a control. Animal survival and changes in body weight were observed daily over 2 weeks in all groups. The highest dose that did not cause toxicity (as defined by a median body weight loss of 10% of the control or abnormal behavior including hunched posture and a rough cough) was defined as the MTD.
Statistical analysis
All experimental data are expressed as the mean ± SD. The significance among groups was determined using a one-way ANOVA, after which post hoc tests with the Bonferroni correction for comparisons between individual groups were performed. Statistical significance was shown at P<0.05.
Results
XlogP and Hansen solubility parameters of NPPA-PTX
As shown in Table 1, the XlogP value of NPPA-PTX was higher than that of PTX, and the values of the Hansen solubility parameters (δd, δp, δh, and δt) of NPPA-PTX were all lower than those of PTX. The ΔXlogP value of NPPA-PTX was 0.57, and the Δδh value of NPPA-PTX was −15.87%. Considering our suggested criterion for SMMDCs self-assembling to form NPs (when the XlogP value of SMMDCs increased more than 1.0-fold compared to that of the parent drug; otherwise, when the δh value of SMMDCs decreased more than 10% compared to the parent drug), we suggest that NPPA-PTX can self-assemble into NPs.
 | Table 1 The values of XlogP and Hansen solubility parameters (δd, δp, δh, and δt) of NPPA-PTX |
The X-ray diffraction spectrum indicated that sharp and intense peaks of NPPA or PTX were present, suggesting that NPPA or PTX was present in crystalline form. In contrast, there was no sharp peak attributable to the crystalline form of NPPA-PTX, suggesting that NPPA-PTX, unlike PTX, was in an amorphous state (Figure 1D), another key factor for SMMDCs self-assembling into NPs.
 | Figure 1 The preparation and characteristics of NPPA-PTX@PEG NPs. |
Preparation and characterization of NPPA-PTX@PEG NPs
NPPA-PTX can self-assemble to form NPPA-PTX NPs (NPPA-PTX@PEG NPs) when the NPPA-PTX mixed DSPE-PEG DMSO solution was added to distilled water, as shown in Figure 1A. The particle size of NPPA-PTX@PEG NPs was ~84.89±0.76 nm. The polydispersity was 0.085±0.006. The value of the zeta potential of NPPA-PTX@PEG NPs was −20.5±0.31, as shown in Table 2. A typical particle size and distribution of NPPA-PTX@PEG NPs are shown in Figure 1B. The TEM image confirmed the spherical shape and uniform size of NPPA-PTX@PEG NPs, as shown in Figure 1C. The X-ray diffraction results indicate that NPPA-PTX@PEG NPs were also in an amorphous state (Figure 1D).
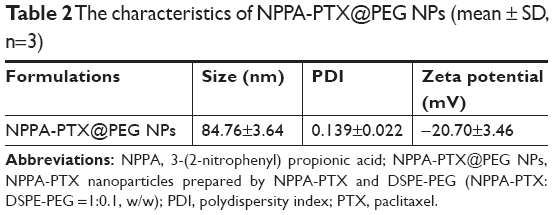 | Table 2 The characteristics of NPPA-PTX@PEG NPs (mean ± SD, n=3) |
In vitro cellular uptake of NPPA-PTX@PEG NPs
The in vitro cellular uptake of NPPA-PTX@PEG NPs was examined in the MDA-MB-231 cell line. As shown in Figure 2, the cellular uptake of NPPA-PTX@PEG NPs at 37°C was clearly higher than that at 4°C (Figure 2A), indicating that the cellular uptake of NPPA-PTX@PEG NPs was energy- and time-dependent. In contrast, the cellular uptake of free PTX at 37°C was almost similar to that at 4°C (Figure 2B), indicating that the cellular uptake of free PTX was by passive diffusion. In addition, NPPA-PTX@PEG NPs exhibited enhanced cellular uptake compared to free PTX after an incubation at 37°C over 2, 4, and 6 hours (~1.9-, 2.9-, and 4.2-fold, respectively), indicating that NPPA-PTX@PEG NPs exhibited a higher uptake into tumor cells. At 4°C condition, the cellular uptake of NPPA-PTX@PEG NPs was almost similar to that of free PTX.
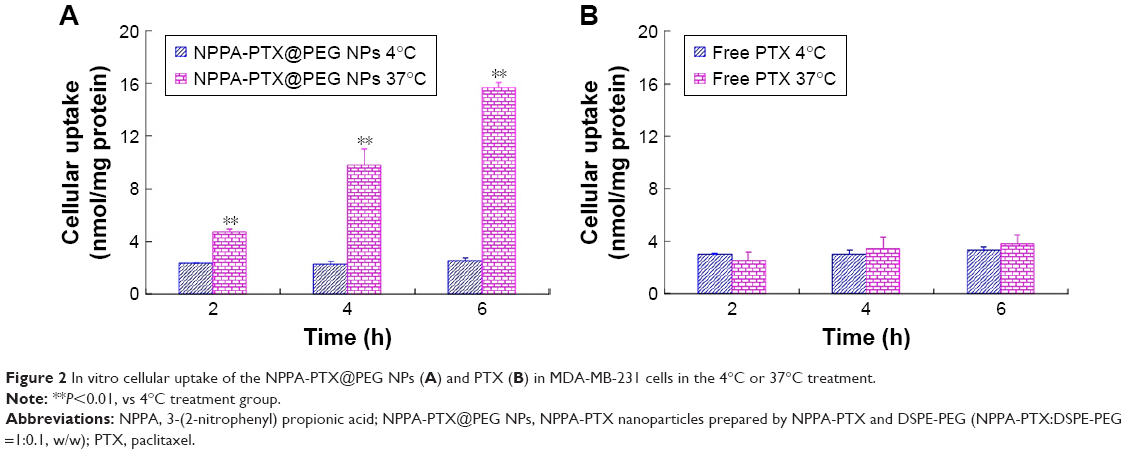 | Figure 2 In vitro cellular uptake of the NPPA-PTX@PEG NPs (A) and PTX (B) in MDA-MB-231 cells in the 4°C or 37°C treatment. |
In vitro cytotoxicity
The in vitro cytotoxicity of NPPA-PTX@PEG NPs was evaluated in the MDA-MB-231 cell line. The calculated IC50 values are shown in Table 3. The results indicate that the in vitro antitumor activity of NPPA-PTX@PEG NPs in the MDA-MB-231 tumor cell line (5.48±0.88 μM) was lower than that of Taxol (1.97±0.34 μM, P<0.01).
 | Table 3 The IC50 values (μM) of NPPA-PTX@PEG NPs in MDA-MB-231 cells (mean ± SD, n=3) |
In vivo imaging
The targeting effect of NPPA-PTX@PEG NPs was investigated using Fluor 680-labeled NPPA-PTX@PEG NPs. The distribution and tumor accumulation of fluorescent Fluor 680 in MDA-MB-231 tumor-bearing nude mice are shown in Figure 3. The results indicate that Fluor 680-labeled NPPA-PTX@PEG NPs have a stronger fluorescence signal in the tumor than glucose injection used as control at all observed times (Figure 3A). The major organs (heart, liver, spleen, lung, and kidneys) and tumor tissues were excised for ex vivo examination at 24 hours postinjection. The results show that a higher fluorescence intensity was found in the tumor tissue compared to that of the other organs in the Fluor 680-labeled NPPA-PTX@PEG NPs treatment group (Figure 3B).
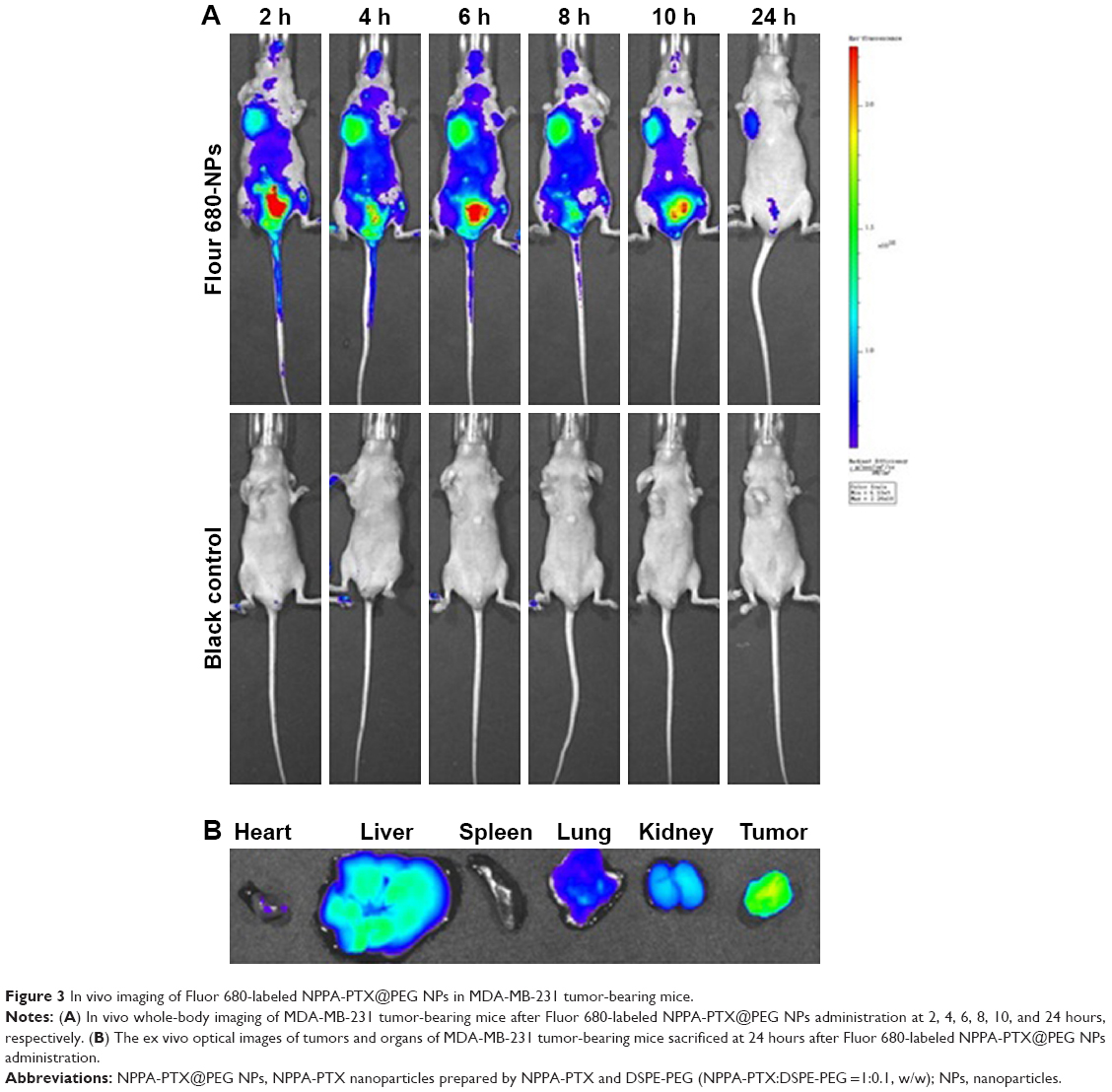 | Figure 3 In vivo imaging of Fluor 680-labeled NPPA-PTX@PEG NPs in MDA-MB-231 tumor-bearing mice. |
Distribution in tumor tissues
As shown in Figure 4A, the dissociated PTX in the MDA-MB-231 tumor tissues was detected at 1 hour as well as at 4 and 8 hours. Compared to the free PTX in the MDA-MB-231 tumor tissues in the Taxol treatment group (Figure 4B), the dissociated PTX in the NPPA-PTX@PEG NPs treatment group was 0.5-, 1.5-, and 1.5-fold compared to that in Taxol treatment group at 1, 4, and 8 hours, respectively. We also examined the level of NPPA-PTX in MDA-MB-231 tumor tissues after the administration of NPPA-PTX@PEG NPs (Figure 4A). The level of NPPA-PTX was only ~9% compared to that of dissociated PTX in the NPPA-PTX@PEG NPs treatment group at 1 hour. At 4 and 8 hours, no NPPA-PTX was detected in the MDA-MB-231 tumor tissues, indicating that NPPA-PTX was completely transformed into dissociated PTX (Figure 4A). Considering that the concentration of dissociated PTX at 4 and 8 hours in the NPPA-PTX@PEG NPs treatment group was significantly higher than that in the Taxol treatment group, we suggest that the in vivo antitumor activity of NPPA-PTX@PEG NPs would be higher than that of Taxol in MDA-MB-231 tumor-bearing mice.
 | Figure 4 (A) The PTX, NPPA-PTX, and dissociated PTX from NPPA-PTX levels (μM) in tumor tissues after a single intravenous administration of NPPA-PTX@PEG NPs at a dose of 18.11 mg/kg (equimolar to 15 mg/kg PTX) or (B) Taxol at a dose of 15 mg/kg to MDA-MB-231 tumor-bearing nude mice. (Means ± SD, n=3). |
In vivo antitumor activity
The in vivo antitumor activity of NPPA-PTX@PEG NPs was evaluated in MDA-MB-231 tumor-bearing nude mice. As shown in Figure 5, the tumor growth was significantly inhibited in the Taxol and NPPA-PTX@PEG NPs treatment groups compared to the 5% glucose treatment group (P<0.01). Interestingly, the NPPA-PTX@PEG NPs treatment group (12 mg/kg, equimolar to 10 mg/kg PTX, q3d; and 36 mg/kg, equimolar to 30 mg/kg PTX, single-dose) significantly inhibited the growth of MDA-MB-231 tumors compared to the PTX treatment group (P<0.01). The average tumor size at day 21 in the Taxol (10 mg/kg PTX, q3d), NPPA-PTX@PEG NPs (12 mg/kg, equimolar to 10 mg/kg PTX, q3d) or NPPA-PTX@PEG NPs (36 mg/kg, equimolar to 30 mg/kg PTX, single-dose) treatment groups were 528±133 mm3, 206±41 mm3, and 200±71 mm3, respectively, compared to 1,844±390 mm3 in the 5% glucose treatment group (P<0.01). The corresponding tumor growth inhibition in the Taxol and NPPA-PTX@PEG NPs-treated groups were ~71.3%, 88.8%, and 89.1%, respectively.
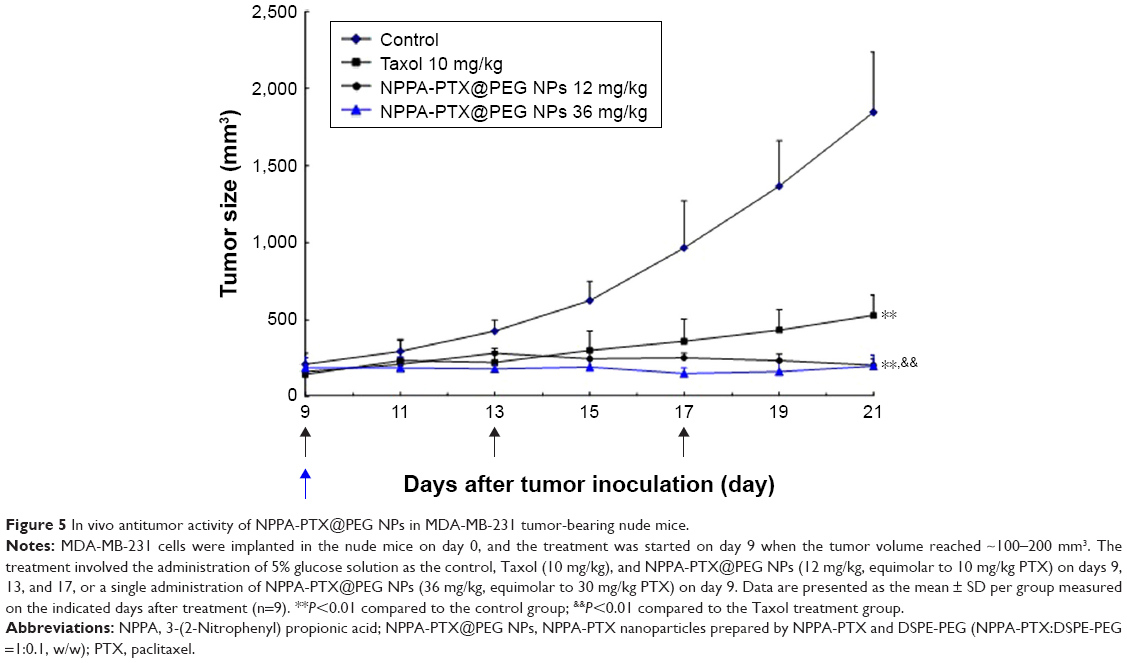 | Figure 5 In vivo antitumor activity of NPPA-PTX@PEG NPs in MDA-MB-231 tumor-bearing nude mice. |
MTD studies
We investigated the MTD of NPPA-PTX@PEG NPs in healthy ICR mice. Considering the toxicity index, defined as the median body weight loss of 10% of the control or abnormal behavior including hunched posture and a rough cough, the results indicate that the MTD of NPPA-PTX@PEG NPs was up to 181 mg/kg (equimolar to 150 mg/kg PTX). Compared to 25 mg/kg for Taxol, we suggest that the safety of NPPA-PTX@PEG NPs is superior to that of Taxol.
Discussion
The prodrug strategy is used to overcome deficiencies in the physicochemical properties of a parent drug, which limit formulation options and result in unacceptable biopharmaceutical performance, to enable its delivery and development.20 Approximately, 10% of small molecular weight drugs approved by the FDA from 2010 to 2016 have been reported to be prodrug.21
Hypoxia is a characteristic feature of the tumor microenvironment.22 Bioreductive prodrug can respond to the hypoxic tumor microenvironment by forming parent drug by the redox potential.3
NPPA-PTX is our previous synthesized bioreductive PTX prodrug which can remain intact and stable in normoxia but be activated in hypoxic tumor tissues.14 The antitumor activity of NPPA-PTX was confirmed by our in vitro and in vivo experiments. However, due to its poor water solubility, NPPA-PTX was dissolved in the mixture of CrEL and dehydrated ethanol (1:1, v/v), similar to that of PTX injection. Interestingly, some SMMDCs can self-assemble into NPs in water without adding a surfactant and organic solvent. Under this circumstance, the safety of these SMMDC NPs might be improved because they possess surfactant-free and organic solvent-free characteristics.12 In the present research, the MTD value of NPPA-PTX@PEG NPs in healthy ICR mice was almost sixfold than that of Taxol, indicating that the safety of NPPA-PTX@PEG NPs was significantly enhanced compared to Taxol.
We previously demonstrated that the XlogP and Hansen solubility parameters are key factors for predicting SMMDCs self-assembling into NPs.13 In the present research, we calculated the XlogP and Hansen solubility parameters of NPPA-PTX. Our results indicate that NPPA-PTX can self-assemble into NPs, as the δh value of NPPA-PTX was decreased more than 10% compared to PTX despite the increased XlogP value of NPPA-PTX being less than 1.0-fold compared to that of PTX. Therefore, we suggest that NPPA-PTX can self-assemble into NPs. In addition, the lower crystallinity of NPPA-PTX observed in the X-ray diffraction experiment also indicates that NPPA-PTX has the potential for self-assembling and forming NPs. The preparation method of NPPA-PTX@PEG NPs is by a simple precipitation, similar to our CLA-PTX@PEG NPs preparation.12 The drug loading of NPPA-PTX in NPPA-PTX@PEG NPs was ~90% (w/w) with CrEL-free and organic solvent-free characteristics. As the ratio of NPPA-PTX: DSPE-PEG was only 1:0.1 (w/w), these as-prepared NPPA-PTX@PEG NPs could be regarded as possessing carrier-free or drug-self carrier characteristics. The characteristics of NPPA-PTX@PEG NPs, such as our previously prepared CLA-PTX@PEG NPs, also meet the requirement of NPs.
In the present research, the enhanced cellular uptake of NPPA-PTX@PEG NPs was observed in the MDA-MB-231 cell line. It is well known that an enhanced cellular uptake of NPs is very important for chemotherapeutic agents loaded in NPs to produce antitumor activity.23 The uptake of NPs by tumor cells was mainly via endocytotic pathways.24 There are five main endocytosis routes, including phagocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, clathrin/caveolin-independent endocytosis, and micropinocytosis.25 The size of NPs plays an important role in cellular uptake pathways.26 In general, larger particles undergo more efficient uptake by phagocytes (>250 nm) and macropinocytosis (0.5–10 μm).27,28 Smaller particles (less than 150 nm) experience more efficient uptake by clathrin-mediated endocytosis (~100 nm), caveolin-mediated endocytosis (60–80 nm), and clathrin/caveolin-independent endocytosis.29,30
Considering the particle size of NPPA-PTX@PEG NPs (~90 nm), we speculated that the cellular uptake pathway of NPPA-PTX@PEG NPs might be clathrin-mediated, caveolin-mediated, or clathrin/caveolin-independent endocytosis. In addition, our results indicate that there was nearly no dissolved PTX detected in our cellular uptake experiment; furthermore, our in vitro release experiment indicated that there was nearly no NPPA-PTX released from NPPA-PTX@PEG NPs (data not shown). Therefore, we suggest that NPPA-PTX@PEG NPs can enter into MDA-MD-231 cells via the endocytotic pathway as intact NPPA-PTX@PEG NPs.
The targeting effect of NPPA-PTX@PEG NPs was investigated using Fluor 680-labeled NPPA-PTX@PEG NPs. The passive targeting effect of NPPA-PTX@PEG NPs is due to the enhanced permeability and retention effect. In fact, when the NPPA-PTX@PEG NPs were modified with a targeting ligand, eg, asparagines-glycine-arginine (NGR)-modified NPPA-PTX@PEG NPs, the targeting effect was further improved (data not shown). This means that these NPPA-PTX@PEG NPs could be further modified by targeting the ligand or antibody for implementing the active targeting activity.
As a bioreductive PTX prodrug, the antitumor activity of NPPA-PTX was confirmed by our in vitro and in vivo experiments. In the present research, the antitumor activity of NPPA-PTX@PEG NPs was also investigated and confirmed in our in vitro and in vivo experiments. The results of the in vivo antitumor activity indicate that the antitumor efficacy of NPPA-PTX@PEG NPs was better than that of Taxol. Interestingly, the single-dose treatment of NPPA-PTX@PEG NPs also produced similar antitumor activity to the multiple-dose administration of NPPA-PTX@PEG NPs, showing a long-term tumor inhibition effect of high-dose administration of NPPA-PTX@PEG NPs as its safety significantly improved.
The results of the tumor tissue distribution indicate that NPPA-PTX@PEG NPs, such as NPPA-PTX, were completely converted into active PTX in MDA-MB-231 tumor tissues. In addition, the concentration of dissociated PTX was significantly higher than that of Taxol in the MDA-MB-231 tumor tissues; this might be a possible reason why the in vivo antitumor activity of NPPA-PTX@PEG NPs was better than that of Taxol in MDA-MB-231 tumor-bearing mice.
Conclusion
In the present research, the XlogP and Hansen solubility parameters of the bioreductive prodrug NPPA-PTX was calculated to predict the possibility of NPPA-PTX self-assembling into NPs. NPPA-PTX@PEG NPs was prepared and characterized. The cellular uptake, in vitro antitumor activity, in vivo targeting effect, tumor distribution, in vivo antitumor activity, and safety of NPPA-PTX@PEG NPs were investigated. The safety and antitumor activity of NPPA-PTX@PEG were confirmed in our in vitro and in vivo experiments. NPPA-PTX@PEG NPs could offer a new way to prepare bioreductive prodrug, self-assembled NPs suitable for antitumor therapy.
Acknowledgments
The authors are grateful for the financial support from the National Key Research and Development Program of China (2017YFA0205600) and the National Natural Science Foundation of China (No. 81573360).
Disclosure
The authors report no conflicts of interest in this work.
References
Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11(6):393–410. | ||
Phillips RM. Targeting the hypoxic fraction of tumours using hypoxia-activated prodrugs. Cancer Chemother Pharmacol. 2016;77(3):441–457. | ||
Guise CP, Mowday AM, Ashoorzadeh A, et al. Bioreductive prodrugs as cancer therapeutics: targeting tumor hypoxia. Chin J Cancer. 2014;33(2):80–86. | ||
Mukerabigwi JF, Ge Z, Kataoka K. Therapeutic Nanoreactors as In Vivo Nanoplatforms for Cancer Therapy. Chemistry. 2018;24(59):15706–15724. | ||
Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew Chem Int Ed. 2014;53(46):12320–12364. | ||
Chen S, Zaifman J, Kulkarni JA, et al. Dexamethasone prodrugs as potent suppressors of the immunostimulatory effects of lipid nanoparticle formulations of nucleic acids. J Control Release. 2018;286:46–54. | ||
Wang G, Wang Z, Li C, et al. RGD peptide-modified, paclitaxel prodrug-based, dual-drugs loaded, and redox-sensitive lipid-polymer nanoparticles for the enhanced lung cancer therapy. Biomed Pharmacother. 2018;106:275–284. | ||
Ling X, Chen X, Riddell IA, et al. Glutathione-Scavenging Poly(disulfide amide) Nanoparticles for the Effective Delivery of Pt(IV) Prodrugs and Reversal of Cisplatin Resistance. Nano Lett. 2018;18(7):4618–4625. | ||
Signorell RD, Luciani P, Brambilla D, Leroux JC. Pharmacokinetics of lipid-drug conjugates loaded into liposomes. Eur J Pharm Biopharm. 2018;128:188–199. | ||
Guo D, Xu S, Wang N, et al. Prodrug-embedded angiogenic vessel-targeting nanoparticle: A positive feedback amplifier in hypoxia-induced chemo-photo therapy. Biomaterials. 2017;144:188–198. | ||
Cheetham AG, Chakroun RW, Ma W, Cui H. Self-assembling prodrugs. Chem Soc Rev. 2017;46(21):6638–6663. | ||
Zhong T, Yao X, Zhang S, et al. A self-assembling nanomedicine of conjugated linoleic acid-paclitaxel conjugate (CLA-PTX) with higher drug loading and carrier-free characteristic. Sci Rep. 2016;6:36614. | ||
Zhong T, Hao YL, Yao X, et al. Effect of XlogP and Hansen Solubility Parameters on Small Molecule Modified Paclitaxel Anticancer Drug Conjugates Self-Assembled into Nanoparticles. Bioconjug Chem. 2018;29(2):437–444. | ||
Song P, Yao X, Zhong T, et al. The anti-tumor efficacy of 3-(2-Nitrophenyl) propionic acid-paclitaxel (NPPA-PTX): a novel paclitaxel bioreductive prodrug. Oncotarget. 2016;7(30):48467–48480. | ||
Zheng XC, Ren W, Zhang S, et al. The theranostic efficiency of tumor-specific, pH-responsive, peptide-modified, liposome-containing paclitaxel and superparamagnetic iron oxide nanoparticles. Int J Nanomedicine. 2018;13:1495–1504. | ||
du R, Zhong T, Zhang WQ, et al. Antitumor effect of iRGD-modified liposomes containing conjugated linoleic acid-paclitaxel (CLA-PTX) on B16-F10 melanoma. Int J Nanomedicine. 2014;9:3091–3105. | ||
Guo Y, Zhong T, Duan XC, et al. Improving anti-tumor activity of sorafenib tosylate by lipid- and polymer-coated nanomatrix. Drug Deliv. 2017;24(1):270–277. | ||
Luo LM, Huang Y, Zhao BX, et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34(4):1102–1114. | ||
Zhao BX, Zhao Y, Huang Y, et al. The efficiency of tumor-specific pH-responsive peptide-modified polymeric micelles containing paclitaxel. Biomaterials. 2012;33(8):2508–2520. | ||
Rautio J, Meanwell NA, di L, Hageman MJ. The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Discov. 2018;17(8):559–587. | ||
Rautio J, Kärkkäinen J, Sloan KB. Prodrugs – Recent approvals and a glimpse of the pipeline. Eur J Pharm Sci. 2017;109:146–161. | ||
Krzywinska E, Stockmann C. hypoxia, metabolism and immune cell function. Biomedicines. 2018;6(2):56. | ||
Chistiakov DA, Myasoedova VA, Orekhov AN, Bobryshev YV. nanocarriers in improving chemotherapy of multidrug resistant tumors: key developments and perspectives. Curr Pharm Des. 2017;23(22):3301–3308. | ||
Yameen B, Choi WI, Vilos C, Swami A, Shi J, Farokhzad OC. Insight into nanoparticle cellular uptake and intracellular targeting. J Control Release. 2014;190:485–499. | ||
Behzadi S, Serpooshan V, Tao W, et al. Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev. 2017;46(14):4218–4244. | ||
Salatin S, Maleki Dizaj S, Yari Khosroushahi A. Effect of the surface modification, size, and shape on cellular uptake of nanoparticles. Cell Biol Int. 2015;39(8):881–890. | ||
Rabinovitch M. Professional and non-professional phagocytes: an introduction. Trends Cell Biol. 1995;5(3):85–87. | ||
Falcone S, Cocucci E, Podini P, Kirchhausen T, Clementi E, Meldolesi J. Macropinocytosis: regulated coordination of endocytic and exocytic membrane traffic events. J Cell Sci. 2006;119(Pt 22):4758–4769. | ||
Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8(3):185–194. | ||
Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377(Pt 1):159–169. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.