Back to Journals » Cancer Management and Research » Volume 12
Antitumor Activity of Cabazitaxel and MSC-TRAIL Derived Extracellular Vesicles in Drug-Resistant Oral Squamous Cell Carcinoma
Authors Qiu Y, Sun J, Qiu J, Chen G, Wang X, Mu Y, Li K, Wang W
Received 19 August 2020
Accepted for publication 3 October 2020
Published 29 October 2020 Volume 2020:12 Pages 10809—10820
DOI https://doi.org/10.2147/CMAR.S277324
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Yongle Qiu,1 Jieming Sun,2 Junping Qiu,2 Guoling Chen,2 Xiao Wang,2 Yaxu Mu,2 Kunshan Li,1 Wenjing Wang1
1Department of Stomatology, Fourth Affiliated Hospital, Hebei Medical University, Shijiazhuang, Hebei 050017, People’s Republic of China; 2Department of Stomatology, Xianghe County People ‘S Hospital, Langfang, Hebei 065400, People’s Republic of China
Correspondence: Kunshan Li Email [email protected]
Introduction: TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) can induce apoptosis in a variety of cancer cells. However, drug resistance of tumor and short half-life seriously affects its clinical targeted therapy. Cabazitaxel (CTX) is a taxane drug, which can induce apoptosis or autophagy by inhibiting the phosphorylation of PI3K/Akt/mTOR and sensitive to some drug-resistant tumors. Therefore, we explored the possibility of developing a mesenchymal stem cell-derived exosomes (MSC-EXO) vector for oral squamous cell carcinoma (OSCC) to deliver CTX/TRAIL combinations.
Methods: After ultracentrifugation and dialysis, CTX/TRAIL loaded exosomes transfected MSC (MSCT)-derived exosome (EXO) (MSCT-EXO/CTX) were isolated and purified. The expression of CD63, CD9 and TRAIL was detected by BCA to confirm the origin of EXO. High-performance liquid chromatography (HPLC) was used to determine the drug loading of VPF and draw the in vitro release profile. MTT assay, flow cytometry and Western blot were used to detect the antitumor effect of MSCT-EXO/CTX in vitro. Subsequently, the antitumor effect of MSCT-EXO/CTX in vivo was verified by mouse model.
Results: The diameter of the membrane particles was about 60– 150 nm. We have proved that the incorporation and release of CTX in MSCT-EXO can inhibit the activation of PI3K, Akt and mTOR, which is a possible synergistic mechanism of CTX. MSCT-EXO and CTX can induce the apoptosis of SCC25 tumor cells in a dose-dependent manner and exert a good synergistic effect in the proportion range of 10:1– 5:1. The inherent activity of MSCT-EXO and the direct effect of MSCT-EXO/CTX on OSCC confirm that MSCT-EXO/CTX makes MSCT-EXO and CTX have an efficient synergistic effect and a highly effective pharmacological inhibition on cancer cells, as verified by the subsequent mouse model. MSCT-EXO/CTX showed the lowest relative tumor volume and the highest tumor inhibition rate (P< 0.05) in vivo.
Conclusion: An MSCT-EXO-based CTX delivery system might be an effective anticancer method.
Keywords: cabazitaxel, extracellular vesicles, mesenchymal stromal cells, oral squamous cell carcinoma
Introduction
The recombinant human soluble TRAIL and TRAIL receptor agonists have shown a good antitumor activity in preclinical studies.1 Among them, soluble TRAIL can induce apoptosis of colon, lung, breast and other tumor cells and inhibit the occurrence and growth of tumor.2 The difficulties in the transformation of preclinical achievements lie in the short cycle half-life of TRAIL (in rodents, the half-life is only a few minutes; in humans, the half-life is approximately 60 min), the insufficient accumulation of drugs at tumor targets, the low activation efficiency of DR4 and DR5 antibodies on the surface of tumor cells, and the tumor cells on TRAIL primary or secondary drug resistance.3,4
As a taxol drug, CTX has the same mechanism as paclitaxel and docetaxel. PI3K, Akt and mTOR participate in the phosphorylation of caspase-9 and ser196 enhance the expression of Bal-xl, reduce the permeability of mitochondria, inhibit the apoptosis of cancer cells and promote cell proliferation.5,6 CTX can inhibit the activation of PI3K, Akt and mTOR pathway by inhibiting the phosphorylation of PI3K, Akt and mTOR.7,8 Due to the excellent antitumor activity, CTX has been used as an alternative for docetaxel and other paclitaxel-resistant patients.9 Due to poor water solubility and high side effects, CTX has derived a series of formulations, among which nanoparticles are one. The literature has proved that the derivatives of nanoparticles have a better curative effect and lower toxicity.10
Extracellular vesicles (EVs) are submicron membrane vesicles released by cells, which are composed of lipids, proteins, and nucleic acids. Evidence shows that EVs mediate intercellular communication by transferring biological molecules from donor cells to nearby or distant receptor cells under physiological and pathological conditions.11–13 As natural nanomaterials, EVs can penetrate tissues and even the blood–brain barrier. Therefore, they have a high application value as drug delivery carriers.14–16 Numerous researchers have used EVs in recent years to deliver natural small molecular compounds, DNA, and proteins related to cancer.17–20
Exosome (EXO) is a kind of extracellular vesicles, which is secreted after the fusion of intracellular multivesicular bodies and cell membrane. Although most cells can release EXO, mesenchymal stromal cell (MSC)-derived EXO (MSC-EXO) shows unique advantages. Compared with the production of other cell types, that of EXO from MSC is more significant and persistent.21 MSC-EXO has good stability in human plasma and can be preserved at −20 °C.22,23 Several studies have shown that EXOs are easy to be surface modified, provided they have high targeting and cell uptake rate.24,25 MSC-EXO can be used as a new drug carrier because it can efficiently load drugs and control their release, and can also protect drugs by shielding the effect of carriers. We speculated that MSC-EXO could be used as a carrier to deliver CTX/TRAIL in the treatment of drug-resistant oral squamous cell carcinoma. It can not only effectively reverse the multidrug resistance of tumor but also improve the sensitivity of chemotherapy drugs, and reduce the toxic and side effects of chemotherapy drugs on normal tissues, which is a new strategy for cancer treatment.
Materials and Methods
Materials
CTX was purchased from MedChemExpress (New Jersey, US) SCC25 cell line from ATCC (Manassas, VA, USA) and cultured in an RPMI-1640 medium containing 10% FBS. Adult mesenchymal stem cells in good condition were purchased from ATCC. MSCTs were established by transfection of MSCs with lentivirus-expressing human TRAIL.26 They were cultured and preserved in an RPMI-1640 medium containing 17% FBS. To isolate EXO derived from MSCTs, cells were cultured in an RPMI-1640 medium containing 10% FBS without EXO. Other cell culture reagents were all purchased from Sigma.
Isolation of MSCT-EXO/CTX
MSCTs were cultured in an RPMI-1640 cell culture medium containing 10% FBS at 37 °C and 5% CO2 to prepare drug-loaded EXO. The medium was changed every 48 h. It was changed to RPMI-1640 without EXO but with 0.3 mg/mL of CTX and 10% FBS for 48 h. It was then collected and centrifuged at 300 g for 10 min and then centrifuged again at 2000 g at 4 °C to remove cells and fragments. Thereafter, the medium was vacuum-filtered using a 0.22-μm filter (Membrana, Germany). It was concentrated with 100 kDa (PES, Sartorius, Germany) for 5 times and finally ultracentrifuged at 1 × 105 g at 4 °C for 2 h. The EXO product was filtered using a 0.22-μm membrane filter and washed with phosphate-buffered saline (PBS) twice. The final product was stored in PBS at −80 °C.
Characterization of EXO and Mass Spectrometry
After centrifugation, EXO samples were resuspended with 10 μL of PBS and then added to a copper net for 2 min precipitation. A filter paper was used to absorb the suspension. At 80–120 kV, a JEM-1400 transmission electron microscope (JEOL, Japan) was used for observation.
A sodium phosphate buffer solution with a pH of 7.4 and 0.5% Tween-80 was prepared. The PBS solution (100 mL) was collected and added with 0.5 g of Tween-80; 2 mL of MSCT-EXO and MSCT-EXO/CTX were also collected and placed into a dialysis bag (MCWO: 8000–14,000). Both ends of the dialysis bag were clamped, and 40 mL of sodium phosphate buffer containing 0.5% (w/v) Tween-80 was placed into the beaker. The solution was used as a dialysate. The beaker was placed into a constant temperature shaker, in which the rotating speed was set to 100 r/min, and the temperature was 37 °C. The CTX standard was used. In accordance with the HPLC method, acetonitrile and water were used as the mobile phase. The flow rate was 1 mL/min, and the wavelength was 230 nm.
In vitro Release Experiment
The solution (100 mL) was prepared in accordance with the preparation method of the sodium phosphate buffer solution, and 0.5 g of Tween-80 was added to the solution. The MSCT-EXO/CTX solution (2 mL) was placed into a dialysis bag (MCWO: 8000–14,000). Both ends of the dialysis bag were clamped, and 40 mL of a sodium phosphate buffer solution containing 0.5% (w/v) Tween-80 was placed into a beaker as the dialysate. The beaker was placed into a constant-temperature shaker, in which the rotating speed was set to 100 r/min, and the temperature was 37.0 °C. LM samples were collected from the dialysate at a predetermined time interval. A buffer solution was added to compose the volume of the dialysate. The sample (1 mL) was centrifugated at 14,000 rpm/min for 10 min. HPLC was used to determine the content of CTX in the dialysate at each time point and the total amount before dialysis.
Cytotoxic and Apoptosis Analysis
SCC25 cells were inoculated into six-well plates with 5 × 105 cells per well and cultured for 24 h. The growth of SCC25 cells converged to approximately 70%. Four groups (CTX, MSCT-EXO, MSCT-EXO:CTX (free MSCT-EXO + free CTX) and MSCT-EXO/CTX) were added to the six-well plates. At the end of administration, the blank wells were set as the negative control group. In the experimental group, cells were digested with trypsin without EDTA, beaten gently, centrifuged, and collected. Then, the cells were resuspended; thus, the cell concentration was 1×106 cells/mL. Cell suspension (100 μL) was absorbed, and 5 μL of tested drugs, including CTX (2μg/mL), MSCT-EXO (20μg/mL), MSCT-EXO:CTX (20μg/mL), MSCT-EXO/CTX (20 μg/mL), and PBS medium, were added. After treatment, floating and adherent cells were collected and stained with AF647, annexin V (Invitrogen), and 2 μg/mL of DAPI (Sigma). Cell death was evaluated via flow cytometry.
Western Blot Analysis
EXO source detection: The samples of MSCT-EXO were collected, and protein content was detected using the BCA method and separated by SDS-PAGE electrophoresis. After electrophoresis, the gel was removed, and the protein was transferred to a PVDF membrane, which was sealed with 5% skimmed milk powder for 1 h. They were incubated with the first antibody of CD9, CD63, CD90, TRAIL, GAPDH, Bcl-2, and Bax (1:1000) prepared using 10% skimmed milk powder at 4 °C for overnight. After washing by using TBS, the corresponding 10% skimmed milk powder was added to prepare the second antibody (1:5000) and incubated at room temperature for 2 h. ECL development was observed after TBS cleaning.
Phosphorylation of PI3K, Akt and mTOR:SCC25 cells were treated with CTX, MSCT-EXO, MSCT-EXO:CTX, MSCT-EXO/CTX for 24 h, then the protein was extracted and quantified. The protein was transferred to a PVDF membrane, which was sealed with 5% skimmed milk powder for 2 h. 5% skim milk seal, PI3K (1:500), p-PI3K (1:400), AKT (1:500), p-AKT (1:400), mTOR (1:500), p-mTOR (1:500) and β-actin (1:500) were added, respectively, overnight at 4 °C. After three times of washing with TBS, the samples were incubated with horseradish peroxidase-labeled secondary antibody (1:1000) for 2 h at room temperature. After washing with TBS, ECL luminescence test was carried out. β-actin was used as the internal control to evaluate the expression level of each sample.
Cell Uptake
MSCTs were seeded in a confocal dish with 5 × 105 cells per well, containing 100 μg of pure EXO. The cells were labeled with DiR (60,017, Biotium) in 1 mL of PBS, ultracentrifugated at 1 × 105 g for 3 h, and resuspended with PBS. They were incubated at 37 °C, 0 °C, 5 °C, 10 °C, 15 °C, or 4 °C (pH 7.4) by using an Opti-MEM medium instead of the original medium. Then, they were washed with cold PBS twice, fixed with 4% paraformaldehyde for 15 min at room temperature, washed with PBS twice, and stained for 15 min with Hoechst 33,342 (63,493, Sigma Aldrich) at 1:2000 at room temperature. Confocal laser scanning showed the cellular uptake of nanoparticles. To measure the uptake of EXO, trypsin was used to digest the cells, PBS was used to wash them, and the DiR signal was analyzed by flow cytometry (Invitrogen Attune NxT, Thermo Fisher, USA).
Establishment of Xenograft Model
SCC25 cells were digested and collected, washed with serum-free medium RPMI-1640 twice, and resuspended with a serum-free medium to make the cell density 5×106 cells/mL. The mice were placed in a biosafety cabinet and anesthetized with an anesthetic breathing device. The anesthetic gas was isoflurane. After anesthesia, 75% ethanol was used to wipe the back of the mice for disinfection, and an insulin needle was used to suck 120 μL of cell suspension, which was injected subcutaneously into the right rear back of the mice. After the modeling, the mice were put back into the cage and then into the feeding room after waking up. The state and size of the transplanted tumor were observed regularly. After 1–2 weeks, the transplanted tumor grew to 100–200 mm3, and then the drug was given. All animal experiments were approved by the animal ethics committee of Hebei Medical University (201,902,007). All experimental procedures were conducted in accordance with the guidelines for the humane treatment of experimental animals (《The Three Rs and the Humanity Criterion》, published in 2009).
Pharmacodynamic Study in Animal Groups
The tumor-bearing mice were randomly divided into four groups with five animals in each group. The groups were the saline, CTX, MSCT-EXO, MSCT-EXO:CTX, and MSCT-EXO/CTX groups. The dosage of MSCT-EXO was 1.59 mg/kg, MSCT-EXO/CTX was 1.61 mg/kg, that of MSCT-EXO:CTX was 1.59 mg/kg±0.25 mg/kg, and that of free CTX was 0.25 mg/kg. The frequency of administration was once every 2 days, the length and width of the transplanted tumor were measured using a vernier caliper before each administration, and the weight data of mice were recorded. After each recording, the tumor volume was calculated using the following formula: tumor volume V (mm3) = length × width2/2.
At the end of the experiment, the animals were euthanized with a carbon dioxide suffocation. After the tumor was removed and PBS was used to clean the residual blood, photos were taken and recorded. Each tissue (tumor) was weighed to calculate the tumor inhibition rate.
Tumor inhibition rate (%) = (tumor weight in the PBS group − tumor weight in the experimental group)/tumor weight in the PBS group × 100%
Statistical Analysis
The data were analyzed using GraphPad Prism 6 software and one-way ANOVA. Dunnett’s post-test or Student’s t-test was used to compare the differences. The significant probability values were *P < 0.05, **P < 0.01, and **P < 0.001.
Results
Isolation and Characterization of MSC-EXO
The MSCT-EXO/CTX was characterized by particle size. The experimental results showed that the particle size of the synthesized MSCT-EXO/CTX EXO was approximately 60–150 nm. Particle size uniformity and narrow size distribution were observed (Figure 1A and B). CD63 and CD9 were typical markers of MSC-EXO (Figure 1C); CD63, CD9, and TRAIL were expressed in MSCT-EXO (Figure 1C and D); CD81 was expressed in MSCs (Figure 1C).
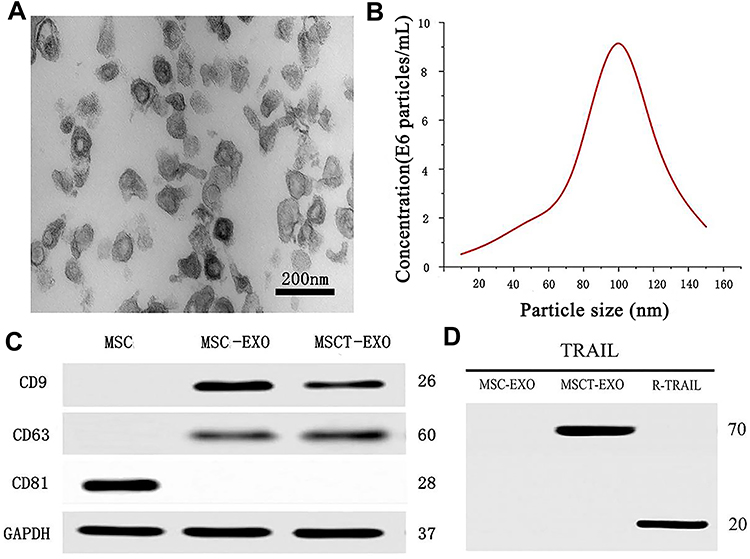 |
Figure 1 (A) TEM image of MSCT-EXO with a scale of 200 nm. (B) Size distribution of MSCT-EXO. (C) Western blot analysis of CD63, CD9, and CD81 in MSC, MSC-EXO, and MSCT-EXO. (D) Western blot analysis of TRAIL in MSC-EXO, MSCT-EXO, and R-TRAIL used as a positive control. |
Drug Loading and in vitro Drug Release
A regression equation prediction model was established by the concentration of CTX standard solution detected by HPLC (Figure 2A). The entrapment efficiency and drug loading were 93.7±1.53% and 15.43±0.44%, respectively. The drug content in MSCT-EXO/CTX was detected by dialysis. The release profile of CTX was detected by dialysis in a phosphoric acid buffer medium containing 0.5% Tween-80. Figure 2B shows that in the release medium, 57.8% of CTX was released from MSCT-EXO/CTX within 10 h, and 82.5% of CTX was released within this period after reaching the final release time point. In the first 15 h, MSCT-EXO/CTX showed a fast release feature. Afterward, the drug release rate slowed down, and the final drug release entered the platform stage. Finally, almost no CTX was released from MSCT-EXO/CTX.
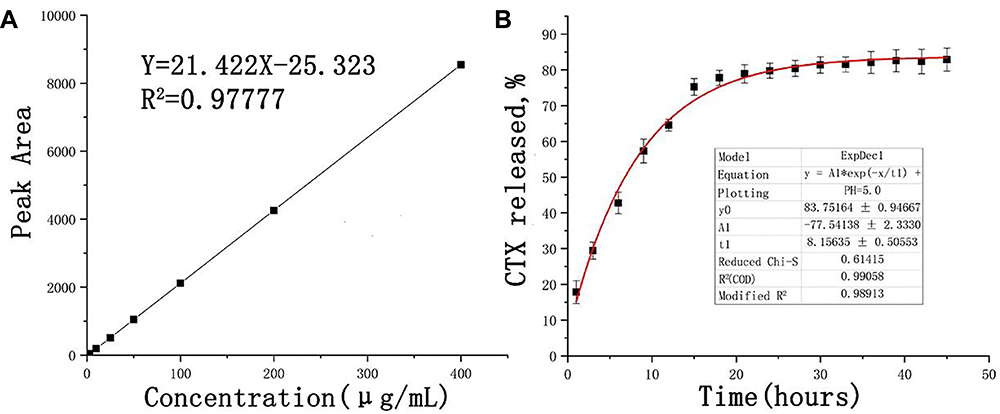 |
Figure 2 (A) Regression curve of CTX based on the HPLC results; (B) Release curve of CTX. |
Cytotoxicity
Cytotoxicity and Half Maximal Inhibitory Concentration (IC50)
Before testing the effect of drugs, whether MSCT-EXO can inhibit the growth of SCC25 cells was determined. The results showed that MSCT-EXO and CTX could inhibit the growth of SCC25 cells in a dose-dependent manner (Figure 3A and B). Figure 3C depicts that the proportion of CTX and MSCT-EXO was closely related to their synergistic killing effect on cancer cells. The IC50 values of free CTX and MSCT-EXO in various proportions were relatively different. MSCT-EXO and CTX exerted a high synergistic effect when the proportional range was 10:1–5:1. MSCT-EXO/CTX and MSCT-EXO:CTX could inhibit the activity of SCC25 cells (Figure 3D). The inhibitory effect of MSCT-EXO/CTX was significantly higher than that of the MSCT-EXO:CTX group (P < 0.05), and the combined index (CI) value was lower (CI = 0.55) at the same proportion. Therefore, we speculated that the MSCT-EXO/CTX group exhibited a good synergistic effect in the range of 10:1–5:1. However, the optimal synergistic ratio could not be tested due to the limitation of the drug loading of MSCT-EXO.
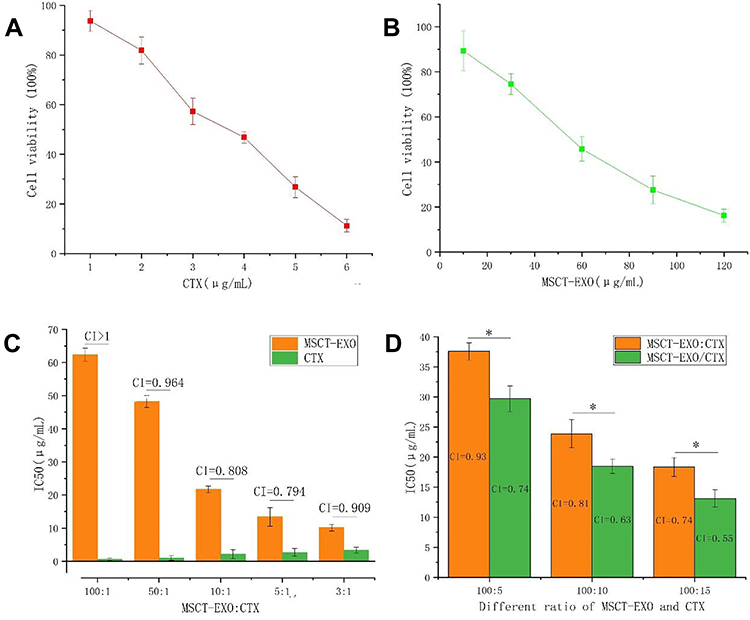 |
Figure 3 (A) Inhibition effects of different concentrations of CTX on SCC25 cells; (B) Inhibition effects of different concentrations of MSCT-EXO on SCC25 cells; (C) IC50 and CI values of different proportions of MSCT-EXO and CTX; (D) IC50 and CI values of MSCT-EXO:CTX and MSCT-EXO/CTX with different proportions of MSCT-EXO and CTX in SCC25 cells. *Comparison between MSCT-EXO:CTX and MSCT-EXO/CTX groups, P < 0.05. |
Apoptosis Induced by MSCT-EXO/CTX
Apoptosis effects were induced by CTX, MSCT-EXO, MSCT-EXO:CTX and MSCT-EXO/CTX on SCC25 cells in a dose-dependent manner, in which CTX had a limited role in apoptosis induction (Table 1). CTX loaded by MSCT-EXO was more effective in killing cancer cells than free MSCT-EXO:CTX (100:15) group, which induced more apoptosis in SCC25 cells compared with free MSCT-EXO:CTX at different concentrations (P<0.05). Annexin V/DAPI was used to confirm the killing capability of MSCT-EXO/CTX to SCC25 cells. The results showed that the apoptosis-inducing effect of MSCT-EXO/CTX on SCC25 cells was higher than that of other groups (P<0.05). These data indicated that MSCT-EXO/CTX could effectively induce cancer cell apoptosis, which could also reduce the dosage of free drugs (Figure 4, Table 1).
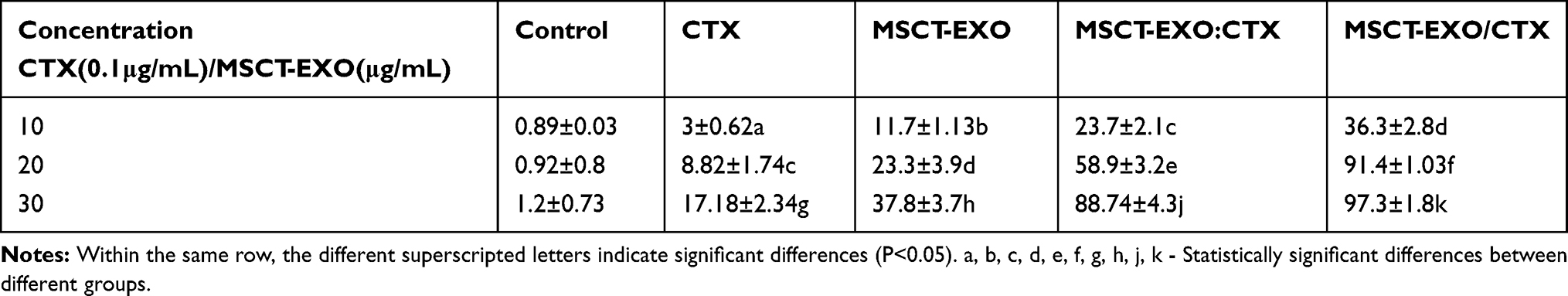 |
Table 1 The Apoptosis Rate in Different Groups (±S, n=4) |
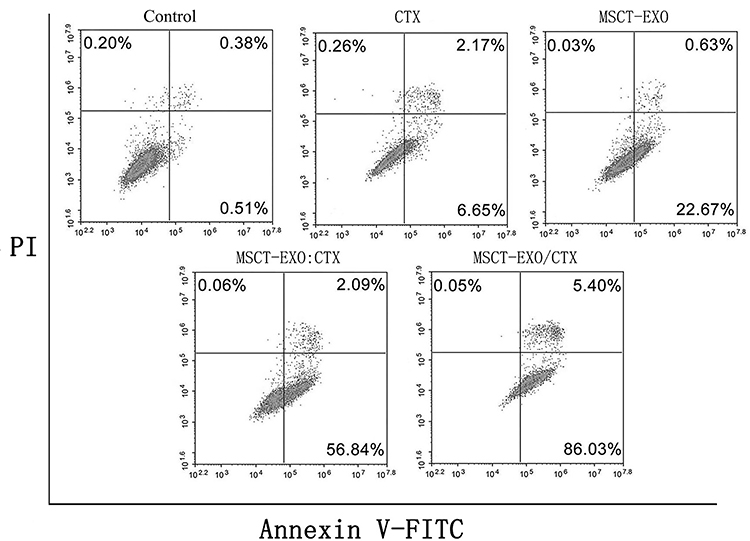 |
Figure 4 Apoptosis detected using annexin V/DAPI. SCC25 cells were treated with CTX (2μg/mL) MSCT-EXO (20μg/mL), MSCT -EXO:CTX (20μg/mL), MSCT-EXO/CTX (20μg/mL). |
Cell Uptake
Human EXO can be internalized by cancer cells to inhibit the proliferation of cancer cells and kill them. To determine whether MSCT-EXO has direct effects on squamous cell carcinoma, we evaluated MSCT-EXO internalization in SCC25 cells. Flow cytometry and confocal microscopy were used to detect the intracellular staining of SCC25 cells and quantify endocytosis in a time-dependent manner. As shown in Figure 5, SCC25 cells effectively ingested EXO in an increasing manner during 15 h. The geometric mean of fluorescence intensity (MFI, SCC25) increased from 827 ± 136 in 5 h to 1362 ± 193 in 10 h and to 2035 ± 324 in 15 h because substantial chromatids were internalized. The MIF of the negative control group at 4 °C was 382 ± 105, which confirmed the active internalization of EXOs to cancer cells. To determine whether the EXOs are certainly internalized or only membrane bound, we confirmed their cytoplasmic localization via confocal microscopy, as shown in Figure 5. The EXOs of confocal microscopy were stained with DiR (yellow). Confocal microscopy confirmed that the EXOs were effective with time-dependent internalization into SCC25 cells.
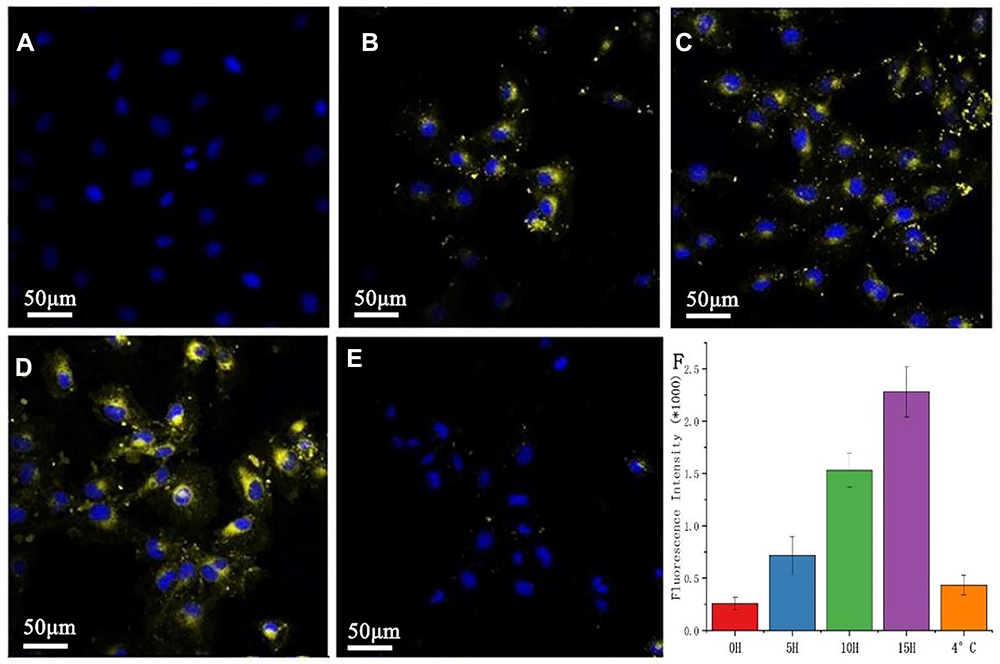 |
Figure 5 Rapid uptake of MSCT-EXO by SCC25 cells. (A–E) represent 0 h, 5 h, 10 h, and 15 h respectively, incubated SCC25 with DiR-labeled EXO observed using a confocal microscope. (F) refers to the quantitative results detected by flow cytometry. Laser confocal microscope scale: 50 μm. The bar graph of the flow cytometer shows the average value, error line, and N = 3. |
Inhibition of PI3K/Akt/mTOR Signaling Pathway Activation
Western blotting showed that SCC25 cells treatment with CTX (2 μg/mL) MSCT-EXO (20 μg/mL), MSCT-EXO:CTX (20 μg/mL) and MSCT-EXO/CTX (20 μg/mL) for 24 h, the phosphorylation levels of PI3K, Akt and mTOR were significantly inhibited (Figure 6, P<0.05), while the expression of total PI3K, Akt and mTOR did not change significantly (P>0.05). The inhibition of PI3K/Akt/mTOR signaling pathway activation by MSCT-EXO/CTX on SCC25 cells was higher than that of other groups (P<0.05).
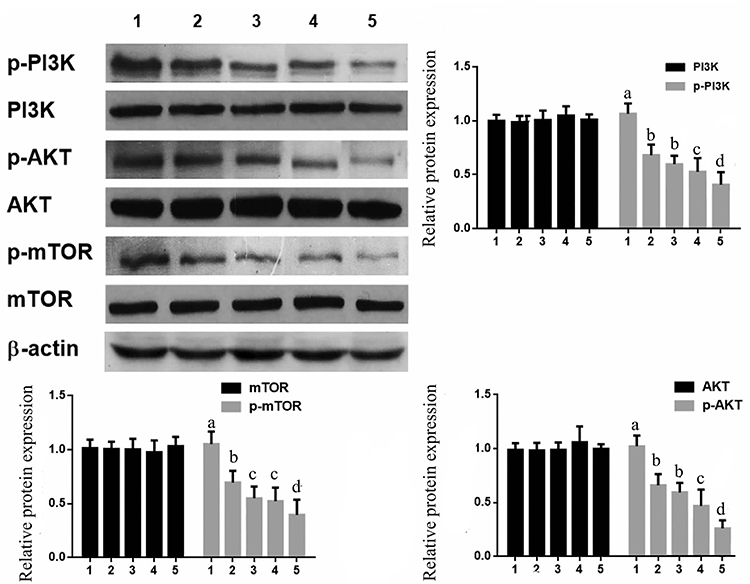 |
Figure 6 Effects of MSCT-EXO/CTX on PI3K/Akt/mTOR signaling pathway in SCC25 cells 1: Control; 2: CTX; 3: MSCT-EXO ;4: MSCT-EXO:CTX; 5: MSCT-EXO/CTX. a, b, c, d- Statistically significant differences between different groups. |
Antitumor Effect of MSCT-EXO/CTX in vivo
At the beginning of tumor growth, the tail vein was injected once every other day, the length and width of tumor were measured with a vernier caliper at each time of administration, and the weight of mice was recorded. The entire experimental period lasted 19 days. At the end of the experiment, the mice were euthanized, and the transplanted tumor was dissected and weighed. The toxicity and side effects of the nanosystem could be evaluated using the body weight change of mice. As showed in Figure 7A, the transplanted tumor grew rapidly in the PBS group. There was no significant change in the weight of mice treated with MSCT-EXO, MSCT-EXO:CTX and MSCT-EXO/CTX (Figure 7B). The weight of mice in CTX group decreased to 93.48%, and no weight recovery was observed at 18 days, which may be due to the non-specific toxicity of CTX. The MSCT-EXO/CTX group showed the highest antitumor effects, and the tumor growth inhabitation was approximately 63.7% (Figure 7C). As shown in Figure 7D, compared with PBS group, the tumor volume of mice treated with CTX, MSCT-EXO, MSCT-EXO:CTX or MSCT-EXO/CTX group decreased significantly (P<0.05). MSCT-EXO/CTX group showed the lowest relative tumor volume and the highest tumor inhibition rate (P < 0.05).
 |
Figure 7 In vivo antitumor effects of the MSCT-EXO/CTX nanosystem (A) Picture of the heterotopic transplanted tumor in mice; (B) Tumor weight curve of each group; (C) Tumor inhibition rate of each group; (D) Tumor volume curve of each group. a, b, c, d - Statistically significant differences between different groups. |
Discussion
CTX is a chemical semisynthetic small-molecule compound of taxanes.27 In June 2010, the US Food and Drug Administration approved CTX to treat cancer patients who have not improved after docetaxel treatment. In the following years, it was listed in the United States, Germany, Britain, Italy, and other countries. As a taxane drug, CTX has the same action mechanism as paclitaxel and docetaxel, which are used to stabilize microtubules. CTX can bind to p-tubulin subunits, which can make cell growth stay at G2/M phase and promote stable microtubules, thus inhibiting cell mitosis and interphase cell function.28 Compared with docetaxel and paclitaxel, CTX has a better antitumor activity when used as a small-molecule chemotherapeutic drug in tumor treatment. Therefore, CTX has been used as an alternative drug for paclitaxel-resistant patients.29 However, as an anticancer chemotherapy drug, it has the same disadvantages, such as hydrophobic property, as other drugs. Thus, Jevtana, a type of commercial product of cabazitaxel, uses polysorbate 80 (Tween 80) as a solvent (containing 60 mg of CTX in 1.5 mL of polysorbate 80), but it has created serious adverse effects in clinic.30,31 To inhibit the disadvantages of poor water solubility and serious side effects, a series of dosage forms is derived from the raw material, among which nanoparticles are one of the best types. Considerable literature has proven that the derivatives of these systems have high efficacy and low toxicity.30
TRAIL has been widely concerned by researchers since its discovery. The specific killing effect on tumor cells has been leading people to explore the clinical application of TRAIL.32 Nevertheless, for a long time, neither the recombinant TRAIL protein with increased stability nor the more specific TRAIL antibody has achieved clinical success. Therefore, the single drug application of TRAIL might not be able to achieve the desired effect, but it might be a way to explore the combination of TRAIL with other drugs.33,34 The study of TRAIL indications should also be conducted in parallel. The drug resistance of tumor cells to TRAIL is a considerable obstacle to its application.35 At present, finding unique biological indicators sensitive to TRAIL is particularly important.
Bone marrow mesenchymal stem cells usually do not express secretory TRAIL.36 Under TNF-α stimulation, bone marrow mesenchymal stem cells express membrane-binding TRAIL but not secretory TRAIL.37 MSCTs can secrete EXO-carrying TRAIL. In addition, the expression of EXO in MSCTs is higher than that in untransfected MSCs or empty body lentivirus-transfected MSCs. For the treatment method of TRAIL, the combination of other drug and nanotechnology can enhance the tumor-targeting effect of TRAIL and reduce the side effects, thereby providing a promising treatment method.
The cell source of EXO is important to judge whether EXOs have antitumor activity or promote tumor growth. For example, EXO derived from human umbilical cord Wharton’s jelly mesenchymal stem cells has been shown to reduce the growth of T24 bladder cancer.38 Similarly, EXO isolated from normal human bone marrow mesenchymal stem cells has been reported to inhibit cell cycle progression and induce the apoptosis of hepatoma (HepG2) and Kaposi cells.39 However, Roccaro et al found that BMSCs from multiple myeloma patients can promote the growth of multiple myeloma tumor cells, whereas BMSCs from healthy patients can inhibit the growth of multiple myeloma tumor cells.40 Therefore, the function of EXO is closely related to the source of MSCs. Choosing appropriate MSCs could avoid possible adverse reactions.
In this study, CTX-induced MSCT-EXO was prepared through ultracentrifugation and 0.22 μm filtration. The results of TEM and DLS showed that EXO nanoparticles had a narrow particle size distribution. The EXO system contained TRAIL and CTX and exerted a good antitumor effect through the synergistic apoptosis-promoting effect. MSCs were first transfected with TRAIL (MSCTs) and then incubated with CTX. High-speed centrifugation could remove the microbubbles with particle sizes higher than 220 nm, followed by further purification via sucrose density gradient ultracentrifugation. The composite EXO exhibited high drug loading with a good release curve. The delivery system showed a good antitumor activity in vitro, and the antitumor mechanism of the codrug-loaded system was also investigated at the cellular level. The results showed that the nanosystem could promote the apoptosis of tumor cells through synergistic effect, which was consistent with the cytotoxic test. PI3K, Akt and mTOR play important roles in many cell processes, including proliferation, apoptosis, cell cycle progression, cell migration and autophagy. To better understand the mechanism of cell cycle arrest and apoptosis induced by MSCT-EXO/CTX, the effect of MSCT-EXO/CTX on OSCC cell targets was studied by Western blot. We demonstrated that MSCT-EXO/CTX effectively reduced the protein levels of p-PI3K, p-AKT and p-mTOR. MSCT-EXO/CTX showed better efficacy than free mixed drug group.
Compared with the MSCT-EXO:CTX group, the MSCT-EXO/CTX group showed higher toxicity and apoptosis rate and a better synergistic effect. This result might be attributed to the fact that the MSCT-EXO/CTX group had a fixed proportion and was a whole, which could enter the cells at a fixed proportion at the same time, thus showing a stronger cytotoxicity and a higher apoptosis rate. This phenomenon is consistent with previous studies, such as those on gemcitabine and paclitaxel loaded in mesoporous silicon41 and cisplatin and metformin-loaded nanocubes.42 Both groups showed higher cytotoxicity or apoptosis induction than the free drug group. In addition to the abovementioned reasons in vitro, the high efficiency in vivo could also be attributed to the cell-homing effect of EXO derived from mesenchymal stem cells and the short half-life of the hydrophobic drug CTX in vivo.
To sum up, we successfully collected CTX-related EXO from MSCT and proved that MSCT-EXO can induce the apoptosis of OSCC partly via the inhibition of PI3K/Akt/mTOR signaling pathway activation. In particular, MSCT-EXO can significantly improve the apoptosis effect of CTX on SCC25. However, the apoptosis-promoting mechanism of MSCT-EXO with CTX and the effect of tumor homing need to be further studied. With the in-depth study of stem cell EXO and the extensive application of synergistic therapy, drug-induced MSCT-EXO is expected to develop into a clinical anticancer treatment method.
Funding
This work was supported by Hebei Key Project Plan of Medical Science Research (No. 20200102).
Disclosure
The authors report no conflict of interest for this work.
References
1. Holland PM. Targeting Apo2L/TRAIL receptors by soluble Apo2L/TRAIL. Cancer Let. 2013;332(2):156–162. doi:10.1016/j.canlet.2010.11.001
2. Belkahla H, Herlem G, Picaud F, et al. TRAIL–NP hybrids for cancer therapy: a review. Nanoscale. 2017;9(18):5755–5768. doi:10.1039/C7NR01469D
3. Dianat-Moghadam H, Heidarifard M, Mahari A. et al. TRAIL in oncology: from recombinant TRAIL to nano-and self-targeted TRAIL-based therapies. Pharm Res;2020. 104716. doi:10.1016/j.phrs.2020.104716
4. Yu B, Zhang X, Yan J, et al. Improved stability, antitumor effect, and controlled release of recombinant soluble trail by combining genetic engineering with coaxial electrospinning. ACS Appl Bio Mater. 2019;2(6):2414–2420.
5. Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314–341. doi:10.1016/j.ejmech.2016.01.012
6. Zhang X, Li S, Zheng M, et al. Effects of the PI3K/Akt signaling pathway on the apoptosis of early host cells infected with Eimeria tenella. Parasitol Res. 2020;1–13.
7. Huo R, Wang L, Liu P, et al. Cabazitaxel-induced autophagy via the PI3K/Akt/mTOR pathway contributes to A549 cell death. Mol Med Rep. 2016;14(4):3013–3020. doi:10.3892/mmr.2016.5648
8. de Porras VR, Laguía F, Marín-Aguilera M, et al. Effect of selumetinib plus AZD8186 treatment on cabazitaxel sensitivity in docetaxel-acquired resistant metastatic prostate cancer cell lines. Ann Oncol. 2019;30:v785. doi:10.1093/annonc/mdz268.077
9. Vrignaud P, Sémiond D, Lejeune P, et al. Preclinical antitumor activity of cabazitaxel, a semisynthetic taxane active in taxane-resistant tumors. Clin Cancer Res. 2013;19(11):2973–2983. doi:10.1158/1078-0432.CCR-12-3146
10. Sun B, Straubinger RM, Lovell JF. Current taxane formulations and emerging cabazitaxel delivery systems. Nano Res. 2018;11(10):5193–5218. doi:10.1007/s12274-018-2171-0
11. Paolicelli RC, Bergamini G, Rajendran L. Cell-to-cell communication by extracellular vesicles: focus on microglia. Neuroscience. 2019;405:148–157. doi:10.1016/j.neuroscience.2018.04.003
12. Zappulli V, Friis KP, Fitzpatrick Z, et al. Extracellular vesicles and intercellular communication within the nervous system. J Clin Investig. 2016;126(4):1198–1207. doi:10.1172/JCI81134
13. Rahman MA, Patters BJ, Kodidela S, et al. Extracellular vesicles: intercellular mediators in alcohol-induced pathologies. J Neuroimmune Pharm. 2019;1–13.
14. Kabanov AV, Batrakova EV. Polymer nanomaterials for drug delivery across the blood brain barrier. J Neuroimmune Pharm. 2017;847–868.
15. Li X, Tsibouklis J, Weng T, et al. Nano carriers for drug transport across the blood–brain barrier. J Drug Target. 2017;25(1):17–28. doi:10.1080/1061186X.2016.1184272
16. Patel MM, Patel BM. Crossing the blood–brain barrier: recent advances in drug delivery to the brain. CNS Drugs. 2017;31(2):109–133. doi:10.1007/s40263-016-0405-9
17. Zhang D, Qin X, Wu T, et al. Extracellular vesicles based self-grown gold nanopopcorn for combinatorial chemo-photothermal therapy. Biomaterials. 2019;197:220–228. doi:10.1016/j.biomaterials.2019.01.024
18. Saari H, Lázaro-Ibáñez E, Viitala T, et al. Microvesicle-and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J Control Release. 2015;220:727–737. doi:10.1016/j.jconrel.2015.09.031
19. Cai J, Wu G, Jose PA, et al. Functional transferred DNA within extracellular vesicles. Exp Cell Res. 2016;349(1):179–183. doi:10.1016/j.yexcr.2016.10.012
20. Sotillo J, Pearson M, Potriquet J, et al. Extracellular vesicles secreted by Schistosoma mansoni contain protein vaccine candidates. Int J Parasitol. 2016;46(1):1–5. doi:10.1016/j.ijpara.2015.09.002
21. Furuta T, Miyaki S, Ishitobi H, et al. Mesenchymal stem cell‐derived exosomes promote fracture healing in a mouse model. Stem Cells Transl Med. 2016;5(12):1620–1630. doi:10.5966/sctm.2015-0285
22. Shi B, Wang Y, Zhao R, et al. Bone marrow mesenchymal stem cell-derived exosomal miR-21 protects C-kit+ cardiac stem cells from oxidative injury through the PTEN/PI3K/Akt axis. PLoS One. 2018;13(2):e0191616. doi:10.1371/journal.pone.0191616
23. Kalra H, Adda CG, Liem M, et al. Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics. 2013;13(22):3354–3364. doi:10.1002/pmic.201300282
24. El-Andaloussi S, Lee Y, Lakhal-Littleton S, et al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. 2012;7(12):2112. doi:10.1038/nprot.2012.131
25. Kooijmans SAA, Aleza CG, Roffler SR, et al. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J Extracell Vesicles. 2016;5(1):31053.
26. Sage EK, Kolluri KK, McNulty K, et al. Systemic but not topical TRAIL-expressing mesenchymal stem cells reduce tumour growth in malignant mesothelioma. Thorax. 2014;69(7):638–647. doi:10.1136/thoraxjnl-2013-204110
27. De Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147–1154. doi:10.1016/S0140-6736(10)61389-X
28. Duran GE, Wang YC, Francisco EB, et al. Mechanisms of resistance to cabazitaxel. Mol Cancer Ther. 2015;14(1):193–201. doi:10.1158/1535-7163.MCT-14-0155
29. Azarenko O, Smiyun G, Mah J, et al. Antiproliferative mechanism of action of the novel taxane cabazitaxel as compared with the parent compound docetaxel in MCF7 breast cancer cells. Mol Cancer Ther. 2014;13(8):2092–2103. doi:10.1158/1535-7163.MCT-14-0265
30. He Z, Schulz A, Wan X, et al. Poly (2-oxazoline) based micelles with high capacity for 3rd generation taxoids: preparation, in vitro and in vivo evaluation. J Controlled Release. 2015;208:67–75. doi:10.1016/j.jconrel.2015.02.024
31. Coors EA, Seybold H, Merk HF, et al. Polysorbate 80 in medical products and nonimmunologic anaphylactoid reactions. Ann Allergy, Asthma Immunol. 2005;95(6):593–599. doi:10.1016/S1081-1206(10)61024-1
32. Azijli K, Weyhenmeyer B, Peters GJ, et al. Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: discord in the death receptor family. Cell Death Differ. 2013;20(7):858–868. doi:10.1038/cdd.2013.28
33. Hellwig CT, Rehm M. TRAIL signaling and synergy mechanisms used in TRAIL-based combination therapies. Mol Cancer Ther. 2012;11(1):3–13. doi:10.1158/1535-7163.MCT-11-0434
34. Lecis D, Drago C, Manzoni L, et al. Novel SMAC-mimetics synergistically stimulate melanoma cell death in combination with TRAIL and Bortezomib. B J Cancer. 2010;102(12):1707–1716. doi:10.1038/sj.bjc.6605687
35. Garofalo M, Di Leva G, Romano G, et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16(6):498–509. doi:10.1016/j.ccr.2009.10.014
36. Subramanian A, Shu‐Uin G, Kae‐Siang N, et al. Human umbilical cord Wharton’s jelly mesenchymal stem cells do not transform to tumor‐associated fibroblasts in the presence of breast and ovarian cancer cells unlike bone marrow mesenchymal stem cells. J Cell Biochem. 2012;113(6):1886–1895. doi:10.1002/jcb.24057
37. Guiho R, Biteau K, Heymann D, et al. TRAIL-based therapy in pediatric bone tumors: how to overcome resistance. Future Oncol. 2015;11(3):535–542. doi:10.2217/fon.14.293
38. Wu S, G Q J, Du T, et al. Microvesicles derived from human umbilical cord Wharton’s jelly mesenchymal stem cells attenuate bladder tumor cell growth in vitro and in vivo. PLoS One. 2013;8(4):e61366. doi:10.1371/journal.pone.0061366
39. Bruno S, Collino F, Deregibus MC, et al. Microvesicles derived from human bone marrow mesenchymal stem cells inhibit tumor growth. Stem Cells Dev. 2013;22(5):758–771. doi:10.1089/scd.2012.0304
40. Roccaro AM, Sacco A, Maiso P, et al. BM mesenchymal stromal cell–derived exosomes facilitate multiple myeloma progression. J Clin Investig. 2013;123(4):1542–1555. doi:10.1172/JCI66517
41. Meng H, Wang M, Liu H, et al. Use of a lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice. ACS Nano. 2015;9(4):3540–3557. doi:10.1021/acsnano.5b00510
42. Saber MM, Al-Mahallawi AM, Nassar NN, et al. Targeting colorectal cancer cell metabolism through development of cisplatin and metformin nano-cubosomes. BMC Cancer. 2018;18(1):822. doi:10.1186/s12885-018-4727-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.