Back to Journals » Infection and Drug Resistance » Volume 13
Antimalarial Drug Resistance and Novel Targets for Antimalarial Drug Discovery
Authors Shibeshi MA, Kifle ZD ![]() , Atnafie SA
, Atnafie SA ![]()
Received 31 August 2020
Accepted for publication 29 October 2020
Published 10 November 2020 Volume 2020:13 Pages 4047—4060
DOI https://doi.org/10.2147/IDR.S279433
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Melkamu Adigo Shibeshi, Zemene Demelash Kifle, Seyfe Asrade Atnafie
Department of Pharmacology, School of Pharmacy, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
Correspondence: Zemene Demelash Kifle
Department of Pharmacology, School of Pharmacy, University of Gondar, P.O. Box: 196, Gondar, Ethiopia
Email [email protected]
Abstract: Malaria is among the most devastating and widespread tropical parasitic diseases in which most prevalent in developing countries. Antimalarial drug resistance is the ability of a parasite strain to survive and/or to multiply despite the administration and absorption of medicine given in doses equal to or higher than those usually recommended. Among the factors which facilitate the emergence of resistance to existing antimalarial drugs: the parasite mutation rate, the overall parasite load, the strength of drug selected, the treatment compliance, poor adherence to malaria treatment guideline, improper dosing, poor pharmacokinetic properties, fake drugs lead to inadequate drug exposure on parasites, and poor-quality antimalarial may aid and abet resistance. Malaria vaccines can be categorized into three categories: pre-erythrocytic, blood-stage, and transmission-blocking vaccines. Molecular markers of antimalarial drug resistance are used to screen for the emergence of resistance and assess its spread. It provides information about the parasite genetics associated with resistance, either single nucleotide polymorphisms or gene copy number variations which are associated with decreased susceptibility of parasites to antimalarial drugs. Glucose transporter PfHT1, kinases (Plasmodium kinome), food vacuole, apicoplast, cysteine proteases, and aminopeptidases are the novel targets for the development of new antimalarial drugs. Therefore, this review summarizes the antimalarial drug resistance and novel targets of antimalarial drugs.
Keywords: antimalarial, drug resistance, novel targets, vaccines
Introduction
Malaria is an infectious, hematologic disease causing death and illness in children and adults, especially in tropical countries1 Malaria control requires an integrated approach, including prevention, primarily vector control, and prompt treatment with effective antimalarial drugs.2 Malaria is among the most devastating and widespread tropical parasitic diseases in which most prevalent in developing countries.3 Malaria is caused by the Plasmodium parasite, which is transmitted by the bite of a mosquito vector. Five species are known to infect humans: P. falciparum, Plasmodium vivax, Plasmodium ovalae, Plasmodium malariae, and Plasmodium knowelsi. The parasite P. falciparum causes the most dangerous, with the highest rates of complications and mortality.3 Antimalarial drug resistance results in a global resurgence of malaria making a major threat to malaria control. Widespread and indiscriminate use of antimalarial drugs contributes to malaria parasites to evolve mechanisms of resistance.4,5
The malaria life cycle is very complex which requires two organisms as host, mosquito, and human being.6 The most common symptoms of malaria (chills, high fever, sweating, malaise, headache, and muscle aches) manifest usually one to four weeks after infection with the parasite; in relapsing Plasmodium parasites it ranges from five to eight years, but these signs and symptoms may also have seen in other diseases.7 Currently, available malaria diagnostic tools for identification of plasmodium species in human blood samples include microscopy (light or fluorescence)-gold standard method, immuno-chromatographic lateral flow assays (also called rapid diagnostic tests, RDTs), serology, nucleic acid amplification techniques (NATs) that include polymerase chain reaction (PCR) and isothermal amplification and others.8,9
According to the 2018 malaria report WHO estimated that approximately, 219 million cases of malaria from 90 countries, an increase of 2 million cases over 2016. Infants and young children in malaria-endemic countries of Africa typically experience several clinical episodes of malaria before they develop partial immunity. This protects against severe disease and death from malaria.10,11 Malaria continues to burden the overstretched health services in the sub-Saharan region and is a serious public health problem. The same report indicated that 3.1 billion US$ was invested for malaria control and elimination program, of which US$ 2.2 billion benefited the WHO African Region, followed by the WHO Southeast Asia Region US$ 0.3 billion. The WHO African region with 200 million cases (92%) in 2017, followed by the WHO Southeast Asia region (5%), especially Sub-Saharan Africa suffers by far the greatest malaria burden worldwide and is currently undergoing a profound demographic change. Almost 93% of all deaths due to malaria in 2017 were from Africa. Globally 266, 000 (61%) malaria deaths were estimated to be in children less than 5 years age.12 In most areas of Africa, P. vivax infection is essentially absent because of the inherited lack of the Duffy antigen receptor for chemokine on the surface of red blood cells that are involved in the parasite invasion of erythrocytes.13 However, in Brazilian Amazon, Madagascar, and Central Sudan implicated that individuals with negative Duffy antigen receptor were infected with p. vivax. P.falciparum species are dominant in Africa and the highest-burden of P. vivax infection is in Southeast Asia and South America14 In Ethiopia, major malaria transmission seasons are from September to December and June to August.15 According to the 2018 federal ministry of health (FMOH) of Ethiopia report many densely populated highland areas are malaria-free including the capital city of Addis Ababa. Health management information system (HMIS) of Ethiopia report between June 2016 and July 2017, 1,530,739 confirmed malaria illnesses (69.24% P. falciparum, 30.76% P. vivax) malaria illnesses from these 356 deaths were reported.16
Drugs Used for the Treatment of Malaria
Currently available antimalarial drugs are broadly categorized into three types. Aryl amino alcohol compounds including quinine, quinidine, halofantrine, lumefantrine, chloroquine, amodiaquine, mefloquine, cycloquine, etc. Antifolate compounds: proguanil, pyrimethamine, trimethoprim, etc. Artemisinin compounds like artemisinin, dihydroartemisinin, artesunate, artemether, arteether, etc.17,18
Most of the antimalarial drugs target the asexual erythrocytic stages of the parasite (blood schizonticidal drugs). Two types either fast-acting (Chloroquine, quinine, and mefloquine) or slow-acting (Pyrimethamine, sulphonamides, and sulphone). Tissue schizonticidal drugs target the hypnozoites (dormant stage of the parasite) in the liver whereas gametocytocidal drugs destroy sexual erythrocytic forms of the parasite in the bloodstream preventing transmission of malaria to the mosquito. Sporontocides prevent or inhibit the formation of malarial oocysts and sporozoites in the infected mosquito.19
Quinolines (affects polymerization of hemozoin), antifolates (block dihydrofolate reductase and dihydropteroate synthetase enzymes of the parasite) and artemisinin (have various mechanisms), administered alone or in combination to treat malaria18 Artemisinin combination therapy is the cornerstone of malaria control in sub-Saharan Africa such as artemether/lumefantrine and artesunate/amodiaquine. Because of the notorious capacities of Plasmodium falciparum to develop drug resistance, many antimalarial programs have recently included dihydroartemisinin/piperaquine (DHA/PPQ) as a second-line antimalarial drug.20
Resistance to Plasmodium vivax and Plasmodium falciparum
Before dealing with resistance to malaria it is better to know the terminologies of recurrence, recrudescence, relapse, and resistance (4R’s). Recurrence is the recurrence of asexual parasitemia following treatment (in P. vivax and P. ovale infections only) or a new infection. Recrudescence is the recurrence of asexual parasitemia after the treatment of the infection with the same infection that caused the original illness. Relapse is the recurrence of asexual parasitemia in P. vivax and P. ovale malaria deriving from persisting liver stages from persisting hypnozoites. Resistance is the ability of a parasite strain to survive and/or multiply despite the proper administration and absorption of an antimalarial medicine in the dose normally recommended.21 Plasmodium vivax is continued to put a substantial burden on the malaria-endemic world with the morbidity and mortality due to its propensity to cause recurrent infections.22 Plasmodium vivax forms dormant liver stages (hypnozoites), which causes relapses of infection weeks to months after the initial attack. Recurrent infections can occur as often as every three weeks, with relapses the main cause of vivax illness. Even though chloroquine is the first-line treatment for P.vivax malaria in most endemic countries resistance is the main problem facing chloroquine in different parts of the world. In Africa and South America chloroquine resistance to plasmodium falciparum first appeared in 1978 and 1996 respectively.23,24
Chloroquine-resistant Plasmodium vivax was first reported in 1989 from Papua New Guinea. High-grade chloroquine-resistant Plasmodium vivax is prevalent in areas such as Indonesia and Oceania (regarded as epicenters of chloroquine resistance).25 Both the acute illness and relapses from hypnozoites can be effectively prevented by the administration of a combination of chloroquine with primaquine (radical cure). Primaquine has activity against both blood and liver stages, including against chloroquine-resistant strains. Severe P. vivax infections can cause cerebral malaria with generalized convulsions and status epilepticus, severe anemia, hepatic dysfunction and jaundice, acute lung injury, pulmonary edema, splenic rupture, acute renal failure, and severe thrombocytopenia with or without bleeding from different parts of the body.26–28
Primaquine has activity against both asexual and sexual blood stages of the parasite as well as against the liver stage schizonts and hypnozoites.29 Primaquine can result in significant hemolysis in people with glucose-6-phosphate dehydrogenase deficiency (G6PDd). G6PD deficiency is the most common heritable enzymopathy in the world, with a prevalence range of 2% to 40%.30 The WHO for radical cure of vivax malaria currently recommends the use of a daily dose of 0.25 mg/kg/day (3.5 mg/kg total dose) primaquine taken with food once daily, which can be either co-administered with chloroquine or artemisinin combination therapy depending on chloroquine sensitivity in the area for radical cure of vivax malaria. Current guidelines recommend a 14-day course of primaquine administered either once or twice daily to reduce the risk of hemolysis and improve tolerability from gastrointestinal disturbance.2
In Ethiopia first report of P. falciparum and P. vivax chloroquine treatment failure in Debre Zeit, was in 1995. The invasion of human red blood cells by the extracellular merozoite form of Plasmodium falciparum is a process central to the pathogenesis of this devastating pathogen. In the present time control of multidrug-resistant P. falciparum malaria has become a very difficult task because endogenous allelic exchanges occurred in P. falciparum have increased the therapeutic failures and significantly increased the levels of resistance worldwide. As evolution is an unending process how the formation of drug-resistant mutant alleles stops is a very concerning question.31 Usually higher mean parasitemia index is seen in infected individuals with P. falciparum but P. vivax infection generally exhibits low parasitemia index due to its preference to invade reticulocytes rather than erythrocytes.32
Mechanism of Antimalarial Drug Resistance
According to the World Health Organization (WHO), antimalarial drug resistance is defined as the ability of a parasite strain to survive and/or to multiply despite the administration and absorption of medicine given in doses equal to or higher than those usually recommended but within the tolerance of the subject, provided drug exposure at the site of action is adequate. Resistance to antimalarial arises because of the selection of parasites with genetic mutations or gene amplifications that confer reduced susceptibility.2
Resistance appears to be caused by a change in the structure, function, or quantity of a protein. The change in the protein is mediated by genetic changes such as single nucleotide polymorphisms (SNP) or gene amplification. Because of antimalarial drug resistance is becoming the most difficult hurdle for the success of antimalarial therapy, so scientists are in continuous move researching to overcome the problem. Resistant parasite strains will always emerge, requiring the continual generation of new molecules. The novel drugs with a new mechanism of action are entering into clinical trials.17
Several factors facilitate the emergence of resistance to existing antimalarial drugs. To mention some factors, the parasite mutation rate, the overall parasite load, the strength of drug selected, the treatment compliance, and poor adherence to malaria treatment guidelines.33 Improper dosing, poor pharmacokinetic properties, fake drugs lead to inadequate drug exposure on parasites.34 Poor-quality antimalarial (falsified antimalarial without active pharmaceutical ingredient (APIs)) may aid and abet resistance by increasing the risk of hyperparasitaemia, recrudescence, and hypergametocyopaenia, wrong APIs such as the use of halofantrine instead of artemisinin which without chemical analysis will be invisible to investigators but not to parasites.35,36 Frequently targeted biological pathways by antimalarial drugs in parasites of plasmodium are heme detoxification (in digestive vacuole) biosynthesis folate and pyrimidine and electron transport (in mitochondrion). Studies done during treatment with aryl amino alcohols quinine, lumefantrine (LMF), and mefloquine (MFQ) from South East Asia showed that copy-number changes in pfmdr1, as well as PfCRT and PfMDR1 sequence variants, can affect the parasite’s susceptibility.32 Unlike other diseases (eg Tuberculosis, AIDS), malaria drug resistance mechanisms are unique, as the parasite is capable of inducing resistance in the exact cellular target of the drug, drug resistance phenotype is mostly induced due to enhanced and non-specific efflux of drugs through induction of multidrug resistance (MDR) transporters. In malaria, MDR transporters are not the primary mechanism of resistance (Table 1).37
 |
Table 1 Summary of Some Antimalarial Drugs, Mechanism of Action, Site of Action, and Mechanism of Resistance |
The antimalarial activity of Artemisinin is due to its unique trioxane structure with an endoperoxide bond. Usually, semi-synthetic derivatives are used clinically (artemether, artesunate, and dihydroartemisinin) because due to the low solubility of artemisinin.38 Artemisinin combination therapy (ACT), currently recommended are artemether + lumefantrine, artesunate + amodiaquine, artesunate + mefloquine, artesunate + sulphadoxine-pyrimethamine, and dihydroartemisinin + piperaquine, the current gold standard for malaria treatment but resistance is emerging in different areas. Resistance to Artemisinin and its derivatives are emerging and troubling phenomena in malaria treatment.39 In the Greater Mekong Subregion of Asia, the Artemisinin-based drug resistance is emerging.40 As resistance to each new malaria drug arises, it becomes necessary to combine two or more component drugs to slow the spread of resistance to reduce the chance of resistance combinations containing an artemisinin derivative that is currently in use.41 Within the malaria parasite-host hemoglobin is degraded by a series of protease enzymes to release peptides and amino acids required for development and to create space within its digestive vacuole in which buildup of hematin occurs which is potentially toxic to the parasite.
Artemisinin and its derivatives have a fast onset of action but are eliminated soon (half-life 0.5–1.4 h) from humans for this reason it is essential to combine with slow clearing drugs to kill residual parasites.42 Artemisinin and its derivatives can be combined with other antimalarial drugs at least for two reasons, first to prolong the half-life of Artemisinin and its derivatives, second, to prevent resistance.43 Recent reports in Equatorial Guinea showed that P. falciparum isolate was resistant to artemisinin.44
Molecular Mechanism of Artemisinin Resistance
Artemisinin possesses a long-acting effect against drug-resistant malaria parasites, and also able to reduce the parasite burden in asymptomatic individuals who serve as reservoirs for malaria transmission.45 Artemisinin and its derivatives (artesunate, artemether, and arteether) are potent and fast-acting drugs that cause a rapid decline in parasitemia during the first days of treatment. Meshnick using mass spectroscopy observed that Artemisinin can alkylate heme resulting in decomposition of the endoperoxide bridge to produce carbon-centered free radicals which are crucial for selectively toxic to malaria parasites.46 Specific protein or enzyme is used as a molecular target for Artemisinin. Invitro studies show that hemoproteins such as catalase, cytochrome c, and hemoglobin but not free globin, is alkylated by Artemisinin.47
Another molecular target is PfATP6; Artemisinin inhibits of a parasite Ca2+ transporting ATPases (SERCA – Sarco/endoplasmic reticulum membrane calcium ATPase). SERCA reduces cytosolic free calcium concentrations by actively concentrating Ca2+ into membrane-bound stores, an activity critical to cellular survival.48 In the parasite membrane, Artemisinin accumulates within neutral lipids and causes parasite membrane damage.49 Artemisinin has shown resistance in P. falciparum cultures50 and P. voelii mouse models51 and in vitro resistance in field isolates.52
P. falciparum Kelch 13 (PfKelch13), the marker for artemisinin resistance in P. falciparum malaria, is not an enzyme or a pump but rather is predicted to be a substrate adapter for a cullin E3 ligase, with a putative substrate of P. falciparum phosphatidylinositol 3-kinase (PfPI3K) and a redox sensor.53 Kelch-like protein K13 is a molecular marker for Artemisinin resistance, but no detectable impact in Africa (except one report with P. falciparum K13-variant infection from western Africa). The reason behind this low impact is the greater degree of acquired immunity there, resulting from repeated exposure to P. falciparum, which builds host immunity to help control drug-resistant infections.44 Mutant K13 results in lowered ART interactions with P. falciparum phosphatidylinositol-3-kinase (PfPI3K).54 In vitro and in vivo studies in many areas of Southeast Asia show that mutations in the K13 propeller gene (PF3D7_1343700 or PF13_0238) are linked to artemisinin resistance.55
Molecular Markers of Antimalarial Drug Resistance
Molecular markers of antimalarial drug resistance are used to screen for the emergence of resistance and assess its spread. It provides information about the parasite genetics associated with resistance, either single nucleotide polymorphisms or gene copy number variations which are associated with decreased susceptibility of parasites to antimalarial drugs. Detection of molecular markers provides a feasible means of tracking the emergence and/or spread of antimalarial drug resistance.56 P. falciparum chloroquine resistance transporter gene (PfCRT), chloroquine accumulates within the DV (digestive vacuole) of the parasites where there is mutant PfCRT, accumulation of chloroquine in parasites is very less as compared to parasites expressing wild type PfCRT,57 as a result, chloroquine-resistant parasites can export chloroquine via active transport,58 implying that mutant and wild type PfCRT have different drug transporting properties. P. falciparum multidrug resistance transporter 1 (PfMDR1) locates in the digestive vacuole of the parasite and function as a general importer sequestering toxic metabolites and drugs into the digestive vacuole (DV). Pfmdr1 indirectly influences drug flux by affecting intracellular ions and PH.59 Studies show that wild type PfMDR1 transports quinine and chloroquine but not halofantrine while mutant PfMDR1 transports halofantrine but not quinine or chloroquine.60
The multidrug resistance-associated protein (PfMRP) is a member of the ATP-binding cassette (ABC) proteins family and ABC transporter C subfamily. Genetic disruption PfMRP leads to increased parasite susceptibility to several antimalarial drugs like chloroquine, quinine, artemisinin, piperaquine, and primaquine and accumulates more glutathione (GSH), chloroquine, and quinine.61 Cytochrome bc1 complex catalyzes the transfer of electrons from ubiquinol to cytochrome c thereby maintaining the membrane potential of mitochondria used to produce ATP by an ATP synthase. Mutations in the Cytochrome bc1 complex leads to atovaquone resistance this is because atovaquone binds to the ubiquinol binding site, thereby disrupting the electron transfer chain.62,63
Imidazolopiperazine (IPZ) class of drug compounds (saturated cyclic amines) has activity in both liver and blood-stage parasites as an antimalarial drug for use in prophylaxis, treatment, and prevention of malaria disease transmission. Whole-genome sequencing done by Prof Paul and his co-workers of these drug-resistant Plasmodium falciparum clones, genes associated with drug resistance are identified, an acetyl-CoA transporter (PFACT) and a UDP-galactose transporter (PFUGT). These mechanisms responsible for resistance are members of a family of membrane transporter proteins (major facilitator superfamily or MFS).MFS is the largest and most ubiquitous secondary transporter family responsible for the translocation of small molecules including metabolites, nucleosides, oligosaccharides, amino acids, oxyanions, and drugs.64,65
Imidazolopiperazine is promising drug candidates with the potential to aid in malaria elimination include KAF156 and KAF179 (currently in Phase II clinical trials). They possess low Nanomolar potency against P. falciparum liver stages, asexual blood stages, and sexual stage gametocytes.66 P. falciparum V-type H+ pyrophosphatase (PFVP2) is located in the DV membrane and increased transcription of pfvp2 has been observed in-vitro when P. falciparum are exposed to chloroquine (10-fold up-regulation) and lumefantrine (2-fold up-regulation). The up-regulation of pfvp2 implies that it could be involved in maintaining the H+ balance in the parasite DV and to compensate for H+ loss caused by the removal of protonated CQ.67,68
Antimalarial Drug Resistance Surveillance
Antimalarial drug resistance surveillance can be performed through in vivo studies such as therapeutic efficacy studies, in vitro/ex vivo studies of cultured malaria parasites, and molecular studies assessing known markers of antimalarial drug resistance. As outcomes have direct clinical relevance in therapeutic efficacy studies it is regarded as the gold standard for informing antimalarial drug resistance and for drug regimen change as well. In malaria-endemic countries, routine monitoring of antimalarial drug efficacy is carried out at sentinel sites by national malaria control programs using a standardized WHO protocol. Treatment response is defined as the absence of parasitemia at follow-up, on day 28 or 42. WHO recommends a switch to another more effective first-line drug if a 10% treatment failure rate is reached.69,70
Novel Targets of Antimalarial Drugs
The existing antimalarial drugs were identified based on the major metabolic pathway differences of the parasite with its host. The Key metabolic pathways of the Plasmodium species, including oxidative stress, heme detoxification, fatty acid synthesis, and nucleic acid synthesis are some of the novel targets for antimalarial drug discovery and development.71,72 Though most of the antimalarial drugs used for many years, presently the use of such drugs is limited as a result of drug resistance. According to previous studies, there are no antimalarial agents recognized to inhibit an identified antimalarial drug targets.73 In its place, the majority of the antimalarial agents were discovered in both in vitro model and animal models (in vivo). Thus, the exact mechanism of action of most antimalarial drugs is not known. In addition, the mechanism of antimalarial drug resistance was not well known for most antimalarial drugs.72 In addition to increasing the need to develop new antimalarial drugs, identifying countermeasures either to delay or minimize the development of resistance against new drugs is an important phenomenon.
Glucose Transporter PfHT1
Glucose is a source of energy for Intra erythrocytic malarial parasites in which infected erythrocytes consume higher energy than normal erythrocytes.74 P. falciparum almost fully depend on glycolysis for energy production, deprived of energy stores; depend on continuous uptake of glucose as a source of energy. The Pyruvate is converted into lactate to yield ATP in the parasite, which necessitates for replicating in the intraerythrocytic site.75 Initially, via GLUT1 transporter Glucose is transported from the blood into the parasitized erythrocyte, which is abundant in the erythrocyte membrane.76 The Plasmodium glucose transporter P. Falciparium Hexose transporter (PFHT) is essential for parasite growth and survival,77 as well as, is the main transporter of glucose.78 GLUT1 transporter can only transport D-glucose, while P. Falciparum Hexose transporter non selectively transports both D-fructose and D-glucose. Thus, the differences between PFHT and GLUT1 in their interaction with the substrates, proposed that selective inhibition of P. Falciparum Hexose transporter is a potential novel target for the discovery of new antimalarial agents.79 In the previous study, Compound 3361 which is a long chain O-3-hexose derivative can hinder the uptake of fructose and glucose by P. Falciparium Hexose transporter nevertheless, it cannot hinders hexose transport by mammalian transporters (GLUT1 and 5). Similarly, Compound 3361 was reported to hinders the glucose uptake by P. vivax of P. Falciparum Hexose transporter.80
Targeting the Parasite Protein Kinases
Kinases are involved in phosphorylation, transcriptional control, post-transcriptional control, and protein degradation in the plasmodium parasite life cycle. So, could be the strategic targets for the development of antimalarial drugs. The most studied Cyclin-dependent kinases (CDKs) in Plasmodium falciparum are P. falciparum protein kinase 5 (PfPK5), 6, and P. falciparum mitogen related kinase (PfMRK). By in-vitro study two compounds, flavopiridol and lomoucine have shown inhibition of PfPK5, by decreasing DNA synthesis and changing total RNA synthesis and parasite growth.81
The P. falciparum kinases play a significant role in the parasite differentiation and growth. Amongst numerous kinases, cyclin-dependent protein kinases (CDKs) are conspicuous targets for the development of drugs, numerous cyclin-dependent protein kinases selective inhibitors were discovered for the management of different diseases such as neurological disorders, infectious diseases, and cancers. Presently, they become a potential novel target in the discovery and development of new antimalarial drugs.82,83 The PfCDPK4 plays a key role in the formation of infectious sporozoites through the sexual phase of the malarial life cycle. Compound 1294 which is PfCDPK4 inhibitor, revealed an antimalarial effect with a novel mechanism of action through preventing the transmission of parasites from mosquitoes to humans.84 Likewise, Imidazopyridine derivatives have shown significant PfCDPK1 inhibitory effect with nanomolar antimalarial activity in both in vitro and in vivo models (Table 2).85
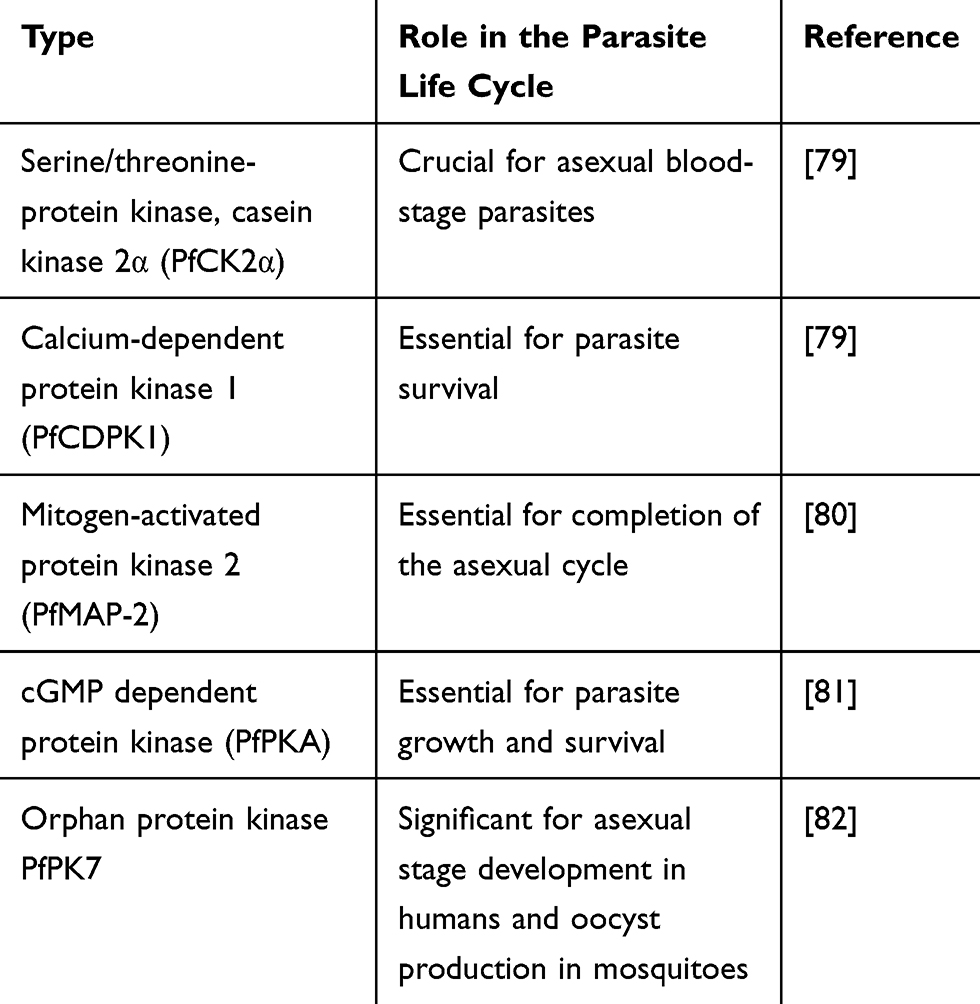 |
Table 2 Role of Kinase in the Plasmodium Parasite Life Cycle |
Food Vacuole as a Drug Target
The blend of digestive vesicles provides a large digestive vacuole/food vacuole through the growth of the malaria parasite inside the human erythrocytes. Food vacuole is accountable for the degradation of 60–80% of the host red cell hemoglobin, which has a key role in the attainment of amino acid which is essential for parasite development and growth. Investigation of this degradation pathway can be a promising method for the discovery and development of novel antimalarial agents. This pathway is started by a series of protease enzymes that digest hemoglobin into small peptides. During proteolysis, heme is released from hemoglobin as a toxic byproduct which is detoxified by conversion into hemozoin. In the previous studies, hemozoin comprises about 95% of the free iron synthesized through hemoglobin digestion.86,87 In addition, two possible mechanisms responsible for the degradation of hemoglobin were reported (degradation by hydrogen peroxide inside the large digestive vacuole and glutathione-dependent degradation within the cytoplasm).88–90
Electron Transport Chain (ETC)
The plasmodial mitochondrial electron transport chain is produced from non-proton motive quinone reductases, such as malate quinone oxidoreductase (MQO), (DHODH), (Alternative Complex I), type II NADH dehydrogenase (NDH2, glycerol 3-phosphate dehydrogenase (G3PDH), dihydroorotate dehydrogenase, and succinate dehydrogenase (SDH, Complex II), and proton motive respiratory complexes, such as ATP synthase (Complex V), cytochrome c oxidase (Complex IV), and bc1 complex (Complex III). The electron transport chain needs cytochrome c1 and ubiquinone (coenzyme Q) which serve as electron carriers between the complexes.91–93 The pool of electron transport chain and carbon metabolism antimalarial targets that have been under the lamp post in recent years, as well as suggest a promising new avenue for the validation of novel drug targets for the treatment of malaria. The interaction between the pathways vital for the parasite, such as aspartate metabolism, mitochondrial tricarboxylic acid cycle, and pyrimidine biosynthesis, is described to create a road map of novel antimalarial agents.94
Apicoplast as Drug Targets
Recently, blocking the P. falciparum ribosome and other parts of the translational machinery accountable for protein synthesis are becoming a promising target for the discovery and development of novel antimalarial agents. The plasmodium species have three genomes: apicoplast, nuclear, and mitochondrial.95 The apicoplast is a chloroplast like organelle of apicomplexan parasites. The apicoplast resulted from endosymbiosis, leading to an organelle that maintains certain specific functions, probably including fatty acid, heme, and amino acid metabolism.96 The apicoplast genome of P. falciparum comprises a 35-kb DNA which is small in size.97 The apicoplast is a non-photosynthetic plastid that is vital for the malarial parasite since it covers a large number of important metabolic biochemical pathways (biosynthesis of fatty acid, isoprenoid precursors, and heme synthesis) for the Plasmodium falciparum survival. Human beings do not have these metabolic biochemical pathways which are important for ideal drug targeting.98,99
Even though the majority of proteins of this organelle are encoded in the nuclear genome and are subsequently transported to the apicoplast, it also encodes a full set of tRNAs, some ribosomal proteins, three genes for the subunits of an oligomeric RNA polymerase, a gene for the elongation factor PfTu and a gene contributing to the Fe–S pathway.100 Since the apicoplast possess unique metabolic pathways such as isoprenoid, heme synthesis, and fatty acid, which are not found in the human,101 it could be a potential drug target for the management of malaria. As reported in the previous study, protein syntheses inhibitors play a key role in the clinical success of potent antibiotics. Azithromycin, Clindamycin, and Doxycycline revealed antimalarial activity since they can inhibit the ribosomes within the apicoplast and Plasmodium species mitochondria, resulting in loss of the normal function of these organelles.95 For prevention of malaria, azithromycin has shown a noticeable protective effect in Kenyan102 and Indonesian103 adults when received daily doses, even though the preventive activity was lower than doxycycline in both trials (protective efficacy in Kenya was 93% for doxycycline vs 83% for azithromycin; in Indonesia 96% vs 72%, respectively). In Kenya, azithromycin preventive activity was fairly deprived when administered weekly (64%). Mass distribution of azithromycin for the control of trachoma was linked with a decrease in malaria parasitemia as compared to controls.104 Azithromycin + piperaquine was well tolerated in pregnant Papua New Guinean women,105 even though preventive efficacy data are not obtainable.
Plasmodium Proteases
Plasmodium proteases are a regulatory and ubiquitous catalytic enzyme that play a significant role in the survival of the plasmodium parasite and responsible for the hydrolysis of the peptide bond (Figure 1).106 The role of plasmodium proteases in the pathogenesis of malaria disease includes activation of inflammation, cell/tissue penetration, invasion of erythrocyte, development of the parasite, immune evasion, autophagy, and hemoglobin and other proteins breakdown.107 Plasmodium proteases such as aspartate, serine, cysteine, metallo, threonine, and glutamate are auspicious drug targets for the treatment of malaria since the disruption of the plasmodium proteases gene inhibits the degradation of hemoglobin and the growth of the parasite in the erythrocyte stages.108
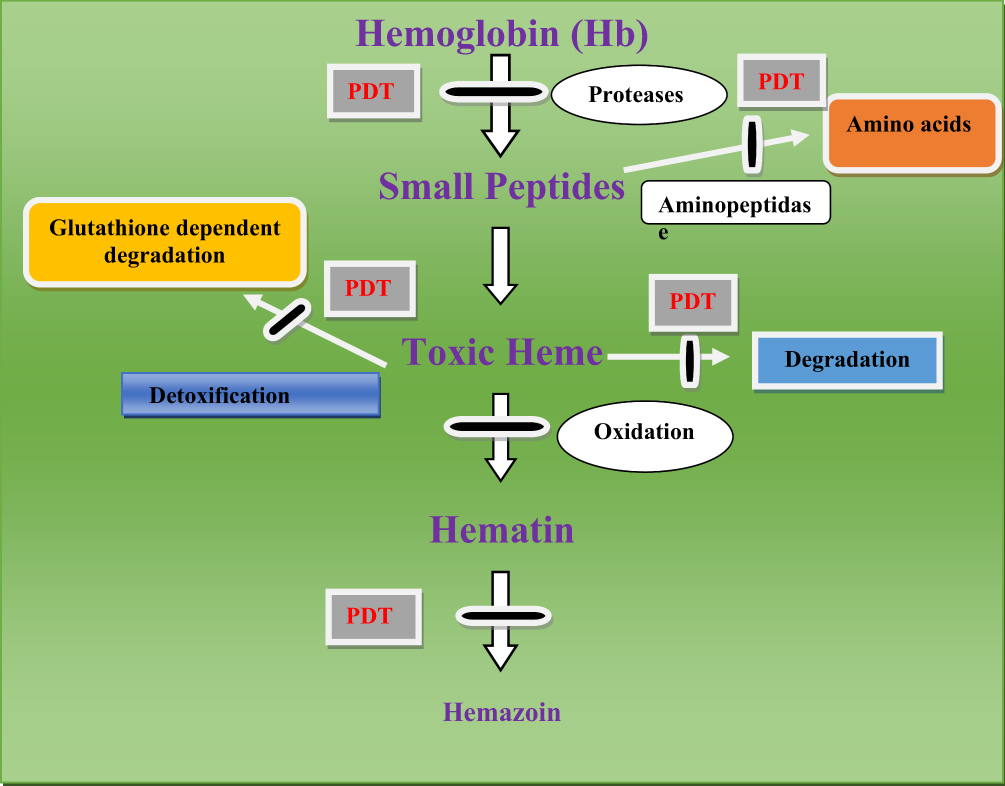 |
Figure 1 Targets of proteases and amino peptidase, malaria parasite detoxification mechanism. |
Proteases are generally used for the rupture and subsequent reinvasion of erythrocytes by merozoite-stage parasites and the degradation of hemoglobin by intraerythrocytic trophozoites. For instance, drugs that inhibit Plasmodium cysteine proteases are the potential targets for malarial treatment and shown potential effects.109 Cysteine proteases have different roles in Plasmodium parasites including hemoglobin hydrolysis (provide amino acids for parasite protein synthesis, maintain the osmotic stability of malaria parasites),110 erythrocytic rupturing, helps merozoites to be released,111 erythrocyte invasion also it has a role on non-erythrocytic parasitic stages (Table 3).
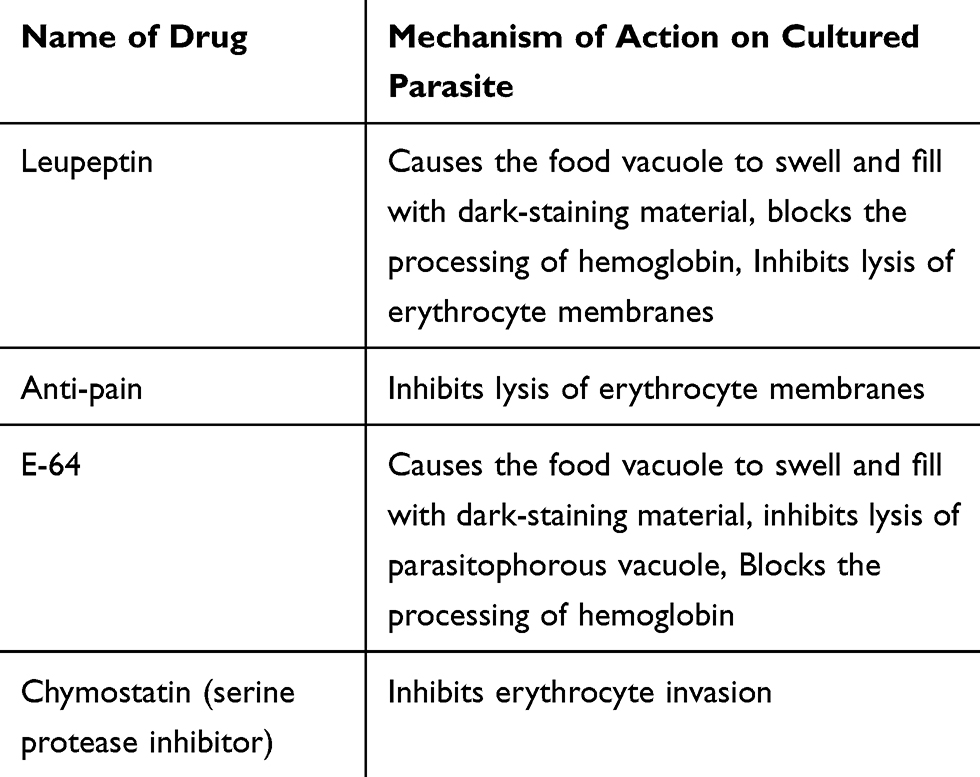 |
Table 3 Mechanism of Action of Protease Inhibitors |
Aminopeptidases
Amino peptidases catalyze the cleavage of amino acids from the amino terminus of peptides and proteins and are distributed widely in prokaryotes and eukaryotes as either integral membrane or cytosolic proteins (Figure 1). They play a role in protein and peptide metabolism, activation/inactivation of biologically active peptides, removal of the N-terminal methionine from newly synthesized proteins, and the trimming of antigens for presentation by the major histocompatibility complex-1 system.112 Bestatin inhibits the growth of P. falciparum in vitro and in vivo and is active against the intraerythrocytic stages. Bestatin appears to inhibit both leucine aminopeptidase (PfLAP) and membrane alanine aminopeptidase, (PfA-M1) by chelating the active metal ions in their metal-binding centers. Inhibitors capable of binding compactly within the active site and chelating the tightly bound metal ions of both PfA-M1 and PfLAP. It has two metal-binding sites, a readily exchangeable site, and a tight binding site may prove more potent and show greater anti-malarial activity.113–115
A Recent Achievement in the Discovery and Development of Antimalarial Agents
Drugs currently in Phase I, II and III trials for blood-stage treatments of malaria includes KAE609 (cipargamin) inhibit Na+-TPase 4 ion channel,116 KAF156/GNF156/(Cyclic amine resistance unknown mechanism of locus (PfCARL) inhibitor),117 Albitiazolium/SAR9727/(Inhibit the transport of choline into the parasite),118 DSM265 (Inhibit dihydroorotate dehydrogenase enzyme),119 Methylene Blue (Prevents haem polymerisation by inhibiting P. falciparum glutathione reductase),120 Sevuparin/DF02/(Anti-adhesive polysaccharide derived Blocks merozoite invasion and sequestration),121 MMV048 (Inhibiting the parasite enzyme phosphoinositol 4-kinase enzyme),122 MMV390048 (Phosphatidylinositol 4-kinase (PfPI4K) inhibitor),123 Fosmidomycin + piperaquine (DOXP pathway), Artefenomel (oz439) + Piperaquine (Synthetic endoperoxide),124 OZ277+ Piperaquine (Inhibit Pf-encoded sarcoplasmic endoplasmic reticulum calcium ATPase), P218 (PfDHFR inhibitor),125 M5717/DDD498/(Protein-making machinery of the malaria parasite, liver- stage P. falciparum),126 SJ733 (The P-type Na+–ATPase transporter),116 and Spiroindolone (cipargamin) inhibits PfATP4, a parasite plasma membrane Na+-ATPase that regulates sodium (maintains low-level Na+ in the cytosol) and osmotic homeostasis. Cipargamin is used in the treatment of falciparum and vivax malaria. Inhibition of PfATP4 increases a Na+ in the cytosol as Na+ moves into the cell, down its electrochemical gradient leading to a concomitant increase in cytosolic pH (PfATP4-mediated acid load). Mutation in PfATP4 results in cipargamin.127 Artefenomel is a new synthetic antimalarial peroxide that clears parasitemia rapidly in both P falciparum and P vivax malaria. It has a good safety profile and long half-life (for a single dose malaria cure).128
Conclusion
In conclusion, present and future therapeutic targets for the discovery and development of novel antimalarial agents were reviewed. The frequently emerging antimalarial drug resistance including combination therapies globally forces the scientists to search and develop antimalarial drugs with novel mechanisms of action. Resistance to two highly dominant species Plasmodium falciparum and Plasmodium vivax is highly predominant in south East Asia, Africa, and South America. The complex life cycle of malaria parasite provoke obstacle in the discovery of new therapeutic agents, nevertheless, the discovery of novel biochemical pathways in the malaria parasite offers new opportunities for development antimalarial agents. Due to the resistance of antimalarial agents globally, searching for novel cellular targets and developing new therapeutic agents targeting old targets is both imperative aspects in fighting drug-resistant malaria. The future antimalarial drug development will better target medicines with a distinctive mechanism of action.
Abbreviations
ABC, ATP binding cassette; ACT, artemisinin combination therapy; ART, artemisinin; ATQ, atovaquone; CDKs, cyclic dependent kinases; CLD, clindamycin; CQ, chloroquine; CYC, cycloguanil; Cytb1, cytochrome b subunit 1; DHA/PPQ, dihydroartemisinin/piperaquine; DHFR, dihydrofoliate reductase; DHPS, dihydropteroate synthase; DV, digestive Vacuole; G6PDd, glucose-6-phosphate dehydrogenase deficiency; GSH, glutathione; IPZ, imizolopiperazine; LMF, lumefantrine’ MFQ, mefloquine; MFS, major facilitator superfamily; NATs, nucleic acid amplification; PAM, pregnancy associated malaria; PCR, polymerase chain reaction; Pfact, Plasmodium falciparum acetyl CoA transport; PfA-M1, Pf membrane alanine aminopeptidase; PfCRT, Pf chloroquine-resistant transporter; PfKelch13, Pf kelch like protein 13; PfLAP, Pf leuci ne aminopeptidase; PfMDR1, pf multidrug resistance 1; Pfmrk, Pf mitogen related kinases; pfMRP, Pf multidrug resistance-associated protein; PfPI3K, Pf phosphatidylinositol-3-kinase; Pfpk5, Plasmodium falciparum protein kinase 5; SERCA, sarco/endoplasmic reticulum Ca2+ ATPase.
Ethical Approval
Not applicable.
Acknowledgment
We would like to acknowledge the University of Gondar for providing materials.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no competing interests.
References
1. Buffet PA, Safeukui I, Deplaine G, et al. The pathogenesis of Plasmodium falciparum malaria in humans: insights from splenic physiology. Blood. 2011;117(2):381–392. doi:10.1182/blood-2010-04-202911
2. Reyburn H. New WHO guidelines for the treatment of malaria. BMJ. 2010;340:c2637. doi:10.1136/bmj.c2637
3. Zhang Y, Xie L, Xie L, Bourne P. The Plasmodium falciparum drugome and its polypharmacological implications. bioRxiv. 2016;042481.
4. Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell. 2006;124(4):755–766. doi:10.1016/j.cell.2006.02.006
5. Lee RS, Waters AP, Brewer JM. A cryptic cycle in haematopoietic niches promotes initiation of malaria transmission and evasion of chemotherapy. Nat Commun. 2018;9.
6. Woldearegai TG Characterization of Plasmodium falciparum mature gametocytes: lifespan, immunogenicity and susceptibility to novel compounds. 2018.
7. Kondilis E, Giannakopoulos S, Gavana M, Ierodiakonou I, Waitzkin H, Benos A. Economic crisis, restrictive policies, and the population’s health and health care: the Greek case. Am J Public Health. 2013;103(6):973–979. doi:10.2105/AJPH.2012.301126
8. Britton S, Cheng Q, McCarthy JS. Novel molecular diagnostic tools for malaria elimination: a review of options from the point of view of high-throughput and applicability in resource limited settings. Malar J. 2016;15(1):88. doi:10.1186/s12936-016-1158-0
9. Trampuz A, Jereb M, Muzlovic I, Prabhu RM. Clinical review: severe malaria. Crit Care. 2003;7(4):315. doi:10.1186/cc2183
10. O’Meara WP, Mangeni JN, Steketee R, Greenwood B. Changes in the burden of malaria in sub-Saharan Africa. Lancet Infect Dis. 2010;10(8):545–555. doi:10.1016/S1473-3099(10)70096-7
11. Sachs J, Malaney P. The economic and social burden of malaria. Nature. 2002;415(6872):680. doi:10.1038/415680a
12. Organization WH. WHO malaria policy advisory committee meeting: meeting report, April 2018. World Health Organization;2018.
13. Guerra CA, Snow RW, Hay SI. Mapping the global extent of malaria in 2005. Trends Parasitol. 2006;22(8):353–358. doi:10.1016/j.pt.2006.06.006
14. Woldearegai TG, Kremsner PG, Kun JF, Mordmüller B. Plasmodium vivax malaria in Duffy-negative individuals from Ethiopia. Trans R Soc Trop Med Hyg. 2013;107(5):328–331. doi:10.1093/trstmh/trt016
15. Deressa W, Olana D, Chibsa S. Magnitude of malaria admissions and deaths at hospitals and health centers in Oromia, Ethiopia. Ethiop Med J. 2004;42(4):237–246.
16. Ethiopia U. President’s malaria initiative Ethiopia-malaria operational plan FY 2017. 2017.
17. Kumar S, Bhardwaj T, Prasad D, Singh RK. Drug targets for resistant malaria: historic to future perspectives. Biomed Pharmacother. 2018;104:8–27. doi:10.1016/j.biopha.2018.05.009
18. Golenser J, Waknine JH, Krugliak M, Hunt NH, Grau GE. Current perspectives on the mechanism of action of artemisinins. Int J Parasitol. 2006;36(14):1427–1441. doi:10.1016/j.ijpara.2006.07.011
19. Alam A, Goyal M, Iqbal MS, et al. Novel antimalarial drug targets: hope for new antimalarial drugs. Expert Rev Clin Pharmacol. 2009;2(5):469–489.
20. Plucinski MM, Talundzic E, Morton L, et al. Efficacy of artemether-lumefantrine and dihydroartemisinin-piperaquine for treatment of uncomplicated malaria in children in Zaire and Uíge provinces, Angola. Antimicrob Agents Chemother. 2015;59(1):437–443.
21. Popovici J, Pierce-Friedrich L, Kim S, et al. Recrudescence, reinfection, or relapse? A more rigorous framework to assess chloroquine efficacy for Plasmodium vivax malaria. J Infect Dis. 2019;219(2):315–322.
22. John GK, Douglas NM, Von Seidlein L, et al. Primaquine radical cure of Plasmodium vivax: a critical review of the literature. Malar J. 2012;11(1):280. doi:10.1186/1475-2875-11-280
23. Fogh S, Jepsen S, Effersøe P. Chloroquine-resistant Plasmodium falciparum malaria in Kenya. Trans R Soc Trop Med Hyg. 1979;73(2):228–229. doi:10.1016/0035-9203(79)90220-7
24. Phillips EJ, Keystone JS, Kain KC. Failure of combined chloroquine and high-dose primaquine therapy for Plasmodium vivax malaria acquired in Guyana, South America. Clin Infect Dis. 1996;23(5):1171–1173. doi:10.1093/clinids/23.5.1171
25. Rieckmann K, Davis D, Hutton D. Plasmodium vivax resistance to chloroquine? Lancet. 1989;334(8673):1183–1184. doi:10.1016/S0140-6736(89)91792-3
26. Mohapatra M, Padhiary K, Mishra D, Sethy G. Atypical manifestations of Plasmodium vivax malaria. Indian J Malariol. 2002;39(1–2):18–25.
27. Rifakis PM, Hernandez O, Fernández CT, Rodriguez‐Morales AJ, Von A, Franco‐Paredes C. Atypical Plasmodium vivax malaria in a traveler: bilateral hydronephrosis, severe thrombocytopenia, and hypotension. J Travel Med. 2008;15(2):119–121. doi:10.1111/j.1708-8305.2007.00178.x
28. Tan LK, Yacoub S, Scott S, Bhagani S, Jacobs M. Acute lung injury and other serious complications of Plasmodium vivax malaria. Lancet Infect Dis. 2008;8(7):449–454. doi:10.1016/S1473-3099(08)70153-1
29. Arnold J, Alving A, Hockwald E, et al. The antimalarial action of primaquine against the blood and tissue stages of falciparum malaria (Panama, PF-6 strain). J Lab Clin Med. 1955;46(3):391–397.
30. Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. 2009;42(3):267–278. doi:10.1016/j.bcmd.2008.12.005
31. Tulu AN, Webber RH, Schellenberg JA, Bradley DJ. Failure of chloroquine treatment for malaria in the highlands of Ethiopia. Trans R Soc Trop Med Hyg. 1996;90(5):556–557. doi:10.1016/S0035-9203(96)90322-3
32. Sidhu ABS, Uhlemann A-C, Valderramos SG, Valderramos J-C, Krishna S, Fidock DA. Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J Infect Dis. 2006;194(4):528–535. doi:10.1086/507115
33. Paget-McNicol S, Saul A. Mutation rates in the dihydrofolate reductase gene of Plasmodium falciparum. Parasitology. 2001;122(5):497–505. doi:10.1017/S0031182001007739
34. Muller O. Challenges for control and elimination in the 21st century. Malaria Afri. 2011;60:193.
35. Newton PN, Green MD, Mildenhall DC, et al. Poor quality vital anti-malarials in Africa-an urgent neglected public health priority. Malar J. 2011;10(1):352. doi:10.1186/1475-2875-10-352
36. Hall KA, Newton PN, Green MD, et al. Characterization of counterfeit artesunate antimalarial tablets from southeast Asia. Am J Trop Med Hyg. 2006;75(5):804–811. doi:10.4269/ajtmh.2006.75.804
37. Goldberg DE, Siliciano RF, Jacobs Jr WR. Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell. 2012;148(6):1271–1283. doi:10.1016/j.cell.2012.02.021
38. Bray P, Ward S, O’neill P. Quinolines and artemisinin: chemistry, biology and history. In: Malaria: Drugs, Disease and Post-Genomic Biology. Springer-Verlag Berlin Heidelberg; 2005:3–38.
39. White NJ. Counter perspective: artemisinin resistance: facts, fears, and fables. Am J Trop Med Hyg. 2012;87(5):785. doi:10.4269/ajtmh.2012.12-0573
40. Burrows JN, Duparc S, Gutteridge WE, et al. New developments in anti-malarial target candidate and product profiles. Malar J. 2017;16(1):26.
41. Baker DA, Drought LG, Flueck C, et al. Cyclic nucleotide signalling in malaria parasites. Open Biol. 2017;7(12):170213. doi:10.1098/rsob.170213
42. Morris CA, Duparc S, Borghini-Fuhrer I, Jung D, Shin C-S, Fleckenstein L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar J. 2011;10(1):263. doi:10.1186/1475-2875-10-263
43. Yeung S, Socheat D, Moorthy VS, Mills AJ. Artemisinin resistance on the Thai–Cambodian border. Lancet. 2009;374(9699):1418–1419. doi:10.1016/S0140-6736(09)61856-0
44. Lu F, Culleton R, Zhang M, et al. Emergence of indigenous artemisinin-resistant Plasmodium falciparum in Africa. N Engl J Med. 2017;376(10).
45. Baragaña B, Hallyburton I, Lee MC, et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522(7556):315. doi:10.1038/nature14451
46. Yang Y-Z, Little B, Meshnick SR Alkylation of proteins by artemisinin: effects of heme, pH, and drug structure. 1994.
47. Yang Y-Z, Asawamahasakda W, Meshnick SR Alkylation of human albumin by the antimalarial artemisinin. 1993.
48. Eckstein-Ludwig U, Webb R, Van Goethem I, et al. Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003;424(6951):957. doi:10.1038/nature01813
49. Hartwig CL, Rosenthal AS, D’angelo J, Griffin CE, Posner GH, Cooper RA. Accumulation of artemisinin trioxane derivatives within neutral lipids of Plasmodium falciparum malaria parasites is endoperoxide-dependent. Biochem Pharmacol. 2009;77(3):322–336. doi:10.1016/j.bcp.2008.10.015
50. Inselburg J. Induction and isolation of artemisinine-resistant mutants of Plasmodium falciparum. Am J Trop Med Hyg. 1985;34(3):417–418. doi:10.4269/ajtmh.1985.34.417
51. Chawira A, Warhurst D, Peters W. Qinghaosu resistance in rodent malaria. Trans R Soc Trop Med Hyg. 1986;80(3):477–480. doi:10.1016/0035-9203(86)90351-2
52. Gay F, Ciceron L, Litaudon M, et al. In-vitro resistance of Plasmodium falciparum to qinghaosu derivatives in West Africa. Lancet. 1994;343(8901):850–851. doi:10.1016/S0140-6736(94)92049-4
53. Haldar K, Bhattacharjee S, Safeukui I. Drug resistance in Plasmodium. Nat Rev Microbiol. 2018;16:156. doi:10.1038/nrmicro.2017.161
54. Bhattacharjee S, Stahelin RV, Haldar K. Host targeting of virulence determinants and phosphoinositides in blood stage malaria parasites. Trends Parasitol. 2012;28(12):555–562. doi:10.1016/j.pt.2012.09.004
55. Carter TE, Boulter A, Existe A, et al. Artemisinin resistance-associated polymorphisms at the K13-propeller locus are absent in Plasmodium falciparum isolates from Haiti. Am J Trop Med Hyg. 2015;92(3):552–554. doi:10.4269/ajtmh.14-0664
56. Plowe CV, Roper C, Barnwell JW, et al. World Antimalarial Resistance Network (WARN) III: molecular markers for drug resistant malaria. Malar J. 2007;6(1):121. doi:10.1186/1475-2875-6-121
57. Yayon A, Cabantchik Z, Ginsburg H. Identification of the acidic compartment of Plasmodium falciparum‐infected human erythrocytes as the target of the antimalarial drug chloroquine. EMBO J. 1984;3(11):2695–2700.
58. Sanchez CP, Rohrbach P, McLean JE, Fidock DA, Stein WD, Lanzer M. Differences in trans‐stimulated chloroquine efflux kinetics are linked to PfCRT in Plasmodium falciparum. Mol Microbiol. 2007;64(2):407–420. doi:10.1111/j.1365-2958.2007.05664.x
59. van Es HH, Renkema H, Aerts H, Schurr E. Enhanced lysosomal acidification leads to increased chloroquine accumulation in CHO cells expressing the pfmdr1 gene. Mol Biochem Parasitol. 1994;68(2):209–219. doi:10.1016/0166-6851(94)90166-X
60. Sanchez CP, Rotmann A, Stein WD, Lanzer M. Polymorphisms within PfMDR1 alter the substrate specificity for anti‐malarial drugs in Plasmodium falciparum. Mol Microbiol. 2008;70(4):786–798.
61. Raj DK, Mu J, Jiang H, et al. Disruption of a Plasmodium falciparum multidrug resistance-associated protein (PfMRP) alters its fitness and transport of antimalarial drugs and glutathione. J Biol Chem. 2009;284(12):7687–7696. doi:10.1074/jbc.M806944200
62. Kessl JJ, Lange BB, Merbitz-Zahradnik T, et al. Molecular basis for atovaquone binding to the cytochrome bc1 complex. J Biol Chem. 2003;278(33):31312–31318. doi:10.1074/jbc.M304042200
63. Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. nature. 2007;446(7131):88. doi:10.1038/nature05572
64. Lim MY-X, LaMonte G, Lee MC, et al. UDP-galactose and acetyl-CoA transporters as Plasmodium multidrug resistance genes. Nat Microbiol. 2016;1(12):16166. doi:10.1038/nmicrobiol.2016.166
65. Chow ED, Lim L, Fidock DA, Diagana TT, Winzeler EA, Bifani P UDP-galactose and acetyl-CoA transporters as Plasmodium multidrug resistance genes. 2016.
66. Kuhen KL, Chatterjee AK, Rottmann M, et al. KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob Agents Chemother. 2014;58(9):5060–5067. doi:10.1128/AAC.02727-13
67. Jiang H, Patel JJ, Yi M, et al. Genome-wide compensatory changes accompany drug-selected mutations in the Plasmodium falciparum crt gene. PLoS One. 2008;3(6):e2484. doi:10.1371/journal.pone.0002484
68. Mwai L, Diriye A, Masseno V, et al. Genome wide adaptations of Plasmodium falciparum in response to lumefantrine selective drug pressure. PLoS One. 2012;7(2):e31623. doi:10.1371/journal.pone.0031623
69. Vestergaard LS, Ringwald P. Responding to the challenge of antimalarial drug resistance by routine monitoring to update national malaria treatment policies. Am J Trop Med Hyg. 2007;77(6_Suppl):153–159. doi:10.4269/ajtmh.2007.77.153
70. Organization WH. Methods for surveillance of antimalarial drug efficacy. 2009.
71. Oyelade J, Isewon I, Aromolaran O, et al. Computational identification of metabolic pathways of plasmodium falciparum using the-shortest path algorithm. Int J Genomics. 2019;2019.
72. Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov. 2004;3(6):509–520. doi:10.1038/nrd1416
73. Comer E, Beaudoin JA, Kato N, et al. Diversity-oriented synthesis-facilitated medicinal chemistry: toward the development of novel antimalarial agents. J Med Chem. 2014;57(20):8496–8502. doi:10.1021/jm500994n
74. Mehta M, Sonawat HM, Sharma S. Glycolysis in Plasmodium falciparum results in modulation of host enzyme activities. J Vector Borne Dis. 2006;43(3):95.
75. Tilley L, Dixon MW, Kirk K. The Plasmodium falciparum-infected red blood cell. Int J Biochem Cell Biol. 2011;43(6):839–842. doi:10.1016/j.biocel.2011.03.012
76. Dickerman BK, Elsworth B, Cobbold SA, et al. Identification of inhibitors that dually target the new permeability pathway and dihydroorotate dehydrogenase in the blood stage of Plasmodium falciparum. Sci Rep. 2016;6:37502. doi:10.1038/srep37502
77. Kraft TE, Armstrong C, Heitmeier MR, Odom AR, Hruz PW. The glucose transporter PfHT is an antimalarial target of the HIV protease inhibitor lopinavir. Antimicrob Agents Chemother. 2015;
78. Kirk K. Membrane transport in the malaria-infected erythrocyte. Physiol Rev. 2001;81(2):495–537. doi:10.1152/physrev.2001.81.2.495
79. Heitmeier MR, Hresko RC, Edwards RL, et al. Identification of druggable small molecule antagonists of the Plasmodium falciparum hexose transporter PfHT and assessment of ligand access to the glucose permeation pathway via FLAG-mediated protein engineering. PLoS One. 2019;14(5):e0216457. doi:10.1371/journal.pone.0216457
80. Meireles P, Sales‐Dias J, Andrade CM, et al. GLUT1‐mediated glucose uptake plays a crucial role during Plasmodium hepatic infection. Cell Microbiol. 2017;19(2):e12646. doi:10.1111/cmi.12646
81. Jirage D, Keenan S,C, Waters N. Exploring novel targets for antimalarial drug discovery: plasmodial protein kinases. Infect Disorders Drug Targets. 2010;10(3):134–146.
82. Lucet IS, Tobin A, Drewry D, Wilks AF, Doerig C. Plasmodium kinases as targets for new-generation antimalarials. Future Med Chem. 2012;4(18):2295–2310. doi:10.4155/fmc.12.183
83. Derbyshire ER, Zuzarte‐Luís V, Magalhães AD, et al. Chemical interrogation of the malaria kinome. Chembiochem. 2014;15(13):1920–1930. doi:10.1002/cbic.201400025
84. Ojo KK, Eastman RT, Vidadala R, et al. A specific inhibitor of Pf CDPK4 blocks malaria transmission: chemical-genetic validation. J Infect Dis. 2014;209(2):275–284. doi:10.1093/infdis/jit522
85. Chapman TM, Osborne SA, Wallace C, et al. Optimization of an imidazopyridazine series of inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (Pf CDPK1). J Med Chem. 2014;57(8):3570–3587. doi:10.1021/jm500342d
86. Banerjee R, Liu J, Beatty W, Pelosof L, Klemba M, Goldberg DE. Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc Natl Acad Sci. 2002;99(2):990–995. doi:10.1073/pnas.022630099
87. Shenai BR, Sijwali PS, Singh A, Rosenthal PJ. Characterization of native and recombinant falcipain-2, a principal trophozoite cysteine protease and essential hemoglobinase of Plasmodium falciparum. J Biol Chem. 2000;275(37):29000–29010. doi:10.1074/jbc.M004459200
88. Eggleson KK, Duffin KL, Goldberg DE. Identification and characterization of falcilysin, a metallopeptidase involved in hemoglobin catabolism within the malaria parasite Plasmodium falciparum. J Biol Chem. 1999;274(45):32411–32417. doi:10.1074/jbc.274.45.32411
89. Combrinck JM, Joanne E, Hearne GR, et al. Fate of haem iron in the malaria parasite Plasmodium falciparum. Biochem J. 2002;365(2):343–347. doi:10.1042/bj20020793
90. Coronado LM, Nadovich CT, Spadafora C. Malarial hemozoin: from target to tool. Biochimica et Biophysica Acta. 2014;1840(6):2032–2041. doi:10.1016/j.bbagen.2014.02.009
91. Vaidya AB, Mather MW. Mitochondrial evolution and functions in malaria parasites. Annu Rev Microbiol. 2009;63:249–267. doi:10.1146/annurev.micro.091208.073424
92. Nixon GL, Pidathala C, Shone AE, et al. Targeting the mitochondrial electron transport chain of Plasmodium falciparum: new strategies towards the development of improved antimalarials for the elimination era. Future Med Chem. 2013;5(13):1573–1591. doi:10.4155/fmc.13.121
93. Nina PB, Morrisey JM, Ganesan SM, et al. ATP Synthase Complex of Plasmodium falciparum dimeric assembly in mitochondrial membranes and resistance to genetic disruption. J Biol Chem. 2011;286(48):41312–41322. doi:10.1074/jbc.M111.290973
94. Lunev S, Batista FA, Bosch SS, Wrenger C, Groves MR. Identification and validation of novel drug targets for the treatment of Plasmodium falciparum malaria: new insights. InTech. 2016.
95. Sheridan CM, Garcia VE, Ahyong V, DeRisi JL. The Plasmodium falciparum cytoplasmic translation apparatus: a promising therapeutic target not yet exploited by clinically approved anti-malarials. Malar J. 2018;17(1):465. doi:10.1186/s12936-018-2616-7
96. Köhler S, Delwiche CF, Denny PW, et al. A plastid of probable green algal origin in Apicomplexan parasites. Science. 1997;275(5305):1485–1489. doi:10.1126/science.275.5305.1485
97. Wilson R. Parasite plastids: approaching the endgame. Biol Rev. 2005;80(1):129–153. doi:10.1017/S1464793104006591
98. Lim L, McFadden GI. The evolution, metabolism and functions of the apicoplast. Biol Sci. 2010;365(1541):749–763. doi:10.1098/rstb.2009.0273
99. Limenitakis J, Soldati-Favre D. Functional genetics in Apicomplexa: potentials and limits. FEBS Lett. 2011;585(11):1579–1588. doi:10.1016/j.febslet.2011.05.002
100. Padmanaban G, Nagaraj VA, Rangarajan PN. Drugs and drug targets against malaria. Curr Sci. 2007;92(11).
101. McFadden GI, Roos DS. Apicomplexan plastids as drug targets. Trends Microbiol. 1999;7(8):328–333. doi:10.1016/S0966-842X(99)01547-4
102. Andersen S, Oloo A, Gordon D, et al. Successful double-blinded, randomized, placebo-controlled field trial of azithromycin and doxycycline as prophylaxis for malaria in Western Ken. Clin Infect Dis. 1998;26(1):146–150. doi:10.1086/516281
103. Taylor WR, Richie TL, Fryauff DJ, et al. Malaria prophylaxis using azithromycin: a double-blind, placebo-controlled trial in Irian Jaya, Indonesia. Clin Infect Dis. 1999;28(1):74–81. doi:10.1086/515071
104. Gaynor BD, Amza A, Kadri B, et al. Impact of mass azithromycin distribution on malaria parasitemia during the low-transmission season in Niger: a cluster-randomized trial. Am J Trop Med Hyg. 2014;90(5):846–851. doi:10.4269/ajtmh.13-0379
105. Moore BR, Benjamin JM, Auyeung SO, et al. Safety, tolerability and pharmacokinetic properties of coadministered azithromycin and piperaquine in pregnant Papua New Guinean women. Br J Clin Pharmacol. 2016;82(1):199–212. doi:10.1111/bcp.12910
106. Teixeira C, Gomes J, Gomes P. Falcipains, Plasmodium falciparum cysteine proteases as key drug targets against malaria. Curr Med Chem. 2011;18(10):1555–1572. doi:10.2174/092986711795328328
107. Roy KK. Targeting the active sites of malarial proteases for antimalarial drug discovery: approaches, progress and challenges. Int J Antimicrob Agents. 2017;50(3):287–302. doi:10.1016/j.ijantimicag.2017.04.006
108. Verma S, Dixit R, Pandey KC. Cysteine proteases: modes of activation and future prospects as pharmacological targets. Front Pharmacol. 2016;7:107. doi:10.3389/fphar.2016.00107
109. Rosenthal PJ. Cysteine proteases of malaria parasites. Int J Parasitol. 2004;34(13–14):1489–1499.
110. McKerrow JH, Sun E, Rosenthal PJ, Bouvier J. The proteases and pathogenicity of parasitic protozoa. Ann Rev Microbiol. 1993;47(1):821–853. doi:10.1146/annurev.mi.47.100193.004133
111. Debrabant A, Delplace P. Leupeptin alters the proteolytic processing of P126, the major parasitophorous vacuole antigen of Plasmodium falciparum. Mol Biochem Parasitol. 1989;33(2):151–158. doi:10.1016/0166-6851(89)90029-7
112. Harris C, Hunte B, Krauss M, Taylor A, Epstein L. Induction of leucine aminopeptidase by interferon-gamma. Identification by protein microsequencing after purification by preparative two-dimensional gel electrophoresis. J Biol Chem. 1992;267(10):6865–6869.
113. Grembecka J, Mucha A, Cierpicki T, Kafarski P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J Med Chem. 2003;46(13):2641–2655. doi:10.1021/jm030795v
114. McGowan S, Porter CJ, Lowther J, et al. Structural basis for the inhibition of the essential Plasmodium falciparum M1 neutral aminopeptidase. Proc Natl Acad Sci. 2009;106(8):2537–2542. doi:10.1073/pnas.0807398106
115. Nankya-Kitaka M, Curley G, Gavigan C, Bell A, Dalton J. Plasmodium chabaudi chabaudi and P. falciparum: inhibition of aminopeptidase and parasite growth by bestatin and nitrobestatin. Parasitol Res. 1998;84(7):552–558. doi:10.1007/s004360050447
116. Zhang R, Suwanarusk R, Malleret B, et al. A basis for rapid clearance of circulating ring-stage malaria parasites by the spiroindolone KAE609. J Infect Dis. 2016;213(1):100–104. doi:10.1093/infdis/jiv358
117. Blascod DL, Wittyd MJ, Doninid C, et al.UCT943, a next generation Plasmodium falciparum PI4K inhibitor preclinical candidate for the treatment of malaria 2. 2018.
118. Schiafino-Ortega S, Baglioni E, Pérez-Moreno G, et al. 1, 2-Diphenoxiethane salts as potent antiplasmodial agents. Bioorg Med Chem Lett. 2018;28(14):2485–2489. doi:10.1016/j.bmcl.2018.05.060
119. Belete TM. Novel targets to develop new antibacterial agents and novel alternatives to antibacterial agents. Human Microbio J. 2019;11:100052. doi:10.1016/j.humic.2019.01.001
120. Almeida MR, Darin JD, Hernandes LC, Ramos M, Antunes LMG, Freitas O. Genotoxicity assessment of Copaiba oil and its fractions in Swiss mice. Genet Mol Biol. 2012;35(3):664–672. doi:10.1590/S1415-47572012005000052
121. Batchvarova M, Shan S, Zennadi R, et al. Sevuparin Reduces Adhesion of Both Sickle Red Cells and Leukocytes to Endothelial Cells in vitro and Inhibits Vaso-Occlusion in Vivo. Washington, DC: American Society of Hematology; 2013.
122. Mathews ES, John ARO. Tackling resistance: emerging antimalarials and new parasite targets in the era of elimination. F1000Research. 2018;7.
123. Rosenthal PJ. Antimalarial drug discovery: old and new approaches. J Exp Biol. 2003;206(21):3735–3744. doi:10.1242/jeb.00589
124. Ogeto T, Ndubi F, Murithi M, et al. Malaria vaccines targeting the pre-erythrocytic stage: a scoping review. F1000Research. 2020;9(680):680.
125. Reis RA, Calil FA, Feliciano PR, Pinheiro MP, Nonato MC. The dihydroorotate dehydrogenases: past and present. Arch Biochem Biophys. 2017;632:175–191. doi:10.1016/j.abb.2017.06.019
126. Rottmann M, Jonat B, Gumpp C, et al. Preclinical antimalarial combination study of M5717, a Plasmodium falciparum elongation factor 2 inhibitor, and pyronaridine, a hemozoin formation inhibitor. Antimicrob Agents Chemother. 2020;64(4).
127. Spillman NJ, Kirk K. The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. Int J Parasitol Drugs Drug Resist. 2015;5(3):149–162.
128. Phyo AP, Jittamala P, Nosten FH, et al. Antimalarial activity of artefenomel (OZ439), a novel synthetic antimalarial endoperoxide, in patients with Plasmodium falciparum and Plasmodium vivax malaria: an open-label Phase 2 trial. Lancet Infect Dis. 2016;16(1):61–69. doi:10.1016/S1473-3099(15)00320-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.