Back to Journals » International Journal of Nanomedicine » Volume 15
Antimicrobial Peptide-Loaded Nanoparticles as Inhalation Therapy for Pseudomonas aeruginosa Infections
Authors Falciani C ![]() , Zevolini F, Brunetti J
, Zevolini F, Brunetti J ![]() , Riolo G
, Riolo G ![]() , Gracia R, Marradi M
, Gracia R, Marradi M ![]() , Loinaz I
, Loinaz I ![]() , Ziemann C, Cossío U, Llop J
, Ziemann C, Cossío U, Llop J ![]() , Bracci L
, Bracci L ![]() , Pini A
, Pini A
Received 17 September 2019
Accepted for publication 27 January 2020
Published 17 February 2020 Volume 2020:15 Pages 1117—1128
DOI https://doi.org/10.2147/IJN.S218966
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Chiara Falciani, 1 Fabrizia Zevolini, 1 Jlenia Brunetti, 1 Giulia Riolo, 2 Raquel Gracia, 3 Marco Marradi, 3 Iraida Loinaz, 3 Christina Ziemann, 4 Unai Cossío, 5 Jordi Llop, 5, 6 Luisa Bracci, 1 Alessandro Pini 1
1Department of Medical Biotechnologies, University of Siena, Siena, Italy; 2Setlance, Siena, Italy; 3CIDETEC, Basque Research and Technology Alliance (BRTA), Donostia-San Sebastián, Spain; 4Fraunhofer Institute for Toxicology and Experimental Medicine ITEM, Hannover, Germany; 5CIC biomaGUNE, Basque Research and Technology Alliance (BRTA), San Sebastian, Spain; 6Centro de Investigación Biomédica en red Enfermedades Respiratorias – CIBERES, Madrid, Spain
Correspondence: Chiara Falciani
Department of Medical Biotechnologies, University of Siena, via Aldo Moro 2, Siena, Italy
Tel +39 0577 232022
Email [email protected]
Introduction: Antibiotic-resistant bacteria kill 25,000 people every year in the EU. Patients subject to recurrent lung infections are the most vulnerable to severe or even lethal infections. For these patients, pulmonary delivery of antibiotics would be advantageous, since inhalation can achieve higher concentration in the lungs than iv administration and can provide a faster onset of action. This would allow for the delivery of higher doses and hence reduce the number of treatments required. We report here about a new nanosystem (M33-NS) obtained by capturing SET-M33 peptide on single-chain dextran nanoparticles. SET-M33 is a non-natural antimicrobial peptide synthesized in branched form. This form gives the peptide resistance to degradation in biological fluids. SET-M33 has previously shown efficacy in vitro against about one hundred of Gram-negative multidrug and extensively drug-resistant clinical isolates and was also active in preclinical infection models of pneumonia, sepsis and skin infections.
Methods: The new nanosystem was evaluated for its efficacy in bacteria cells and in a mouse model of pneumonia. Toxicity and genotoxicity were also tested in vitro. Biodistribution and pharmacokinetic studies in healthy rats were carried out using a radiolabeled derivative of the nanosystem.
Results: The M33-nanosystem, studied here, showed to be effective against Pseudomonas aeruginosa in time-kill kinetic experiments. Cytotoxicity towards different animal cell lines was acceptable. Lung residence time of the antimicrobial peptide, administered via aerosol in healthy rats, was markedly improved by capturing SET-M33 on dextran nanoparticles. M33-NS was also efficient in eradicating pulmonary infection in a BALB/c mouse model of pneumonia caused by P. aeruginosa.
Discussion: This study revealed that the encapsulation of the antimicrobial peptide in dextran nanoparticles markedly improved lung residence time of the peptide administered via aerosol. The result has to be considered among the aims of the development of a new therapeutic option for patients suffering recurrent infections, that will benefit from high local doses of persistent antimicrobials.
Keywords: antimicrobial peptides, nanoparticles, multi-drug resistance
Introduction
According to the European Medicines Agency (EMA), antibiotic-resistant bacteria kill 25,000 people every year in the EU. The imminent emergency is now not only perceived by the scientific community, but also by economists and governments. In 2014, it was estimated that the global problem of antimicrobial resistance would cost up to $100 trillion by 2050 and that mortality from superbugs would exceed deaths from cancer.1 The WHO has identified the four most dangerous bacterial species to be Pseudomonas aeruginosa, Acinetobacter baumannii, Klebsiella pneumoniae and Escherichia coli, because of their widespread nature and frequency of drug-resistance.2
Patients subject to recurrent lung infections, especially those with cystic fibrosis, are the most vulnerable to severe or lethal infections. For these patients, pulmonary delivery of antibiotics could be an interesting solution from the viewpoints of patient compliance and local delivery. However, pulmonary administration is technically challenging, because oral deposition can be high and the amount of antibiotic actually administered to the deep lung strongly depends on the inhalation technique used. Interestingly, the application of nanotechnology to antibiotic aerosols has added advantages and increased their effectiveness. Some nano-based formulations are already commercially available, such as Arikayce, an amikacin liposome inhalation suspension; others are in the clinical phase, such as Quinsair, a formulation of levofloxacin in PLGA nanoparticles.3
Here we report the design and use of a nanosystem (M33-NS), composed of dextran nanoparticles as carriers for an antimicrobial peptide, SET-M33, for pulmonary administration. SET-M334–9 is a non-natural cationic antimicrobial peptide built in branched form (Figure 1). This branched form confers resistance to degradation in biological fluids to the peptide,10,11 and produces multivalent binding.10 SET-M33 has shown efficacy against about 100 Gram-negative multidrug- and extensively drug-resistant clinical isolates in vitro4,5,7,12 and also efficacy in eradication of biofilms.13 Its efficacy has also been established in preclinical infection models of pneumonia, sepsis and skin infections.7 The peptide lacked immunogenicity4 and hemolytic activity and its toxicity in human cells and in mice was acceptable.7 Like other antimicrobial peptides,14 SET-M33 also features anti-inflammatory activity.8 A study dealing with the antimicrobial mechanism of action of SET-M33 demonstrated that after an initial binding to the bacterial cell wall lipopolysaccharide, the peptide interacts with the bacterial membrane and acquires an amphipathic helix structure. This structure may be partially embedded into the membrane, thereby destroying its function and eventually the cell itself.15 Notably, this mechanism does not allow the emergence of bacterial resistance.15
 |
Figure 1 Structures of the nanocarrier NC, SET-M33 and the nanosystem M33-NS. |
The new nanosystem reported here was obtained by capturing SET-M33 peptide on single-chain dextran nanoparticle. We compared the efficacy and toxic and mutagenic potential of the new M33-nanosystem with the free SET-M33 peptide.
Materials and Methods
Peptide Synthesis
SET-M33 was prepared as reported previously.5,7,12 In brief, solid-phase synthesis by standard Fmoc chemistry was carried out on a Syro multiple peptide synthesizer (MultiSynTech, Witten, Germany). The side chain protecting groups were 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl for R, t-butoxycarbonyl for K and t-butyl for S (Iris Biotech GmbH, Marktredwitz, Germany). The final products were cleaved from the solid support, de-protected by treatment with trifluoroacetic acid (TFA) containing triisopropylsilane and water (95/2.5/2.5), and precipitated with diethyl ether. Final peptide purity and identity was confirmed by reverse-phase chromatography on a Phenomenex Jupiter C18 analytical column (300 Å, 250 x 4.6 mm) and by mass spectrometry.
SET-M33 ((KKIRVRLSA)4K2KβA-OH) was synthesized on a Fmoc4-Lys2-Lys-β-Ala Wang resin (Iris Biotech GmbH, Marktredwitz, Germany). The crude peptide, released as carboxylic acid, was purified by reverse-phase chromatography on a Phenomenex Jupiter C18 analytical column (300 Å, 250 x 10 mm) in a linear gradient, using water with 0.1% TFA as eluent A and acetonitrile as eluent B (from 82% to 75% of A in 60 min). The purified peptide was obtained as a trifluoroacetate salt and exchanged with acetate using a quaternary ammonium resin (AG1-X8, 100–200 mesh, 1.2 meq/mL capacity). The resin-to-peptide ratio was 2000:1. Resin and peptide were stirred for 1 h, the resin filtered off and washed extensively and the peptide finally recovered and freeze-dried. The compound was characterized on a MALDI-TOF mass spectrometer (Ultraflex III Bruker Daltonics): (KKIRVRLSA)4K2KβA-OH MALDI-MS: 4682.48 [M+H]+; RP-HPLC: tR = 21.10 min, purity >99%. SET-M33 solubility, water ≥20 mg/mL, saline ≥20 mg/mL, PBS ≥15 mg/mL.
For the synthesis of SET-M33-Tyr ((KKIRVRLSA)4K2K-peg-Tyr), Fmoc-Tyr(tBu)-OH was used in the first coupling step followed by Fmoc-NH-PEG(4)-COOH; then, two consecutive couplings of Fmoc-Lys(Fmoc)-OH were used to build the branched core. The rest of the processes was carried out as above. (KKIRVRLSA)4K2K-peg-Tyr MALDI-MS: 5374.68 [M+H]+; RP-HPLC: tR = 24.10 min, purity >99%.
M33-Nanosystem
Dextran nanoparticles (DXT-NPs) were prepared by controlled addition of the dithiol cross-linker to the methacrylate-functionalized dextran precursor polymer. Further functionalization of the DXT-NPs surface with carboxylic groups was achieved by quenching the non-reacted methacrylate groups with an excess of 3-mercaptopropionic acid. The final product was achieved after purification by dialysis against distilled water (MWCO 3500Da).16
To prepare the M33-nanosystem, SET-M33 peptide was conjugated with DXT-NPs, using non-covalent interactions at an adequate pH. The maximum loading capacity of the nanoparticles was obtained by titration until neutral zeta potential of the nanosystem was reach. In a standard procedure, 6 mg SET-M33 peptide were incubated with 43 mg DXT-NPs in 1 mL saline solution at pH= 7.2 for 5 h at room temperature. The crude reaction mixture was then purified by membrane ultrafiltration using Vivaspin-500 10kDa MWCO (GE Healthcare, Chicago, USA) to ensure the absence of any unbound SET-M33. At this concentration, free SET-M33 was not recovered out from the membrane. M33-NS maximum concentration in water was [NS]=45,6 mg/mL; [SET-M33]=5,6 mg/mL.
Dynamic light scattering (DLS) and zeta potential analyses were conducted using a Zetasizer Nano ZS (ZEN3600 Model, Malvern Instruments, Malvern, UK). Size and polydispersity index measurements were performed in disposable sizing cuvettes at a laser wavelength of 633 nm and a scattering angle of 173°, while the zeta potential measurements were performed in disposable zeta potential cells (pH 7.4, 25 ºC). Before measurement, the samples were dispersed in saline solution (0.9 wt % NaCl for size measurements and 1 mM NaCl for zeta potential measurements) at a concentration of 1 mg/mL. Each measurement was performed in triplicate at 25°C.
Minimum Inhibitory Concentration – MIC Assay
Antimicrobial susceptibility was assessed by determining the “Minimum Inhibitory Concentration” (MIC) of M33-NS, SET-M33 and NC, using the broth microdilution technique, according to the 2014 EUCAST guidelines. The MIC assay measures visible inhibition of bacterial growth after 24 h of exposure of bacteria to the respective antimicrobial in MH-II broth. The concentration range of the two antimicrobials used was 0.125–64 µg/mL, referring to the peptide content. P. aeruginosa PAO1 strain was used. Antibiogram of PAO1 data indicated that the strain used was susceptible to piperacillin/tazobactam, ceftazidime, cefepime, meropenem, amikacin, gentamicin, ciprofloxacin, and levofloxacin.
Time-Kill Kinetic Assay
The concentration- and time-dependent killing capacity of M33-NS and SET-M33 was determined using time-kill kinetic (TKK) assays, as previously described.15 Briefly, stationary-phase MH-II broth cultures of P. aeruginosa(PAO1) were diluted in 25 mL of Tryptic Soy Broth (TBS) to a density of approximately 5 × 105 colony-forming units per mL (CFU per mL). Bacterial cultures were exposed to antimicrobials for 24 h at 2-fold increasing concentrations (0.5–250 µg/mL) for both M33-NS and SET-M33. Incubation was performed at 37°C under shaking conditions at 96 rpm. Next, 1 mL samples were taken at 0, 2, 4, 6 and 24 h of antimicrobials exposure and centrifuged at 12,500 x g for 5 min to pellet the cells, which were then resuspended in sterile PBS and plated on agar plates. Agar plates were then incubated for 24 h at 37°C and the resulting number of CFU was determined.
Mouse Lymphoma Tk Assay
Mutagenicity testing was based both on the OECD Guideline for the Testing of Chemicals n. 49017 and ICH guideline S2(R1).18 The Mouse Lymphoma Tk Assay (MLA) investigates forward mutations at the thymidine kinase (Tk) gene locus. Tk-deficient (Tk-/-) cells survive exposure to the selective toxic nucleotide analogue trifluorothymidine (TFT), whereas Tk+/- cells, which contain Tk, cannot proliferate. The enzyme Tk is not essential for the survival of mutants, as thymidine nucleotides can be synthesized de novo via an alternative pathway. Cultures of L5178Y/Tk+/-, clone 3.7.2C, cells were prepared as already described in detail.19 Different concentrations of M33-NS were used with or without metabolic activation. 500 µg/mL was chosen as maximum concentration.
In brief, 10 x 106 L5178Y/Tk+/- cells in 20 mL R5 (cell growth medium with 5% heat-inactivated horse serum, HS) were exposed to M33-NS and to the reference items, ie, free nanocarrier (NC), free SET-M33 peptide, methyl methanesulfonate (MMS; positive control without metabolic activation) and cyclophosphamide monohydrate (CP; positive control with metabolic activation), for 4 or 24 h without, or for 4 h with S9-mix as metabolic activation system (Envigo, Huntingdon, UK).20 Cells were subsequently sub-cultured for 48 h in cell culture medium with 10% heat-inactivated horse serum (R10) with daily cell population adjustment to determine cytotoxicity (Suspension Growth, SG) and to allow 48 h phenotypic expression prior to mutant selection. For determination of SG, cells were counted after treatment (cell count after treatment), cell number was adjusted to 6 x 106 per 30 mL R10 (cell setup after treatment), cells were cultured for a further 24 h, counted again (cell count day 1), adjusted again to 6 x 106 cells per 30 mL R10 (cell setup day 1), grown for additional 24 h, and finally counted again (cell count day 2). SG was subsequently calculated [SG 4 h treatment = (cell count day 1/cell setup after treatment) x (cell count day 2/cell setup day 1); SG 24 h = (cell counts after treatment/cell setup start of treatment) x (cell count day 1/cell setup after treatment) x (cell count day 2/cell setup day 1)]. As an additional measure of cytotoxicity, “Relative Total Growth” (RTG) was finally calculated from SG and “Plating Efficiency” (PE) of untreated (SG negative control; PE negative control) and treated cultures (SG treatment; PE treatment), using the formula: RTG [%] = (SG treatment/SG negative control) × (PE treatment/PE negative control) × 100. PE of untreated and treated cells was therefore determined by plating approximately 1.6 cells per well of a 96-well plate in R20 (culture medium with 20% HS) without selective agent. Colonies were counted 6–7 days later and PE after the phenotypic expression period was calculated [PE = P/1.6 where P = -ln (number of empty wells/total number of wells plated)]. Mutant frequency of cells (MF) was determined after the phenotypic expression period by seeding approximately 2 x 103 cells per well in 96-well plates, using restrictive, TFT-containing (3 µg/mL) R20 for selection of the mutant phenotype. After a 10–11-days selection period, mutant colonies were counted and sized by light microscopy to determine potential mechanisms of action. Large colonies represent intragenic lesions (gene mutations), whereas small ones are derived from severe genetic alterations in the autosomal TK-locus (chromosomal aberrations). Microscopy examination identified “large colonies” as those covering more than 1/4 of the well surface. Large colonies were generally not more than one or two cells thick. “Small colonies” covered less than 1/4 of the well surface, showed a compact growth, and were more than two cells thick. Finally, the mutant frequency MF [MF = (PE mutant/PE viable) x 10−6] was finally derived from the PE of mutant colonies in selective TFT-medium [PE mutant = -ln (number of empty wells/total number of wells plated)/2000] and the PE of colonies in non-selective medium [PE viable = -ln (empty wells/total wells plated)/1.6]. Existence of a major increase in MF was determined, using the so-called “Global Evaluation Factor” (GEF), as proposed by the IWGT.21 For the microtiter version of the MLA used in this study, GEF, based on historical control data of a broad range of laboratories, was set at 126. To draw a final conclusion with regard to mutagenicity, the validity of the study, major increases in MF and concentration-dependencies were considered.
Cytotoxicity
T24 human urinary bladder epithelial carcinoma (ATCC) cells and RAW264.7 mouse macrophages (ATCC) were plated at a density of 2.5 x 103 cells per well of a 96-well plate. 16HBE14o-human bronchial epithelial cells (ATCC) were plated at a density of 2.5 x 103 per well of 96-well microplates, pre-coated with a specific coating solution, consisting of 88% LHC basal medium, 10% bovine serum albumin, 30 µg/mL bovine collagen type I and 1% human fibronectin. All cells were incubated at 37°C and 5% CO2 in a humidified atmosphere.
Different concentrations of M33-NS and SET-M33 peptide (250 ± 0.50 µg/mL) and the corresponding concentrations of free NC (2025 ± 4,05 µg/mL) were added 24 h after plating. After 72 h of incubation, growth inhibition was assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test. The results underwent nonlinear regression analysis, using GraphPad Prism 5.03 software, with a built-in analysis (log[inhibitor] vs response–variable slope).
Animal Experiments
The experiments conducted at CIC biomaGUNE were approved by the Ethical Committee of CIC biomaGUNE and local authorities (project number: AE-biomaGUNE-0615/PRO-AE-SS-052) and were performed in accordance with the Spanish policy for animal protection (RD53/2013), which meets the requirements of the European Union directive 2010/63/UE regarding the protection of animals used in experimental procedures.
The experiments conducted at the Toscana Life Sciences Animal Care Facility by the University of Siena were approved by the Local Ethical Committee of Toscana Life Sciences and by the Italian Ministry of Health (authorization n34/2016-PR).
Radiolabeling of SET-M33 and M33-NS for in vivo Imaging Studies
Radio-iodination of SET-M33 peptide was carried out by aromatic substitution on the tyrosine residues. To do so, a solution of the peptide SET-M33-Tyr (50 µg/50 μL) was incubated with Na[124I]I (Perkin Elmer, Waltham, MA, USA) in 0.02 M sodium acetate buffer solution (50 µL, pH 5.5) for 2 h at 25°C in the presence of Iodo-beads (Thermo Fisher Scientific, Waltham, MA, USA). The crude of reaction was purified by size exclusion chromatography (SEC), using Illustra™ Nap™-5 Sephadex™ columns G-25 DNA grade (GE Healthcare, Chicago, USA), preconditioned with a sodium acetate buffer solution (10 mL, 0.02 M, pH 5.5). Chemical and radiochemical purity were determined by radio-HPLC, using a Mediterranean Sea 18 column (4.6 x 150 mm, 5 μm) as the stationary phase and 0.1% solution of TFA in water (A) and pure methanol (B) as the mobile phase. The following gradient was used; initial: A-60% B-40%; 4 min: A-60% B-40%; 14 min: A-20% B-80%; 18 min: A-60% B-40%; 20 min: A-60% B-40%. Injected volume was 20 µL.
Radiochemical stability of the labelled peptide was assessed by incubation in different media at 37°C. Samples were withdrawn at different times and analyzed by HPLC using the experimental conditions described above. Radiochemical stability was directly calculated from chromatographic profiles.
The formation of radiolabeled M33-NS ([124I]M33-NS) was achieved by combining previously radiolabeled [124I]SET-M33 peptide with dextran nanoparticles (NC) by electrostatic interactions. In brief, 50 μg of [124I]SET-M33 peptide were incubated with 0.5 g of NC in 1 mL of ultrapure water at 25°C for 15 h. To eliminate unbound peptide, the crude of reaction was purified by filtration, using Millipore Amicon® Ultra filters (5000 MWCO). Radiochemical yields were calculated as the ratio between the amount of [124I]M33-NS and the starting amount of [124I]SET-M33. The stability of [124I]M33-NS was determined by incubation in water at 37°C for 24 h. The solution was then transferred to a 5000 MWCO filter (Millipore Amicon® Ultra filter) and centrifuged for 20 min at 13,400 rpm. The stability was calculated as the ratio between the amount of radioactivity in the filter ([124I]M33-NS) and the total amount of radioactivity (filter + filtrate, the latter corresponding to unbound [124I]SET-M33).
Biodistribution and Pharmacokinetic Study in Healthy Rats Using Positron Emission Tomography
The labeled peptide ([124I]SET-M33) and the peptide-loaded nanosystem ([124I]M33-NS) were both administered to anesthetized rats using the Penn-Century MicroSprayer® nebulizer (Penn-Century, Wyndmoor, PA, USA). Immediately after aerosol administration, dynamic whole body positron emission tomography (PET) images were acquired for 40 min using an eXplore Vista PET-CT system (GE Healthcare, Chicago, USA). Static imaging sessions were repeated at 3, 6, 9, 12 and 24 h after application. Images were reconstructed by filtered back projection and analyzed using PMOD analysis tool (version 3.4, PMOD Technologies, Zürich, Switzerland). Volumes of interest (VOIs) were delineated manually in the whole lungs, and the concentration of radioactivity in the lungs was determined for each compound and time point.
Efficacy of M33-NS in a Pseudomonas aeruginosa Pneumonia Model
After acclimatization for four days, BALB/c female mice (19–21 g; Charles River) were anesthetized with Zoletil + Xylazine (10–40 mg/kg + 0.4–4 mg/kg), placed on a mouse holder and infected intratracheally with Pseudomonas aeruginosa PAO1 (1 x 107 CFU/mouse in a volume of 20 µL) with a blunt tip Hamilton syringe and with the help of a laryngoscope. After infection, all animals were divided as follows: group 1 (n=8 mice; control, ie, saline only treatment); group 2 (n=10 mice; SET-M33, 2.5 mg/kg); group 3 (n=5 mice; SET-M33, 5 mg/kg); group 4 (n=5 mice; M33-NS, 2.5 mg/kg, calculated as SET-M33 content) group 5 (n = 5 mice; M33-NS, 5 mg/kg, calculated as SET-M33 content). One hour and 20 h post infection animals of all groups were anesthetized again with Zoletil + Xylazine, placed on the mouse holder and treated intratracheally with saline or the antimicrobials, using a blunt tip syringe and a laryngoscope. Twenty-four hours post treatment all animals were anesthetized again with isofluorane and sacrificed. The lungs were harvested and homogenized and the homogenate was plated on agar-MHB plates. After 24 h of incubation at 37°C Pseudomonas aeruginosa CFU were counted. Animals showing severe signs of distress were humanely sacrificed and not included in the results.
Results
Set-Up and Characterization of M33-NS
The surface of dextran-based nanoparticles (DXT-NPs) was rendered negatively charged by covalently binding mercaptopropionic acid, to be suitable for electrostatic interaction with positive charges of SET-M33. SET-M33 peptide was added to a water dispersion of nanoparticles. The addition of SET-M33 peptide to DXT-NPs produced a decrease in zeta potential value and the maintenance of negative potential at pH 7.4 was chosen as an indication of the presence of free carboxylate groups at nanoparticles surface. Positive zeta potential values were assumed as an indication of neutralization of the negatively charged NP surface by the positively charged peptide. SET-M33 was loaded into the dextran nanoparticles (DXT-NPs) at 0.14 mg of SET-M33 per mg dextran NC (Figure 1). This loading dose of SET-M33 was found to be the maximum that did not cause any agglomeration and flocculation of the M33-NS. The particle size measured by DLS was 18 nm with a polydispersity index of 0.3. Moreover, zeta potential measurements confirmed SET-M33 peptide loading of the DXT-NPs with a decrease in surface charge from −22 mV for isolated DXT-NPs to −13 mV for M33-NS, indicating electrostatic interactions between peptide and nanoparticle. The maximum concentration of the nanosystem M33-NS in water was 45.6 mg/mL, which corresponded to 5.6 mg/mL of peptide. The DLS measurement of M33-NS after one month in saline solution did not show any significant change.
Efficacy of M33-NS Against Pseudomonas aeruginosa
To determine the antimicrobial effect of M33-NS, the efficacy of M33-NS against P. aeruginosa was measured in time-kill-kinetic (TKK) experiments. The TKK assay takes the effects of exposure time and concentration of the test item into account. P. aeruginosa was killed (≥99.9%) after 2, 4 and 6 h of exposure to ≥4 µg/mL M33-NS. This was followed by bacterial re-growth up to the level of non-exposed bacteria after 24 h exposure to 4 µg/mL. The 8 µg/mL concentration gave 80% regrowth after 24 h, with respect to non-exposed bacteria and at 16 µg/mL, no significant regrowth of P. aeruginosa was observed (Figure 2). This result was in line with the MIC of M33-NS, which was 16 µg/mL after 24 h of incubation. 16 µg/mL was also the MIC of the free SET-M33 peptide against P. aeruginosa. SET-M33 peptide gave no regrowth with any of the tested concentrations (Figure 1).15
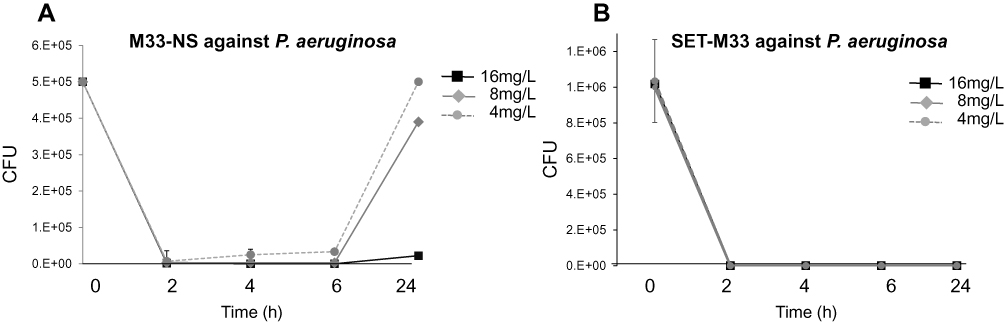 |
Figure 2 Time-kill kinetics. (A) Time-kill kinetics of M33-NS against P. aeruginosa. Concentrations are given as SET-M33 content in M33-NS. (B) Time-kill kinetics of SET-M33 against P. aeruginosa. |
Cytotoxicity in Lung Epithelial Cells, Bladder Cells and Macrophages
To determine in vitro cytotoxicity of M33-NS, compared to the free SET-M33 peptide and the nanocarrier, immortalized 16HBE14o-human bronchial epithelial cells22 and T24 human urinary bladder epithelial carcinoma cells23 were used as model systems. The bronchial epithelium is clearly the tissue most exposed to aerosol administration, whereas in case of absorption and subsequent systemic availability, urinary clearance also has to be kept in mind, as urinary clearance was observed in a previous study.7 Furthermore, macrophages, ie, RAW264.7 cells, with their special aptitude for engulfing foreign substances, were used as an additional model system for in vitro cytotoxicity experiments, because M33-NS has particle morphology and could therefore damage macrophages after phagocytosis/uptake.
M33-NS was slightly more toxic than the free SET-M33 peptide in all three cell types (Figure 3), however without reaching statistical significance. Overall cytotoxicity of M33-NS was considered acceptable since at 32 µg/mL, which is twice the MIC, viability ranged from 79%, for RAW264.7 to 65% for T24 and 16HBE14o-. Interestingly, at higher concentrations M33-NS seemed to be slightly less toxic than the free SET-M33 peptide, particularly in bronchial epithelial cells and macrophages. NC was very little toxic, showing a minimum of 40% of viability at the highest concentration in T24 cell line.
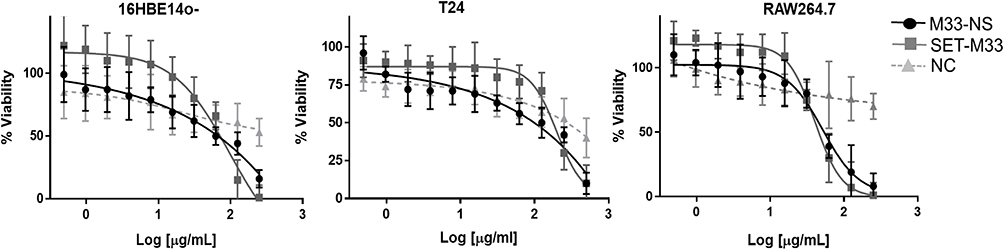 |
Figure 3 Cytotoxicity. Cytotoxicity of M33-NS compared to SET-M33 and nude NC, in 16HBE14o-, T24 and RAW264.7. |
Genotoxicity of M33-NS
In vitro mutagenicity testing of M33-NS, SET-M33 and nude NC was performed according to OECD no. 490 and ICH S2(R1) guidelines.17,18
L5178Y/Tk+/- mouse lymphoma cells were exposed to the test items and to the positive (methylmethane sulfonate, MMS) and negative reference materials (vehicle) for 4 h with, and 4 and 24 h without S9-mix. All experiments fulfilled the main validity criteria, described in OECD no. 490, with regard to the number of experiments, exposed cells and concentrations, spontaneous mutant frequencies (50–170 x 10−6) and effects of positive controls.
After 4 h of treatment without S9-mix, M33-NS, SET-M33 and nude NC showed no substantial mutagenic potential, according to the Global Evaluation Factor concept and the corresponding limit 188.3 x10−6 (126 x 10−6 + 62.3 x 10−6), above which mutagenic potential has to be declared. The positive control MMS (10 µg/mL) mediated a major increase of MF upto 572.5 x 10−6, based on total colony count, compared to the corresponding negative control (Table 1). Cytotoxicity of the free SET-M33 peptide was observed at the very high concentration of 70 µg/mL (more than four times the MIC), with an RTG value of 1%. Interestingly, cytotoxicity of M33-NS was only observed at 500 µg/mL, which corresponded to 70 µg/mL of SET-M33, but cytotoxicity was slightly lower (RTG: 24%), than that of SET-M33 alone. There was no obvious cytotoxicity or mutagenicity for the free NC.
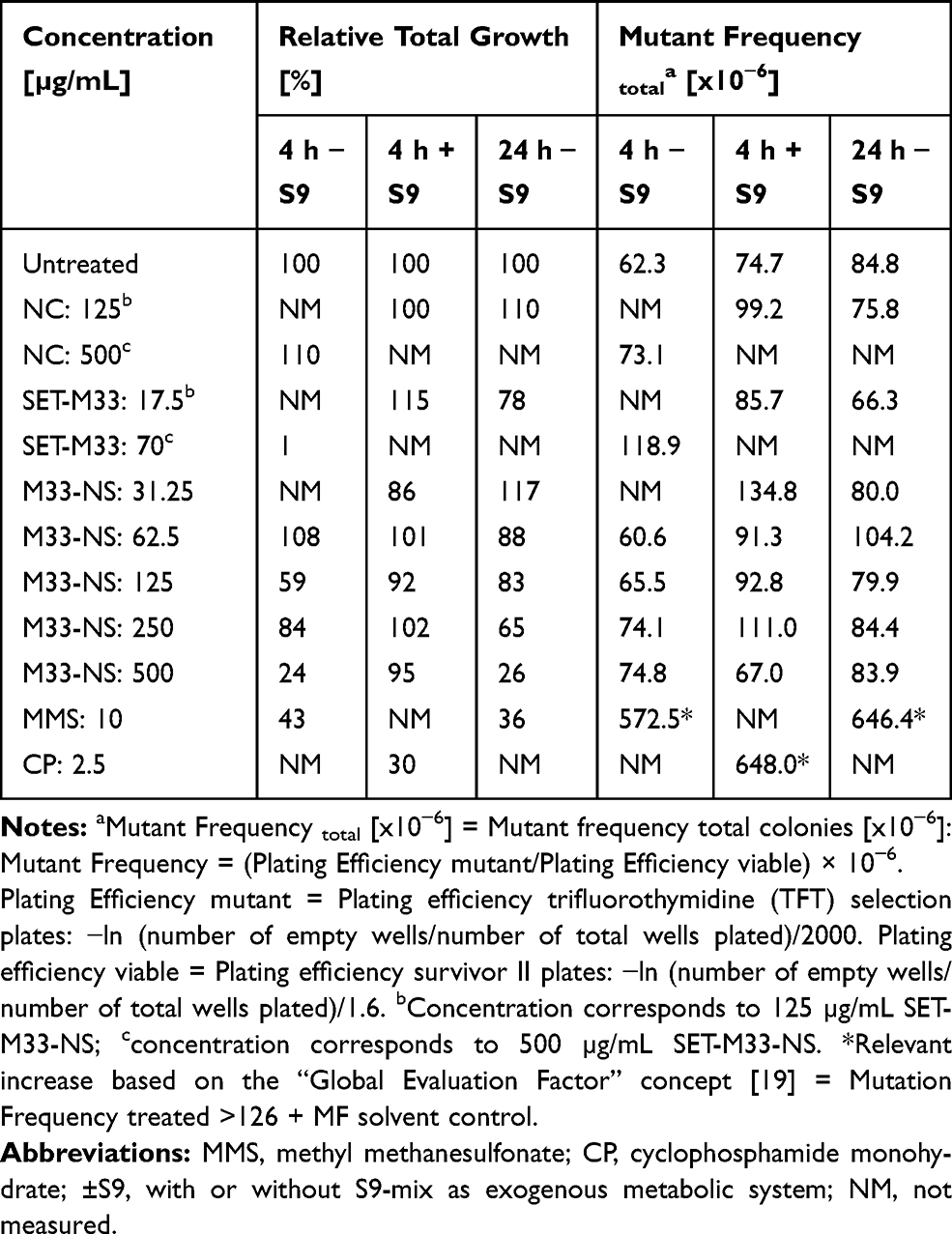 |
Table 1 Genotoxicity of M33-NS. MLA with M33-NS, 4 h Treatment with and Without S9-Mix, and 24 h Treatment Without S9-Mix |
After 4 h of incubation with S9-mix, none of the tested items showed cytotoxic potential. This may be due to metabolic inactivation and/or binding to the protein fraction of the S9-mix (Table 1). Based on the “Global Evaluation Factor” concept, no substantial mutagenicity was observed for any of the test and reference compounds, as all MFs were below the limit of 200.7 x 10−6 (126 x 10−6 + 74.7 x 10−6), whereas the positive control CP (2.5 µg/mL) determined a major increase in MF to 648 x 10−6. The assay was therefore performed correctly and the S9-mix exhibited sufficient metabolizing activity.
After 24 h of treatment without S9-mix, again 33-NS did not show any mutagenic potential, but a slight concentration-dependent inhibition of cell growth, that was maximum at the limit concentration of 500 µg/mL (RTG: 26%). The free SET-M33 peptide at 17.5 µg/mL also showed some inhibitory effect on cell growth (RTG: 78%), but no mutagenic potential. The NC tested at 125 µg/mL did not show neither cytotoxic nor any mutagenic potential (Table 1). In contrast, the positive control MMS (10 µg/mL) was quite effective (MF: 646.4 x 10−6).
In conclusion, in the MLA, M33-NS and SET-M33 did not show evidence of any substantial, substance-specific induction of gene or chromosome mutations and can therefore be judged not to be mutagenic in mammalian cells. The result is in line with our previous observations on SET-M33 in an in vitro micronucleus test in human lymphocytes, where the peptide again showed no genotoxic potential.5 Interestingly, M33-NS was less cytotoxic in mouse lymphoma cells than the free SET-M33 peptide, and the cytotoxicity of M33-NS and SET-M33 was lost in the presence of S9-mix.
Radiolabeling of SET-M33 and M33-NS for in vivo Imaging Studies
SET-M33 was efficiently labelled with Iodine-124 with a decay-corrected radiochemical yield of 36 ± 9% in an overall synthesis time of 3 h. The molar activity was within the range of 0.5–1.5 GBq/µmol at the end of the synthesis. Stability studies showed that [124I]SET-M33 was stable in moderately acidic conditions.
Incorporation of [124I]SET-M33 into the dextran nanoparticles was achieved with a radiochemical yield of more than 50%. Stability studies in water did not show any significant dissociation of [124I]SET-M33 after 24 h.
Biodistribution/Pharmacokinetic Study in Healthy Rats After Pulmonary Administration
PET-CT image sequences were obtained for [124I]SET-M33 and [124I]M33-NS (Figure 4). The images showed a steady clearance of radioactivity from the lungs over 24 h and a longer residence time of [124I]M33-NS in the lungs. The elimination pathways of [124I]M33-NS suggest excretion mainly via the gastrointestinal tract and some renal elimination of the free SET-M33 peptide.
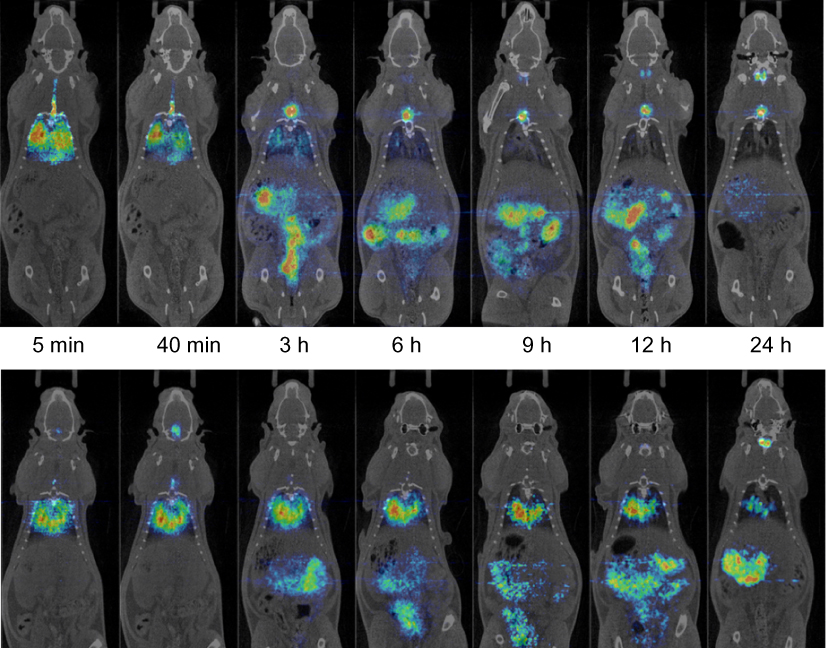 |
Figure 4 Biodistribution. Positron emission tomography (PET) images (coronal projections) obtained at different time points after intratracheal instillation of [124I]SET-M33 (top panel) and [124I]M33-NS (bottom panel) into wild-type rats. PET images (in color code) show the distribution of the radioactive signal, and have been co-registered with representative computerized tomography (CT) images (coronal slices) of the same animal for unequivocal localization of the radioactive signal. |
Quantification of radioactivity in the lungs confirmed the trends observed by visual inspection of the images. To evaluate the biological half-lives of the radiolabeled compounds in rat lungs, the concentration of radioactivity in the lungs at different times were fitted to a mono-exponential or bi-exponential decay equation (Figure 5). The residence time in the lungs of free [124I]SET-M33 was found to be 1.21 h. Notably, the radioactivity values of [124I]M33-NS, obtained at different times, did not fit a mono-exponential decay equation (red line fitting in Figure 5), but a bi-functional equation. This was a two-phase decay system where the half-life of the first phase was 1.07 h, and therefore almost equivalent to that obtained for free SET-M33, while values for the second (slow) phase were close to 13 h. These results suggest that a fraction of the peptide is loosely bound to the NC and behaves almost as “free” peptide, while another fraction is more tightly attached to the NC, resulting in prolonged residence time.
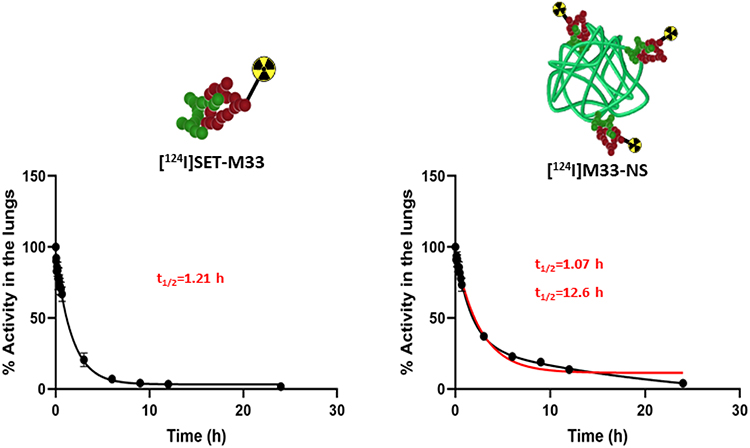 |
Figure 5 Pharmacokinetics. Decay curves for [124I]SET-M33 and [124I]M33-NS, obtained from the in vivo quantification results. Half-life values are shown in red. |
Efficacy of M33-NS Against Pseudomonas aeruginosa in a Pneumonia Model
The antibacterial efficacy of the nanosystem M33-NS was finally determined in a pneumonia model induced by intratracheal instillation of Pseudomonas aeruginosa PAO1 (1x107) into healthy BALB/c mice. Infected animals developed a severe infection, which, if left untreated, led to death. After infection, animals were randomized into five groups: group 1 (control, saline treatment only); group 2 (SET-M33, 2.5 mg/kg); group 3 (SET-M33, 5 mg/kg); group 4 (M33-NS, 2.5 mg/kg, calculated as SET-M33 content); group 5 (M33-NS, 5 mg/kg, calculated as SET-M33 content). One hour and 20 h after infection, animals were treated again using the same route as for infection, to mimic intrapulmonary administration, in a reproducible protocol. Twenty four hours after infection, the animals were sacrificed and the lungs harvested to count CFUs (Figure 6). All animals showed signs of distress after infection (ie, changes in respiration and rough coat). In 70% of cases, signs of distress worsened (ie, hunched posture and reduced motility) randomly among groups, just after the treatments. The signs were scored as non-observable, mild and severe. Mice showing severe signs were sacrificed and not included in the graph. Signs observed showed no dose-dependency, and have been tentatively ascribed to the route of administration, which is a repeated intratracheal instillation that might generate more distress to animals if compared to aerosol administration.
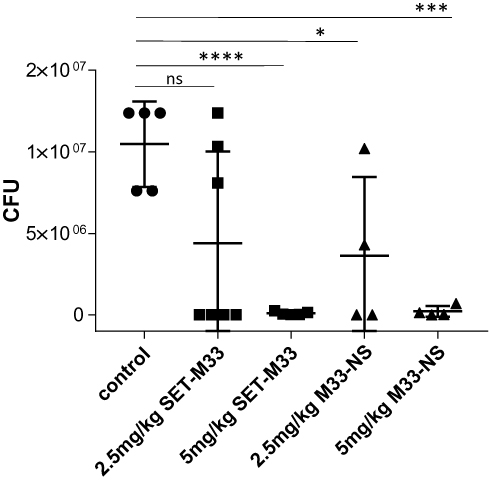 |
Figure 6 Pneumonia model of infection. Efficacy of SET-M33 and M33-NS in a pneumonia model of Pseudomonas aeruginosa-infected BALB/c mice. The median was calculated for each group. Data were analyzed with Graph-pad using an unpaired t test and comparing each group with the controls (*p=0.0291; ***p=0.0001; ****p<0.0001). |
CFU counts of the lungs showed a dose-dependent efficacy of intrapulmonary SET-M33, where only the higher dose (5 mg/kg) mediated complete eradication of the infection (Figure 6). M33-NS too, at the higher dose (5 mg/kg), produced eradication of the infection; hence, a difference between SET-M33 and M33-NS cannot be observed. At the lower dose, M33-NS gave a slightly higher efficacy compared to the free peptide at the same concentration, and though not statistically significant has to be considered an encouraging trend.
Conclusions
Nanosystems that increase lung persistence of antibiotic drugs are of great interest and much needed,23 particularly for those patients who suffer from recurrent pulmonary infections and require high local concentrations and low systemic distribution of the drug. Polymeric particles are used to overcome solubility issues that impair absorption and can cause drug aggregation and associated toxicity.24 Nanoparticles are also considered useful in reducing the onset of resistance, by modulating the interaction of the drug with bacteria and by promoting intracellular uptake.25 When used for lung treatments, high drug concentrations can be achieved directly at the site of infection, overcoming biodistribution issues and reducing toxicity.26
Single-chain polymer nanoparticles are soft-matter biocompatible and biodegradable systems, they show controllable size and shape and have great potential as nanocarriers. In particular, dextran-based single-chain polymer nanoparticles (DXT-NPs) are water-dispersible, biocompatible and easily functionalized.16
SET-M33 is an antibacterial peptide that is currently being developed in preclinical studies, for the treatment of Gram-negative bacteria infections.4–13 Encapsulation of SET-M33 in a nanosystem could theoretically enable its use in aerosols for pulmonary administration with improved bioavailability. To create such a nanosystem the peptide was electrostatically conjugated with dextran nanoparticles to produce M33-NS, 18 nm in size, with acceptable polydispersity and no tendency to aggregate.
M33-NS was shown to be effective against Pseudomonas aeruginosa in a time-kill kinetic experiment. Both M33-NS and the free SET-M33 peptide proved not to be mutagenic in a MLA experiment performed according to OECD guideline no. 490. Cytotoxicity towards animal cell lines was acceptable, since viability reached an average of 70% at a concentration (32 µg/mL) which is twice the MIC, in T24, RAW264.7 and 16HBE14o−. At higher concentrations, M33-NS showed lower toxicity towards macrophages and epithelial cells, than SET-M33 at the same doses, calculated on M33 content. In mouse lymphoma cells, toxicity of the peptide was slightly mitigated by using the M33-NS.
The lung residence time of the antimicrobial peptide administered via aerosol was also greatly improved by encapsulation of SET-M33 in dextran nanoparticles. This was shown by a biodistribution experiment with a radiolabeled analog, where lung residence time for a fraction of the labeled peptide (the fraction bound to the nanocarrier) was about 12-fold with respect to the free SET-M33 peptide. Finally, M33-NS was very efficient in eradicating lung infection in a BALB/c mouse model of pneumonia caused by P. aeruginosa. This study shows the benefits, in terms of higher local concentration and persistence, of using a nanosystem for intrapulmonary administration. Dextran polymers are reported not to be toxic in animals upon iv administration, mice did not show any signs for seven days after treatment, there was no change in the biomarkers of liver and kidney and no apparent histopathological abnormalities.27 The dextran nanocarrier used in this study proved promising in the preliminary in vitro efficacy and toxicity experiments. In the mouse model of pneumonia, the nanosystem showed the same efficacy of the free drug at the eradicating dose and slightly higher efficacy than the free drug at a lower dose, most probably referable to the longer persistence of the nanosystem in the lungs.
Acknowledgments
We thank Silvia Scali for the synthesis of the peptide, Stefano Bindi for animal handling and Hartmut Rahmer for the mouse lymphoma assay. The study received funding from EU FP7, grant N. 604434 PNEUMO-NP.
The current affiliation for Marradi Marco is Department of Chemistry “Ugo Schiff”, University of Florence, via della Lastruccia 3-13, 50019 Sesto Fiorentino (Firenze), Italy.
Disclosure
Professors Chiara Falciani and Alessandro Pini report a patent, WO20101038220, licensed to Setlance. Dr Iraida Loinaz, Dr Raquel Gracia and Professor Marco Marradi report a patent, EP3215545B1, licensed to Kusudama Therapeutics. At the time of the study, Giulia Riolo was employed by SetLance srl, and Marco Marradi by CIDETEC. The authors report no other conflicts of interest in this work.
References
1. Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. WHO newsletter; 2014. Available from: https://www.who.int/antimicrobial-resistance/news/newsletter/en/. Accessed February 6, 2020.
2. Antibiotic resistance: multi-country public awareness survey. WHO documents; 2015. ISBN: ISBN 978 92 4 150981 7. Available from: http://www.who.int/drugresistance/documents/baselinesurveynov2015/en/.
3. Cystic Fibrosis Foundation. Drug Development Pipeline. Available from: https://www.cff.org/trials/pipeline.
4. Pini A, Giuliani A, Falciani C, et al. Characterization of the branched antimicrobial peptide M6 by analyzing its mechanism of action and in vivo toxicity. J Pept Sci. 2007;13(6):393–399. doi:10.1002/psc.858
5. Pini A, Falciani C, Mantengoli E, et al. A novel tetrabranched antimicrobial peptide that neutralizes bacterial lipopolysaccharide and prevents septic shock in vivo. FASEB J. 2010;24:1015–1022. doi:10.1096/fj.09-145474
6. Pini A, Lozzi L, Bernini A, et al. Efficacy and toxicity of the antimicrobial peptide M33 produced with different counter-ions. Amino Acids. 2012;43:467–473. doi:10.1007/s00726-011-1103-z
7. Brunetti J, Falciani C, Roscia G, et al. In vitro and in vivo efficacy, toxicity, bio-distribution and resistance selection of a novel antibacterial drug candidate. Sci Rep. 2016;6:26077. doi:10.1038/srep26077
8. Brunetti J, Roscia G. Lampronti I et al. Immunomodulatory and anti-inflammatory activity in vitro and in vivo of a novel antimicrobial candidate. J Biol Chem. 2019;291:25742–25748. doi:10.1074/jbc.M116.750257
9. Pollini S, Brunetti J, Sennati S, et al. Synergistic activity profile of an antimicrobial peptide against multidrug-resistant and extensively drug-resistant strains of gram-negative bacterial pathogens. J Pept Sci. 2017;23:329–333. doi:10.1002/psc.2978
10. Bracci L, Falciani C, Lelli B, et al. Synthetic peptides in the form of dendrimers become resistant to protease activity. J Biol Chem. 2003;278:46590–46595. doi:10.1074/jbc.M308615200
11. Brunetti J, Falciani C, Bracci L, Pini A. Branched peptides as bioactive molecules for drug design. Pept Sci. 2018;110(5):e24089. doi:10.1002/pep2.v110.5
12. van der Weide H, Vermeulen-de Jongh DMC, van der Meijden A, et al. Antimicrobial activity of two novel antimicrobial peptides AA139 and SET-M33 against clinically and genotypically diverse Klebsiella pneumoniae isolates with differing antibiotic resistance profiles. Int J Antimicrob Agents. 2019;
13. Falciani C, Lozzi L, Pollini S, et al. Isomerization of an antimicrobial peptide broadens antimicrobial spectrum to gram-positive bacterial pathogens. PLoS One. 2012;7:e46259. doi:10.1371/journal.pone.0046259
14. Mansour SC, Pena OM, Hancock RE. Host defense peptides: front-line immunomodulators. Trends Immunol. 2014;35:443–450. doi:10.1016/j.it.2014.07.004
15. van der Weide H, Brunetti J, Pini A, et al. Investigations into the killing activity of an antimicrobial peptide active against extensively antibiotic-resistant K. pneumoniae and P. aeruginosa. Biochim Biophys Acta Biomembr. 2017;1859(10):1796–1804. doi:10.1016/j.bbamem.2017.06.001
16. Gracia R, Marradi M, Cossío U, et al. Synthesis and functionalization of dextran-based single-chain nanoparticles in aqueous media. J Mater Chem B. 2017;5:1143–1147. doi:10.1039/C6TB02773C
17. OECD. Test No. 490: In vitro Mammalian Cell Gene Mutation Tests Using the Thymidine Kinase Gene. OECD Publishing; 2015.
18. Guidance on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use. ICH; June; 2012. Available from: https://www.ich.org/. Accessed February 6, 2020.
19. Volk J, Ziemann C, Leyhausen G, Geurtsen W. Genotoxic and mutagenic potential of camphorquinone in L5178/TK± mouse lymphoma cells. Dent Mater. 2018;34(3):519–530. doi:10.1016/j.dental.2017.12.013
20. Maron DM, Ames BN. Revised methods for the Salmonella mutagenicity test. Mutat Res. 1983;113(3–4):173–215. doi:10.1016/0165-1161(83)90010-9
21. Moore MM, Honma M, Clements J, et al. Mouse lymphoma thymidine kinase gene mutation assay: follow-up meeting of the International Workshop on Genotoxicity Testing—Aberdeen, Scotland, 2003—assay acceptance criteria, positive controls, and data evaluation. Environ Mol Mutagen. 2006;47:1–5. doi:10.1002/(ISSN)1098-2280
22. Cozens AL, Yezzi MJ, Kunzelmann K, et al. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10(1):38–47. doi:10.1165/ajrcmb.10.1.7507342
23. Suzuki S, Cohen SM, Arnold LL, et al. Orally administered nicotine effects on rat urinary bladder proliferation and carcinogenesis. Toxicology. 2018;398–399:31–40. doi:10.1016/j.tox.2018.02.008
24. Xiong MH, Bao Y, Yang XZ, Zhu YH, Wang J. Delivery of antibiotics with polymeric particles. Am J Adv Drug Deliv. 2014;78:63–76. doi:10.1016/j.addr.2014.02.002
25. Kumar M, Curtis A, Hoskins C. Application of nanoparticle technologies in the combat against anti-microbial resistance. Pharmaceutics. 2018;10(1):E11. doi:10.3390/pharmaceutics10010011
26. Ungaro F, d’Angelo I, Coletta C, et al. Dry powders based on PLGA nanoparticles for pulmonary delivery of antibiotics: modulation of encapsulation efficiency, release rate and lung deposition pattern by hydrophilic polymers. J Control Release. 2012;157:149–159. doi:10.1016/j.jconrel.2011.08.010
27. Li J, Zhang K, Ruan L, et al. Block copolymer nanoparticles remove biofilms of drug-resistant gram-positive bacteria by nanoscale bacterial debridement. Nano Lett. 2018;18(7):4180–4187. doi:10.1021/acs.nanolett.8b01000
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.