")
Back to Journals » International Journal of Nanomedicine » Volume 12
Antitumor activity of a folate receptor-targeted immunoglobulin G-doxorubicin conjugate
Authors Yang T, Xu L, Li B, Li W, Ma X, Fan L, Lee RJ, Xu C , Xiang G
Received 24 October 2016
Accepted for publication 13 February 2017
Published 30 March 2017 Volume 2017:12 Pages 2505—2515
DOI https://doi.org/10.2147/IJN.S125591
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Tan Yang,1,* Ling Xu,1,* Bin Li,1 Weijie Li,1 Xiang Ma,1 Lingling Fan,2 Robert J Lee,3 Chuanrui Xu,1 Guangya Xiang1
1Department of Biopharmaceuticals, School of Pharmacy, 2Stem Cell Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, People’s Republic of China; 3Division of Pharmaceutics and Pharmaceutical Chemistry, College of Pharmacy, The Ohio State University, Columbus, OH, USA
*These authors contributed equally to this work
Abstract: Development of antibody-drug conjugates (ADCs) is a promising therapeutic strategy for cancer therapy. In this study, folate was conjugated via a polyethyleneglycol (PEG) linker to immunoglobulin G (IgG), which was linked to doxorubicin (DOX), to form a novel ADC folate-PEG-IgG-DOX (FA-PEG-IgG-DOX). The FA-PEG-IgG-DOX showed high targeting efficiency in HeLa and KB cells and significantly improved the uptake and retention of DOX compared with IgG-DOX about 10-fold. Subsequently, FA-PEG-IgG-DOX was shown to have at least 8 times higher antitumor activity than IgG-DOX both in HeLa and KB cells and also induced more apoptosis in those cells than IgG-DOX. Moreover, FA-PEG-IgG-DOX had a 2 times longer circulating time than FA-IgG-DOX, but did not increase the DOX distribution in mouse hearts. Importantly, FA-PEG-IgG-DOX treatment significantly inhibited tumor growth in xenograft mice. Together, our results indicate that FA-PEG-IgG is an effective ADC carrier for delivery of chemotherapeutic agents and that conjugating tumor targeting ligands to antibodies is a promising strategy for producing ADC drugs.
Keywords: folate, immunoglobulin G, doxorubicin, antibody-drug conjugate, cancer
Introduction
Since the 1980s, two generations of antibody–drug conjugates (ADCs) have been developed. First-generation ADC products did not achieve widespread clinical application due to unstable linkage between the antibody and the drug.1 Second-generation ADCs circumvented this barrier using novel linkers to improve the stability of ADCs in the bloodstream and maintain the integrity of ADCs until they reach tumor cells.2
Another barrier to ADC development is the lack of effective receptors, which is critical for the recognition and internalization of ADC drugs by targeting cells. Since ADC/antigen complex are typically internalized by receptor-mediated endocytosis, the internalization efficiency of ADCs depends at least in part on the identity of the target antigen.3 Inefficient internalization will result in insufficient concentration of cytotoxicity and lead to low treatment effects. Unfortunately, most cell-surface targets are internalized at moderate rates.4 Therefore, identifying appropriate targeting antigens still presents an urgent need in designing an ADC strategy.
Folate receptor (FR) is a glycosyl phosphatidylinositol (GPI)-anchored membrane protein that is overexpressed in over 90% ovarian carcinomas and in other epithelial cancers to varying degrees.5–9 Overexpression of FR generally promotes proliferation and resistance of cancer cells to chemotherapy.8 Conversely, the expression levels of FR in normal tissues are much lower than tumor tissues.9,10 Differential expression of FR in normal tissue and malignant tissue makes it an ideal target for drug delivery. As a natural ligand of FR, folate shows highly selective affinity to FR.11–13 Folate is required for survival and rapid proliferation of tumor cells. Tumor cells take up folate by internalizing folate or folate-conjugates via receptor-mediated endocytosis,14,15 and this process is rapid and efficient. Hence, FR may serve as an appropriate target for ADCs design and folate can be an ideal recognition ligand.
In this study, we designed and synthesized a novel ADC drug, folate-polyethylene glycol-immunoglobulin G-doxorubicin (FA-PEG-IgG-DOX). In this conjugate, FA-PEG-IgG was designed as a FR-targeting antibody and has the potential to display antibody-dependent cytotoxicity (ADCC) compared with albumin or other protein carriers. We explored its targeting effects and internalization efficiency of FA-PEG-IgG-DOX and found it has better tumor cell targeting effects and internalization efficiency than IgG-DOX. In addition, FA-PEG-IgG-DOX showed higher antitumor activity than IgG-DOX in vitro, but significantly lower toxicity than DOX in tumor-bearing nude mice. Our data support the concept of using FA-PEG-IgG as an ADC carrier to promote antitumor activity and reduce side effects of chemotherapeutic agents.
Materials and methods
Chemicals
Doxorubicin hydrochloride (DOX·HCl), folate, 1-ethyl-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), L-glutamate, polyethylene glycol 3350-bis-amine (NH2-PEG-NH2), 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT), Sephadex G-75 chromatography media, and folate-free Roswell Park Memorial Institute (RPMI) 1640 cells culture medium were purchased from Sigma-Aldrich Chemical Co (St Louis, MO, USA). Dulbecco’s Modified Eagle’s Medium (DMEM) high-glucose medium was purchased from the Hyclone of Thermo Scientific (Chicago, IL, USA). Trypsin and bicinchoninic acid (BCA) protein assay kit were purchased from Beyotime Institute of Biotechnology (Wuhan, People’s Republic of China). PD-10 desalting columns (G25) were purchased from GE Healthcare Biosciences (Pittsburgh, PA, USA). All reagents and solvents were of analytical or high-performance liquid chromatography grade and were used without further purification.
Cell culture
HeLa and KB cells are from the China Center for Type Culture Collection at Wuhan University (Wuhan, People’s Republic of China). The cells were cultured with folate-free RPMI 1640 or high-glucose DMEM medium supplemented with penicillin, streptomycin, and 10% FBS at 37°C in a 5% CO2 incubator.
Preparation of FA-PEG-IgG-DOX
The synthetic procedure is shown in Figure 1. For the synthesis of IgG-DOX, DOX (30 mg) was resolved in PBS buffer (3 mL) and then reacted with NaIO4 (12 mg) at 25°C for 1 h in the dark. Then glycerol (160 μL) was added to a final concentration of 0.05 M, and the reaction mixture was stirred at 25°C for 0.5 h. Subsequently, the solution of IgG (105 mg) in carbonate buffer (2 mL pH 9.5) was added and the reaction solution was stirred at room temperature for 1 h in the dark. Next, NaCNBH3 (2.0 mg) was added to a final concentration of 0.3 mg/mL, and the reaction was stirred at 37°C in the dark for 2 h. The reaction mixture was then centrifuged (6,000 rpm, 10 min) and the supernatant was passed through a Sephadex G-25 column. Product concentration in the effluent was determined with a UV-Vis spectrophotometer, and the product was freeze-dried and stored at 4°C.
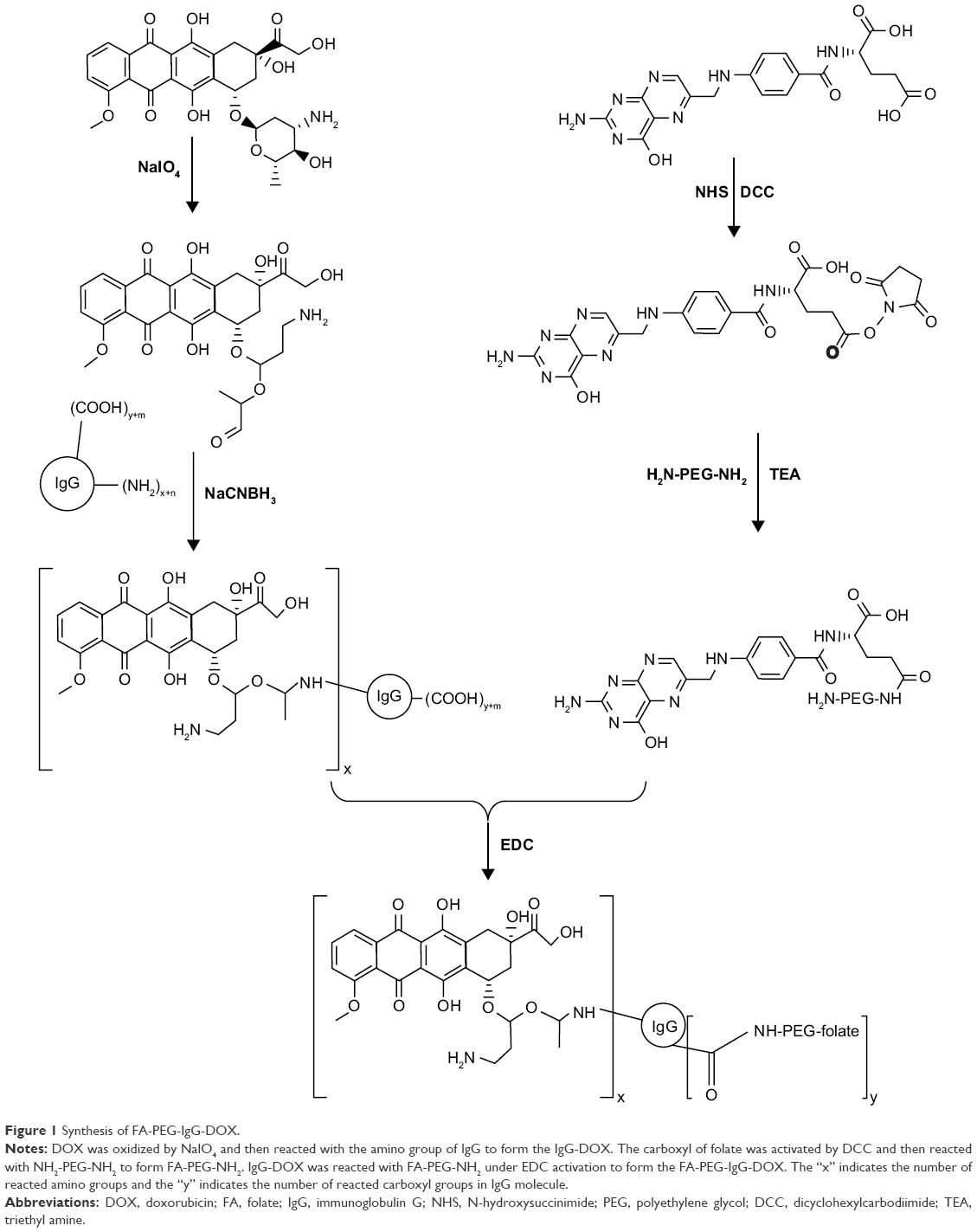 | Figure 1 Synthesis of FA-PEG-IgG-DOX. |
For the synthesis of FA-PEG, folate (26.5 mg) was dissolved in DMSO (1 mL) (pretreated with calcium chloride to remove moisture) in an oil bath. Then, NH2-PEG-NH2 (167.5 mg), DCC (15.5 mg), NHS (8.63 mg), and triethylamine (35 μL) were sequentially added. The reaction was carried out at 30°C in an oil bath overnight. The reaction solution was centrifuged at 8,000 rpm for 10 min. Then, two volumes of sodium carbonate solution (50 mM) were added and the solution was centrifuged at 8,000 rpm for 10 min. The supernatant was then passed through a Sephadex G-25 column and the early fractions were collected. The collected product was freeze dried.
For the synthesis of FA-PEG-IgG-DOX, IgG-DOX (40 mg) was dissolved in MES buffer (2 mL) and then stirred with EDC (120 mg) and NHS (60 mg) for 1 h at room temperature in the dark. Then FA-PEG (30 mg) was added and the reaction proceeded for 2 h at room temperature in the dark. The reaction mixture was passed through a G-75 gel column and the effluent was collected and detected by spectral scanning.
Coupling ratio of DOX and folate in FA-PEG-IgG-DOX
Coupling ratio of DOX/IgG was calculated by the equation MDOX/MIgG = CDOX ×150,000/(CIgG ×580), and the coupling ratio of FA/IgG was calculated by the equation MFA/MIgG = CFA ×150,000/(CIgG ×441). This was based on the molecular weights of IgG (150,000), DOX (580), and folate (441). Concentrations of DOX and FA were determined according to its absorption at 495 nm and 363 nm, respectively. Concentration of IgG was determined using a BCA method according to the user’s manual.
Cellular uptake of FA-PEG-IgG-DOX
For microscopy examination, HeLa and KB cells grown in a monolayer were washed once with fresh folate-free RPMI 1640 medium and then were incubated with FA-PEG-IgG-DOX, FA-IgG-DOX, or IgG-DOX at a DOX concentration of 5 μg/mL for 1 h at 37°C. For folate blocking group, 1 mM free folate was added to the incubation media prior to the addition of folate conjugates. After incubation for 1 h, the cells were rinsed with PBS 3 times. For the microscopy observation, the cells were fixed with 4% paraformaldehyde solution (Thermo Scientific) for 15 min and then observed under an Olympus fluorescence microscope (Tokyo, Japan).
For FACS assay, the cells grown in a monolayer were trypsinized and aliquoted to 1.5 mL EP tubes with 3×105 cells each. Then, the cells were incubated with FA-PEG-IgG-DOX, FA-IgG-DOX, or IgG-DOX at a DOX concentration of 5 μg/mL for 1 h at 37°C. For folate blocking group, 1 mM free folate was added to the incubation media prior to the addition of folate conjugates. After incubation for 1 h, the cells were rinsed with PBS 3 times and then examined by flow cytometry on a cytometer from BD Biosciences (San Jose, CA, USA).
Pharmacokinetic and distribution analysis
Male Kunming mice (body weight ~25 g, 6–8 weeks old) were obtained from Laboratory Animal Center of the Huazhong University of Science and Technology. IgG-DOX, FA-IgG-DOX, FA-PEG-IgG-DOX, or free DOX were injected as a single intravenous bolus via the lateral tail vein at a dose of 5 mg/kg DOX. At 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h, 24 h, and 48 h after the injection, blood of 500 μL was collected in heparin-treated tubes and then centrifuged (5,000 rpm, 5 min) to obtain plasma. Plasma aliquots of 100 μL were added with 1 mL ethanol and 100 μL deionized water, followed by addition of 25 μL 20% sodium dodecyl sulfate (SDS). Then the plasma was vortexed for 30 s to extract the DOX from the proteins. DOX concentration in the plasma was measured on a fluorescence spectrophotometer (Hitachi F-2700, software FL solution, Tokyo, Japan) at 495 nm. The concentration of DOX in each sample was calculated using a calibration curve, with DOX itself as the internal standard. Pharmacokinetic variables including AUC, Cmax, t1/2, and CL were calculated using pharmacological software Drug and Statistics (version 2.0, Hefei, People’s Republic of China).
For tissue distribution, IgG-DOX, FA-IgG-DOX, FA-PEG-IgG-DOX, or free DOX was administered at a dose of 5 mg/kg. The tissues from the heart, liver, spleen, and kidney were collected at 30 min and 3 h after injection. Tissue samples of 0.1 mg were homogenized in a buffer (300 μL EtOH, 15 μL 37% HCl, and 185 μL H2O), and the homogenate was then centrifuged (12,000 rpm, 15 min) to collect the supernatant. Concentration of DOX was determined on a fluorescence spectrophotometer (Hitachi F-2700, software FL solution).
Microscopy
Microscopic examination of cells or tissue section slides was performed with an Olympus SZX12 fluorescence microscope equipped with digital camera and connected to a PC running MagnaFire 2.0 camera software from Optronics (Goleta, CA, USA). Pictures were taken at equal exposure time for each sample.
Cytotoxicity
MTT assays were used to measure cytotoxicity. HeLa and KB cells cultured in folate-free medium were seeded in 96-well culture plates at a density of 5×103 cells per well. After 24 h growth, used medium was discarded and 200 μL fresh medium containing a serial dilutions (1.56, 3.12, 6.25, 12.50, 25.00 μg/mL DOX) of free DOX, IgG-DOX, FA-IgG-DOX, or FA-PEG-IgG-DOX was added. The cells were incubated with drugs for 2 h at 37°C and then cultured in fresh DMEM medium without drugs for 72 h. Then, 20 μL MTT (5 mg/mL) was added into each well and the plates were incubated for additional 4 h. After that, the medium was removed again and 200 μL DMSO was added into each well to dissolve the blue formazan crystal with shaking for 5 min. Cell viability was assessed on a microplate reader (MK3 Multiskan) at 490 nm.
Apoptosis assay
For the apoptosis assay, the KB and HeLa cells in six well plates (4×105 cells per well) were cultured 24 h and then incubated with IgG-DOX, FA-IgG-DOX, FA-PEG-IgG-DOX, FA-IgG-DOX + FA, or FA-PEG-IgG-DOX + FA at a DOX concentration of 3 μg/mL. For FR blocking, 1 mM free folate was added prior to addition of drugs. Then, both trypsinized and suspended cells were harvested and incubated with annexin V-FITC and PI according to the manufacturer’s instructions (Liankebio, Nanjing, People’s Republic of China). For FACS assay, 20,000 cells were examined for each sample. Forward versus side scatter gating was used to remove cell debris from the total population.
Xenograft tumor mouse model
Female BALB/c nude mice (6–8 weeks of age, 16–18 g) were obtained from Huafukang Corporation (Beijing, People’s Republic of China) and kept in filter-topped cages with standard rodent chow and water available ad libitum and a 12 h light/dark cycle. Animal experiments were performed according to Regulations for the Administration of Affairs Concerning Experimental Animals in China, and the experiment was approved by the Committee on Ethical Animal Experiment at Huazhong University of Science and Technology. Subcutaneous KB tumors were seeded by inoculation of 2.5×106 KB cells in the front armpit of the mice. At a tumor volume of IgG, immunoglobulin G; Fa, folate. 100 mm3, mice received IgG conjugated DOX or free DOX by tail vein injection at a single dose of 8 mg/kg. Tumor size was determined every 4 days and calculated as the function: volume = length × width2 ×0.52.
Statistical analysis
Statistical analysis of all the data was performed using Student’s t-test or ANOVA analysis followed by Dunnett’s t-test (Statistical Package for the Social Sciences (SPSS) Software, Chicago, IL, USA). A value of P<0.05 was considered significant, and P<0.01 was considered highly significant compared to the corresponding control.
Results
Synthesis of FA-PEG-IgG-DOX
FA-PEG-IgG-DOX was synthesized, as described in methods and illustrated in Figure 1. Coupling of either FA-PEG or folate to IgG-DOX was accomplished by amide linkages using EDC and NHS as coupling reagents. The products were confirmed by ultraviolet spectrometer with a characteristic absorption peak of folate at 363 nm. The uncoupled folate and DOX were removed through Sephadex G-75 gel column separation. According to its UV-Vis spectrum, purified IgG-DOX showed absorption peaks at 495 nm (DOX) and 280 nm (IgG) (Figure 2A), whereas FA-PEG-IgG-DOX showed absorption at 495 nm (DOX), 280 nm (IgG), and 363 nm (FA) (Figure 2B). We determined the final concentrations of FA, IgG, and DOX in FA-PEG-IgG-DOX solution using a BCA method or the UV-Vis spectroscopy and found that each FA-PEG-IgG-DOX contained 7 FA and 8 DOX molecules on average (Figure 1). This result demonstrated that FA-PEG was successfully conjugated to the IgG-DOX.
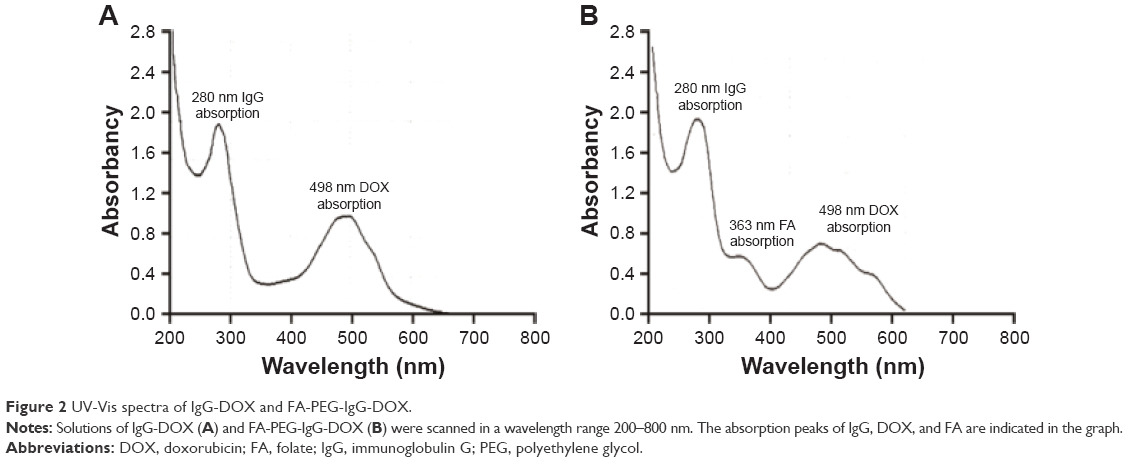 | Figure 2 UV-Vis spectra of IgG-DOX and FA-PEG-IgG-DOX. |
In vitro cellular uptake
After the preparation of FA-PEG-IgG-DOX, we evaluated whether it could be taken up by cancer cells. Uptake of IgG-DOX, FA-IgG-DOX, and FA-PEG-IgG-DOX was determined in both KB and HeLa cells, which expressed high level and medium level of folate receptors, respectively. Before drug treatment, KB and HeLa cells were cultured in RPMI 1640 medium deprived of folate for 48 h. In both KB and HeLa cells, the fluorescence intensity following treatment of FA-IgG-DOX and FA-PEG-IgG-DOX were apparently higher than IgG-DOX (Figure 3A). Almost all the cells treated with FA-IgG-DOX or FA-PEG-IgG-DOX showed robust uptake of DOX (Figure 3A). Uptake of FA-IgG-DOX or FA-PEG-IgG-DOX in KB and HeLa cells could be blocked by 1 mM folate pretreatment (Figure 3A).
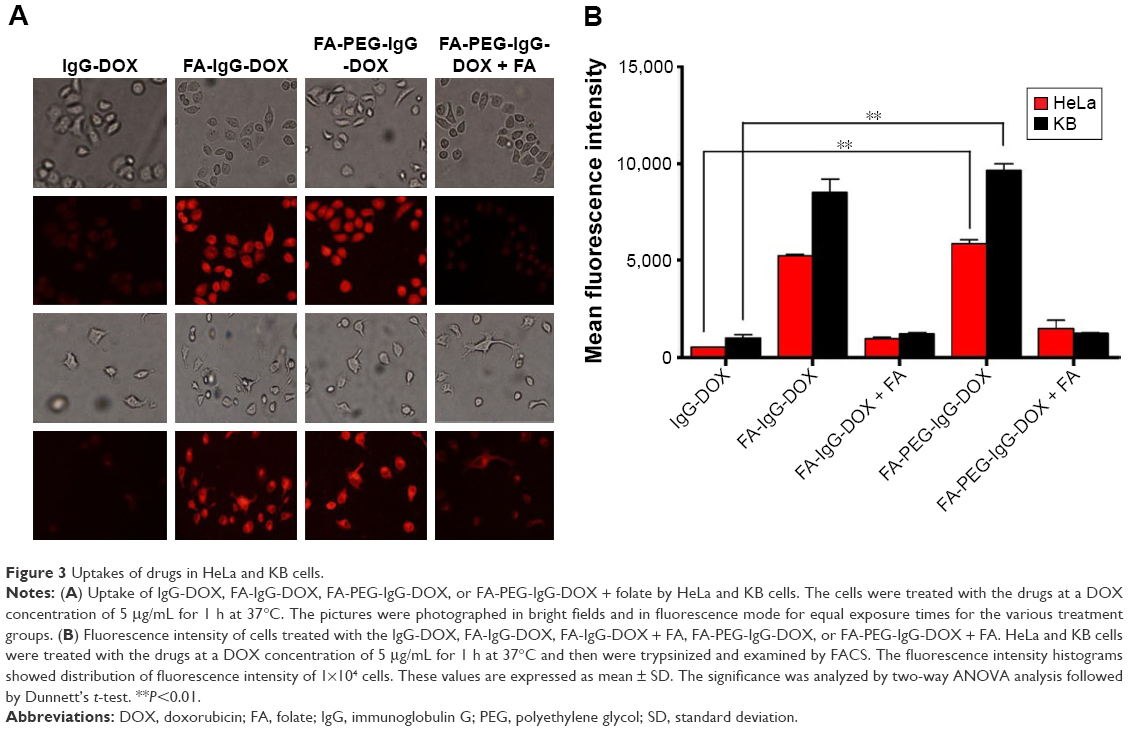 | Figure 3 Uptakes of drugs in HeLa and KB cells. |
Quantitative analysis by FACS confirmed this result. The fluorescence intensities of FA-IgG-DOX and FA-PEG-IgG-DOX group were 10.6- and 10.8-fold higher than that of the IgG-DOX in treated HeLa cells and 6.5- and 7.3-fold higher than that of IgG-DOX in treated KB cells, respectively (Figure 3B). Uptake of FA-IgG-DOX and FA-PEG-IgG-DOX was reduced to 1.5- to 3-folds of that of IgG-DOX by preincubation with 1 mM free folate (Figure 3B). These results clearly indicate that folate conjugation improved the uptake of IgG-DOX in KB and HeLa cells via FR-mediated internalization.
In vitro cell cytotoxicity
The cytotoxicities of IgG-DOX, FA-IgG-DOX, and FA-PEG-IgG-DOX were evaluated in HeLa and KB cells by MTT assay. Among all the formulations, free DOX showed the greatest cytotoxicity with a IC50 of 0.2 μg/mL in HeLa cells (Figure 4A; Table 1) and 0.18 μg/mL in KB cells (Figure 4B; Table 1). IgG-DOX had a much lower cytotoxicity, with the IC50 23.9 μg/mL in HeLa cells (Figure 4A; Table 1) and 20.6 μg/mL in KB cells (Figure 4B; Table 1), respectively. FA-IgG-DOX and FA-PEG-IgG-DOX, in contrast, had relatively high cytotoxicity, with the IC50 3.85 and 3.22 μg/mL in HeLa cells and 2.65 and 1.78 μg/mL in KB cells, respectively (Figure 4; Table 1). The IC50s of FA-PEG-IgG-DOX was about 1/8 of that of IgG-DOX in HeLa cells (Figure 4A; Table 1) and about 1/12 of IgG-DOX in KB cells (Figure 4B; Table 1). These data suggest that the IgG conjugation retarded penetration of DOX into the cells, whereas FA conjugation improved the uptake and cytotoxicity of IgG-DOX in vitro.
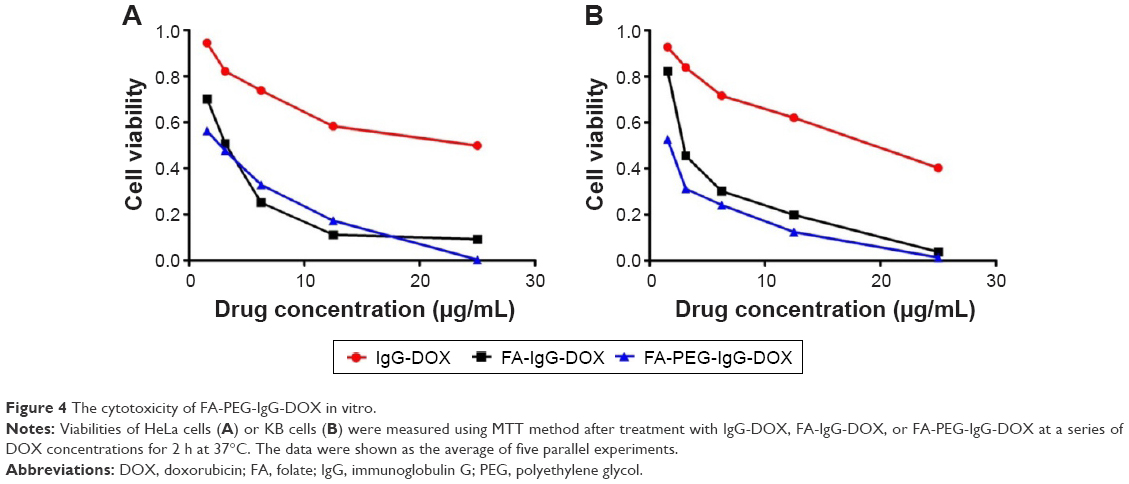 | Figure 4 The cytotoxicity of FA-PEG-IgG-DOX in vitro. |
 | Table 1 IC50 of DOX formulations in HeLa and KB cells |
FA-PEG-IgG-DOX promotes apoptosis in KB and HeLa cells
DOX is reported to kill tumor cells by inducing mitotic arrest and consequent apoptosis. To further examine the antitumor effects of FA-PEG-IgG-DOX, we assayed the apoptosis induced by IgG-DOX, FA-IgG-DOX, FA-PEG-IgG-DOX, FA-IgG-DOX + folate, and FA-PEG-IgG-DOX + folate. Compared with IgG-DOX, FA-IgG-DOX induced more apoptosis both in HeLa and KB cells (Figure 5A and B). The apoptosis rates induced by FA-IgG-DOX were 3.3 times more than IgG-DOX in HeLa cells and about 5.5 times more in KB cells. FA-PEG-IgG-DOX had a similar effect with FA-IgG-DOX (Figure 5A and B). Pretreatment with 1 mM folate significantly diminished the proapoptotic effect of FA-PEG-IgG-DOX (Figure 5A and B). These results indicate that the FA conjugation not only improved the uptake of IgG-DOX but also increased the apoptosis induction.
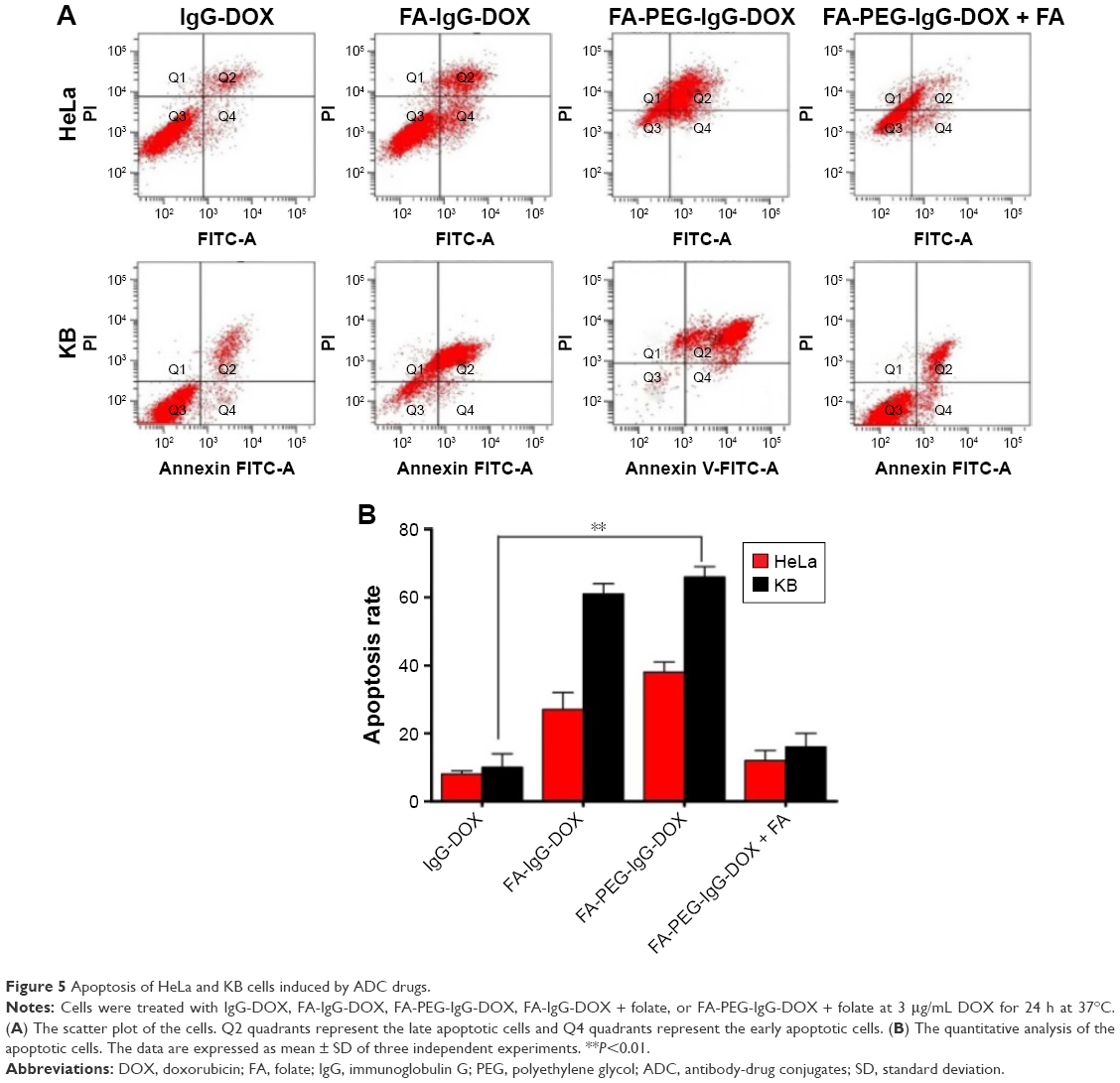 | Figure 5 Apoptosis of HeLa and KB cells induced by ADC drugs. |
Pharmacokinetic analysis
To investigate the impact of FA conjugation and PEGylation on the pharmacokinetics of IgG-DOX, we assayed the plasma concentration of DOX in mice injected with free DOX, IgG-DOX, FA-IgG-DOX, and FA-PEG-IgG-DOX. As expected, free DOX was cleared rapidly from the plasma in mice and was almost undetectable after 30 min, with a t1/2z of only 0.681 h (Figure 6; Table 2). IgG-DOX showed significantly prolonged circulation with a t1/2 of about 4 h (Figure 6; Table 2). FA-IgG-DOX had a t1/2 of about 3 h, and FA-PEG-IgG-DOX had the longest t1/2 of about 6 h. Clearly, PEGylation significantly prolonged the circulation of IgG-DOX, while FA conjugation had a limited effect as well.
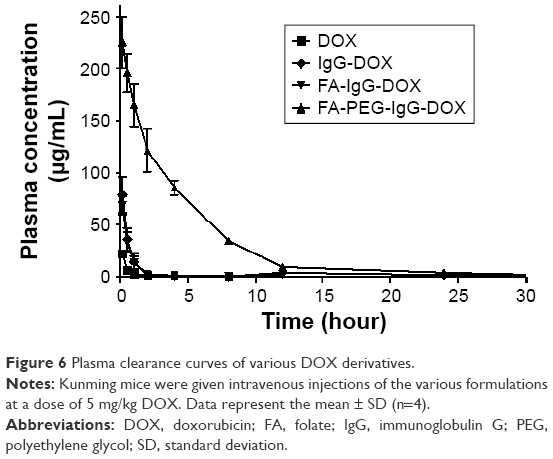 | Figure 6 Plasma clearance curves of various DOX derivatives. |
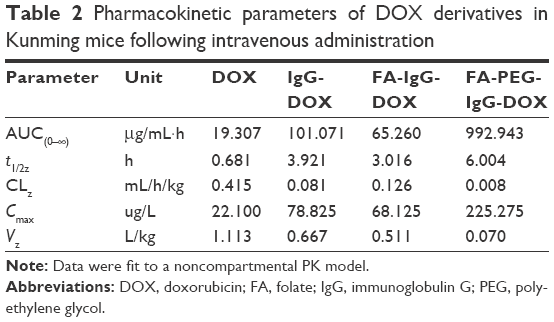 | Table 2 Pharmacokinetic parameters of DOX derivatives in Kunming mice following intravenous administration |
In addition, plasma clearance kinetics also demonstrated that the AUC of IgG-DOX was increased by PEGylation and FA conjugation. Free DOX injection had an AUC of only 19.307 μg/mL·h, while IgG-DOX and FA-IgG-DOX had increased AUCs of 101.071 and 65.260 μg/mL·h (Figure 6; Table 2), respectively. FA-PEG-IgG-DOX showed the largest AUC of 992.943 μg/mL·h (Figure 6; Table 2). Consistently, FA-PEG-IgG-DOX also showed the lowest clearance rate but highest Cmax among those formulations (Figure 6; Table 2). This experiment demonstrated that FA-PEG-IgG-DOX achieved long circulation and high plasma concentration through FA conjugation and PEGylation.
Tissue distribution
The tissue distributions of free DOX, IgG-DOX, FA-IgG-DOX, and FA-PEG-IgG-DOX were evaluated at 0.5 and 3 h after intravenous administration at a DOX concentration of 5 mg/kg. At 0.5 h, all these 4 formulations showed high DOX accumulation in the liver, although the FA-PEG-IgG-DOX showed slightly lower DOX concentration compared with other 3 formulations (Figure 7). The DOX concentrations of all 4 formulations in spleens, kidneys, and hearts were significantly lower than in the livers (Figure 7). At 3 h, both DOX, IgG-DOX, and FA-IgG-DOX injections showed decreased DOX distribution in the liver, but FA-PEG-IgG-DOX injection showed increased DOX concentration (Figure 7). In other three mouse organs, no significant change of DOX concentration was observed (Figure 7). Of note, DOX concentration in the heart by IgG-DOX, FA-PEG-IgG-DOX, or FA-PEG-IgG-DOX administration was at the same level with that following free DOX injection. These results demonstrated that the PEGylation did not alter the tissue distribution pattern of DOX in mice while it increased the circulation time of DOX. Especially, long-circulating FA-PEG-IgG-DOX did not show higher distribution in the heart compared with free DOX, and hence should not increase the cardiac toxicity of DOX.
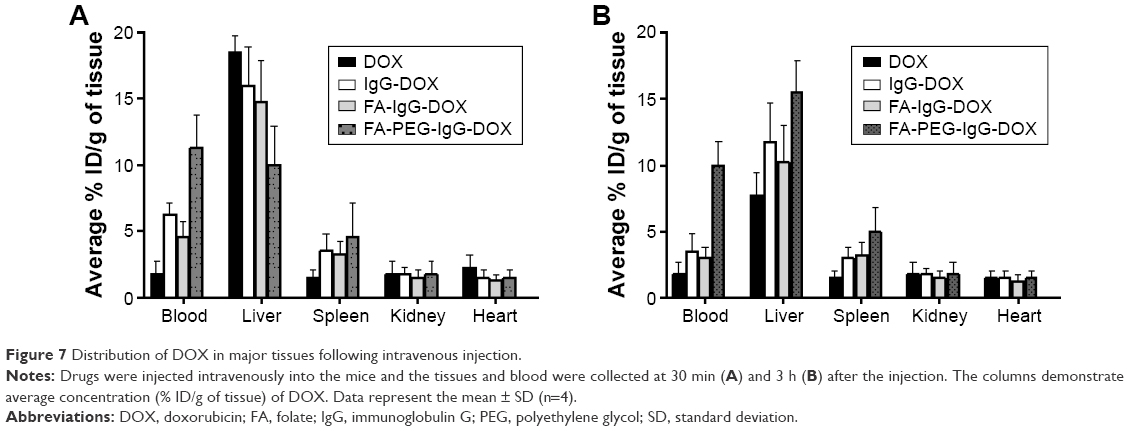 | Figure 7 Distribution of DOX in major tissues following intravenous injection. |
Tumor inhibition in vivo
We then investigated the antitumor effect of FA-PEG-IgG-DOX in tumor-bearing mice. All the DOX formulations were intravenously injected at a dose of 8 mg/kg DOX when the tumor reached 50 mm3. Compared with saline injection, DOX, IgG-DOX, and FA-IgG-DOX administration all inhibited the tumor growth at a similar strength (Figure 8A). At the day 25 after injection, the average tumor size of saline group reached 2,407 mm3, while the tumor sizes of DOX, IgG-DOX, and FA-IgG-DOX groups were 1,300, 1,265, and 1,177 mm3, respectively (Figure 8A). Significantly, FA-PEG-IgG-DOX showed higher antitumor effect than all other formulations. On day 25 after injection, the average tumor size of FA-PEG-IgG-DOX-treated mice was only 553 mm3. While tumors treated with DOX or IgG-DOX restored their rapid growth from the day 12 to day 25 (Figure 8A), the tumors in mice treated with FA-PEG-IgG-DOX maintained a very low growth rate (Figure 8A). Of note, although DOX treatment had similar tumor inhibition effect as IgG-DOX and FA-IgG-DOX, it led to great body weight loss in treated mice (Figure 8B). However, FA-PEG-IgG-DOX and FA-IgG-DOX treatment resulted in minor body weight reduction in the first half period of this experiment (Figure 8B). In addition, FA-PEG-IgG-DOX prolonged the survival of treated mice compared with DOX, but did not show superiority compared with IgG-DOX and FA-IgG-DOX (Figure 8C). This result clearly showed that the long-circulating FA-PEG-IgG-DOX enhanced the antitumor effects of IgG-DOX and confirmed that FA-PEG-IgG is a safe and effective carrier to deliver antitumor drugs in FR positive tumors.
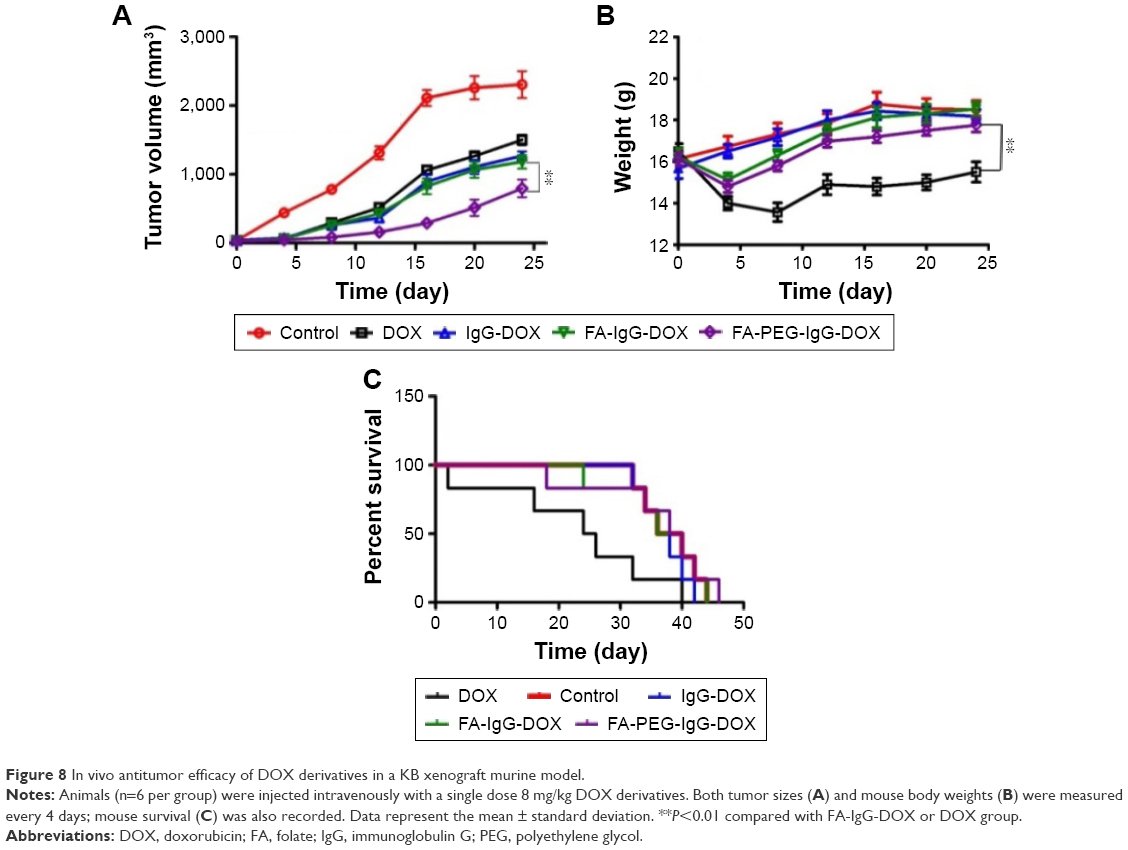 | Figure 8 In vivo antitumor efficacy of DOX derivatives in a KB xenograft murine model. |
Discussion
For two decades, antibodies have been extensively explored as the carriers to deliver various drugs. However, this attempt has been greatly impeded by the lack of antibodies with high affinity to tumor cells. Thus, there has been a continuing search for better antibody-derived alternatives or reconstructed antibodies.16 In this study, we designed and synthesized a novel ADC drug, FA-PEG-IgG-DOX. In this drug, DOX was the cytotoxic moiety, and FA-PEG-IgG was used a folate-targeting antibody carrier. Our results demonstrated that FA-PEG-IgG-DOX could deliver DOX into tumor cells efficiently and promote apoptosis of tumor cells. In addition, FA conjugation can significantly promote the uptake and cytotoxicity of IgG-DOX in FR expressing KB and HeLa cells, suggesting that FA-PEG-IgG-DOX can function as an ADC.
Performance of ADCs are adversely affected by short plasma half-life and immunogenicity. PEGylation has been shown having capability to address these problems.17–19 In this study, we found that PEGylation could significantly increase the half-life and reduce the clearance of IgG-DOX. These data suggest that PEGylation can also be utilized to prolong the circulation life of those new type of ADC drugs. Moreover, given that PEGylation can reduce the phagocytosis of ADCs by macrophages, it should reduce the immunogenecity of FA-PEG-IgG-DOX as well.
Our study also confirmed the importance of long circulation and tumor types to the antitumor effect of ADC drugs. Both FA-IgG-DOX and FA-PEG-IgG-DOX showed excellent uptake and cytotoxicity in KB and HeLa cells. However, in tumor mice, FA-PEG-IgG-DOX showed significantly greater antitumor effect than FA-IgG-DOX, suggesting that long circulation of FA-PEG-IgG-DOX contributed to its efficacy. Another finding was that the cytotoxicity of FA-PEG-IgG-DOX was highly dependent on the expression of FR in tumor cells. For example, the apoptosis in KB cells induced by FA-IgG-DOX was higher than in HeLa cells. This could be explained by that fact that KB cells express higher level of FR than HeLa cells. In vivo data also confirmed the role of FR targeting in improved efficacy. FA-IgG-DOX-treated mice showed significant greater tumor inhibition than IgG-DOX treated mice. These results collectively support that FR targeting improves the efficacy of IgG based ADC drugs in vivo and that FR targeting is effective against tumors with elevated levels of FR.
Doubtless, we will continue to explore other potential advantages of FA-PEG-IgG compared with human serum albumin (HSA) or other protein carrier. First, FA-PEG-IgG may have even particle size and good stability. Drugs were generally loaded to HSA or other proteins by surfaces adsorption or by incorporation into the particle matrix.20 These loading strategies normally resulted in uneven particle sizes ranging from 150 nm to 500 nm and instability in plasma.20,21 FA-PEG conjugation may prevent the aggregation of IgG proteins and thus ensure the even distribution of particle sizes. On the other hand, FA-PEG-IgG is expected to show ADCC effect in mice or other mammalian animals. In the tumor tissues, IgG can be released from the FA-PEG-IgG-DOX conjugates through amidase hydrolysis. Released IgG should have the capability to induce ADCC antitumor effect. Since the ADCC is dependent on intact immune system and nude mice are immune-deficient, we did not evaluate the antitumor effect of FA-PEG-IgG itself in the xenograft tumor model. However, currently we are establishing primary tumor models in wild-type mice. The superior antitumor activity of FA-PEG-IgG compared with other protein carrier will be evaluated in the primary tumor models.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No 81370058, 81572723, 81402435 and 81673368) and Innovation Foundation of Higher Education of China (2016JCTD109). The authors also thank Yongkang Gai for his assistance in FA-PEG-IgG-DOX preparation and Yong Liu for his assistance in animal experiments.
Disclosure
The authors report no conflicts of interest in this work.
References
Hughes B. Antibody-drug conjugates for cancer: poised to deliver? Nat Rev Drug Discov. 2010;9(9):665–667. | ||
Hamilton GS. Antibody-drug conjugates for cancer therapy: the technological and regulatory challenges of developing drug-biologic hybrids. Biologicals. 2015;43(5):318–332. | ||
Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21(1):5–13. | ||
Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer. 2001;1(2):118–129. | ||
Elwood PC. Molecular cloning and characterization of the human folate-binding protein cDNA from placenta and malignant tissue culture (KB) cells. J Biol Chem. 1989;264(25):14893–14901. | ||
Lacey SW, Sanders JM, Rothberg KG, Anderson RG, Kamen BA. Complementary DNA for the folate binding protein correctly predicts anchoring to the membrane by glycosyl-phosphatidylinositol. J Clin Invest. 1989;84(2):715–720. | ||
Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem. 2005;338(2):284–293. | ||
Lemos C, Kathmann I, Giovannetti E, Calhau C, Jansen G, Peters GJ. Impact of cellular folate status and epidermal growth factor receptor expression on BCRP/ABCG2-mediated resistance to gefitinib and erlotinib. Br J Cancer. 2009;100(7):1120–1127. | ||
Sudimack J, Lee RJ. Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev. 2000;41(2):147–162. | ||
Huang Y, Yang T, Zhang W, et al. A novel hydrolysis-resistant lipophilic folate derivative enables stable delivery of targeted liposomes in vivo. Int J Nanomedicine. 2014;9:4581–4595. | ||
Leamon CP. Folate-targeted drug strategies for the treatment of cancer. Curr Opin Investig Drugs. 2008;9(12):1277–1286. | ||
Zhao XB, Lee RJ. Tumor-selective targeted delivery of genes and antisense oligodeoxyribonucleotides via the folate receptor. Adv Drug Deliv Rev. 2004;56(8):1193–1204. | ||
Pan XQ, Zheng X, Shi G, Wang H, Ratnam M, Lee RJ. Strategy for the treatment of acute myelogenous leukemia based on folate receptor beta-targeted liposomal doxorubicin combined with receptor induction using all-trans retinoic acid. Blood. 2002;100(2):594–602. | ||
Pan X, Lee RJ. Tumour-selective drug delivery via folate receptor-targeted liposomes. Expert Opin Drug Deliv. 2004;1(1):7–17. | ||
Hilgenbrink AR, Low PS. Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J Pharm Sci. 2005;94(10):2135–2146. | ||
Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2(5):347–360. | ||
Tosatti S, De Paul SM, Askendal A, et al. Peptide functionalized poly(L-lysine)-g-poly(ethylene glycol) on titanium: resistance to protein adsorption in full heparinized human blood plasma. Biomaterials. 2003;24(27):4949–4958. | ||
Aimetti AA, Machen AJ, Anseth KS. Poly(ethylene glycol) hydrogels formed by thiol-ene photopolymerization for enzyme-responsive protein delivery. Biomaterials. 2009;30(30):6048–6054. | ||
Xu FJ, Liu LY, Yang WT, Kang ET, Neoh KG. Active protein-functionalized poly(poly(ethylene glycol) monomethacrylate)-Si(100) hybrids from surface-initiated atom transfer radical polymerization for potential biological applications. Biomacromolecules. 2009;10(6):1665–1674. | ||
Chuang VT, Kragh-Hansen U, Otagiri M. Pharmaceutical strategies utilizing recombinant human serum albumin. Pharm Res. 2002;19(5):569–577. | ||
Dreis S, Rothweiler F, Michaelis M, Cinatl J Jr, Kreuter J, Langer K. Preparation, characterisation and maintenance of drug efficacy of doxorubicin-loaded human serum albumin (HSA) nanoparticles. Int J Pharm. 2007;341(1–2):207–214. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.